

Abstract
Gastrin is a gastrointestinal peptide hormone, secreted by the gastric G cells and can exist as a fully processed amidated form (G17) or as unprocessed forms. All forms of gastrin possess trophic properties towards the gastrointestinal mucosa. An understanding of the signaling pathways involved is important to design therapeutic approaches to target gastrin-mediated cellular events. The studies described here were designed to identify the signaling pathways by which amidated gastrin (G17) mediates cancer cell migration. These studies indicated a time- and dose-dependent increase in gastric cancer cell migration after G17 stimulation, involving cholecystokinin 2 receptor. G17-induced migration was preceded by activation of MAPK pathways and was antagonized after pretreatment with SP600125, a pharmacological inhibitor of c-Jun-NH2-terminal kinase (JNK) pathway. Knockdown of endogenous JNK1 expression via small interference RNA (JNK1-siRNA) inhibited G17-induced phosphorylation of c-Jun and migration, and overexpression of wild-type JNK1 or constitutive active JNK1 promoted G17-induced migration. Studies designed to identify the MAPK kinase kinase member mediating JNK activation indicated the involvement of mixed lineage kinase-3 (MLK3), which was transiently activated upon G17 treatment. Inhibition of MLK3 pathway via a pan-MLK inhibitor or knockdown of MLK3 expression by MLK3-siRNA antagonized G17-induced migration. Incubation with G17 also resulted in an induction of matrix metalloproteinase 7 promoter activity, which is known to mediate migration and invasion pathways in cancer cells. Modulation of MLK3, JNK1, and c-Jun pathways modulated G17-induced matrix metalloproteinase 7 promoter activation. These studies indicate that the MLK3/JNK1 axis mediates G17-induced gastric cancer cell migration, which can be targeted for designing novel therapeutic strategies for treating gastric malignancies.
The gastrointestinal peptide hormone gastrin induces migration and induces MMP7 promoter activation in gastric cancer cells via activation of MLK3, JNK1 and c-Jun pathways.
Gastrin is a gastrointestinal (GI) peptide hormone, produced by the gastric G cells and is involved in gastric acid secretion. Gastrin mediates its effects via activation of the CCK2 receptor (CCK2R), formerly known as CCKBR (1). Studies over the past two decades have demonstrated a distinct link between gastrin and GI cancer (2, 3). Mature amidated gastrin (G17), as well as its nonamidated precursor, glycine-extended gastrin (G-Gly), can induce growth and expression of growth-promoting genes in various GI cancer cell lines (4, 5, 6). The link between gastrin and GI cancer has been later validated by the creation of transgenic animal models overexpressing various forms of gastrin (7). Patients infected with Heliobacter pylori show an increase in their circulating gastrin levels (particularly G17), suggesting a possible participation of this peptide in gastric cancer. In the studies with INS-GAS transgenic mice producing high levels of amidated gastrin, Wang et al. (8) demonstrated an increase in proliferation and hypertrophy of gastric mucosa, which when combined with H. pylori, produced highly invasive gastric cancer at a faster rate. More recent studies have linked gastrin-mediated CCK2R activation to cell invasion and scattering (9), associated with loss of membrane localization of adherens junction proteins, involving Janus family of tyrosine kinase 2/phosphatidylinositol-3 kinase pathway (10). In gastric cancer cells, gastrin stimulated an invasive pathway downstream of matrix metalloproteinase (MMP)9 induction (11), and a migratory pathway (12), both of which involved activation of protein kinase C and MAPK pathways. The P21-activated kinase 1 (PAK1) pathway also mediates G17 and G-Gly-induced migration in gastric cells (13) via modulation of β-catenin. In other studies G-Gly was shown to induce migration in gastric epithelial cells involving phosphatidylinositol-3 kinase and MAPK pathways (14), and through activation of small G protein Rho (15).
The MAPK family of kinases are known to regulate a variety of cellular processes that include cell proliferation, differentiation, inflammation, and migration and are involved in cancer (16). The MAPK family consists of ERK, c-Jun-NH2-terminal kinase (JNK), and p38 kinase, of which the JNK pathway acts as a crucial regulator of cell migration and epithelial morphogenesis (17). The JNK subgroup consists of JNK1, JNK2, and JNK3. JNK1 and JNK2 are ubiquitously expressed, whereas JNK3 is expressed primarily in the heart, testis, and brain (18, 19). The JNK pathway is activated by inflammatory cytokines and environmental stress (20), via MKK4 and MKK7 (21), which then phosphorylate its downstream substrates c-Jun, Jun-D, activating transcription factor, leading to an induction of activator protein 1 transcriptional activity (22, 23). In Drosophila, mutation of the gene (basket) that encodes Drosophila JNK leads to a developmental migration defect termed “dorsal open” phenotype, suggesting a critical role of JNK in epithelial cell migration (24). In addition, MEK kinase 1, an upstream activator of JNK, is essential for migration and eyelid closure (25, 26). Similarly, Drosophila mixed lineage kinase (slpr) also functions as JNK3K, mediating JNK activation during dorsal closure (27). JNK activation is linked with the migration pathways initiated by epidermal growth factor (28), and in the process of epithelial-to-mesenchymal transition, which is crucial for the promotion of tissue fibrosis and metastasis (29). Gene knockout studies by Javelaud et al. (30) showed that JNK-mediated c-Jun phosphorylation is involved in fibroblast cell migration. A recent study has demonstrated JNK1 to be a critical regulator of gastric tumorigenesis (31) and indicated frequent JNK activation in gastric cancer.
Several studies have linked activation of MAPK pathways (ERK, p38, and JNK) with the growth-promoting effects of gastrin (32, 33). The mechanism by which gastrin activates the ERK pathway has been studied extensively and involves the Src family of tyrosine kinase leading to the activation of Ras/Raf/MEK/ERK cascade (34, 35). Despite a role of JNK in mediating gastrin-induced growth, the detailed mechanism and the upstream kinases that regulate gastrin-induced JNK activation are still unknown. This is particularly important because JNK can be activated via multiple upstream kinases acting as MAPK kinase kinases (MAP3Ks), which include mixed lineage kinase (MLK), LZK, TAK, ASK, MEKK, and TPL depending upon the specific agonist added. Identification of the specific MAP3K/JNK axis that regulates gastrin-mediated events is critical for developing rational, therapeutic strategies to target the gastrin pathway. The studies described here were aimed to determine whether G17-induced migration in gastric cancer cells involves the JNK pathway, and to delineate the mechanism by which G17 activates JNK. Our studies indicated that G17 can induce significant migration in the gastric cancer cells, associated with an increase in MMP-7 promoter activity, both of which involve activation of the JNK1 pathway. G17-induced activation of JNK1 as well as migration was mediated via MLK3, and MLK3 showed a transient activation upon G17 treatment.
Results
Amidated gastrin (G17) induces migration and MAPK pathway activation in gastric cancer cells
To understand the role of G17 in mediating migration, wound-healing assays were designed with the CCK2R overexpressing gastric cancer (AGSE) cells as described earlier (36). Incubation of AGSE cells with G17 induced a potent migratory response in a time- (Fig. 1A) and dose- (Fig. 2A) dependent manner. This migratory response was mediated via activation of the CCK2R, as pretreatment with the receptor antagonist L365,260 abolished migration (Fig. 2B).
Fig. 1.
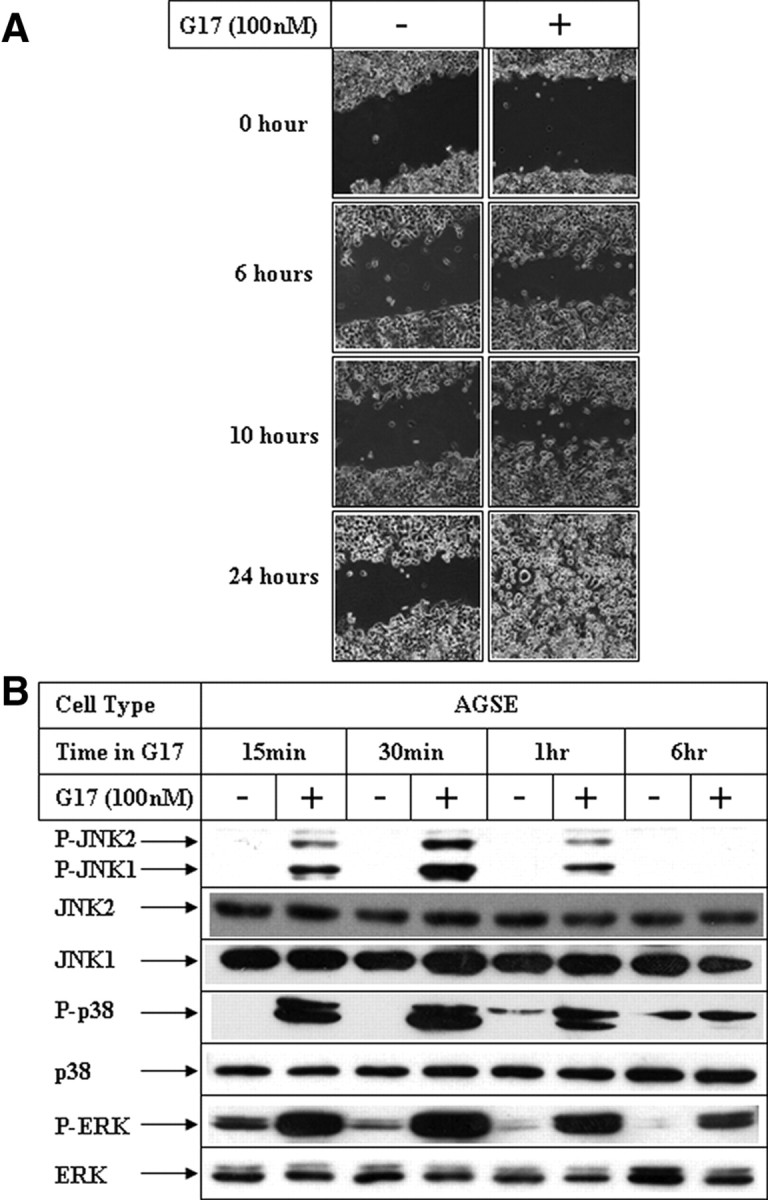
Effect of G17 on MAPK activation and migration in gastric cancer cells. A, Confluent AGSE cells were wounded linearly using a small pipette tip followed by treatment in the absence (−) or presence (+) of 100 nm G17. Phase-contrast microscopic pictures of the migratory cells were obtained at the indicated time points using inverted phase-contrast microscopy (Axiovert 200 inverted microscope; Zeiss), interfaced with a camera (Axiocam). B, AGSE cells treated as in panel A were harvested at different time points, and total protein was extracted. Western blot analyses of the cell extracts were then performed with the indicated antibodies.
Fig. 2.

Effect of increasing concentrations of G17 on MAPK activation and migration in gastric cancer cells. A, AGSE cells were treated with increasing concentrations of G17, and wound-healing assays were performed as described in Fig. 1A. Pictures were obtained after 24 h of G17 treatment. B, AGSE cells were treated with 100 nm G17, after an overnight pretreatment with varying concentrations of CCK2R antagonist (L365,260) and subjected to wound-healing assays as in panel A. C, AGSE cells treated as in panel A or (D) treated as in panel B above were harvested 30 min after G17 treatment and analyzed by Western blot analysis with the antibodies indicated. conc., Concentration.
To elucidate the signaling pathway(s) mediating G17-induced migration, studies were designed first to determine the effect of G17 on MAPK pathway, because members of this pathway are known to mediate migration in various cells (37). Time course studies indicated an early activation of all three MAPK members (ERK, p38, and JNK), which was maximal after 30 min of G17 stimulation (Fig. 1B), preceding G17-induced migration (Fig. 1A). Studies performed with increasing concentrations of G17 indicated a G17 concentration-dependent activation of JNK and p38, which were maximal at 100 nm (Fig. 2C, phospho-JNK and phospho-p38 panels). ERK activation, however, was maximal at 1 nm G17 and showed no further increase with increasing G17 concentration (Fig. 2C, phospho-ERK panel). In addition, G17-induced JNK activation was antagonized by 1 μm L365,260 (Fig. 2D, phospho-JNK panel; compare lanes 2 and 3), suggesting involvement of CCK2R pathway. L365,260, however, was unable to inhibit p38 activation (phospho-p38 panel; compare lane 2 with lanes 3–5) and inhibited ERK at a higher concentration (phospho ERK panel; compare lanes 2 and 5), suggesting the possible involvement of different receptor-mediated pathways for various MAPK activations.
Pharmacological inhibition of JNK inhibits G17-induced gastric cancer cell migration
To understand the participation of MAPKs in G17-induced migration, wound-healing assays were designed after pretreatment with the known pharmacological inhibitors of different MAPK pathways. In these studies pretreatment with the MEK inhibitor U0126 (upstream activator of ERK) produced slight effects toward inhibiting G17-induced migration (Fig. 3A), although U0126 potently inhibited G17-induced ERK phosphorylation (Supplemental Fig. 1A published on The Endocrine Society’s Journals Online web site at http://mend.endojournals. org; compare lanes 2 and 4). Similarly, pretreatment with p38 inhibitor SB203580 was unable to antagonize G17-induced migration (Fig. 3B), despite inhibiting p38 pathway activation as shown by a decrease in phospho-CREB levels (Supplemental Fig. 1B, phospho-CREB panel; compare lanes 2, 8, and 10). However, pretreatment with the JNK inhibitor (SP600125) abrogated G17-induced phosphorylation of JNK (Supplemental Fig. 1B, phospho-JNK panel; compare lanes 2, 4, and 6) and its downstream c-Jun (phospho-c-Jun panel; compare lanes 2, 4, and 6) and resulted in a complete inhibition of G17-induced migration (Fig. 3C). Studies designed to determine the minimal concentration of SP600125 required for antagonizing G17-induced events indicated that as low as 5 μm of the inhibitor could antagonize G17-induced c-Jun phosphorylation (Supplemental Fig. 2B; compare lanes 2 and 4) and migration (Supplemental Fig. 2A).
Fig. 3.

Effect of inhibition of MAPK pathways on G17-induced migration. AGSE cells were treated with 100 nm G17 after a 1-h pretreatment with MEK inhibitor U0126 (A), p38 inhibitor SB203580 (B), or JNK inhibitor SP600125 (C). Wound-healing assays were performed as described in Fig. 1A.
G17-induced migration is mediated via JNK1
To establish the participation of JNK in this pathway, more in-depth molecular studies were designed after ectopic overexpression of JNK1 (wild type) and its mutants. The expression profile of ectopic hemagglutinin-tagged JNK1 expression vectors is shown in Supplemental Fig. 3B. To determine any synergistic effect of JNK1 overexpression on G17-induced migration, these pictures were obtained at a 6-h time point after G17 addition, when EV didn’t show significant migratory response (Fig. 4A). Overexpression of JNK1-wild type (JNK1-WT) and constitutive active JNK1 (JNK1-CA) induced spontaneous migration even in the absence of G17 (top panel, G17 for 0 h) and produced synergistic effects when treated with G17 (bottom panel, G17 for 6 h). However, overexpression of JNK1-dominant negative (JNK1-DN) was unable to induce any migration even in the presence of G17 (JNK1-DN panel). Quantitation of the average gap following overexpression of different JNK1 forms is shown in Supplemental Fig. 3A. To confirm that JNK1 mediates G17-induced migration, studies were also designed after knockdown of JNK1 with small interference RNA (siRNA). Transfection of JNK1-siRNA resulted in a significant decrease in endogenous JNK1 expression (Fig. 4C, JNK1 panel) in these cells and resulted in an antagonism of G17-induced activation of JNK (phospho JNK panel; compare lanes 2 and 4) and its downstream c-Jun (phospho c-Jun panel). Knockdown of JNK1 expression significantly attenuated G17-induced migration (Fig. 4B), as indicated from the absence of migratory cells in the JNK1-siRNA treated cells (bottom right) compared with the control-siRNA treated ones (bottom left). These studies demonstrated that G17-induced migration in gastric cancer cells involves JNK1.
Fig. 4.
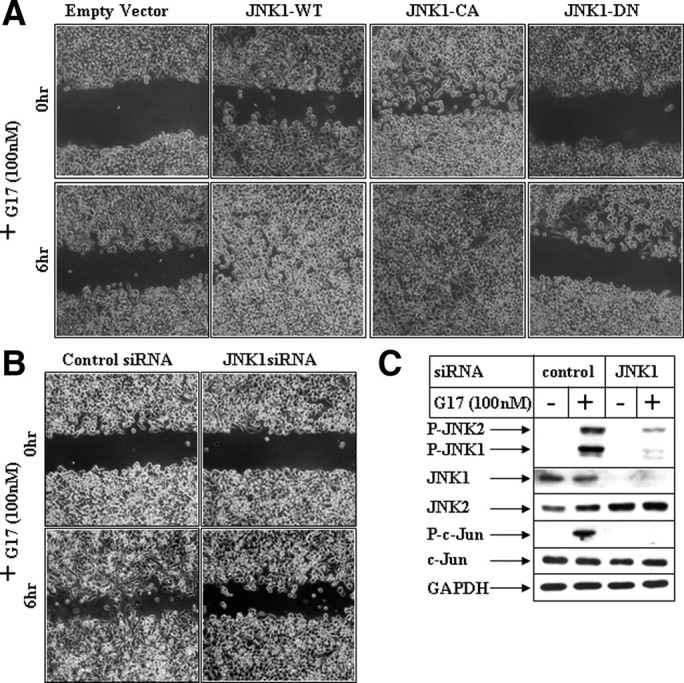
Effect of modulation of JNK1 pathway on G17-induced migration. A, Subconfluent AGSE cells were transiently transfected with an empty vector, a JNK1-WT vector, a JNK1-CA vector, or JNK1-DN vector. The cells were wounded 48 h post-transfection and, following an overnight recovery after wounding, they were incubated with G17 and pictures were obtained at the indicated times. B, Subconfluent AGSE cells were transiently transfected with 100 nm of either control-siRNA or JNK1-siRNA, followed by wounding and G17 treatment after 48 h. C, Western blot analysis of AGSE cells transfected with either control-siRNA or JNK1-siRNA, followed by G17 treatment for 30 min. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase.
G17-induced migration involves MLK3
To obtain an insight on the mechanism how G17 activated JNK pathway, studies were focused on the MAP3K member MLK3, which is known to activate the JNK pathway (38). Pretreatment with a pan-MLK inhibitor CEP11004 significantly inhibited G17-induced phosphorylation of JNK (Supplemental Fig. 4, phospho-JNK panel) and its downstream c-Jun (phospho-c-Jun panel) to the same extent as JNK inhibitor SP600125 (compare lanes 2, 4, and 6). CEP11004 pretreatment also antagonized G17-induced migration (Fig. 5A), suggesting the participation of MLK group of kinases in G17 pathway. Furthermore, knockdown of endogenous MLK3 expression by MLK3-siRNA attenuated G17-induced JNK activation (Fig. 5C, phospho-JNK panel; compare lanes 2 and 4), c-Jun phosphorylation (phospho-c-Jun panel), and migration (Fig. 5B), establishing the participation of MLK3 in this migratory response.
Fig. 5.

Effect of inhibition of MLK3 pathway on G17-induced JNK activation and migration. A, AGSE cells were treated with 100 nm G17 after a pretreatment with 1 μm pan-MLK inhibitor CEP11004. Wound-healing assays were performed next and pictures obtained as described in Fig. 1A. B, Subconfluent AGSE cells were transiently transfected with either 50 nm control-siRNA or 50 nm MLK3-siRNA, followed by wounding and G17 treatment as described in Fig. 4B. C, Subconfluent AGSE cells were transiently transfected with either control-siRNA or MLK3-siRNA, followed by G17 treatment for 30 min. Western blot analyses were performed with the antibodies indicated. D, Endogenous MLK3 immunoprecipitated from AGSE cells treated in the absence (−) or presence (+) of G17 for the indicated periods of time was subjected to in vitro kinase assay using recombinant SEK1 (K-R) as substrate and [γ32P]ATP. The samples were fractionated by SDS-PAGE and transferred to membranes, and the radiolabeled bands were detected by phosphoimaging screens. Western blots of the immunoprecipitates were shown in the bottom to indicate equal input. DMSO, Dimethylsulfoxide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IPs, immunoprecipitations.
To determine whether G17 can activate MLK3, in vitro kinase assays were designed after immunoprecipitation of endogenous MLK3 from the AGSE cells treated in the presence or absence of G17. These studies indicated a transient induction of MLK3 kinase activity up to 60 min after G17 addition (Fig. 5D, compare − and + lanes).
G17 induces MMP7 transcriptional activity via MLK3/JNK1/c-Jun axis
MMP7 is a matrix metalloproteinase, which is involved in migration and invasion of gastric cells (39). To understand whether MMP7 was induced during G17-mediated migration, changes in MMP7 transcriptional activity were estimated by utilizing the MMP7-luciferase reporter obtained from Dr. Howard Crawford (Stony Brook University, Stony Brook, NY) (40). Treatment with G17 resulted in a significant induction of MMP7 promoter activity (Fig. 6A; compare lanes 1 and 2), which was completely inhibited after pretreatment with CCK2R antagonist L365,260 (compare lanes 2 and 4), suggesting the involvement of CCK2R. In addition, pretreatment with JNK inhibitor SP600125 abolished G17-induced MMP7 promoter activation (compare lanes 2 and 6). Knockdown of JNK1 expression via JNK1-siRNA also resulted in complete antagonism of G17-activated MMP7 transcription (Fig. 6B; compare lanes 2 and 4). In addition, ectopic overexpression of JNK1-WT and JNK1-CA resulted in increased MMP7 promoter activity in the absence of G17, with synergistic effects when treated with G17 (Supplemental Fig. 5). These results confirmed the involvement of JNK1 in G17-induced MMP7 transcriptional activation. In addition, pretreatment with pan-MLK inhibitor CEP-11004 completely antagonized G17-induced MMP7 transcription (Fig. 6C, compare lanes 2 and 4), suggesting the involvement of the MLK group of kinases in mediating this. Knockdown of MLK3 expression by an MLK3-siRNA abolished MMP7 transcriptional activation (Fig. 6D; compare lanes 2 and 4), thus confirming that MLK3 participates in G17-induced activation of MMP7 transcription. To determine the transcription factors involved in activating MMP7 promoter after G17 stimulation, studies were designed after modulation of c-Jun, which is a known downstream mediator of JNK pathway. siRNA-mediated knockdown of c-Jun expression resulted in a significant decrease in endogenous c-Jun expression (Fig. 7B; compare lanes 1 and 2 with lanes 3 and 4, c-Jun panel) and was associated with a dramatic reduction in G17-induced MMP7 promoter activation (Fig. 7A; compare lanes 2 and 4). To confirm the role of c-Jun, studies were also designed after overexpression of two dominant-negative c-Jun mutants DBM-3 (DNA-binding domain mutant) (41) or TAM-67 (deletion mutant lacking the transactivation domain). The DBM-3 mutant expressed as full-length c-Jun mutant protein (Fig. 7D, lanes 3 and 4, c-Jun panel) although is highly phosphorylated by G17 (Fig. 7D; compare lanes 2 and 4, phospho-c-Jun panel), inhibits MMP7 promoter activation after G17 addition (Fig. 7C; compare lanes 2 and 4). The TAM67 mutant expressed as a truncated c-Jun protein (Fig. 7D) serves as a potent dominant-negative mutant because it inhibits endogenous c-Jun phosphorylation by G17 (Fig. 7D; compare lanes 2 and 6, phospho-c-Jun panel) and thus significantly attenuates G17-induced MMP-7 promoter activation (Fig. 7C; compare lanes 2 and 6). These results indicate that G17-induced MMP7 promoter induction is mediated via activation of the MLK3/JNK1/c-Jun axis.
Fig. 6.

Role of MLK3 and JNK1 pathways on G17-induced MMP7 transcription. A, Subconfluent AGSE cells were transiently transfected with the MMP7-luciferase vector along with a β-Gal vector (for normalization of transfection). Forty-eight hours post-transfection, cells were treated with G17 after a pretreatment with either none (lanes 1 and 2), or with CCK2R antagonist L365,260 (lanes 3 and 4), or JNK inhibitor SP600125 (lanes 5 and 6), and luciferase and β-Gal assays were performed. The RLU/β-Gal values were represented as percent control, considering the corresponding untreated samples as control. B, AGSE cells were transfected as in panel A along with either control siRNA (lanes 1 and 2) or JNK1 siRNA (lanes 3 and 4). Luciferase and β-Gal assays were performed after G17 treatment as in panel A. C, Cells transfected with MMP7-luc and β-Gal vectors were treated with G17 after pretreatment with pan-MLK inhibitor CEP11004. Luciferase and β-Gal assays were performed next. D, AGSE cells were transfected with MMP7-luc or β-Gal vectors along with either control siRNA (lanes 1 and 2) or MLK3 siRNA (lanes 3 and 4), followed by G17 treatment. Luciferase and β-Gal assays were performed next. Each transfection (A–D) was performed in triplicate, and the data represent the mean ± sd of at least two independent experiments.
Fig. 7.

Role of c-Jun on G17-induced MMP7 transcription. A, Subconfluent AGSE cells were transiently transfected with the MMP7-luciferase and β-Gal vectors along with either control siRNA (lanes 1and 2) or c-Jun siRNA (lanes 3 and 4). Luciferase and β-Gal assays were performed after G17 treatment as described in Fig. 6A. B, AGSE cells were transiently transfected with either control-siRNA or c-Jun-siRNA, followed by G17 treatment for 30 min. Western blot analyses were performed with the antibodies indicated. C, AGSE cells were transfected with MMP7-luc and β-Gal vectors along with either an empty vector (lanes 1 and 2), or c-Jun-DBM3 (containing mutations in the DNA binding domain) mutant vector (lanes 3 and 4), or c-Jun-TAM67 (deletion mutant of c-Jun lacking the transactivation domain) mutant vector (lanes 5 and 6). Luciferase and β-Gal assays were performed after G17 treatment as described in Fig. 6A. D, AGSE cells were transiently transfected with either empty vector, C-Jun DBM3 mutant, or c-Jun TAM67 mutant vectors, followed by G17 treatment for 30 min. Western blot analyses were performed with antibodies against phospho-c-Jun, total c-Jun (against DNA-binding domain), and glyceraldehyde-3-phosphate dehydrogenase. Each transfection (A and C) was performed in triplicate, and the data represent the mean ± sd of at least two independent experiments.
Discussion
The GI peptide hormone gastrin is known to regulate various cellular events, which include growth, apoptosis, migration, and invasion (2, 3). Although the signaling pathways involved in the growth-promoting effects of gastrin have been relatively well characterized, those regulating cellular migration and invasion are still poorly understood. The JNK pathway regulates cellular migration during development (24) as well as after growth factor stimulation (28). Recent studies by Shibata et al. (31) have demonstrated a link of JNK1 pathway in the development of gastric cancer. The participation of JNK pathway in G17-induced growth has also been shown in earlier studies (32). The studies described here were undertaken to determine any role of JNK pathway in mediating G17-induced cancer cell migration. Elucidation of the detailed signaling pathway involved in G17-induced migration will provide novel information, which can be targeted for designing rational therapeutic strategies to control gastrin-linked malignancies. Our studies indicated a time- and concentration-dependent activation of JNK pathway after stimulation by G17, which was mediated via CCK2R activation. Activation of JNK preceded G17-induced migration. In addition, inhibition of JNK pathway by a pharmacological inhibitor (SP600125) or siRNA-mediated knockdown of JNK1 expression abolished G17-induced migration, suggesting the involvement of JNK1 in mediating these events. Inhibition of the other MAPK pathways (ERK and p38), however, was unable to antagonize this migration significantly. Overexpression of constitutive active JNK1 induced spontaneous migration, with synergistic effects when combined with G17. These indicated that JNK1 plays an important role in mediating gastric cancer cell migration after G17 stimulation.
MMP7 is known to be involved in promoting the migratory pathway in cancer cells (39). Incubation of the gastric cancer cells with G17 also activated MMP7 promoter significantly, indicating a possible involvement of this metalloproteinase in G17-mediated events. In addition, inhibition of the JNK pathway using SP600125 or JNK1-siRNA transfection resulted in an antagonism of G17-induced MMP7-luciferase reporter activity. Similarly, overexpression of constitutively active JNK1 promoted G17-induced MMP7 promoter activation. Studies were also designed after knockdown of c-Jun expression (by c-Jun-siRNA) and overexpression of two dominant-negative mutants of c-Jun (DBM-3 and TAM67) to determine any role of the transcription factor c-Jun (JNK downstream target) in mediating G17-induced MMP7 promoter induction. Knockdown of c-Jun expression or overexpression of c-Jun dominant-negative mutants significantly attenuated MMP7 promoter activation after G17 stimulation. These studies established that G17-induced activation of JNK1 and c-Jun pathways mediate MMP7 induction and cellular migration.
Despite an involvement of JNK1 in mediating G17-induced migration, a key question that still remains is the mechanism by which G17 activates the JNK pathway. MLKs were identified as dual-specificity kinases because they possess signature motifs for both serine/threonine and tyrosine kinases (42). MLK3, a member of the mixed lineage kinase subfamily of serine/threonine protein kinases, is a MAP3K member that regulates the activity of JNK (38). MLK3 is the most intensively studied member of this family of kinases, which is known to mediate cell-death pathway in various cancer cells (43, 44), as well as cellular migration, and the latter in a novel kinase-independent manner (45). In addition, MLK3 plays a role in mitogen-induced cell proliferation and ERK activation (46). A very recent study has described mutations of MLK3 in GI tumors (47). The role of MLK3 in regulating other cellular processes is not clearly defined and is still emerging.
Our studies showed that MLK3 is expressed in gastric (Fig. 5C, MLK3 panel) and other cancer cells (data not shown), suggesting that this MAP3K might be involved in mediating specific cellular events in cancer cells. The facts that JNK1 plays an active role in mediating G17-induced migration in the gastric cancer cells (Fig. 3C and Fig. 4, A and B), and that MLK3 can activate JNK (38) strongly indicated that MLK3 might play an active role in G17-induced JNK activation and cellular migration. As shown in Figs. 5A and Supplemental Fig. 4, pharmacological inhibition of all MLK members by a pan-MLK inhibitor CEP11004 can antagonize G17-induced migration and activation of JNK1, respectively. More mechanistic studies designed to pinpoint the role of MLK3 showed that siRNA-mediated knockdown of endogenous MLK3 expression significantly attenuates G17-induced migration (Fig. 5B) and JNK activation (Fig. 5C), suggesting the involvement of MLK3 in the G17 pathway. G17 treatment also resulted in a transient activation of MLK3 in these gastric cancer cells (Fig. 5D). MMP7-luciferase assays designed after incubation with G17 showed that inhibition of MLK3 pathway, either by CEP11004 or by knockdown of MKL3 expression by MLK3-siRNA, strongly inhibits G17-induced MMP7-luciferase activity (Fig. 6, C and D).
The downstream mediators of G17/MLK3/JNK1-induced migration pathway are yet to be identified in detail, although our studies indicate one of them to be c-Jun. In recent studies, He et al. (13) demonstrated that G17- and G-Gly-induced migration of gastric cells involve the PAK1 pathway. In the same studies the authors also demonstrated that PAK1 was involved in gastrin-induced phosphorylation of β-catenin and activation of the β-catenin pathway, which might eventually lead to increased migration. Participation of β-catenin in G17- induced cyclin D1 transcription has been demonstrated by us earlier (6), suggesting that β-catenin might be a possible target of these migratory events as well. In fact, JNK has also been shown recently to phosphorylate β-catenin at specific serine residues and increase its nuclear translocation (48). It is thus tempting to postulate that MLK3/JNK1-mediated modulation of β-catenin and c-Jun pathways might play a role toward G17-induced migration.
The studies described here provide the first evidence that MLK3 functions as a MAP3K to activate JNK pathway during G17-induced migration and that G17 increases MLK3 kinase activity. The mechanism by which G17 activates MLK3 is still unknown and needs to be investigated. More elaborate studies are required to determine whether the MLK3/JNK1 axis also plays a role in G17-induced growth and other cellular events. Identification of the detailed signaling axis mediating specific events in the G17 pathway and characterization of their downstream targets are critical in the design of rational therapeutic approaches targeting these pathways to control GI malignancies.
Materials and Methods
Reagents
DMEM, LipofectAMINE 2000, and β-galactosidase (β-Gal) assay kit were purchased from Invitrogen (Carlsbad, CA); amidated gastrin (G17) was from Bachem (King of Prussia, PA), and the luciferase assay kit was from Promega Corp. (Madison, WI). The antibodies used were obtained from the following sources: total JNK1, JNK2, ERK, and c-Jun (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), total MLK3 (Epitomics, Burlingame, CA), phospho-JNK (Promega), glyceraldehyde-3-phosphate dehydrogenase (Ambion, Austin, TX), HA.11 (Covance Laboratories, Inc., Berkeley, CA); all other antibodies were from Cell Signaling Technology (Danvers, MA). The MEK inhibitor U0126 was from Promega, p38 inhibitor SB203580 was supplied by Calbiochem (San Diego, CA), JNK inhibitor SP600125 was from Alexis Biochemicals, Axxora (San Diego, CA), and Pan-MLK inhibitor CEP11004 was a gift from Cephalon (Frazer, PA). The AGSE cells stably overexpressing the CCK2R were obtained from Dr. Timothy C. Wang (Columbia University Medical Center, New York, NY) as described earlier (6). These cells were tested for CCK2R overexpression by Northern blotting and by gastrin responsiveness (36). The MMP-7-luciferase promoter construct was obtained from Dr. Howard Crawford (Stony Brook University, Stony Brook, NY) (40), and the hemagglutinin-tagged JNK1 expression vectors (JNK1-WT, JNK1-constitutive active, and JNK1-dominant negative) were from Dr. Anning Lin (University of Chicago, Ben May Department for Cancer Research, Chicago, IL) (49). The c-Jun dominant-negative mutants (DBM3 and TAM67) were obtained from Dr. Michael Birrer (National Institutes of Health, Bethesda, MD) (41).
Cell culture
The AGSE cells were maintained in DMEM supplemented with 10% fetal bovine serum and 100 IU/ml penicillin. In experiments with G17, confluent populations of cells were treated with 100 nm G17 in serum-deficient media and subjected to either Western blot analysis or migration assays. In the studies with various kinase inhibitors, cells were pretreated with the specific inhibitors followed by G17 treatment.
Transient transfection and luciferase assays
Luciferase assays were performed as described earlier (6). Briefly, subconfluent AGSE cells were transfected with MMP-7-luciferase-reporter construct (40) along with β-Gal vector (to correct for transfection efficiency) using lipofectAMINE 2000 as per manufacturer’s instructions. Each transfection was performed in triplicate, and each experiment was repeated at least twice. After 48 h of recovery in the growth medium, the transfected cells were treated with either vehicle or G17 for 24 h. Luciferase and β-Gal assays were performed using a luminometer (Centro XS (3) LB 960; Berthold Technologies, Bad Wildbad, Germany) and a plate reader (Power Wave XS; BioTek, Highland Park, IL), respectively. The results obtained were calculated as the ratio of relative light units (RLU) to β-Gal values (RLU/β-Gal) and expressed as percent increase compared with controls.
Western blot analysis
Western blot analysis was performed after treatment with G17 using procedures described previously (6). Briefly, cells were extracted with radioimmune precipitation assay buffer, and equal amounts of total protein were fractionated by SDS-PAGE, transferred to polyvinylidene difluoride membranes, and subjected to Western blot analysis using various antibodies.
siRNA
JNK1siRNA (sense 5′-UCACAGUCCUGAAACGAUAdTdT-3′) (50) and MLK3 siRNA (sense 5′-GGGCAGUGACGUCUGGAGUUUdTdT-3′) (46) were synthesized from Dharmacon (Lafayette, CO). The c-Jun-siRNA (51) was from Santa Cruz Biotechnology, and control-siRNA was from Ambion. siRNA transfection was performed using lipofectAMINE 2000 as per manufacturer’s instructions following protocols described earlier (52). G17 treatment was performed after 48 h of siRNA transfection, and the cells were then subjected to Western blot or migration assays.
Wound-healing assay
Confluent populations of cells plated on six-well plates were wounded linearly using a small pipette tip, washed with PBS to remove cell debris, and treated with the various agents in serum-deficient media for the indicated periods of time. In the experiments involving transient transfections (with JNK1), cells were wounded 48 h after transfection, and allowed to recover overnight before adding G17. This was necessary to allow the cells to settle down after transfection and also to determine the effects of JNK1 expression on spontaneous migration. The migratory cells were visualized and photographed using inverted phase-contrast microscopy (Axiovert 200 inverted microscope; Carl Zeiss, Inc., Thornwood, NY), interfaced with a camera (Axiocam) and the image analyzer software (Axiovision, Zeiss). To estimate relative migration, the unclosed distances at three points in each scratch were measured at each time point through the microscope, and their average was calculated and plotted as average gap.
In vitro kinase assay
Equal amounts of total protein extracted from cells treated with G17 for various lengths of time were immunoprecipitated with an antibody against MLK3 as described elsewhere (53). The immunoprecipitates were subjected to in vitro kinase assay in the presence of SEK1 (K-R) as MLK3 substrate and [γ32P]ATP. The samples were then fractionated by SDS-PAGE, and 32P incorporation in SEK1 (K-R) was quantified using a phosphor imager (STORM 820; GE Healthcare Bio-Sciences Corp., Piscataway, NJ).
Acknowledgments
We thank Dr. Timothy Wang (Columbia University Medical Center, New York, NY) for providing the AGSE cells; Dr. Anning Lin (University of Chicago, Ben May Department for Cancer Research, Chicago, IL) for the JNK1 constructs; Dr. Howard Crawford (Stony Brook University, Stony Brook, NY) for the MMP-7 luciferase reporter; and Dr. Michael Birrer (National Institutes of Health, Bethesda, MD) for the c-Jun mutant vectors (DBM3 and TAM67). We also thank Drs. Shannon Glaser (Texas A&M University-Health Science Center, Temple, TX) and Ajay Rana (Loyola University Chicago, Chicago, IL) for providing reagents and critical suggestions.
Footnotes
This work was supported by awards from the Department of Veterans Affairs (Merit and VISN17) to B. R.
Disclosure Summary: The authors have nothing to disclose.
First Published Online February 11, 2010
Abbreviations: β-Gal, β-Galactosidase; CCK, cholecystokinin; CCK2R, CCK2 receptor; G17, amidated gastrin; G-Gly, glycine-extended gastrin; GI, gastrointestinal; JNK, c-Jun NH2-terminal kinase; JNK1-WT, JNK1-wild type; JNK1-CA, JNK1-constitutive active; JNK1-DN, JNK1-dominant negative; MAP3K, MAPK kinase kinase; MLK, mixed lineage kinase; PAK1, P21-activated kinase 1; MMP, matrix metalloproteinase; RLU, relative light unit; siRNA, small interference RNA.
References
- 1.Dufresne M, Seva C, Fourmy D2006. Cholecystokinin and gastrin receptors. Physiol Rev 86:805–847 [DOI] [PubMed] [Google Scholar]
- 2.Rozengurt E, Walsh JH2001. Gastrin, CCK, signaling, and cancer. Annu Rev Physiol 63:49–76 [DOI] [PubMed] [Google Scholar]
- 3.Ferrand A, Wang TC2006. Gastrin and cancer: a review. Cancer Lett 238:15–29 [DOI] [PubMed] [Google Scholar]
- 4.Kusyk CJ, McNiel NO, Johnson LR1986. Stimulation of growth of a colon cancer cell line by gastrin. Am J Physiol 251:G597–G601 [DOI] [PubMed]
- 5.Seva C, Dickinson CJ, Yamada T1994. Growth-promoting effects of glycine-extended progastrin. Science 265:410–412 [DOI] [PubMed] [Google Scholar]
- 6.Pradeep A, Sharma C, Sathyanarayana P, Albanese C, Fleming JV, Wang TC, Wolfe MM, Baker KM, Pestell RG, Rana B2004. Gastrin-mediated activation of cyclin D1 transcription involves β-catenin and CREB pathways in gastric cancer cells. Oncogene 23:3689–3699 [DOI] [PubMed] [Google Scholar]
- 7.Wang TC, Koh TJ, Varro A, Cahill RJ, Dangler CA, Fox JG, Dockray GJ1996. Processing and proliferative effects of human progastrin in transgenic mice. J Clin Invest 98:1918–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang TC, Dangler CA, Chen D, Goldenring JR, Koh T, Raychowdhury R, Coffey RJ, Ito S, Varro A, Dockray GJ, Fox JG2000. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 118:36–47 [DOI] [PubMed] [Google Scholar]
- 9.Bierkamp C, Kowalski-Chauvel A, Dehez S, Fourmy D, Pradayrol L, Seva C2002. Gastrin mediated cholecystokinin-2 receptor activation induces loss of cell adhesion and scattering in epithelial MDCK cells. Oncogene 21:7656–7670 [DOI] [PubMed] [Google Scholar]
- 10.Ferrand A, Kowalski-Chauvel A, Bertrand C, Pradayrol L, Fourmy D, Dufresne M, Seva C2004. Involvement of JAK2 upstream of the PI 3-kinase in cell-cell adhesion regulation by gastrin. Exp Cell Res 301:128–138 [DOI] [PubMed] [Google Scholar]
- 11.Wroblewski LE, Pritchard DM, Carter S, Varro A2002. Gastrin-stimulated gastric epithelial cell invasion: the role and mechanism of increased matrix metalloproteinase 9 expression. Biochem J 365:873–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noble PJ, Wilde G, White MR, Pennington SR, Dockray GJ, Varro A2003. Stimulation of gastrin-CCKB receptor promotes migration of gastric AGS cells via multiple paracrine pathways. Am J Physiol Gastrointest Liver Physiol 284:G75–G84 [DOI] [PubMed]
- 13.He H, Shulkes A, Baldwin GS2008. PAK1 interacts with β-catenin and is required for the regulation of the β-catenin signalling pathway by gastrins. Biochim Biophys Acta 1783:1943–1954 [DOI] [PubMed] [Google Scholar]
- 14.Hollande F, Choquet A, Blanc EM, Lee DJ, Bali JP, Baldwin GS2001. Involvement of phosphatidylinositol 3-kinase and mitogen-activated protein kinases in glycine-extended gastrin-induced dissociation and migration of gastric epithelial cells. J Biol Chem 276:40402–40410 [DOI] [PubMed] [Google Scholar]
- 15.He H, Pannequin J, Tantiongco JP, Shulkes A, Baldwin GS2005. Glycine-extended gastrin stimulates cell proliferation and migration through a Rho- and ROCK-dependent pathway, not a Rac/Cdc42-dependent pathway. Am J Physiol Gastrointest Liver Physiol 289:G478–G488 [DOI] [PubMed]
- 16.Dhillon AS, Hagan S, Rath O, Kolch W2007. MAP kinase signalling pathways in cancer. Oncogene 26:3279–3290 [DOI] [PubMed] [Google Scholar]
- 17.Xia Y, Karin M2004. The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol 14:94–101 [DOI] [PubMed] [Google Scholar]
- 18.Dérijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ1994. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76:1025–1037 [DOI] [PubMed] [Google Scholar]
- 19.Kallunki T, Su B, Tsigelny I, Sluss HK, Dérijard B, Moore G, Davis R, Karin M1994. JNK2 contains a specificity-determining region responsible for efficient c-Jun binding and phosphorylation. Genes Dev 8:2996–3007 [DOI] [PubMed] [Google Scholar]
- 20.Weston CR, Davis RJ2002. The JNK signal transduction pathway. Curr Opin Genet Dev 12:14–21 [DOI] [PubMed] [Google Scholar]
- 21.Davis RJ2000. Signal transduction by the JNK group of MAP kinases. Cell 103:239–252 [DOI] [PubMed] [Google Scholar]
- 22.Karin M1995. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem 270:16483–16486 [DOI] [PubMed] [Google Scholar]
- 23.Minden A, Lin A, Smeal T, Dérijard B, Cobb M, Davis R, Karin M1994. c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol Cell Biol 14:6683–6688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riesgo-Escovar JR, Jenni M, Fritz A, Hafen E1996. The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes Dev 10:2759–2768 [DOI] [PubMed] [Google Scholar]
- 25.Xia Y, Makris C, Su B, Li E, Yang J, Nemerow GR, Karin M2000. MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc Natl Acad Sci USA 97:5243–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Wang W, Hayashi Y, Jester JV, Birk DE, Gao M, Liu CY, Kao WW, Karin M, Xia Y2003. A role for MEK kinase 1 in TGF-β/activin-induced epithelium movement and embryonic eyelid closure. EMBO J 22:4443–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stronach B, Perrimon N2002. Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev 16:377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hauck CR, Sieg DJ, Hsia DA, Loftus JC, Gaarde WA, Monia BP, Schlaepfer DD2001. Inhibition of focal adhesion kinase expression or activity disrupts epidermal growth factor-stimulated signaling promoting the migration of invasive human carcinoma cells. Cancer Res 61:7079–7090 [PubMed] [Google Scholar]
- 29.Alcorn JF, Guala AS, van der Velden J, McElhinney B, Irvin CG, Davis RJ, Janssen-Heininger YM2008. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-β1. J Cell Sci 121:1036–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Javelaud D, Laboureau J, Gabison E, Verrecchia F, Mauviel A2003. Disruption of basal JNK activity differentially affects key fibroblast functions important for wound healing. J Biol Chem 278:24624–24628 [DOI] [PubMed] [Google Scholar]
- 31.Shibata W, Maeda S, Hikiba Y, Yanai A, Sakamoto K, Nakagawa H, Ogura K, Karin M, Omata M2008. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Res 68:5031–5039 [DOI] [PubMed] [Google Scholar]
- 32.Dehez S, Bierkamp C, Kowalski-Chauvel A, Daulhac L, Escrieut C, Susini C, Pradayrol L, Fourmy D, Seva C2002. c-Jun NH(2)-terminal kinase pathway in growth-promoting effect of the G protein-coupled receptor cholecystokinin B receptor: a protein kinase C/Src-dependent-mechanism. Cell Growth Differ 13:375–385 [PubMed] [Google Scholar]
- 33.Dehez S, Daulhac L, Kowalski-Chauvel A, Fourmy D, Pradayrol L, Seva C2001. Gastrin-induced DNA synthesis requires p38-MAPK activation via PKC/Ca2+ and Src-dependent mechanisms. FEBS Lett 496:25–30 [DOI] [PubMed] [Google Scholar]
- 34.Daulhac L, Kowalski-Chauvel A, Pradayrol L, Vaysse N, Seva C1999. Src-family tyrosine kinases in activation of ERK-1 and p85/p110-phosphatidylinositol 3-kinase by G/CCKB receptors. J Biol Chem 274:20657–20663 [DOI] [PubMed] [Google Scholar]
- 35.Daulhac L, Kowalski-Chauvel A, Pradayrol L, Vaysse N, Seva C1997. Ca2+ and protein kinase C-dependent mechanisms involved in gastrin-induced Shc/Grb2 complex formation and P44-mitogen-activated protein kinase activation. Biochem J 325:383–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raychowdhury R, Fleming JV, McLaughlin JT, Bulitta CJ, Wang TC2002. Identification and characterization of a third gastrin response element (GAS-RE3) in the human histidine decarboxylase gene promoter. Biochem Biophys Res Commun 297:1089–1095 [DOI] [PubMed] [Google Scholar]
- 37.Huang C, Jacobson K, Schaller MD2004. MAP kinases and cell migration. J Cell Sci 117:4619–4628 [DOI] [PubMed] [Google Scholar]
- 38.Rana A, Gallo K, Godowski P, Hirai S, Ohno S, Zon L, Kyriakis JM, Avruch J1996. The mixed lineage kinase SPRK phosphorylates and activates the stress-activated protein kinase activator, SEK-1. J Biol Chem 271:19025–19028 [DOI] [PubMed] [Google Scholar]
- 39.Wroblewski LE, Noble PJ, Pagliocca A, Pritchard DM, Hart CA, Campbell F, Dodson AR, Dockray GJ, Varro A2003. Stimulation of MMP-7 (matrilysin) by Helicobacter pylori in human gastric epithelial cells: role in epithelial cell migration. J Cell Sci 116:3017–3026 [DOI] [PubMed] [Google Scholar]
- 40.Crawford HC, Fingleton B, Gustavson MD, Kurpios N, Wagenaar RA, Hassell JA, Matrisian LM2001. The PEA3 subfamily of Ets transcription factors synergizes with β-catenin-LEF-1 to activate matrilysin transcription in intestinal tumors. Mol Cell Biol 21:1370–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown PH, Kim SH, Wise SC, Sabichi AL, Birrer MJ1996. Dominant-negative mutants of cJun inhibit AP-1 activity through multiple mechanisms and with different potencies. Cell Growth Differ 7:1013–1021 [PubMed] [Google Scholar]
- 42.Gallo KA, Mark MR, Scadden DT, Wang Z, Gu Q, Godowski PJ1994. Identification and characterization of SPRK, a novel src-homology 3 domain-containing proline-rich kinase with serine/threonine kinase activity. J Biol Chem 269:15092–15100 [PubMed] [Google Scholar]
- 43.Mishra R, Barthwal MK, Sondarva G, Rana B, Wong L, Chatterjee M, Woodgett JR, Rana A2007. Glycogen synthase kinase-3β induces neuronal cell death via direct phosphorylation of mixed lineage kinase 3. J Biol Chem 282:30393–30405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim KY, Kim BC, Xu Z, Kim SJ2004. Mixed lineage kinase 3 (MLK3)-activated p38 MAP kinase mediates transforming growth factor-β-induced apoptosis in hepatoma cells. J Biol Chem 279:29478–29484 [DOI] [PubMed] [Google Scholar]
- 45.Swenson-Fields KI, Sandquist JC, Rossol-Allison J, Blat IC, Wennerberg K, Burridge K, Means AR2008. MLK3 limits activated Gαq signaling to Rho by binding to p63RhoGEF. Mol Cell 32:43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chadee DN, Kyriakis JM2004. MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nat Cell Biol 6:770–776 [DOI] [PubMed] [Google Scholar]
- 47.Velho S, Oliveira C, Paredes J, Sousa S, Leite M, Matos P, Milanezi F, Ribeiro AS, Mendes N, Licastro D, Karhu A, Oliveira MJ, Ligtenberg M, Hamelin R, Carneiro F, Lindblom A, Peltomaki P, Castedo S, Schwartz Jr S, Jordan P, Aaltonen LA, Hofstra RM, Suriano G, Stupka E, Fialho AM, et al.2010. Mixed lineage kinase 3 gene mutations in mismatch repair deficient gastrointestinal tumours. Hum Mol Genet 19:697–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F2008. Rac1 activation controls nuclear localization of β-catenin during canonical Wnt signaling. Cell 133:340–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng C, Xiang J, Hunter T, Lin A1999. The JNKK2-JNK1 fusion protein acts as a constitutively active c-Jun kinase that stimulates c-Jun transcription activity. J Biol Chem 274:28966–28971 [DOI] [PubMed] [Google Scholar]
- 50.Shen C, Chen Y, Liu H, Zhang K, Zhang T, Lin A, Jing N2008. Hydrogen peroxide promotes Aβ production through JNK-dependent activation of γ-secretase. J Biol Chem 283:17721–17730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Han S, Ritzenthaler JD, Sitaraman SV, Roman J2006. Fibronectin increases matrix metalloproteinase 9 expression through activation of c-Fos via extracellular-regulated kinase and phosphatidylinositol 3-kinase pathways in human lung carcinoma cells. J Biol Chem 281:29614–29624 [DOI] [PubMed] [Google Scholar]
- 52.Senthivinayagam S, Mishra P, Paramasivam SK, Yallapragada S, Chatterjee M, Wong L, Rana A, Rana B2009. Caspase-mediated cleavage of β-catenin precedes drug-induced apoptosis in resistant cancer cells. J Biol Chem 284:13577–13588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma C, Pradeep A, Wong L, Rana A, Rana B2004. Peroxisome proliferator-activated receptor γ activation can regulate β-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J Biol Chem 279:35583–35594 [DOI] [PubMed] [Google Scholar]