

Abstract
Kisspeptin and its receptor, GPR54, are major regulators of the hypothalamic-pituitary-gonadal axis as well as regulators of human placentation and tumor metastases. GPR54 is a Gq/11-coupled G protein-coupled receptor (GPCR), and activation by kisspeptin stimulates phosphatidy linositol 4, 5-biphosphate hydrolysis, Ca2+ mobilization, arachidonic acid release, and ERK1/2 MAPK phosphorylation. Physiological evidence suggests that GPR54 undergoes agonist-dependent desensitization, but underlying molecular mechanisms are unknown. Furthermore, very little has been reported on the early events that regulate GPR54 signaling. The lack of information in these important areas led to this study. Here we report for the first time on the role of GPCR serine/threonine kinase (GRK)2 and β-arrestin in regulating GPR54 signaling in human embryonic kidney (HEK) 293 cells, a model cell system for studying the molecular regulation of GPCRs, and genetically modified MDA MB-231 cells, an invasive breast cancer cell line expressing about 75% less β-arrestin-2 than the control cell line. Our study reveals that in HEK 293 cells, GPR54 is expressed both at the plasma membrane and intracellularly and also that plasma membrane expression is regulated by cytoplasmic tail sequences. We also demonstrate that GPR54 exhibits constitutive activity, internalization, and association with GRK2 and β- arrestins-1 and 2 through sequences in the second intracellular loop and cytoplasmic tail of the receptor. We also show that GRK2 stimulates the desensitization of GPR54 in HEK 293 cells and that β-arrestin-2 mediates GPR54 activation of ERK1/2 in MDA-MB-231 cells. The significance of these findings in developing molecular-based therapies for treating certain endocrine-related disorders is discussed.
GPR54 exhibits constitutive activity, internalization and association with GRK2 and β-arrestins-1 and 2. Kisspeptin-stimulated GPR54 undergoes GRK2-dependent desensitization and β-arrestin-dependent activation of ERK1/2.
In 2003, the neuropeptide kisspeptin (Kp) and its cognate receptor, G protein-coupled receptor 54 (GPR54) were positioned at the forefront of the neuroendocrine control of the hypothalamic-pituitary-gonadal (HPG) axis (1, 2). Although the functions of Kp originally were believed to be restricted to metastasis suppression, loss-of-function mutations in GPR54 were found to cause an absence of puberty and hypogonadotropic hypogonadism, a condition characterized by an absence of sexual maturation and low levels of gonadotropic hormones (LH and FSH), in humans. Mice with targeted deletions of GPR54 also have a hypogonadotropic phenotype, confirming the important role of the Kp/GPR54 signaling system in the control of puberty and reproductive function (2).
GPR54 is a Gq/11-coupled GPCR and activation by Kp stimulates PIP2 hydrolysis, Ca2+ mobilization, arachidonic acid release, and ERK1/2 and p38 MAPK phosphorylation (3). GPR54 signaling is known to be regulated at the level of Kp expression, and a rapidly growing body of literature describes the spatial and temporal characteristics of hypothalamic Kp expression in detail (4). GPR54 activity is also believed to be regulated at the level of receptor desensitization, internalization or sequestration, and degradation. To date, although very little has been directly reported on these early regulatory events at the molecular level, there is strong physiological evidence that suggests GPR54 undergoes agonist-dependent desensitization.
In 2005, Messager et al. (5) reported that intracerebroventricular infusion of Kp-10 over 4 h to adult ovariectomized ewes treated with estradiol implants led to a decline in LH levels by the end of the treatment period in some animals, thus suggesting for the first time that a continuous administration of Kp desensitizes gonadotropin release. Shortly after, Thompson et al. (6) reported that continuous peripheral administration of Kp-54 causes a desensitization of the HPG axis in male rats. These initial studies were then followed by other investigations specifically designed to examine the prolonged and continuous administration of Kp on gonadotropin release. The first of these studies demonstrated that in both agonadal juvenile and adult male monkeys, continuous administration of human Kp-54 abolishes the LH response after an initial, acute stimulatory effect (7, 8). Most recently, it has also been reported that continuous intracerebral administration of Kp-10 in female rats also induces a desensitization of gonadotropin release (9). The study by Seminara et al. (7) was particularly noteworthy because the authors clearly demonstrated that the abolished LH response was due to the desensitization and/or down-regulation of GPR54 and not to diminished GnRH stores in hypothalamic neurons or weakened GnRH receptor signaling in the pituitary.
A major mechanism for switching off many GPCRs is homologous desensitization, a two-step process that involves the coordinated actions of two families of proteins, the GPCR serine/threonine kinases (GRKs) and the arrestins (10, 11, 12). Homologous desensitization is perhaps best understood for the β2-adrenergic receptor, and, based on this, the following prototypical model has been proposed. The receptor binds its ligand and adopts a conformation that allows it to bind one or more of the GRKs (of which there are seven) and, in doing so, becomes phosphorylated at residues on its intracellular loops and carboxyl terminus. Phosphorylation of the receptor promotes the high-affinity binding of the arrestin family of proteins [the visual arrestins and the nonvisual arrestins (β-arrestins-1 and -2)] to the receptor, which physically interdicts further coupling to G proteins. β-Arrestins also target many GPCRs for internalization in clathrin-coated vesicles via a direct interaction of the carboxyl-terminal tail domain of β-arrestins with both β-adaptin and clathrin. For a number of GPCRs, β-arrestins also act as molecular adapters and recruit signaling proteins to the agonist-occupied GPCR (reviewed in Refs. 13 and 14). In all of these established roles, β-arrestin therefore serves as a critical regulator of early GPCR activity.
Here we report on the roles of GRK-2 and β-arrestin in regulating GPR54 signaling in two cell systems. We demonstrate that GPR54 is constitutively associated with GRK2 and β-arrestins-1 and -2 and that these interactions are mediated through sequences in the second intracellular loop and cytoplasmic tail (Ct) of the receptor. We also show that GRK2 stimulates the desensitization of GPR54 in human embryonic kidney (HEK) 293 cells and that β-arrestin-2 mediates GPR54 activation of ERK1/2 in MDA MB-231 cells.
Results
Wild-type GPR54 is expressed at the plasma membrane and undergoes rapid agonist-dependent and -independent internalization
HEK 293 cells were used to study the molecular regulation of wild-type GPR54 (Fig. 1A) and one of its naturally occurring mutants R331X (Fig. 1B). We first demonstrated by RT-PCR that HEK 293 cells express GPR54 but not KISS1, thus allowing us to examine the effects of exogenous Kp-10 on GPR54 activity (Fig. 1C). The R331X mutant suffers a deletion of most of its Ct (68 residues) and only retains the two most membrane-proximal amino acid residues based on predicted structure (Fig. 1B). In this study, we used amino-terminally FLAG-tagged receptors to visualize the receptor in the cell due to the absence of commercially available robust anti-GPR54 antibodies. The FLAG epitope was selected after we demonstrated the FLAG-tagged wild-type GPR54 maintained the characteristics of the untagged receptor in several biochemical and molecular tests such as Kp-10-dependent inositol phosphate (IP) formation (data not shown).
Fig. 1.

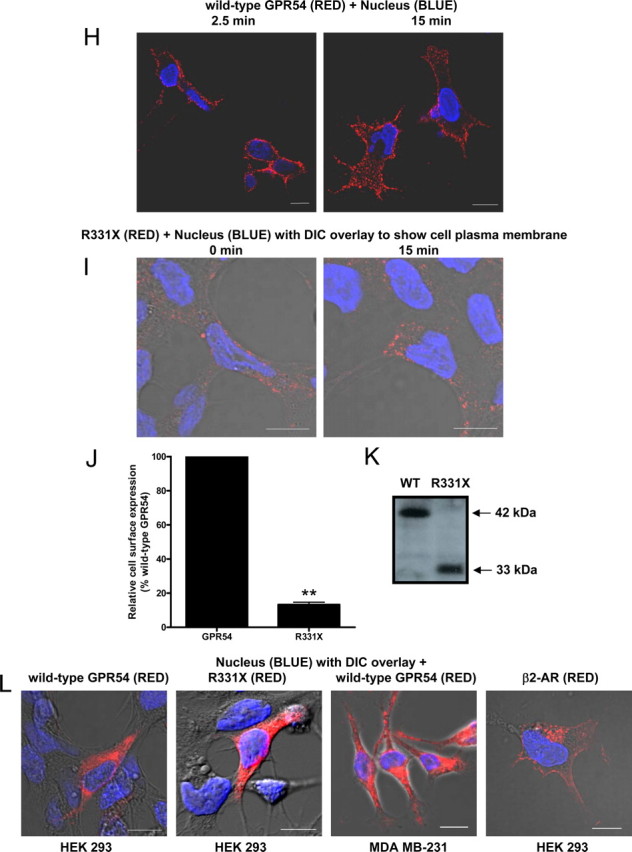
The expression, localization, and internalization of GPR54. Schematic of wild-type GPR54 (A) and R331X (B) showing absence of most of the Ct, except for two residues based on the predicted model. C, RT-PCR analysis of GPR54 and KISS1 expression in HEK 293 and MDA-MB-231 cells. Third lane shows water control. D, HEK 293 cells transiently expressing FLAG-GPR54 were surface labeled at 4 C (0 min) using rabbit anti-FLAG antibody followed by agonist treatment (100 nm Kp-10) at 37 C for 2.5 and 15 min. After fixation, surface GPR54 was detected by Alexa Fluor 568-conjugated antirabbit IgG in the nonpermeabilized cells. E, HEK 293 cells transiently transfected with 5 μg FLAG-GPR54 and 5 μg clathrin light chain-GFP (left panel) or 5 μg FLAG-GPR54 only (right panel) were labeled at 4 C using rabbit anti-FLAG primary antibody. Cells transfected with receptor only were also incubated with transferrin conjugate-488. After agonist treatment (100 nm Kp-10) at 37 C for 2.5 (left panel) and 15 mins (right panel), cells were fixed and surface GPR54 was detected by Alexa Fluor 568-conjugated antirabbit IgG. F, Time course for GPR54 basal or constitutive (closed circle) and Kp-10-stimulated (open circle) internalization. The data represent the mean ± se of nine independent experiments. HEK 293 cells transiently expressing GPR54 were surface labeled at 4 C using mouse anti-FLAG antibody. Cells were left untreated or incubated with 100 nm Kp-10 at 37 C for the indicated times and then fixed. Internalization was calculated as the percentage of loss of cell surface immunofluorescence over time and measured by flow cytometry. G, Bar chart showing receptor internalization of HEK 293 cells transiently transfected with FLAG-GPR54 or FLAG β2-AR after 5 min of treatment with 100 nm Kp-10 or 10 μm isoproterenol at 37 C. The data represent the mean ± se of three independent experiments. ***, P < 0.001 vs. GPR54. H, HEK 293 cells transiently expressing FLAG-GPR54 were surface labeled at 4 C (0 min) using rabbit anti-FLAG antibody followed by incubation in the absence of agonist at 37 C for 2.5 and 15 min. After fixation, surface GPR54 was detected by Alexa Fluor 568-conjugated antirabbit IgG in the nonpermeabilized cells. I, HEK 293 cells transiently expressing FLAG-R331X were surface labeled at 4 C (0 min) using rabbit anti-FLAG antibody followed by agonist treatment (100 nm Kp-10) at 37 C for 15 min. After fixation, surface receptor was detected by Alexa Fluor 568-conjugated antirabbit IgG in the nonpermeabilized cells. **, P < 0.01 vs. GPR54. J, Analysis of wild-type FLAG-GPR54 and R331X surface expression in nonstimulated cells. HEK 293 cells transiently expressing the receptors were surface labeled at 4 C using rabbit anti-FLAG antibody and fixed, and cell surface immunofluorescence was measured by flow cytometry. Data represent mean ± se for three independent experiments. K, Western blot analysis of wild-type (WT) FLAG-GPR54 and R331X. Protein was detected with the rabbit anti-FLAG primary antibody (L) HEK 293 cells transfected with FLAG-GPR54 (first panel), R331X (second panel), and FLAG β2-AR (fourth panel), and MDA-MB-231 cells transfected with FLAG-GPR54 (third panel) were fixed, permeabilized, and subjected to indirect immunofluorescent staining using rabbit anti-FLAG antibody followed by Alexa Fluor 568-conjugated antirabbit IgG. Scale bars, 10 μm. DIC, Differential interference contrast; WT, wild type.
We began our study by conducting a spatial and temporal analysis of wild-type GPR54 localization in the presence and absence of Kp-10 in HEK 293 cells. Our studies revealed that after 30 min of serum starvation, wild-type GPR54 was strongly and evenly localized to the plasma membrane of nonpermeabilized (detects surface GPR54 expression only) and nonstimulated cells that were kept at 4 C (Fig. 1D, left panel). However, after Kp-10 treatment at 37 C, we observed a rapid (within 2.5 min) redistribution of GPR54 into distinct punctate structures along the plasma membrane (Fig. 1D, middle panel). Through coexpression studies, using a green fluorescent protein (GFP)-tagged clathrin light chain molecule and FLAG-GPR54, we subsequently demonstrated that these punctuate structures were clathrin-coated pits (CCPs) and that GPR54 had indeed colocalized to them after agonist treatment (Fig. 1E, left panel). After this redistribution to CCPs, GPR54 internalized very rapidly (Fig. 1D, right panel) with approximately 80% of the cell-surface signal removed after 5 min (Fig. 1F). Using transferrin, a well-established marker for clathrin-mediated endocytosis (15, 16), we demonstrated through colocalization studies that the majority of GPR54 internalized via CCPs (Fig. 1E, right panel). The rapid loss of cell surface receptor after agonist treatment led us to compare the kinetics of internalization to that of the highly studied β2-adrenergic receptor (β2-AR). Our results revealed that after 5 min of isoproterenol (10 μm) treatment, only about 30% of β2-AR had internalized (vs. 80% internalized GPR54, P < 0.0001) (Fig. 1G), a value similar to that previously reported (17). In a simultaneous experiment, we also examined the effect of Kp-10-independent internalization of GPR54 and, much to our surprise, found that GPR54 displayed a high rate of basal internalization in HEK 293 cells (Fig. 1, F and H). However, the rate of Kp-10-dependent internalization was significantly higher than the basal internalization rate (P < 0.05) (Fig. 1F).
We also compared the spatial and temporal properties of R331X (Fig. 1B) localization to wild-type GPR54 in the presence and absence of Kp-10 in HEK 293 cells. Our data clearly revealed that visually, after 30 min of serum starvation, in nonstimulated cells which were kept at 4 C, R331X was weakly localized to the plasma membrane compared with wild-type GPR54 (Fig. 1I; in this figure the intensity of the red was increased relative to Fig. 1D after image acquisition, to allow us to demonstrate that there was some detectable expression of R331X at the plasma membrane). This was confirmed quantitatively where we determined that the mutant receptor displayed approximately 90% less expression, compared with wild-type GPR54, at the plasma membrane (P < 0.05) (Fig. 1J). Although R331X was weakly localized to the plasma membrane, we were able to visually detect a loss of cell-surface receptor after Kp-10 treatment (Fig. 1I; in this figure the intensity of the red was increased relative to Fig. 1D after image acquisition, to allow us to demonstrate that there was internalization of the R331X). To confirm that the reduced plasma membrane expression of R331X was not simply due to an overall reduced expression of the mutant, we next assayed for total receptor content under basal conditions by both visual observation (conducted on serum-starved cells that were subsequently permeabilized at 4 C) and by Western blotting for the receptor in cell lysate. Here we observed visually that both the wild-type receptor and R331X were highly expressed intracellularly at similar levels, and Western blot analysis also revealed similar expression levels with no indication of degradation, under conditions that would not promote cell-surface receptor internalization (Fig. 1K). The high level of intracellular expression of wild-type GPR54 (FLAG-tagged) in HEK 293 cells was unexpected, and this led us to compare its intracellular expression to that of the identical receptor in the invasive breast cancer cell line MDA-MB-231 and to that of the unrelated FLAG-tagged β2-AR in HEK 293 cells under identical conditions. Our data revealed that compared with wild-type GPR54 and R331X, under conditions that do not promote receptor internalization, GPR54 is also highly expressed intracellularly in the MDA-MB-231 cells whereas β2-AR is weakly localized intracellularly in HEK 293 cells (Fig. 1L). Instead, under these conditions, β2-AR is mostly found at the plasma membrane (Fig. 1L).
GRK2 interacts with wild-type GPR54 and R331X
To determine whether GRK2 interacts with wild-type GPR54, we assessed whether FLAG-GPR54 and myc-GRK2 can form an immunocomplex. Here we observed that FLAG-GPR54 strongly coimmunoprecipitated with myc-GRK2 both in the absence and presence of Kp-10 (Fig. 2, A and B) and that the level of interaction between R331X and GRK2 was similar (Fig. 2, A and B). To delineate the specific regions on the receptor that mediate this interaction with GRK2, glutathione-S-transferase (GST) precipitation assays were conducted with GST-GPR54 intracellular domains and myc-GRK2. The GST-tagged intracellular domains consisted of loop 1 (GST-IL1, corresponding to amino acid positions 67-78 on GPR54), loop 2 (GST-IL2, corresponding to amino acid positions 139-157 on GPR54), loop 3 (GST-IL3, corresponding to amino acid positions 227-263 on GPR54), and the Ct (GST-Ct, corresponding to amino acid positions 329-398 on GPR54) (Fig. 1A). Escherichia coli-expressed GST-tagged GPR54 intracellular domains were isolated and purified, and their expression was determined by SDS-PAGE (Fig. 2C). Despite using an approximately equal number of recombinant E. coli cells for purifying each peptide, we observed that GST-IL2 and GST-Ct were consistently weakly expressed compared with GST-IL1 and GST-IL3 (Fig. 2C). GST protein levels were therefore adjusted, and equal amounts of peptides were used in the GST precipitation assay. The results of this assay clearly revealed that GRK2 interacts strongly with GPR54 via sequences in the receptor’s second intracellular loop and Ct (Fig. 2D).
Fig. 2.

GPR54 interacts with GRK2. Densitometric analysis (A) and representative autoradiograph (B) showing the coimmunoprecipitation of GRK2 with wild-type FLAG-GPR54 and R331X. HEK 293 cells were transfected with 5 μg of myc-GRK2 and 10 μg of FLAG-GPR54 or empty FLAG vector control. Cells were left untreated (gray bars) or stimulated (black bars) with 100 nm Kp-10 for 5 min at 37 C. Lysates were prepared, immunoprecipitated with mouse anti-FLAG antibody, and immunoblotted with mouse anti-myc antibody. The expression of myc-GRK2 in 50 μg of total protein from the corresponding HEK 293 cell lysates is also shown. The data represent the means ± se for three independent experiments. C, Purified peptides corresponding to GST-tagged GPR54 intracellular domains (GST-IL1, GST-IL2, GST-IL3, GST-Ct) were analyzed by Western blotting using a rabbit anti-GST-antibody. D, GRK2 overexpresed in HEK 293 cells was tested for its ability to coprecipitate with GST, GST-IL1, GST-IL2, GST-IL3, GST-Ct. Interaction was determined by Western blotting using a mouse anti-myc antibody. The expression of myc-GRK2 in 50 μg of total protein from the corresponding HEK 293 cell lysates is also shown. AU, Arbitrary units; IB, immunoblotting; IP, immunoprecipitation; WT, wild type.
GRK2 mediates the desensitization of GPR54
To assess whether GRK2 mediates the desensitization of GPR54, we examined the effect of myc-GRK2 overexpression on the Kp-10-dependent IP formation by GPR54. Our results showed that coexpression of myc-GRK2 resulted in a downward shift of the agonist dose-dependent curve suggesting that GRK2 mediated the desensitization of GPR54 (P < 0.0001) (Fig. 3A).
Fig. 3.

GPR54 undergoes GRK2-dependent desensitization (A) FLAG-GPR54-stimulated IP formation in response to increasing concentrations of Kp-10 at 37 C in either the absence (closed circles) or the presence (open circles) of wild-type GRK2 or K220R (open triangles). The data points represent the mean ± se for three to six independent experiments. B, Representative autoradiograph demonstrating the whole-cell phosphorylation of FLAG-GPR54 treated with 100 nm Kp-10 for 10 min in the presence and absence of GRK2 or K220R. C, Basal IP formation in nontransfected (NT) vs. FLAG-GPR54-expressing HEK 293 cells. Data are expressed as a percentage of the maximal Kp-10-stimulated response observed in HEK 293 cells expressing FLAG-GPR54. The data represent the mean ± se for nine independent experiments. ***, P < 0.0001 vs. NT (D) The effect of GRK2 and K220R on basal GPR54 activity in HEK 293 cells. The data represent the mean ± se for three to six independent experiments. ***, P < 0.001 vs. control; ΔΔΔ, P < 0.001 vs. GRK2. E, Relative cell surface expression of FLAG-GPR54 in HEK 293 cells in the presence or absence of GRK2. Cell surface expression was determined by flow cytometry and represents the mean cell surface fluorescence. The data represents the mean ± se for three independent experiments. F, Time course for GPR54 basal or constitutive (closed symbols) and Kp-10-stimulated (open symbols) internalization in the absence (circles) or presence (triangles) of GRK2. The data represent the mean ± se of three to nine independent experiments. In all of the experiments, HEK 293 cells were transiently transfected with 10 μg of receptor and with 5 μg of either empty plasmid vector or plasmid cDNA encoding GRK2 or K220R.
K220R mediates the desensitization of GPR54
To determine whether the GRK2-dependent desensitization of Kp-10-stimulated GPR54 activity could also occur in the absence of GRK2 kinase activity, we assessed the effect of coexpressing the catalytically inactive GRK2 expression construct, K220R (18), on the Kp-10-dependent IP formation by GPR54. Our results revealed that overexpression of K220R resulted in the desensitization of GPR54 (P < 0.0001) (Fig. 3A). To further test this we compared the phosphorylation of GPR54 both in the absence and presence of coexpressed wild-type GRK2 and K220R. The results showed that in the presence of both GRK2 and K220R, receptor phosphorylation was similar to that observed in the control cells expressing GPR54 only (Fig. 3B). Taken together, the data suggest that GRK2-dependent desensitization of Kp-10-activated GPR54 is occurring, at least in part, by a phosphorylation independent-mechanism.
Regulation of basal and Kp-10-stimulated GPR54 activity by GRK2: the role of phosphorylation
Because we had demonstrated that GPR54 displays a high rate of basal internalization in HEK 293 cells (Fig. 1F), we assessed whether there was also basal IP formation. Our data revealed that overexpression of GPR54 led to an approximately 6-fold increase in IP formation in the absence of exogenous Kp-10 relative to nontransfected cells (P < 0.0001); this was approximately equivalent to 5% of the maximal Kp-10-dependent IP formation response (Fig. 3C). We then demonstrated that overexpression of GRK2 was able to significantly reduce this GPR54-dependent basal IP formation by about 40% (P < 0.001) (Fig. 3D). This was not surprising given the observation that GRK2 strongly interacted with GPR54 under basal conditions (Fig. 2, A and B). Interestingly, however, overexpression of the catalytically inactive K220R had the opposite effect where we observed that its overexpression led to a significant increase in basal IP formation relative to when GPR54 was overexpressed together with GRK2 (P < 0.001) or alone (P < 0.001) (Fig. 3D). The data therefore suggest that regulation of basal GPR54 activity by GRK2 is phosphorylation dependent, whereas that of Kp-10 stimulated GPR54 activity is not. GRK2 kinase activity, would account, at least in part, for the observed basal phosphorylation of GPR54 (Fig. 3B).
GRK2 does not alter GPR54 cell surface expression or internalization
Next, we determined whether overexpression of GRK2 modulated GPR54 cell surface expression or GPR54 internalization under basal and Kp-10-stimulated conditions. The data revealed that GRK2 had no effect on the cell surface expression of GPR54 or the basal and Kp-10-stimulated internalization of GPR54 (Fig. 3, E and F). Noteworthy, even in the presence of GRK2, the internalization of GPR54 was significantly higher after Kp-10 stimulation (P < 0.05) (Fig. 3F).
Activated GPR54 recruits β-arrestin-1 and -2 to the plasma membrane
Once we had established a role for GRK2 in the desensitization of GPR54, we next assessed whether β-arrestin-1 and -2 were responsive to GPR54 activation and might therefore play a role in regulating receptor signaling. To do this, we conducted live cell confocal-based imaging to assess whether activation of GPR54 in HEK 293 cells can stimulate a redistribution of β-arrestin-GFP in the cell. Our results showed that after agonist treatment, within 2–3 min the cells displayed robust ruffling (an indication that the receptor is activated), and this was immediately followed by the translocation of β-arrestin-1 and -2 from the cytoplasm to the plasma membrane (Fig. 4, A and B). Whereas translocated β-arrestin-1-GFP accumulated evenly at the plasma membrane within the ruffles (Fig. 4A), β-arrestin-2-GFP rapidly localized to distinct puncta at the plasma membrane (Fig. 4B) where it colocalized with the receptor (Fig. 4C). Although we did not show colocalization between GPR54, β-arrestin-2-GFP, and CCPs, based on our previous data (Fig. 1E) we believe that these punctate structures that GPR54 and β-arrestin-2-GFP colocalized to are CCPs. We observed, 10 min after Kp-10 treatment, that GPR54 colocalized with β-arrestin-2-GFP on internal vesicles (Fig. 4C).
Fig. 4.

Activation of GPR54 stimulates the translocation of cytoplasmic β-arrestin-1 and -2 to the plasma membrane in HEK 293 cells. HEK 293 cells coexpressing FLAG-GPR54 and GFP-tagged β-arrestin-1 (A) or -2 (B) were stimulated with 100 nm Kp-10 and analyzed for the translocation of β-arrestin using live-cell confocal imaging. C, HEK 293 cells transfected with 10 μg FLAG-GPR54 and 5 μg β-arrestin-2-GFP were surface labeled at 4 C (0 min) using rabbit anti-FLAG antibody followed by agonist treatment (100 nm Kp-10) at 37 C for 5 or 10 min. After fixation, the localization of GPR54 was detected by Alexa Fluor 568 conjugated to antirabbit IgG and imaged by confocal microscopy. The overlay images show the localization of FLAG-GPR54 (red) and β-arrestin-2-GFP (green). Colocalization of both molecules is shown as yellow puncta. Scale bars, 10 μm.
β-Arrestin-1 and -2 interact with wild-type GPR54 and R331X
Once we had determined that β-arrestin-1 and -2 translocate to the plasma membrane after GPR54 activation, we next determined whether β-arrestin-1 and -2 also interact with wild-type GPR54 and the R331X mutant. Through the use of coimmunoprecipitation assays, we demonstrated that β-arrestin-1-GFP and β-arrestin-2-GFP interacted strongly with wild-type GPR54 both in the absence and presence of agonist (Fig. 5, A and B); the level of interaction with R331X was again highly similar (Fig. 5, A and B). To delineate the specific regions on the receptor that mediate this interaction with β-arrestin, GST precipitation assays were conducted with GST-GPR54 intracellular domains, GST-IL1, GST-IL2, GST-IL3, and GST-Ct (Fig. 1A) and β-arrestin-2-GFP. The results of this assay clearly revealed that β-arrestin-2 interacted strongly with GPR54 via sequences in the receptor’s second intracellular loop and Ct (Fig. 5C).
Fig. 5.

GPR54 interacts with β-arrestin-1 and -2. Densitometric analysis and representative autoradiograph showing the coimmunoprecipitation of β-arrestin-1 (A) and β-arrestin-2 (B) with wild-type FLAG-GPR54 and R331X. HEK 293 cells were transfected with 10 μg of β-arrestin-1 or -2-GFP and 10 μg of FLAG-GPR54. Cells were left untreated (gray bars) or stimulated (black bars) with 100 nm Kp-10 for 5 min at 37 C. Lysates were prepared, immunoprecipitated with mouse anti-FLAG antibody, and immunoblotted with mouse anti-GFP antibody. The expression of β-arrestin-1 or -2-GFP in 50 μg of total protein from the corresponding HEK 293 cell lysates is also shown. The data represent the mean ± se for three independent experiments. C, β-Arrestin-2-GFP overexpressed in HEK 293 cells was tested for its ability to coprecipitate with purified GST, GST-IL1, GST-IL2, GST-IL3, and GST-Ct peptides. Interaction was determined by Western blotting using a mouse anti-GFP antibody. The expression of β-arrestin-2-GFP in 50 μg of total protein from the corresponding HEK 293 cell lysates is also shown. AU, Arbitrary units. IB, Immunoblotting; IP, immunoprecipitation; WT, wild type.
β-Arrestin-2 regulates the signaling of GPR54 to ERK1/2
To investigate the functional consequence of the interaction between β- arrestin-2 and endogenous GPR54 at the plasma membrane, we examined the effect of Kp-10 treatment on the activation of ERK1/2 on overnight serum-starved scrambled short hairpin RNA (shRNA) control and β-arrestin-2 shRNA knockdown MDA-MB-231 cells. MDA-MB-231 is an invasive breast cancer human cell line that endogenously expresses β-arrestin-1 and -2 (19) as well as GPR54 and KISS1 (Fig. 1C). Western blot analysis of these 231 cells revealed that the β-arrestin-2 shRNA silenced 231 cells express about 25% of the wild-type level seen in the control line (P < 0.05) (Fig. 6, A and B). The results from this study showed that Kp-10 treatment induced the rapid (within 2.5 min) and biphasic phosphorylation of ERK1/2 in wild-type cells over a 30-min period (Fig. 6, C and D). This ERK1/2 response was evidence of an active endogenous GPR54 signaling system within MDA-MB-231 cells. However, in the β-arrestin-2 shRNA silenced cells, we observed that after agonist treatment ERK1/2 activity was reduced relative to basal conditions and stayed suppressed at almost undetectable levels during the assay period (Fig. 6C). The results therefore suggest that β-arrestin-2 is required for GPR54 signaling to ERK1/2.
Fig. 6.

β-Arrestin-2 mediates GPR54 signaling to ERK1/2 MAPK. Densitometric analysis (A) and representative autoradiograph (B) showing the expression of β-arrestin-2 in a β-arrestin-2 shRNA-down-regulated and scrambled shRNA MDA-MB-231 cell line. β-Actin is used as a control for loading. Western blot analyses were done using a polyclonal anti-β-arrestin-2 and monoclonal anti-β-actin-antibodies. *, P < 0.05 vs. scrambled control. The data represent the mean ± se of three independent experiments. C, Densitometric analysis and representative autoradiographs showing the expression of total and activated ERK1/2 after 100 nm Kp-10 stimulation (for the indicated time points) of the β-arrestin-2 shRNA-down-regulated (black bars) and scrambled shRNA (gray bars) MDA-MB-231 cell lines. Western blot analyses were done using monoclonal anti-ERK1/2 and antiphospho ERK1/2 antibodies. The data represent the mean ± se of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. respective control. AU, Arbitrary units; β-arr, β-arrestin.
Discussion
GPR54 has emerged as a GPCR of immense clinical importance and through mechanisms that modulate its signaling capacity, the potential exists for treating certain cancers and endocrine disorders. To develop such therapies, however, it is important that we understand mechanisms that regulate GPR54 signaling, particularly those that regulate activity immediately after receptor activation. For that reason we have chosen to focus our attention on the roles of GRK2 and β-arrestin-1 and -2, proteins that regulate GPCR signaling immediately after receptor activation. Specifically, we focused on GRK2 because, like GPR54, GRK2 is also expressed in a wide variety of mammalian tissues (20).
We initiated this study by examining the spatial and temporal distribution of the FLAG-tagged receptor in HEK 293 cells. In this study we selected the HEK 293 cell system to conduct most of our investigations on the roles of GRK2 and β-arrestin-1 and -2 regulation of GPR54 activity because 1) HEK 293 cells display no detectable expression of KISS1 and would therefore allow us to examine the effects of exogenously applied Kp-10; 2) HEK 293 cells also express GRK2 (21) as well as β-arrestin-1 and -2 (22); and 3) it is one of the most widely used cell models for studying GPCR signaling and by employing it in our studies it has allowed direct comparison of our results to that generated by numerous other laboratories. Despite the utility of the HEK 293 cell model, we must acknowledge that there might be cell-specific differences in GPR54 regulation, and ultimately this study needs to be conducted in other cell systems of specific physiological interest. In the case of GPR54 as 1) a regulator of the HPG axis, such a system can be the immortalized mouse hypothalamic neuronal cell system, GT1–7, that endogenously expresses GPR54 (23, 24) or as 2) a regulator of embryo implantation and placentation, an appropriate cell system would be the human extravillous trophoblast cell system HTR-8/SVneo (25, 26). In our investigations, we found that GPR54 is strongly expressed at the plasma membrane of HEK 293 cells and undergoes rapid Kp-dependent internalization. Kp-10-dependent internalization of GPR54 was observed to be very high with approximately 80% of the cell surface receptors internalized by 15 min of Kp-10 treatment. This compared with about 40% for the β2-AR (17) and about 90% for the ghrelin receptor (27) after 15 min treatment by their respective agonists.
Because it has been demonstrated that Ct sequences of GPCRs play a role in potentiating receptor surface expression (28) and efficiency of G protein-coupling (29), we examined the cellular expression of the naturally occurring loss-of-function GPR54 mutant R331X and found that compared with the wild-type receptor, surface expression was diminished by almost 90%. R331X has a highly truncated Ct and is predicted to retain just two amino acid residues. We therefore suggest that the predicted 70-residue long tail encodes sequences that regulate GPR54 surface expression.
During the course of our investigations, we observed that GPR54 displays a high rate of basal internalization. This appears to be true basal internalization because we were unable to detect the expression of the KISS1 gene in HEK 293 cells. The high basal internalization rate for GPR54 was much higher (∼70% after 5 min) than that observed for the metabotropic glutamate receptor 1a (mGluR1a) at an early time point (∼50% of cell surface receptor internalized after 5 min (17), a receptor well known for its constitutive internalization in heterologous cell cultures and primary neurons (17, 30, 32, 33). However, after 15 min of internalization under basal conditions, approximately 80% of GPR54 had internalized, and this compared with about 65% for mGluR1a (17) and about 75% for the ghrelin receptor (27).
Another striking observation we made while addressing the spatial and temporal properties of wild-type GPR54 and its mutant R331X was the high level of receptor molecules localized intracellularly in HEK 293 cells, under conditions that do not promote GPCR internalization. To highlight this observation, we compared the intracellular distribution of GPR54 and R331X in HEK 293 cells to that of GPR54 in the breast cancer cell line, MDA-MB-231, and the well-studied β2-AR in HEK 293 cells under identical conditions. We demonstrated that, unlike the β2-AR, GPR54 is expressed at high levels intracellularly. The high intracellular content of GPR54 under basal conditions, however, appears to be similar to that described for the relaxin receptor RXFP1, which under nonstimulated conditions is mostly localized intracellularly (∼60–70% of total receptors) (34, 35). RXFP1 also displays constitutive internalization in HEK 293 cells, albeit much slower than GPR54 within 15 min at 37 C. Nevertheless, after 90 min, constitutive internalization was much greater where approximately 65% of the receptor had internalized (35). The significance of the high basal internalization rate and large endogenous pool of GPR54 is presently unclear, but perhaps, as suggested for the ghrelin receptor (27), this may provide a quiescent intracellular pool of GPR54 that is protected from desensitization and allows for the rapid delivery of GPR54 to the surface of cells when required.
We next examined whether GRK2 interacts with and desensitizes GPR54. We found that both the wild-type receptor and R331X interacted strongly with GRK2; therefore, it was not surprising to find that this interaction was mediated, in part, via the second intracellular loop in addition to the Ct. GRK is a serine-threonine kinase that has been shown to interact with GPCRs through various intracellular domains. For the α2AAR, GRK2 binds the second and third intracellular loops (36) whereas for mGluR1a, binding occurs via the second intracellular loop and Ct (37). In both cases, the authors determined that basic residues within these domains were critical in mediating receptor-GRK2 interactions. The second intracellular loop and Ct of GPR54 contain several basic residues that can potentially mediate this interaction.
Once we had shown that GRK2 and GPR54 interact with each other, we demonstrated that overexpression of GRK2 leads to the homologous desensitization of Kp-10-stimulated IP formation. Subsequently, through the combined use of the kinase-deficient GRK2-K220R expression construct and whole-cell phosphorylation assays, we demonstrated that GRK2 regulation of Kp-10-stimulated GPR54 activity occurs, at least in part, via a phosphorylation-independent mechanism. The N-terminal domain of GRK2 (residues 1–190) exhibits approximately 20% identity to the RGS domain encoded by members of the family of RGS proteins and has been previously demonstrated to effectively inhibit Gαq/11-mediated activation of phospholipase Cβ, possibly through the binding to and sequestration of activated Gαq (38). In our study, we also show that GRK2 interacts strongly with the second intracellular loop of GPR54, a loop that has been shown to mediate GPR54 binding to Gαq (39). Taken together, we suggest that the Kp-10-dependent desensitization of GPR54 might occur through a GRK2- dependent disruption of the interaction between Gq and GPR54, leading to the attenuation of Kp-10-stimulated GPR54 signaling in the absence of phosphorylation. A similar mechanism of GRK2 phosphorylation-independent desensitization after agonist stimulation has been suggested for other GPCRs including the mGluR1a (37, 40) and calcium-sensing receptor (41).
Finally, in this study, we only explored the role of GRK2 in the desensitization of GPR54. We chose GRK2 to initiate this study because, among all the GRK isoforms, it is widely expressed throughout the body and along with GRK3, and to a lesser extent GRK5 and 6, is reported as the primary regulator of agonist-dependent receptor desensitization (42, 43, 44, 45). Thus, it would not be unexpected to also find roles for GRK3, -5, and -6 as regulators of GPR54 activity in HEK 293 cells. However, depending on the physiological and cellular context in which GPR54 is being studied, other GRK isoforms, in addition to GRK2, should be included in future studies.
Our data also strongly suggest that GPR54 displays constitutive activity, and this finding was reinforced by the observation that HEK 293 cells do not express KISS1. At the present time we cannot confirm the existence of constitutive activity because an inverse agonist is not yet available to test this. But given the observations that we have made and the fact that constitutive activity has been described for more than 60 wild-type GPCRs (46), it should not be surprising to find that GPR54 does indeed display constitutive activity. Further support of constitutive GPR54 activity comes from in vivo studies performed on the Kiss1−/− and Gpr54−/− animals (47). In this study, the authors document a phenotypic variability observed among Kiss1 knockout female mice and suggest that one likely explanation for it can be as a result of modest constitutive GPR54 activity. In our study, we report that maximum basal activity is approximately 5% of the maximum Kp-10-induced IP formation. If the observations by Lapatto et al. (47) are explained by constitutive activity, it appears that this level of activity is physiologically relevant. Furthermore, because there is evidence of molecular regulation of this basal activity, as seen through the strong basal interaction between GPR54 and GRK2 and β- arrestin as well as the observation that GRK2 regulates basal IP formation, the existence of basal receptor activity appears to be of physiological importance.
We next explored the role of β-arrestin-1 and -2 in regulating the signaling of GPR54. It is well established that β-arrestins mediate the desensitization of GPCRs as well as couple the receptor to other signaling pathways, of which the ERK1/2 MAPK cascade is the best studied (reviewed in Ref. 14). Two studies (48, 49) have identified β-arrestin-binding determinants common to the second intracellular loops of rhodopsin family G protein-coupled receptors. These determinants are the DRY (48) and PLR (49) motifs. A survey of the GPR54 sequence revealed that the DRY motif is represented as a DRW motif whereas the PLR motif is fully conserved in GPR54; thus GPR54 was predicted to bind β-arrestins. As expected, we subsequently found that wild-type GPR54, as well as R331X, interacted strongly with β-arrestin-1 and -2. In addition to the role of intracellular loop 2 in mediating this binding, β-arrestin-2 was also demonstrated to bind the Ct of GPR54.
We also demonstrated that activation of GPR54 leads to the translocation of cytosolic β-arrestins to the plasma membrane, an indication that β-arrestins are involved in regulating GPR54 signaling. Because we provide evidence that after Kp-10 stimulation GPR54 does not undergo additional phosphorylation above basal in HEK 293 cells, it is not clear what triggers the translocation of β-arrestins to the plasma membrane. Even if it is not due to a Kp-10-dependent phosphorylation of GPR54, it does not exclude the possibility that β-arrestin can still be recruited to the Kp-10-stimulated receptor because it has previously been documented that β-arrestin translocates to the agonist-stimulated leukotriene B4 and corticotrophin-releasing type I receptors in a phosphorylation-independent manner (50, 51).
β-Arrestins have well-established roles in mediating GPCR desensitization, internalization, and coupling of some GPCRs to G protein-independent signaling pathways (13). In this study, we explored the role of β- arrestins in mediating the desensitization and internalization of GPR54 in HEK 293 cells but were unable to obtain clear data that β-arrestins had any effect on these processes (data not shown), except for the observation that GPR54 and β-arrestin-2 cointernalized on the same vesicles. Our caution in not placing significance on this data lies in the fact that others have previously observed that overexpression of wild-type and mutant β-arrestin expression constructs do not always confer a detectable phenotype, an indication that β-arrestin is expressed at very high levels in many cell types (52), possibly including HEK 293 cells. It must be noted, however, there are several cases in which β-arrestins do not regulate GPCR desensitization and internalization. For example, β-arrestin-independent desensitization has been noted for the GnRH receptor (53) and the N-formyl peptide receptor (54), and β-arrestin-independent internalization has been described for the α2a -adrenergic receptor (55).
The spatial redistribution of β-arrestin in the cell after receptor activation by agonist would imply that after Kp-10 treatment, β-arrestin interacts with GPR54. Yet our coimmunoprecipitation data show that β-arrestin interacts strongly with GPR54 both in the presence and absence of agonist. We suggest that, based on our accumulating evidence of GPR54 basal activity, a fraction of GPR54 is constitutively associated with and regulated by β-arrestin as well as GRK2. However, upon Kp-10 treatment, as a greater quantity of GPR54 becomes activated (a conclusion based on basal and Kp-10-stimulated IP formation data), a very robust and visually detectable cellular redistribution of β-arrestin occurs.
Through our spatial localization studies, we demonstrated that after Kp-10 treatment, GPR54 and β-arrestin-2 as well as GPR54 and clathrin colocalized at the plasma membrane. This occurred within the same time frame that GPR54 was observed to undergo rapid internalization (∼2–5 min). We next demonstrated that GPR54 and β-arrestin-2 colocalized on internalized vesicles, and, in an independent study, we demonstrated that GPR54 internalized mostly via CCPs. Based on this limited data we suggest that GPR54 internalizes via a β-arrestin-2/clathrin-dependent mechanism. Although we did not directly demonstrate that GPR54 colocalized with β-arrestin-2 in CCPs, the punctate pattern and timing of colocalization between GPR54 and β-arrestin-2 and GPR54 and CCPs strongly suggest that this is indeed the case. Finally, we noted that 10 min after Kp-10 stimulation, GPR54 was observed on a large number of internalized β-arrestin-2 and clathrin-positive vesicles. It therefore appears that GPR54 is a Class B receptor, i.e. a receptor that stably associates and cointernalizes with β-arrestin into the same endosomal compartments where the receptor appears to be retained and recycled very slowly if at all (56, 57).
To continue to explore the significance of this interaction between GPR54 and β-arrestin, we determined what effect a loss of β-arrestin expression had on GPR54 signaling to ERK1/2. To perform this study we created a β-arrestin-2 shRNA knockdown MDA-MB-231 cell line. We chose these knockdown cells because we had previously characterized them extensively after expression of the shRNA sequence (19) and also because MDA-MB-231 expresses GPR54 endogenously as well as the KISS1 mRNA (Ref. 58 , as well as our data). Furthermore, these cells are easy to manipulate in vitro and therefore allowed us to look at the role of β-arrestin expression on endogenous GPR54 signaling. With the β-arrestin-2 shRNA knockdown MDA-MB-231 cell line, we demonstrated that compared with wild-type 231 cells, GPR54 was unable to stimulate ERK1/2 activity, suggesting that β- arrestin-2 is required for GPR54 signaling to ERK1/2. The mechanism by which β-arrestin-2 couples GPR54 to ERK1/2 was not a focus of this study. However, because our data show that GRK2-dependent GPR54 desensitization, GPR54 and β-arrestin colocalization, and GPR54-dependent activation of ERK1/2 all occurred within the same time period, it is possible that β-arrestin-2 is specifically coupling the Gq-desensitized GPR54 to ERK1/2 in a manner previously described for the β2-AR (59).
Our study is the first to describe a major mechanism by which GPR54 undergoes Kp-dependent desensitization and is also the first to describe a β-arrestin-dependent mechanism for GPR54 signaling. With respect to desensitization, we specifically describe the role for GRK2 in mediating the desensitization of GPR54. Because GnRH can be used to activate the HPG axis in hypogonadotropic states and GnRH receptor agonists and antagonists can be employed in the selective, reversible suppression of the axis in certain cancers, gynecological disorders, and precocious puberty, a Kp-dependent attenuation of GPR54 signaling upstream of GnRH/GnRH-R in the HPG axis offers novel therapeutic possibilities for the treatment of reproductive disorders. This is of heightened importance given the lack of pharmacological antagonists of GPR54 and the observation that this peptide can be administered to men without significant toxic effects (60).
In this study, we have presented several scenarios whereby Kp/GPR54 signaling can be perturbed to either increase or decrease signaling of this system. For example, decreasing the level of GPR54 surface expression provides one mechanism whereby receptor signaling can be attenuated, and this may prove to be an effective strategy in treating a subspectrum of GPR54-based disorders. Here we show that the predicted 70-residue long tail of GPR54 encodes sequences that regulate GPR54 surface expression. Sequences in the Ct of GPR54 can therefore be targeted using small cell-permeable molecules that might potentially antagonize their role in potentiating GPR54 surface expression. In another example, we show that both GRK2 and β-arrestin are critical regulators of early GPR54 signaling, and we identify receptor domains that mediate GPR54 interactions with GRK2 and β-arrestin. This information is important because it helps build an understanding of the mechanism underlying desensitization and signaling to multiple pathways and may lead to the development of molecular-based therapies that can stabilize or destabilize these interactions and, consequently, attenuate or augment the signaling potential of the receptor.
A specific example of an endocrine disorder that might one day be treated with the molecular therapies we describe is precocious puberty. Precocious puberty can possibly arise from the untimely activity or abnormally prolonged activity of GPR54, as suggested for the naturally occurring GPR54 mutant R386P (61). R386P appears to be associated with the phenotype of central precocious puberty (61). Knowing how we can rapidly attenuate R386P activity may therefore be a means of treating precocious puberty that results from this and other similar activating mutations.
Materials and Methods
Materials
Restriction enzymes were obtained from New England Biolabs, Inc. (Pickering, Ontario, Canada). Kp-10 was purchased from Phoenix Pharmaceuticals (Burlingame, CA), [3H]myo-inositol and [32P]orthophosphate were purchased from PerkinElmer (Waltham, MA), and Dowex 1-X8 (formate form) 200–400 mesh anion exchange resin was obtained from Bio-Rad Laboratories, Inc. (Hercules, CA). Rabbit and mouse anti-FLAG antibody were purchased from Sigma Aldrich, Inc. (Oakville, Ontario, Canada); mouse anti-myc antibody was obtained from Millipore (Billerica, MA); rabbit anti-GST and rabbit anti-β-arrestin-2 antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); and mouse anti-β-actin, rabbit monoclonal anti-ERK1/2, and antiphospho-ERK1/2 antibodies were from Cell Signaling Technology (Pickering, Ontario, Canada). Alexa Fluor 568-conjugated antirabbit IgG secondary antibody, transferrin-conjugate-488, and Hoechst dye were acquired from Invitrogen (Burlington, Ontario, Canada). The HEK 293 and breast cancer MDA-MB-231 cell lines were obtained from American Type Culture Collection (Manassas, VA). Fetal bovine serum and all other cell culture reagents were purchased from Invitrogen. All other reagents were purchased from BioShop Canada Inc. (Burlington, Ontario, Canada), Fisher Scientific (Ottawa, Ontario, Canada), VWR International (Mississauga, Ontario, Canada), GE Healthcare (Piscataway, NJ), and Corning, Inc. (Lowell, MA).
Plasmid constructs
A 1607-bp cDNA clone encoding human GPR54 was purchased from OriGene Technologies (Rockville, MD) (NM_032551.3) and used as a template to amplify the 1197-bp open reading frame by PCR. The FLAG-epitope, engineered into 5′-primer sequences, was introduced at the amino terminus of GPR54 by PCR. FLAG-GPR54 was then cloned into the NheI and NotI sites of a homemade mammalian expression vector derived from the pEGFP-C vector backbone (Invitrogen). The R331X mutant was made as previously described (2) using the QuikChange Site Directed Mutagenesis kit (Stratagene, La Jolla, CA) and FLAG-GPR54 as a template. The GST-tagged GPR54 intracellular domain constructs were generated by PCR and cloned into the EcoRI-XhoI sites of the E. coli expression vector, pGEX-4T (Amersham Biosciences, GE Healthcare). The PCR-generated sequences contained intracellular loop 1 (GST-IL1, corresponding to amino acid positions 67-78 on GPR54), loop 2 (GST-IL2, corresponding to amino acid positions 139-157 on GPR54), loop 3 (GST-IL3, corresponding to amino acid positions 227-263 on GPR54), and the Ct (GST-Ct, corresponding to amino acid positions 329-398 on GPR54). All constructs were subjected to automated DNA sequencing to confirm sequence integrity.
Cell culture and confocal microscopy
HEK 293 cells were cultured in MEM supplemented with 10% (vol/vol) fetal bovine serum, 1% (vol/vol) nonessential amino acids, and gentamicin (5 μg/ml) and maintained at 37 C in a humidified atmosphere containing 5% CO2. MDA-MB-231 cells were cultured at 37 C with 5% CO2 in RPMI 1640. HEK 293 cells were transiently transfected with 5–10 μg of DNA using a modified calcium phosphate method as previously described (17). HEK 293 cells were serum starved, 42–44 h after transfection, for 30 min in Hanks’ balanced salt solution (HBSS: 1.2 mm KH2PO4, 5 mm NaHCO3, 20 mm HEPES, 11 mm glucose, 116 mm NaCl, 4.7 mm KCl, 1.2 mm MgSO4, 2.5 mm CaCl2, pH 7.4). To stain for cell surface receptor expression, cells were prelabeled with primary anti-FLAG polyclonal antibody (Sigma; 1:500) and kept on ice or treated with or without agonist for different periods of time at 37 C. In some experiments, cells were incubated with transferrin conjugate-488 (Invitrogen) before agonist treatment. Next, they were fixed with 4% formaldehyde and permeabilized with 0.2% Triton X-100. HEK cells were then incubated with goat antirabbit Alexa Fluor 568 (1:1200) antibody and later counterstained with Hoechst (1:50,000) to detect DNA. To stain for cell surface and intracellular receptor expression, the cells were incubated with anti-FLAG primary polyclonal antibody after fixation and permeabilization, and this was then followed with goat antirabbit Alexa Fluor 568 (1:1200) antibody and later counterstained with Hoechst (1:50,000) to detect DNA. Confocal analysis was performed on a Fluoview 1000 laser scanning confocal microscope (Olympus Canada, Markham, Ontario, Canada) using the ×60 Plan Apochromat 1.42 oil objective or the ×100 Plan superapochromat 1.4 oil objective. Colocalization studies were performed using multiple excitation (405, 488, 559) and emission (band pass 425–475, 500–545 nm, and 575–675 nm for Hoechst, AlexaFluor 488, and AlexaFluor 568, respectively) filter sets. Multicolor images were acquired in the sequential acquisition mode to avoid cross-excitation.
β-Arrestin translocation assay
HEK 293 cells transiently transfected with FLAG-GPR54 and β-arrestin-1-GFP or -2-GFP were stimulated with 100 nm Kp-10 and imaged in real time at 37 C or fixed in 4% formaldehyde. Fixed cells were then processed to visualize FLAG-GPR54 using the polyclonal anti-FLAG antibody and Alexa Fluor 568 as described above. Cells displaying agonist-dependent β-arrestin translocation to the cell membrane were identified and recorded.
IP formation
HEK 293 cells were transfected with various DNA constructs as described in the figure legends. Inositol lipids were radiolabeled by incubating cells overnight with 1 μCi/ml [3H]myo-inositol in DMEM. Unincorporated [3H]myo-inositol was removed by washing the cells with HBSS. Cells were preincubated for 1 h in HBSS at 37 C and then preincubated in 500 μl of the same buffer containing 10 mm LiCl for an additional 10 min at 37 C. Next, the cells were incubated in either the absence or the presence of multiple concentrations of Kp-10, 3 × 10−11 nm to 3.0 × 10−5 for 4 h at 37 C. The reaction was stopped on ice by adding 500 μl of 0.8 m perchloric acid and then neutralized with 400 μl of 0.72 m KOH, 0.6 m KHCO3. The total [3H]inositol incorporated into the cells was determined by counting the radioactivity present in 50 μl of the cell lysate. Total IP was purified from the cell extracts by anion exchange chromatography using Dowex 1-X8 (formate form) 200–400 mesh anion exchange resin. [3H]IP formation was determined by liquid scintillation using a Wallac LKB 1211 RackBeta liquid scintillation counter (Perkin Elmer). The means ± se are shown for the number of independent experiments indicated in the figure legends.
Coimmunoprecipitation
Transiently transfected HEK 293 cells were serum starved for 30 min in HBSS at 37 C and then stimulated with 100 nm Kp-10 for 5 min. Cells were then solubilized in lysis buffer (25 mm HEPES, pH 7.5; 300 mm NaCl; 1.5 mm MgCl2; 0.2 mm EDTA; and 0.1% Triton X-100) containing protease inhibitors. FLAG-tagged proteins were immunoprecipitated from 750 μg of total protein with FLAG-agarose beads (Sigma). The proteins were analyzed by SDS-PAGE and Western blotting as described in the figure legends. Immunoblots were visualized by chemiluminescence using an ECL kit (Amersham Biosciences, GE Healthcare).
GST-pull-down assays
GST-GPR54 peptides were produced in recombinant DH5α E. coli bacteria at 37 C to an A600 of 0.6–1.0. Gene expression was induced for 1 h with 1 mm isopropyl 1-thio-β-d-galactopyranoside, and the bacteria were pelleted and resuspended in PBS containing protease inhibitors. The cells were then lysed by sonication followed by incubation with 0.2% Triton X-100 for 30 min. Bacteria lysates were centrifuged at 23,000 × g for 20 min to remove cellular debris and supernatants were applied to glutathione-Sepharose 4B (Sigma) overnight at 4 C. Total bacterial lysate (500 μg) was used in each pull-down assay. Beads were washed extensively the following day with PBS. HEK 293 cell lysates containing overexpressed GRK2 or β-arrestin 2 were added to the beads and incubated overnight at 4 C. Beads were washed extensively the following day and analyzed by SDS-PAGE and Western blotting as described in the figure legends.
Whole-cell phosphorylation
Whole-cell phosphorylation experiments were performed as described previously (18). In brief, HEK 293 cells transfected with either 5 μg of empty FLAG-tagged vector; FLAG-GPR54, FLAG-GPR54 and myc-GRK2, and FLAG-GPR54 and myc-K220R were reseeded in six-well dishes. The intracellular ATP pools were labeled with [32P]orthophosphate (100 μCi/ml) in phosphate-free HEPES-buffered salt solution for 1 h at 37 C. Subsequently, the cells were incubated in either the absence or the presence of 100 nm Kp-10 for 10 min at 37 C. The cells were solubilized in radioimmune precipitation buffer (150 mm NaCl; 50 mm Tris; 5 mm EDTA; 10 mm NaF; 10 mm Na2 pyrophosphate; 1% Nonidet P-40; 0.5% deoxycholate; 0.1% sodium dodecyl sulfate; 0.1 mm phenylmethysulfonyl fluoride; 10 μg/ml leupeptin; 5 μg/ml aprotinin; 1 μg/ml pepstatin A, pH 7.4). FLAG-GPR54 was immunoprecipitated with FLAG-agarose beads (Sigma) and subsequently subjected to SDS-PAGE followed by autoradiography.
GPR54 internalization assay
FLAG-GPR54 internalization was measured by flow cytometry as described previously (17). Cell surface epitope-tagged receptors were prelabeled with primary antimouse FLAG antibody (Sigma; 1:500) on ice for 45 min. Cells were then warmed to 37 C in the absence or presence of agonist for the times indicated in the figure legends. Cells were then transferred back on ice and labeled with secondary fluorescein isothiocyante-conjugated antimouse IgG antibody (Sigma; 1:500) for 45 min. Under these conditions, receptors are able to undergo only a single round of internalization. Receptor internalization is defined as the fraction of total cell receptors lost from the cell surface and thus is not available for labeling with the secondary antibody.
β-Arrestin gene knockdown and ERK activity assay
Knockdown of the β-arrestin-2 gene in MDA-MB-231 cells was conducted using four different shRNAs (OriGene Technologies). However, data presented in this paper were generated using the cell line expressing the β-arrestin-2 shRNA: 5′-CAGAATCTTCCATGCTCCGTCACACTGCA-3′. A control scrambled shRNA sequence, was also used: 5′-TGACCACCCTGACCTACGTCGTGCAGTGC-3′. Sequences were introduced into cells by electroporation (Gene Pulser Xcell, Bio-Rad) according to manufacturer’s instructions (250 V, 950 μF). A heterogeneous population of stable transfectants was selected by using media containing 1 μg/ml puromycin. Cell lines were established and knockdown of β-arrestin-2, relative to scramble shRNA control cell line, was verified and semiquantified by Western blot analysis. These cell lines were used in ERK1/2 activation assays. Assays were conducted by treating overnight serum-starved wild-type or knockdown cells with Kp-10 for the indicated times (see figure legends). Immediately after cells were solubilized in lysis buffer (150 mm NaCl; 50 mm Tris; 5 mm EDTA; 10 mm NaF; 10 mm Na2 pyrophosphate; 1% Nonidet P-40; 0.5% deoxycholate; 0.1% sodium dodecyl sulfate; 0.1 mm phenylmethysulfonyl fluoride; 10 μg/ml leupeptin; 5 μg/ml aprotinin; 1 μg/ml pepstatin A, pH 7.4) and 50 μg of total protein were subsequently analyzed by SDS-PAGE and Western blotting as described in the figure legends. Immunoblots were visualized by chemiluminescence using an ECL kit (Amersham).
Data analysis
The means ± se are shown for values obtained for the number of independent experiments indicated in the figure legends. GraphPad Prism software (Graph Pad, San Diego, CA) was used to analyze data for statistical significance, as well as to analyze and fit dose-response and time course data. The statistical significance was determined by analysis of variance or t test.
Acknowledgments
We thank Ms. Cindy Pape for technical assistance.
Footnotes
Research reported in this study was supported by a grant from the Canadian Institutes of Health Research (CIHR) MOP 81383. The following are recipients of a salary awards: Dr. Andy Babwah: CIHR/CIHR’s Institute of Gender and Health/ Ontario Women’s Health Council New Investigator Award; Dr. Moshmi Bhattacharya: National Sciences and Engineering Research Council of Canada University Faculty Award; Natasha Camuso: CIHR Graduate Student Award; Jay Taylor: Department of Obstetrics and Gynaecology, University of Western Ontario, Studentship; Jacob Szereszewski: Department of Obstetrics and Gynaecology, University of Western Ontario, Studentship and CIHR Graduate Student Award; and Mateusz Zajac: Ontario Graduate Scholarship, Ontario Government.
Disclosure Summary: The authors have nothing to disclose.
First Published Online October 21, 2009
M.P. and N.C. contributed equally to this study.
Abbreviations: AR, Adrenergic receptor; CCP, clathrin-coated pit; GFP, green fluorescent protein; GPCR, G protein-coupled receptor; GRKs, GPCR serine/threonine kinases; GST, glutathione-S-transferase; HBSS, Hanks’ balanced salt solution; HEK, human embryonic kidney; HPG, hypothalamic-pituitary-gonadal; Kp, kisspeptin; shRNA, short hairpin RNA.
References
- 1.de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E2003. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA 100:10972–10976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno Jr JS, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, Zahn D, Dixon J, Kaiser UB, Slaugenhaupt SA, Gusella JF, O'Rahilly S, Carlton MB, Crowley Jr WF, Aparicio SA, Colledge WH2003. The GPR54 gene as a regulator of puberty. N Engl J Med 349:1614–1627 [DOI] [PubMed] [Google Scholar]
- 3.Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, Brézillon S, Tyldesley R, Suarez-Huerta N, Vandeput F, Blanpain C, Schiffmann SN, Vassart G, Parmentier M2001. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem 276:34631–34636 [DOI] [PubMed] [Google Scholar]
- 4.Popa SM, Clifton DK, Steiner RA2008. The role of kisspeptins and GPR54 in the neuroendocrine regulation of reproduction. Annu Rev Physiol 70:213–238 [DOI] [PubMed] [Google Scholar]
- 5.Messager S, Chatzidaki EE, Ma D, Hendrick AG, Zahn D, Dixon J, Thresher RR, Malinge I, Lomet D, Carlton MB, Colledge WH, Caraty A, Aparicio SA2005. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc Natl Acad Sci USA 102:1761–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thompson EL, Murphy KG, Patterson M, Bewick GA, Stamp GW, Curtis AE, Cooke JH, Jethwa PH, Todd JF, Ghatei MA, Bloom SR2006. Chronic subcutaneous administration of kisspeptin-54 causes testicular degeneration in adult male rats. Am J Physiol Endocrinol Metab 291:E1074–E1082 [DOI] [PubMed]
- 7.Seminara SB, Dipietro MJ, Ramaswamy S, Crowley Jr WF, Plant TM2006. Continuous human metastin 45–54 infusion desensitizes G protein-coupled receptor 54-induced gonadotropin-releasing hormone release monitored indirectly in the juvenile male Rhesus monkey (Macaca mulatta): a finding with therapeutic implications. Endocrinology 147:2122–2126 [DOI] [PubMed] [Google Scholar]
- 8.Ramaswamy S, Seminara SB, Pohl CR, DiPietro MJ, Crowley Jr WF, Plant TM2007. Effect of continuous intravenous administration of human metastin 45–54 on the neuroendocrine activity of the hypothalamic-pituitary-testicular axis in the adult male rhesus monkey (Macaca mulatta). Endocrinology 148:3364–3370 [DOI] [PubMed] [Google Scholar]
- 9.Roa J, Vigo E, Garcia-Galiano D, Castellano JM, Navarro VM, Pineda R, Diéguez C, Aguilar E, Pinilla L, Tena-Sempere M2008. Desensitization of gonadotropin responses to kisspeptin in the female rat: analyses of LH and FSH secretion at different developmental and metabolic states. Am J Physiol Endocrinol Metab 294:E1088–E1096 [DOI] [PubMed]
- 10.Freedman NJ, Lefkowitz RJ1996. Desensitization of G protein-coupled receptors. Recent Prog Horm Res 51:319–351; discussion 352–353 [PubMed] [Google Scholar]
- 11.Krupnick JG, Benovic JL1998. The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu Rev Pharmacol Toxicol 38:289–319 [DOI] [PubMed] [Google Scholar]
- 12.Pitcher JA, Freedman NJ, Lefkowitz RJ1998. G protein-coupled receptor kinases. Annu Rev Biochem 67:653–692 [DOI] [PubMed] [Google Scholar]
- 13.Luttrell LM, Lefkowitz RJ2002. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci 115:455–465 [DOI] [PubMed] [Google Scholar]
- 14.Barki-Harrington L, Rockman HA2008. β-arrestins: multifunctional cellular mediators. Physiologist 23:17–22 [DOI] [PubMed] [Google Scholar]
- 15.Subtil A, Hémar A, Dautry-Varsat A1994. Rapid endocytosis of interleukin 2 receptors when clathrin-coated pit endocytosis is inhibited. J Cell Sci 107:3461–3468 [DOI] [PubMed] [Google Scholar]
- 16.Paterson AD, Parton RG, Ferguson C, Stow JL, Yap AS2003. Characterization of E-cadherin endocytosis in isolated MCF-7 and chinese hamster ovary cells: the initial fate of unbound E-cadherin. J Biol Chem 278:21050–21057 [DOI] [PubMed] [Google Scholar]
- 17.Bhattacharya M, Babwah AV, Godin C, Anborgh PH, Dale LB, Poulter MO, Ferguson SS2004. Ral and phospholipase D2-dependent pathway for constitutive metabotropic glutamate receptor endocytosis. J Neurosci 24:8752–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhami GK, Anborgh PH, Dale LB, Sterne-Marr R, Ferguson SS2002. Phosphorylation-independent regulation of metabotropic glutamate receptor signaling by G protein-coupled receptor kinase 2. J Biol Chem 277:25266–25272 [DOI] [PubMed] [Google Scholar]
- 19.Li TT, Alemayehu M, Aziziyeh AI, Pape C, Pampillo M, Postovit LM, Mills GB, Babwah AV, Bhattacharya M2009. β-Arrestin/Ral signaling regulates lysophosphatidic acid-mediated migration and invasion of human breast tumor cells. Mol Cancer Res 7:1064–1077 [DOI] [PubMed] [Google Scholar]
- 20.Reiter E, Lefkowitz RJ2006. GRKs and β-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab 17:159–165 [DOI] [PubMed] [Google Scholar]
- 21.Teli T, Markovic D, Levine MA, Hillhouse EW, Grammatopoulos DK2005. Regulation of corticotropin-releasing hormone receptor type 1α signaling: structural determinants for G protein-coupled receptor kinase-mediated phosphorylation and agonist-mediated desensitization. Mol Endocrinol 19:474–490 [DOI] [PubMed] [Google Scholar]
- 22.Ménard L, Ferguson SS, Zhang J, Lin FT, Lefkowitz RJ, Caron MG, Barak LS1997. Synergistic regulation of β2-adrenergic receptor sequestration: intracellular complement of β-adrenergic receptor kinase and β-arrestin determine kinetics of internalization. Mol Pharmacol 51:800–808 [PubMed] [Google Scholar]
- 23.Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI1990. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5:1–10 [DOI] [PubMed] [Google Scholar]
- 24.Mayer CM, Fick LJ, Gingerich S, Belsham DD2009. Hypothalamic cell lines to investigate neuroendocrine control mechanisms. Front Neuroendocrinol 30:405–423 [DOI] [PubMed] [Google Scholar]
- 25.Graham CH, Hawley TS, Hawley RG, MacDougall JR, Kerbel RS, Khoo N, Lala PK1993. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp Cell Res 206:204–211 [DOI] [PubMed] [Google Scholar]
- 26.Cavanagh PC, Dunk C, Pampillo M, Szereszewski JM, Taylor JE, Kahiri C, Han V, Lye S, Bhattacharya M, Babwah AV2009. Gonadotropin-releasing hormone-regulated chemokine expression in human placentation. Am J Physiol Cell Physiol 297:C17–C27 [DOI] [PubMed]
- 27.Holliday ND, Holst B, Rodionova EA, Schwartz TW, Cox HM2007. Importance of constitutive activity and arrestin-independent mechanisms for intracellular trafficking of the ghrelin receptor. Mol Endocrinol 21:3100–3112 [DOI] [PubMed] [Google Scholar]
- 28.Janovick JA, Ulloa-Aguirre A, Conn PM2003. Evolved regulation of gonadotropin-releasing hormone receptor cell surface expression. Endocrine 22:317–327 [DOI] [PubMed] [Google Scholar]
- 29.Castro-Fernández C, Janovick JA, Brothers SP, Fisher RA, Ji TH, Conn PM2002. Regulation of RGS3 and RGS10 palmitoylation by GnRH. Endocrinology 143:1310–1317 [DOI] [PubMed] [Google Scholar]
- 30.Sallese M, Salvatore L, D'Urbano E, Sala G, Storto M, Launey T, Nicoletti F, Knöpfel T, De Blasi A2000. The G-protein-coupled receptor kinase GRK4 mediates homologous desensitization of metabotropic glutamate receptor 1. FASEB J 14:2569–2580 [DOI] [PubMed] [Google Scholar]
- 31.Dale LB, Bhattacharya M, Seachrist JL, Anborgh PH, Ferguson SS2001. Agonist-stimulated and tonic internalization of metabotropic glutamate receptor 1a in human embryonic kidney 293 cells: agonist-stimulated endocytosis is β-arrestin1 isoform-specific. Mol Pharmacol 60:1243–1253 [DOI] [PubMed] [Google Scholar]
- 32.Fourgeaud L, Bessis AS, Rossignol F, Pin JP, Olivo-Marin JC, Hémar A2003. The metabotropic glutamate receptor mGluR5 is endocytosed by a clathrin-independent pathway. J Biol Chem 278:12222–12230 [DOI] [PubMed] [Google Scholar]
- 33.Pula G, Mundell SJ, Roberts PJ, Kelly E2004. Agonist-independent internalization of metabotropic glutamate receptor 1a is arrestin- and clathrin-dependent and is suppressed by receptor inverse agonists. J Neurochem 89:1009–1020 [DOI] [PubMed] [Google Scholar]
- 34.Kern A, Agoulnik AI, Bryant-Greenwood GD2007. The low-density lipoprotein class A module of the relaxin receptor (leucine-rich repeat containing G-protein coupled receptor 7): its role in signaling and trafficking to the cell membrane. Endocrinology 148:1181–1194 [DOI] [PubMed] [Google Scholar]
- 35.Kern A, Bryant-Greenwood GD2009. Characterization of relaxin receptor (RXFP1) desensitization and internalization in primary human decidual cells and RXFP1-transfected HEK293 cells. Endocrinology 150:2419–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pao CS, Benovic JL2005. Structure/function analysis of alpha2A-adrenergic receptor interaction with G protein-coupled receptor kinase 2. J Biol Chem 280:11052–11058 [DOI] [PubMed] [Google Scholar]
- 37.Dhami GK, Babwah AV, Sterne-Marr R, Ferguson SS2005. Phosphorylation-independent regulation of metabotropic glutamate receptor 1 signaling requires G protein-coupled receptor kinase 2 binding to the second intracellular loop. J Biol Chem 280:24420–24427 [DOI] [PubMed] [Google Scholar]
- 38.Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL, Kozasa T1999. Selective regulation of Galpha(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J Biol Chem 274:34483–34492 [DOI] [PubMed] [Google Scholar]
- 39.Wacker JL, Feller DB, Tang XB, Defino MC, Namkung Y, Lyssand JS, Mhyre AJ, Tan X, Jensen JB, Hague C2008. Disease-causing mutation in GPR54 reveals the importance of the second intracellular loop for class A G-protein-coupled receptor function. J Biol Chem 283:31068–31078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dhami GK, Dale LB, Anborgh PH, O'Connor-Halligan KE, Sterne-Marr R, Ferguson SS2004. G Protein-coupled receptor kinase 2 regulator of G protein signaling homology domain binds to both metabotropic glutamate receptor 1a and Gαq to attenuate signaling. J Biol Chem 279:16614–16620 [DOI] [PubMed] [Google Scholar]
- 41.Lorenz S, Frenzel R, Paschke R, Breitwieser GE, Miedlich SU2007. Functional desensitization of the extracellular calcium-sensing receptor is regulated via distinct mechanisms: role of G protein-coupled receptor kinases, protein kinase C and β-arrestins. Endocrinology 148:2398–2404 [DOI] [PubMed] [Google Scholar]
- 42.Tiberi M, Nash SR, Bertrand L, Lefkowitz RJ, Caron MG1996. Differential regulation of dopamine D1A receptor responsiveness by various G protein-coupled receptor kinases. J Biol Chem 271:3771–3778 [DOI] [PubMed] [Google Scholar]
- 43.Gainetdinov RR, Bohn LM, Walker JK, Laporte SA, Macrae AD, Caron MG, Lefkowitz RJ, Premont RT1999. Muscarinic supersensitivity and impaired receptor desensitization in G protein-coupled receptor kinase 5-deficient mice. Neuron 24:1029–1036 [DOI] [PubMed] [Google Scholar]
- 44.Aiyar N, Disa J, Dang K, Pronin AN, Benovic JL, Nambi P2000. Involvement of G protein-coupled receptor kinase-6 in desensitization of CGRP receptors. Eur J Pharmacol 403:1–7 [DOI] [PubMed] [Google Scholar]
- 45.Willets JM, Challiss RA, Nahorski SR2002. Endogenous G protein-coupled receptor kinase 6 Regulates M3 muscarinic acetylcholine receptor phosphorylation and desensitization in human SH-SY5Y neuroblastoma cells. J Biol Chem 277:15523–15529 [DOI] [PubMed] [Google Scholar]
- 46.Smit MJ, Vischer HF, Bakker RA, Jongejan A, Timmerman H, Pardo L, Leurs R2007. Pharmacogenomic and structural analysis of constitutive g protein-coupled receptor activity. Annu Rev Pharmacol Toxicol 47:53–87 [DOI] [PubMed] [Google Scholar]
- 47.Lapatto R, Pallais JC, Zhang D, Chan YM, Mahan A, Cerrato F, Le WW, Hoffman GE, Seminara SB2007. Kiss1−/− mice exhibit more variable hypogonadism than Gpr54−/− mice. Endocrinology 148:4927–4936 [DOI] [PubMed] [Google Scholar]
- 48.Barak LS, Wilbanks AM, Caron MG2003. Constitutive desensitization: a new paradigm for g protein-coupled receptor regulation. Assay Drug Dev Technol 1:339–346 [DOI] [PubMed] [Google Scholar]
- 49.Marion S, Oakley RH, Kim KM, Caron MG, Barak LS2006. A β-arrestin binding determinant common to the second intracellular loops of rhodopsin family G protein-coupled receptors. J Biol Chem 281:2932–2938 [DOI] [PubMed] [Google Scholar]
- 50.Jala VR, Shao WH, Haribabu B2005. Phosphorylation-independent β-arrestin translocation and internalization of leukotriene B4 receptors. J Biol Chem 280:4880–4887 [DOI] [PubMed] [Google Scholar]
- 51.Oakley RH, Olivares-Reyes JA, Hudson CC, Flores-Vega F, Dautzenberg FM, Hauger RL2007. Carboxyl-terminal and intracellular loop sites for CRF1 receptor phosphorylation and β-arrestin-2 recruitment: a mechanism regulating stress and anxiety responses. Am J Physiol Regul Integr Comp Physiol 293:R209–R322 [DOI] [PMC free article] [PubMed]
- 52.Kohout TA, Lin FS, Perry SJ, Conner DA, Lefkowitz RJ2001. β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Natl Acad Sci USA 98:1601–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heding A, Vrecl M, Hanyaloglu AC, Sellar R, Taylor PL, Eidne KA2000. The rat gonadotropin-releasing hormone receptor internalizes via a β-arrestin-independent, but dynamin-dependent, pathway: addition of a carboxyl-terminal tail confers β-arrestin dependency. Endocrinology 141:299–306 [DOI] [PubMed] [Google Scholar]
- 54.Potter RM, Maestas DC, Cimino DF, Prossnitz ER2006. Regulation of N-formyl peptide receptor signaling and trafficking by individual carboxyl-terminal serine and threonine residues. J Immunol 176:5418–5425 [DOI] [PubMed] [Google Scholar]
- 55.DeGraff JL, Gurevich VV, Benovic JL2002. The third intracellular loop of α 2-adrenergic receptors determines subtype specificity of arrestin interaction. J Biol Chem 277:43247–43252 [DOI] [PubMed] [Google Scholar]
- 56.Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG1999. Association of β-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem 274:32248–32257 [DOI] [PubMed] [Google Scholar]
- 57.Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS2000. Differential affinities of visual arrestin, β arrestin1, and β arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem 275:17201–17210 [DOI] [PubMed] [Google Scholar]
- 58.Prentice LM, Miller MA, Turbin DA, Aparicio SAJR, Huntsman DG, Expression of GPR54 and KiSS1 in human breast cancer. Proc 97th Annual Meeting of the American Association for Cancer Research, Washington, DC, 2006. (Abstract 1013-c-1014)
- 59.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ1999. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science 283:655–661 [DOI] [PubMed] [Google Scholar]
- 60.Dhillo WS, Chaudhri OB, Patterson M, Thompson EL, Murphy KG, Badman MK, McGowan BM, Amber V, Patel S, Ghatei MA, Bloom SR2005. Kisspeptin-54 stimulates the hypothalamic-pituitary gonadal axis in human males. J Clin Endocrinol Metab 90:6609–6615 [DOI] [PubMed] [Google Scholar]
- 61.Teles MG, Bianco SD, Brito VN, Trarbach EB, Kuohung W, Xu S, Seminara SB, Mendonca BB, Kaiser UB, Latronico AC2008. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med 358:709–715 [DOI] [PMC free article] [PubMed] [Google Scholar]