

Abstract
The ATP-sensitive potassium (KATP) channel is a key molecule involved in glucose-stimulated insulin secretion. The activity of this channel regulates β-cell membrane potential, glucose- induced [Ca2+]i signals, and insulin release. In this study, the rapid effect of physiological concentrations of 17β-estradiol (E2) on KATP channel activity was studied in intact β-cells by use of the patch-clamp technique. When cells from wild-type (WT) mice were used, 1 nm E2 rapidly reduced KATP channel activity by 60%. The action of E2 on KATP channel was not modified in β-cells from ERα−/− mice, yet it was significantly reduced in cells from ERβ−/− mice. The effect of E2 was mimicked by the ERβ agonist 2,3-bis(4-hydroxyphenyl)-propionitrile (DPN). Activation of ERβ by DPN enhanced glucose-induced Ca2+ signals and insulin release. Previous evidence indicated that the acute inhibitory effects of E2 on KATP channel activity involve cyclic GMP and cyclic GMP-dependent protein kinase. In this study, we used β-cells from mice with genetic ablation of the membrane guanylate cyclase A receptor for atrial natriuretic peptide (also called the atrial natriuretic peptide receptor) (GC-A KO mice) to demonstrate the involvement of this membrane receptor in the rapid E2 actions triggered in β-cells. E2 rapidly inhibited KATP channel activity and enhanced insulin release in islets from WT mice but not in islets from GC-A KO mice. In addition, DPN reduced KATP channel activity in β-cells from WT mice, but not in β-cells from GC-A KO mice. This work unveils a new role for ERβ as an insulinotropic molecule that may have important physiological and pharmacological implications.
ERβ is involved in the rapid regulation of KATP channel activity by E2. Rapid activation of ERβ potentiates glucose-induced Ca2+ signals and insulin secretion. These actions require the presence of the Atrial Natriuretic Peptide receptor.
Blood glucose homeostasis is a complex process that involves intertissue communication between skeletal muscle, white adipose tissue, the liver and the endocrine pancreas (1). The islet of Langerhans is the physiological unit of the endocrine pancreas and it is composed of four different types of cells, the most abundant being insulin-containing β-cells and glucagon-containing α-cells.
The main function of the pancreatic β-cell is the biosynthesis and release of insulin, the only hormone able to decrease blood glucose levels. Insulin promotes glucose uptake by skeletal muscle, white adipose tissue, and the liver, a process that is regulated by estrogen receptors, ERα and ERβ (2, 3, 4, 5, 6).
In mouse β-cells, both estrogen receptors ERα and ERβ are expressed, although their roles are still largely unknown (7, 8). It was recently reported that long-term stimulation of ERα by 17β-estradiol (E2), in synergy with stimulatory glucose concentrations, increases pancreatic insulin biosynthesis and potentiates glucose-induced insulin release without altering pancreatic β-cell mass (7). In addition, ERα mediates the E2-induced increment of β-cell survival after oxidative stress and protects from streptozotocin-induced diabetes (9). However, no role for ERβ in β-cell function has been described so far.
The secretory response of β-cells depends on their electrical activity. This consists of oscillations of the membrane potential that range from electrically silent periods to depolarized plateaus from which Ca2+ action potentials originate (10). A primary event in the classical stimulus-secretion coupling that drives insulin release is the closure of ATP-sensitive potassium (KATP) channels by the increase of ATP/ADP ratio due to glucose metabolism (11). The KATP channel closure induces membrane depolarization, and then, an oscillatory membrane potential is established that elicits [Ca2+]i oscillations (12, 13). Insulin secretion mirrors Ca2+ signals being produced in a pulsatile manner (14, 15, 16).
It has been demonstrated in β-cells that physiological concentrations of E2 rapidly increase cyclic GMP (cGMP) levels (17) and decrease KATP channel activity in a cGMP/cGMP-dependent protein kinase (PKG)-dependent manner (17, 18). Thus, the closure of KATP channels contributes to the rapid E2 potentiation of glucose-induced Ca2+ signals and insulin secretion observed in vitro and in vivo (19, 20, 21, 22). In the present work, we have used pharmacological tools and genetically modified mice to demonstrate that the estrogen receptor ERβ and the cGMP-generating atrial natriuretic peptide receptor (guanylyl cyclase A, GC-A) are involved in the rapid regulation of KATP channel activity by E2. This may represent a new molecular pathway activated by ERβ.
Results
E2 action on KATP channel activity is abolished in β-cells from ERβ−/− mice
ERα and ERβ are expressed in β-cells and located extracellularly (7, 8). Location of both receptors is mainly cytoplasmic, and immunocytochemical studies indicate that they are not mainly located in mitochondria (7). To study the role of ERα and ERβ in E2-induced rapid regulation of KATP channels at the molecular level, cell-attached patch recordings were obtained in β-cells from wild-type (WT), ERα−/− and ERβ−/− mice, in the absence of extracellular glucose. When E2 was applied, a substantial decrease to 46 ± 10% of the initial KATP channel activity was observed within 7 min of E2 application in β-cells from WT animals (Fig. 1, A and B). The reduction in KATP channel activity was not as large as that achieved by 8 mm glucose (activity = 14 ± 9%) (Fig. 1, A and B) and maximal activity was obtained in the presence of the KATP opener diazoxide (185 ± 50%) (Fig. 1A). In cells from ERα−/− mice, response to E2 was similar to that seen in WT cells (activity = 45 ± 8%) (Fig. 1, E and F). However, in β-cells from ERβ−/− mice E2 triggered a very small reduction in KATP channel activity (activity = 81 ± 26%) (Fig. 1, C and D). The effect of 8 mm glucose and diazoxide was similar in all three genotypes. Glucose decreased KATP activity to 11 ± 7% in ERβ−/− cells and 14 ± 6 in ERα−/− cells (Fig. 1, D and F). Diazoxide increased KATP activity to 216 ± 40% of baseline in ERβ−/− cells and to 184 ± 21% in ERα−/− cells. Bar diagrams do not include diazoxide data for clarity.
Fig. 1.

E2 regulation of KATP channel activity is abolished in β-cells from ERβ−/− mice. A, E2 at 1 nm decreased KATP channel activity in intact pancreatic islet cells from WT mice. The records show the KATP channel activity before application of E2, 7 min after application of E2, 8 min after application of 8 mm glucose, and 3 min after application of 100 μm diazoxide. B, Percentage of activity of the KATP channels elicited by vehicle (n =7), 1 nm E2 (n =3), and 8 mm glucose (n =4). C, 1 nm E2 had no significant effect on KATP channel activity in intact pancreatic β-cells from ERβ−/− mice. As in A, the records show KATP channel activity before application of E2, 7 min after application of E2, 8 min after application of 8 mm glucose, and 3 min after application of 100 μm diazoxide. D, Percentage of activity of the KATP channels elicited by vehicle (control) (n =4), 1 nm E2 (n =4), and 8 mm glucose (n =3). **, P < 0.01 Student’s t test comparing 8 mm glucose with control. E, The same experiment as in A but using β-cells from ERα−/− mice. F, Percentage calculated as in B and D. Note that 1 nm E2 decreases KATP channel activity to the same extent as in WT mice (n =4). **, P < 0.01 Student’s t test comparing E2 with control and 8 mm glucose with control.
Effects of ERα and ERβ agonists on stimulus-secretion coupling
The results described above indicate that ERβ, but not ERα, is involved in E2-triggered KATP channel closure. To demonstrate that activation of ERβ initiates the regulation of KATP channels, we analyzed the effect of the ERβ-specific agonist 2,3-bis(4-hydroxyphenyl)-propionitrile (DPN) (23, 24) and compared it with that of the specific agonist of ERα, propylpyrazole-triol (PPT) (23, 24).
The results described in Fig. 2 show that activation of ERβ by DPN produced a reduction of KATP channel activity. Figure 2A shows an illustrative record of the reduction of KATP channel activity by 1 nm DPN. The maximal inhibition by DPN was similar to that elicited by E2 (40 ± 20%; see Fig. 1). Note that the ERα agonist PPT had no significant effect (Fig. 2B). To further verify that the effect of DPN was specifically mediated through ERβ, we demonstrated the lack of effect in cells from ERβ−/− mice (supplemental Material 1).
Fig. 2.

The ERβ agonist DPN mimics E2 action on KATP channel activity. A, 1 nm DPN decreased KATP channel activity in intact pancreatic islet cells from WT mice. The records show KATP channel activity before DPN application and 5 min after application. B, Percentage of KATP channel activity in the presence of vehicle, before application of stimuli (n =12), elicited by 1 nm DPN (n =3) and the ERα agonist PPT in a similar experiment (n =9). *, P < 0.05 Student’s t test compared with control.
It is known that E2 rapidly enhances [Ca2+]i signals and insulin secretion, processes that depend on electrical activity (18). To investigate whether activation of ERβ by DPN potentiated glucose-induced [Ca2+]i signals and insulin secretion, we performed the experiments described in Fig. 3. Once a steady calcium signal was obtained in the presence of 7 mm glucose, application of 1 nm DPN induced oscillatory increases of [Ca2+]i in islets that did not show glucose-induced [Ca2+]i oscillations in 7 mm glucose (Fig. 3A). In addition, DPN enhanced insulin release stimulated by 7 mm glucose, whereas the ERα agonist PPT had no effect under these conditions (Fig. 3B). To study whether there was a cooperative effect of ERβ activation and glucose, the experiment in Fig. 3C was performed. This experiment showed that 1 nm DPN enhanced glucose-induced insulin release in the presence of 7 and 16 mm glucose, but the DPN effect was lower when applied in the presence of a low glucose concentration, 3 mm.
Fig. 3.
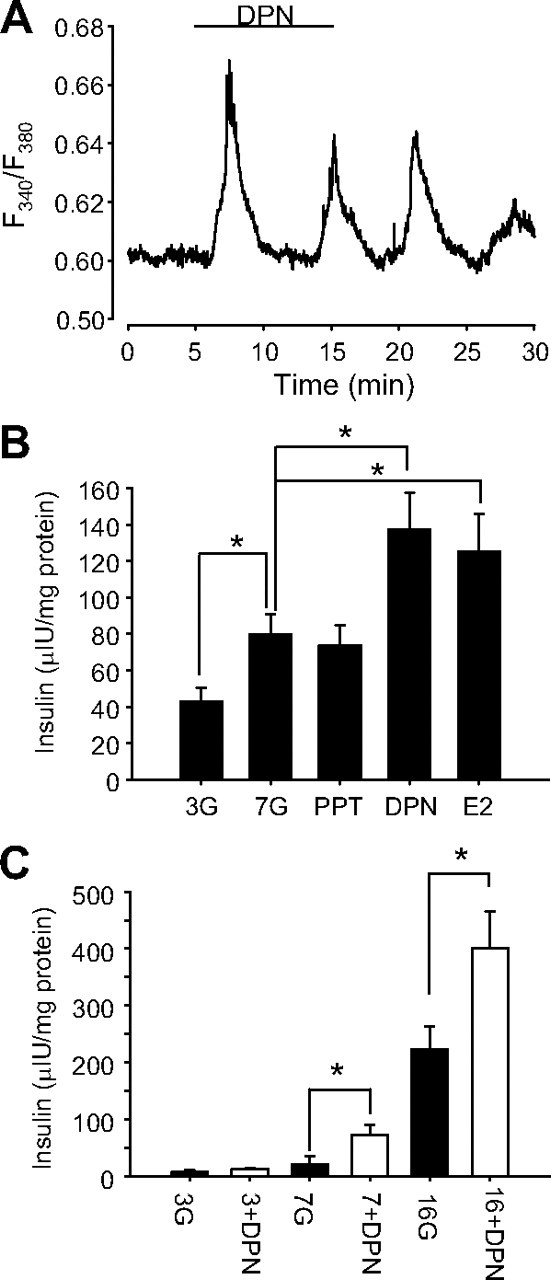
Activation of ERβ by DPN enhanced glucose-induced [Ca2+]i signals and insulin secretion. A, [Ca2+]i response of a typical islet of Langerhans in the presence of 7 mm glucose to 1 nm DPN applied for the period indicated by the bar. The Ca2+-dependent fluorescence of fura 2 is expressed as the ratio F340/F380 (see Materials and Methods). Note that [Ca2+]i oscillations are generated on a nonoscillatory plateau produced by 7 mm glucose. Representative of 11 islets. B, Glucose-induced insulin secretion from islets exposed to 3 mm glucose (3G), 7 mm glucose (7G), 7 mm glucose and 1 nm PPT (PPT), 7 mm glucose and 1 nm DPN (DPN), 7 mm glucose and 1 nm E2 (E2), or 1 nm E2 for 1 h (n = 10 mice). C, DPN-induced insulin secretion from islets exposed to 3 mm, 7 mm, and 16 mm glucose for 1 h (n = 6 mice). DPN was applied at 1 nm. Note that DPN action was significant only when stimulatory glucose concentrations were used. *, P < 0.05 Student’s t test. All stimuli have the same amount of vehicle 10−5% DMSO.
These experiments strongly indicate that activation of ERβ triggers the closure of KATP channels, and it consequently enhances glucose-induced [Ca2+]i oscillations and insulin release in a cooperative manner with glucose.
The GC-A receptor (atrial natriuretic peptide receptor) is involved in E2 blockade of KATP channel activity
It is known that E2 rapidly increases cGMP levels and decreases KATP channel activity in mouse pancreatic β-cells in a cGMP mediated, PKG-dependent manner (17, 18). Our previous results indicated that membrane guanylyl cyclase receptors might be involved in these effects, because the soluble guanylate cyclase (sGC) inhibitor LY83583 had no effect on E2-induced KATP blockade (17). To further investigate a possible role of GC-A, the membrane receptor for atrial natriuretic peptide (ANP), cell-attached patch recordings were obtained as described above. The sGC inhibitor ODQ had no substantial effect on basal KATP activity (83 ± 3%) or on the inhibitory action of E2 (activity = 35 ± 7%) (Fig. 4A). When results were pooled, the maximal inhibition of KATP activity obtained by E2 in the presence of ODQ was similar to that described previously for E2 (Figs. 1 and 4B). These results indicated that sGC was not involved in the inhibitory effect of E2 on KATP activity.
Fig. 4.

The sGC blocker ODQ did not affect E2 action on KATP channel activity. A, 10 μm ODQ modified basal KATP channel activity slightly, yet 1 nm E2 elicited a typical reduction of KATP channel activity in the presence of 10 μm ODQ. B, Percentage of activity in the presence of vehicle (control), 10 μm ODQ, and 10 μm ODQ and 1 nm E2. n =3, *, P < 0.05, Student’s t test.
Besides sGC, membrane GC-A is also expressed in β-cells and might represent a source of cGMP. Hence, pancreatic β-cells respond to ANP with intracellular increases of cGMP, indicating that they express the GC-A (25, 26). Immunocytochemistry performed in whole islets confirmed the expression of GC-A in our preparation (Fig. 5). To study the involvement of GC-A in the E2 regulation of KATP channel activity, patch-clamp recordings in the cell-attached configuration were performed in β-cells from WT and GC-A-deficient mice (GC-A KO). E2 decreased KATP channel activity in β-cells obtained from WT mice (Fig. 6, A and B), but it had no effect on β-cells from GC-A KO mice (Fig. 6, C and D). Note that the basal activity of KATP channels in β-cells from GC-A KO mice was smaller than that of β-cells from WT mice. This is probably due to a decreased expression of KATP channels in GC-A KO β-cells (our manuscript in preparation). Channels, however, were fully functional in GC-A KO mice as demonstrated by an almost complete blockade of KATP channels produced by glucose and the sensitivity of these channels to diazoxide (activity = 120.7 ± 33% of basal) (Fig. 6, C and D).
Fig. 5.
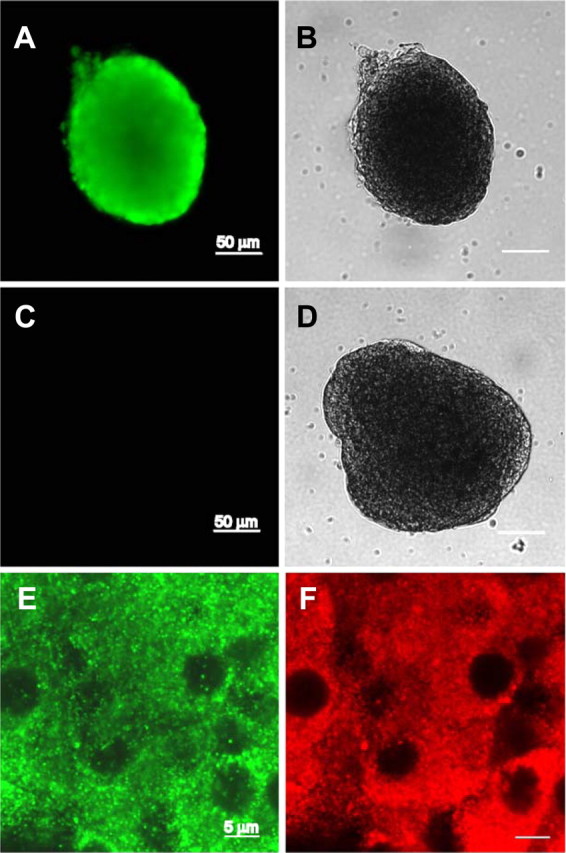
GC-A receptor is expressed in islet of Langerhans. A, Immunostaining of an islet with the antibody against GC-A. B, Transmission image of the islet in A. C, Fluorescence image of an islet immunostained in the absence of the primary antibody anti-GC-A (negative control). D, Transmission image of the islet in C. E and F, Fluorescence images of an islet labeled with the anti-GC-A and the anti-insulin antibodies, respectively.
Fig. 6.

E2-induced reduction of KATP channel activity is abolished in β-cells from GC-A KO mice. A, E2 1 nm decreased KATP channel activity in intact pancreatic islet cells from WT mice. The records show KATP channel activity before application of E2, 7 min after application of E2, 8 min after application of 8 mm glucose, and 3 min after application of 100 μm diazoxide. B, Percentage of activity of the KATP channels elicited by vehicle (control) (n =13), 1 nm E2 (n =5), and 8 mm glucose (n =4). C, 1 nm E2 had no significant effect on KATP channel activity in intact pancreatic islet cells from GC-A KO mice. As in A, the records show KATP channel activity before application of E2, 7 min after application of E2, 8 min after application of 8 mm glucose, and 3 min after application of 100 μm diazoxide. D, Percentage of activity of the KATP channels elicited by vehicle (n =8), 1 nm E2 (n =8), and 8 mm glucose (n =3). **, P < 0.01 Student’s t test comparing 8 mm glucose with control.
Role of GC-A in rapid E2-induced insulin secretion
Because closure of KATP channels is a key step in glucose-stimulated insulin secretion, we investigated whether the rapid stimulatory action of E2 on insulin release involved GC-A. Experiments described in Fig. 7A show that E2 in 1 h increased insulin release in the presence of 7 mm glucose in islets from WT mice. In contrast, this action of E2 was completely abolished in islets from GC-A-deficient islets (Fig. 7B). It is noteworthy that GC-A KO mice respond vigorously to stimulatory glucose concentrations (Fig. 7B). Results presented so far are consistent with a role for GC-A in the rapid actions of E2 on KATP channel activity and insulin release.
Fig. 7.

Rapid E2 action of glucose-induced insulin secretion is abolished in islets from GC-A KO mice. A, Glucose-induced insulin secretion from islets isolated from WT mice exposed to 7 mm glucose (Vehicle), 7 mm glucose and 10 nm E2 (E2), and 16 mm glucose (16 Gluscose) for 1 h (n = 6 mice). B, The same experiment as in A, but using islets from GC-A KO mice. Note that E2 action was completely abolished (n =4 mice). *, P < 0.05 Student’s t test. All stimuli have the same amount of vehicle 10−5% dimethyl sulfoxide.
DPN action on KATP channel activity was abolished in β-cells from GC-A KO mice
The experiments described above suggested the involvement of both ERβ and GC-A in the rapid regulation of KATP channel activity. Thus, we examined the possibility that the activation of ERβ was regulating GC-A activity and that this was responsible for the regulation of the KATP channels. If GC-A was involved, then the ERβ agonist DPN would have no effect on β-cells from GC-A KO mice. In agreement with this notion, DPN had very little effect on KATP channel activity in cells from GC-A KO mice (Fig. 8, C and D), whereas a 70% reduction of the activity was recorded in experiments with WT β-cells in the presence of DPN (Fig. 8, A and B). Note that, as mentioned above, the action of DPN was specifically mediated through ERβ because it is abolished in ERβ−/− mice (supplemental Material 1, which is published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org).
Fig. 8.

DPN had no effect of KATP channel activity in GC-A KO mice. A, 1 nm DPN decreased KATP channel activity in intact pancreatic islet cells from WT mice. The records show KATP channel activity before DPN application and 5 min after application. B, Percentage of KATP channel activity in the presence of vehicle, before application of stimuli (vehicle) (n =7), in the presence of 1 nm DPN (n =5) and 8 mm glucose (n =3). C, The same experiment as in A, but using β-cells from GC-A KO mice. D, Percentage of activity of similar experiments performed in A and C using β-cells from GC-A mice. Note that 1 nm DPN had no effect, whereas 8 mm glucose had a normal action (n =4). *, P < 0.05; **, P < 0.005. Student’s t test compared with control.
Discussion
The findings of this work using β-cells from ERβ−/− mice demonstrate that ERβ is involved in the rapid inhibition of KATP channel activity elicited by physiological concentrations of E2. ERα does not mediate this action because E2 blocks KATP channel activity in β-cells from ERα−/− mice to the same extent as in WT cells. Activation of ERβ by the specific agonist DPN reduced KATP channel activity, an effect absent in cells from ERβ−/− mice. In addition, DPN enhanced glucose-induced [Ca2+]i signals and insulin release, yet the ERα agonist PPT was inactive when tested under the same conditions. Therefore, the activation of ERβ regulates stimulus-secretion coupling in β-cells and elicits a rapid insulinotropic action, similar to the effects previously described for E2 (18). Rapid E2 actions in β-cells involve cGMP and PKG (17). Our new data indicate that the membrane GC-A receptor participates in this process. We show that both E2 and DPN effects are abolished in cells from GC-A KO mice. The mechanism for the interaction between ERβ and GC-A receptors is still undetermined. Figure 9 shows a proposed hypothetical model for the rapid E2 action on β-cell stimulus-secretion coupling. In this model, we propose that, in synergy with glucose, when E2 binds to ERβ, it activates the GC-A receptor through an as yet unknown mechanism. As a consequence, KATP channel activity is decreased in a cGMP/PKG-dependent manner, potentiating calcium signals and insulin secretion. KATP channels control the resting membrane potential and determine the electrical resistance of β-cells. When KATP channels are open and the membrane resistance is low, small currents will affect the plasma membrane potential (and therefore, insulin release) only minimally. However, when KATP channels are largely closed and membrane resistance is high, small currents will elicit the depolarization of the membrane, electrical activity, Ca2+ signals, and insulin release (27). This will explain the reason why a regulator of KATP channel activity like E2 produces an effective secretagogue action only when glucose concentrations are high and most KATP channels are already closed (see Fig. 3). Nevertheless, we cannot rule out other actions of E2 via ERβ or other receptors that may provoke similar effects on insulin secretion.
Fig. 9.

Proposed model for the action of E2 on β-cell stimulus secretion coupling. Glucose enters the pancreatic β-cell when plasma concentration is increased. Intracellular glucose metabolism raises the ATP/ADP ratio, which closes KATP channels. The subsequent depolarization opens calcium channels, increasing [Ca2+]i and promoting insulin secretion. In synergy with glucose, when E2 binds to ERβ, it activates the GC-A receptor through a yet unknown mechanism. As a consequence, KATP channel activity is decreased in a cGMP/PKG-dependent manner, potentiating calcium signals and insulin secretion.
The molecular characterization of the receptor that triggers E2 rapid actions in pancreatic β-cells is still a matter of debate. A membrane estrogen binding site with a pharmacological profile different from ERα and ERβ was shown in β-cells (8). The activation of this membrane binding site by E2 produced a rapid potentiation of glucose-induced calcium signals (8). The pharmacological profile, as well as the absence of effect of the antiestrogen ICI182,780 on Ca2+ signals, suggested that a nonclassical membrane estrogen receptor might be involved (8, 28). The apparent contradiction of this suggestion with the results presented in the current study may have several explanations. First, the pharmacology of classical ERs outside the nucleus may be somewhat different depending on the cell type (29). Second, although the antiestrogen ICI182,780 has been demonstrated to block ERα-mediated rapid effects (29), the lack of effect of ICI182,780 does not necessarily indicate that a nonclassical estrogen receptor is involved. Binding of ICI182,780 to classical ERs might not block all rapid signaling mechanisms (30); extranuclear ERβ may mediate some ICI182,780-insensitive, E2-triggered actions previously attributed to other membrane ERs.
The results presented in this work using genetically modified mice indicate that ERβ is involved in rapid E2 actions in β-cells. Therefore, ERβ mediates, at least in part, the E2 actions previously attributed to the alleged nonclassical membrane ER (ncmER) (21, 22, 28). This does not necessarily exclude the presence of other ncmERs in β-cells. In fact, two membrane proteins have been described to behave as ncmERs in β-cells: the sulfonylurea receptor (SUR1) expressed in β-cells and the G protein-coupled receptor (GPR30). However, both respond to pharmacological rather than physiological concentration of E2. Binding to SUR1 and regulation of apoptosis was demonstrated for E2 concentrations as high as 100 μm (31). Therefore, a physiological role for SUR1 as a nonclassical estrogen receptor is, at the moment, uncertain. GPR30 has been proposed as a novel estrogen receptor (32, 33). It has been demonstrated to be present in β-cells and to mediate rapid E2-induced insulin release, although again only at supraphysiological concentrations of E2 (5 μm) (34). An interesting possibility that has not yet been tested is the direct action of E2 on L-type Ca2+ channels, recently demonstrated in neurons (35). However, it would not explain the effects on KATP channel activity. In conclusion, our results demonstrate that ERβ mediates the E2 inhibition of KATP channels at physiological concentrations and is therefore involved in the insulinotropic effect of E2.
ERα and ERβ have been described to exert their effects from different cell locations: the nucleus, the cytoplasm, and the plasma membrane (36, 37). Activation of extranuclear ERs, at or near the plasma membrane, regulates other membrane proteins like growth factors and G proteins, initiating important signaling pathways (38, 39). In the cytoplasm, ERα directly interacts with the tyrosine kinase Src and the p85 subunit of phosphatidylinositol 3-kinase, activating key downstream signaling kinases such as MAPK and AKT (40, 41, 42, 43, 44). Extranuclear ERα in β-cells is implicated in rapid activation of ERK1/2 and insulin biosynthesis. ERβ is expressed as well in these cells and it is located within and outside the nucleus (7). Previous results demonstrate that E2 rapidly increased cGMP and inhibited KATP activity in a PKG-dependent manner (17). cGMP levels increase after activation of sGC or the membrane guanylyl cyclases. The absence of effect of two sGC blockers on E2 action, LY83583 (17) and ODQ (present work), indicated that the action is mediated by a membrane guanylyl cyclase. Several other reports have described an E2-elicited effect that might be mediated by membrane guanylyl cyclase. In LHRH-releasing GT1-7 cells, E2 reduces carbachol-induced Ca2+ signals via PKG phosphorylation of inositol triphosphate receptors. This effect was imitated by C-type natriuretic peptide (which activates membrane-bound guanylyl cyclase B) and was not inhibited by either the sGC blocker LY83583 or the antiestrogen ICI182,780 (45). In human coronary smooth muscle cells, E2 increases cGMP, an action that was inhibited by the antiestrogen ICI182,780 and was not blocked by the sGC inhibitor ODQ (46). ERβ plays an important role in E2 inhibition of cardiac hypertrophy, in a process that involves E2 stimulation of ANP and brain natriuretic peptide production and may involve GC-A receptors (47).
Rodent islets of Langerhans respond to ANP with an increase of cGMP, indicating that the GC-A receptor is expressed (25, 48). Immunocytochemistry shown in the present study confirms these data. Here, we have used β-cells from WT and GC-A KO mice to molecularly demonstrate the role of the GC-A receptor in E2 regulation of KATP channel activity and in the potentiation of glucose-induced insulin release. The ERβ agonist DPN reduces KATP channel activity and stimulates insulin secretion in β-cells from WT mice but not in cells from GC-A KO mice, which strongly suggests that GC-A activation occurs downstream of ERβ activation.
The actions described here for ERβ may have important implications for the physiology of pancreatic β-cells. The islet of Langerhans is a plastic tissue; it has the capacity to adapt to changes in the organism, in particular, during pregnancy and obesity to counteract peripheral insulin resistance. In both cases, islets adapt to these situations by increasing insulin biosynthesis, insulin secretion, and pancreatic β-cell mass. In addition to the important role that prolactin receptors have in the adaptation of islets to pregnancy (49), estrogen receptors may have an important implication in this process. In β-cells, ERα mediates E2 stimulation of insulin biosynthesis and insulin release without altering β-cell mass (7). In the present work we demonstrate that, in addition to ERα, the estrogen receptor ERβ is a key molecule in the rapid potentiation of glucose-stimulated insulin secretion by E2. It is possible that ERα and ERβ integrate information from E2 and nutrients to provide the insulin biosynthesis and release needed to compensate for the increased insulin resistance generated during pregnancy and puberty. Although more speculative at present, E2 signaling may also play a role in the adaptation of the endocrine pancreas to obesity, at least in some circumstances such as obese postmenopausal women. In postmenopausal women, significant increases in E2 plasma levels are associated with increasing body mass index (50, 51). In these women the major source of estrogen biosynthesis is the adipose tissue, which converts androgens into estrogens by aromatase (50, 52). Therefore, E2 released by adipocytes in obese postmenopausal women may be a signal for β-cells to counteract the insulin resistance produced by obesity. Finally, from a pharmacological point of view, this work opens the possibility of a new treatment for non-insulin-dependent diabetes mellitus based on the use of new ERβ agonists as modulators of insulin secretion.
Materials and Methods
Materials
Fura 2-AM was obtained from Molecular Probes (Invitrogen, Barcelona); ODQ, DPN, and PPT were from Tocris Cookson Ltd (Avonmouth, United Kingdom). Other substances were obtained from Sigma (Madrid, Spain).
Animals
Adult Swiss albino OF1 female mice were used. All animals were kept under standard housing conditions. A committee on internal animal care and use reviewed and approved the method used.
ERα−/−, ERβ−/−, and GC-A KO
ERβ−/− mice were generated as described in Krege et al. (53) and supplied by Jan-Ake Gustafsson’s laboratory. ERα−/− mice were purchased from Taconic Europe. Mice with global deletion of GC-A (GC-A KO) and corresponding wild-type mice were generated by the group of D.L. Garbers (University of Texas Southwestern Medical Center, Dallas, TX) (54). Islets of Langerhans from ERα−/−, ERβ−/−, and GC-A KO mice were treated as described below for islets from OF-1 mice.
Islet and islet cells isolation
Pancreatic islets of Langerhans were isolated by collagenase (Sigma) digestion as previously described (18) and used according to the kind of experiment to be performed. When required, islets were dispersed into single cells and cultured in RPMI 1640 without phenol red, containing 11 mm glucose (Cambrex, Verviers, Belgium) at 37 C in a humidified atmosphere of 95% O2 and 5% CO2. The medium was supplemented with 10% charcoal dextran-treated fetal bovine serum (HyClone Laboratories, Logan, UT), 2 mm l-glutamine, 200 U/ml penicillin, and 0.2 mg/ml streptomycin. The cells were used within 1 d of culture.
Patch-clamp recordings
Islets were dispersed into single cells and cultured as previously described (55). KATP channel activity was recorded by use of standard patch-clamp recording procedures. Currents were recorded by use of an Axopatch 200B patch-clamp amplifier (Axon Instruments Inc., Union City, CA). Patch pipettes were pulled from borosilicate capillaries (Sutter Instrument Co., Novato, CA) using a flaming/brown micropipette puller P-97 (Sutter Instrument Co.) with resistance between 3 and 5 MΩ when filled with the pipettes solutions as specified below.
Bath solution contained 5 mm KCl, 135 mm NaCl, 2.5 mm CaCl2, 10 mm HEPES, and 1.1 mm MgCl2 (pH 7.4) and supplemented with glucose as indicated.
The pipette solution contained 140 mm KCl, 1 mm MgCl2, 10 mm HEPES, and 1 mm EGTA (pH 7.2). The pipette potential was held at 0 mV throughout the recording process. KATP channel activity was quantified by digitizing 60-sec sections of the current record, filtered at 1 kHz, sampled at 10 kHz by a Digidata 1322A (Axon Instruments Inc.), and calculating the mean NPo during the sweep. Channel activity was defined as the product of N, the number of functional channels, and Po, the open-state probability. Po was determined by dividing the total time channels spent in the open state by the total sample time. Values of NPo were normalized relative to the channel activity measured in control conditions, before application of different substances. Data sampling was started 1min before (control) and 5 min after application of test substances.
Experiments were carried out at room temperature (20–24 C). Data are expressed as mean ± se.
Insulin secretion measurement
Pancreatic islets of Langerhans were isolated and cultured in the same medium as islet cells described above. After 24 h in culture, islets were incubated at 37 C in groups of 5 islets in a buffer solution containing 120 mm NaCl, 25 mm NaHCO3, 5 mm KCl, 2.5 mm CaCl2, 1 mm MgCl2, and 3 mm d-glucose, final pH = 7.35. After 2 h, the solution was replaced by the different stimuli and the islets were in the incubator for 1 h at 37 C. Afterward, the medium was collected and insulin was measured in duplicate samples by RIA using a Coat-a-Count kit (DPC, Los Angeles, CA). Protein concentration was measured by the Bradford dye method.
Recording [Ca2+]i
Isolated islets of Langerhans were loaded with 5 μm fura 2-AM for at least 1 h at room temperature. Calcium records in the whole islet of Langerhans were obtained by imaging intracellular calcium under an inverted epifluorescence microscope (Axiovert 200; Carl Zeiss GmbH, Jena, Germany). Images were acquired every 2 sec with an extended Hamamatsu Digital Camera C4742-95 (Hamamatsu Photonics, Barcelona, Spain) using a dual-filter wheel (Sutter Instrument Co.) equipped with 340 and 380 nm, 10-nm bandpass filters (Omega Optics, Madrid, Spain). Data were acquired using Aquacosmos software from Hamamatsu (Hamamatsu Photonics, Barcelona, Spain). Fluorescence changes are expressed as the ratio of fluorescence at 340 and 380 nm (F340/F380). Results were plotted and analyzed with use of commercially available software (Sigmaplot, SPSS Inc., Chicago, IL).
Immunocytochemistry
Islets isolated as previously described were fixed with Bouin’s solution for 5 min and washed with PBS. Then, they were dehydrated with 30, 50, and 70% ethanol, for 3 min each, and washed with PBS. After this, the islets were treated with 0.5% Triton X-100 for 15 min and then washed with PBS. The nonspecific staining was blocked with PBS supplemented with 0.1% Triton X-100 and 5% serum from the same host as the secondary antibodies used. After the islets had been incubated for 1 h at room temperature, primary antibodies were added to the blocking solution. These were anti-Natriuretic Peptide Receptor A (rabbit polyclonal, 1:150, ab14356; Abcam, Cambridge, MA) and anti-insulin (mouse monoclonal, 1:200, Sigma, Madrid). The islets were incubated overnight with the primary antibodies at 4 C. Secondary antibodies (Alexa Fluor, Molecular Probes, Eugene, OR) were used at 1:500 in PBS plus 1% serum from the same host as the secondary antibodies, for 1 h at room temperature. A confocal Zeiss Pascal 5 microscope and a Zeiss 40× objective (numerical aperture = 1.3) were used. The images were analyzed using LSM Zeiss software (Carl Zeiss).
Acknowledgments
We thank Ms. M. Luisa Navarro and Ana B. Rufete for their excellent technical assistance.
NURSA Molecule Pages:
Ligands: 17β-estradiol;
Nuclear Receptors: ER-β.
Footnotes
This work was supported by Ministerio de Ciencia e Innovación Grants BFU2008-01492 and BFU2007-67607 and by the Generalitat Valencia grant, GV/2009/056. Centro de Investigación Biomédica en Red de Diabetes y Enfermedades Metabólicas Asociadas is an initiative of Instituto de Salud Carlos III.
Disclosure Summary: The authors have nothing to disclose.
First Published Online October 23, 2009
Abbreviations: ANP, Atrial natriuretic peptide; cGMP, cyclic GMP; DPN, 2,3-bis(4-hydroxyphenyl)-propionitrile; E2, 17β-estradiol; ER, estrogen receptor; GC-A, guanylyl cyclase A;GC-A KO, GC-A-deficient; KATP, ATP-sensitive potassium; ncmER, nonclassical membrane ER; PKG, cGMP-dependent protein kinase; PPT, propylpyrazole-triol; sGC, soluble guanylate cyclase; WT, wild type.
References
- 1.Kahn SE, Hull RL, Utzschneider KM2006. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444:840–846 [DOI] [PubMed] [Google Scholar]
- 2.Barros RP, Gabbi C, Morani A, Warner M, Gustafsson JA2009. Participation of ERα and ERβ in glucose homeostasis in skeletal muscle and white adipose tissue. Am J Physiol Endocrinol Metab 297:E124–E133 [DOI] [PubMed]
- 3.Barros RP, Machado UF, Gustafsson JA2006. Estrogen receptors: new players in diabetes mellitus. Trends Mol Med 12:425–431 [DOI] [PubMed] [Google Scholar]
- 4.Bryzgalova G, Gao H, Ahren B, Zierath JR, Galuska D, Steiler TL, Dahlman-Wright K, Nilsson S, Gustafsson JA, Efendic S, Khan A2006. Evidence that oestrogen receptor-alpha plays an important role in the regulation of glucose homeostasis in mice: insulin sensitivity in the liver. Diabetologia 49:588–597 [DOI] [PubMed] [Google Scholar]
- 5.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS2000. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc Natl Acad Sci USA 97:12729–12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ropero AB, Alonso-Magdalena P, Quesada I, Nadal A2008. The role of estrogen receptors in the control of energy and glucose homeostasis. Steroids 73:874–879 [DOI] [PubMed] [Google Scholar]
- 7.Alonso-Magdalena P, Ropero AB, Carrera MP, Cederroth CR, Baquié M, Gauthier BR, Nef S, Stefani E, Nadal2008. A pancreatic insulin content regulation by the estrogen receptor ER α. PLoS One 3:e2069 [DOI] [PMC free article] [PubMed]
- 8.Nadal A, Ropero AB, Laribi O, Maillet M, Fuentes E, Soria B2000. Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor α and estrogen receptor β. Proc Natl Acad Sci USA 97:11603–11608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, Tsai MJ, Mauvais-Jarvis F2006. Estrogens protect pancreatic β cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci USA 103:9232–9237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rorsman P, Eliasson L, Renström E, Gromada J, Barg S, Göpel S2000. The cell physiology of biphasic insulin secretion. News Physiol Sci 15:72–77 [DOI] [PubMed] [Google Scholar]
- 11.Ashcroft FM, Harrison DE, Ashcroft SJ1984. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature 312:446–448 [DOI] [PubMed] [Google Scholar]
- 12.Nadal A, Quesada I, Soria B1999. Homologous and heterologous asynchronicity between identified α-, β- and δ-cells within intact islets of Langerhans in the mouse. J Physiol 517 (Pt 1):85–93 [DOI] [PMC free article] [PubMed]
- 13.Santos RM, Rosario LM, Nadal A, Garcia-Sancho J, Soria B, Valdeolmillos M1991. Widespread synchronous [Ca2+]i oscillations due to bursting electrical activity in single pancreatic islets. Pflugers Arch 418:417–422 [DOI] [PubMed] [Google Scholar]
- 14.Barbosa RM, Silva AM, Tome AR, Stamford JA, Santos RM, Rosario LM1998. Control of pulsatile 5-HT/insulin secretion from single mouse pancreatic islets by intracellular calcium dynamics. J Physiol 510 (Pt 1):135–143 [DOI] [PMC free article] [PubMed]
- 15.Dyachok O, Idevall-Hagren O, Sågetorp J, Tian G, Wuttke A, Arrieumerlou C, Akusjärvi G, Gylfe E, Tengholm A2008. Glucose-induced cyclic AMP oscillations regulate pulsatile insulin secretion. Cell Metab 8:26–37 [DOI] [PubMed] [Google Scholar]
- 16.Gilon P, Shepherd RM, Henquin JC1993. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J Biol Chem 268:22265–22268 [PubMed] [Google Scholar]
- 17.Ropero AB, Fuentes E, Rovira JM, Ripoll C, Soria B, Nadal A1999. Non-genomic actions of 17β-oestradiol in mouse pancreatic β-cells are mediated by a cGMP-dependent protein kinase. J Physiol 521(Pt 2):397–407 [DOI] [PMC free article] [PubMed]
- 18.Nadal A, Rovira JM, Laribi O, Leon-quinto T, Andreu E, Ripoll C, Soria B1998. Rapid insulinotropic effect of 17β-estradiol via a plasma membrane receptor. FASEB J 12:1341–1348 [DOI] [PubMed] [Google Scholar]
- 19.Adachi T, Yasuda K, Mori C, Yoshinaga M, Aoki N, Tsujimoto G, Tsuda K2005. Promoting insulin secretion in pancreatic islets by means of bisphenol A and nonylphenol via intracellular estrogen receptors. Food Chem Toxicol 43:713–719 [DOI] [PubMed] [Google Scholar]
- 20.Alonso-Magdalena P, Morimoto S, Ripoli S, Fuentes E, Nadal A2006. The estrogenic effect of bisphenol A disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environ Health Perspect 114:106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nadal A, Ropero AB, Fuentes E, Soria B, Ripoll C2004. Estrogen and xenoestrogen actions on endocrine pancreas: from ion channel modulation to activation of nuclear function. Steroids 69:531–536 [DOI] [PubMed] [Google Scholar]
- 22.Ripoll C, Ropero AB, Alonso-Magdalena P, Quesada I, Fuentes E, Nadal A2008. Rapid regulation of pancreatic α- and β-cell signalling systems by estrogens. Infect Disord Drug Targets 8:61–64 [DOI] [PubMed] [Google Scholar]
- 23.Cruz MN, Douglas G, Gustafsson JA, Poston L, Kublickiene K2006. Dilatory responses to estrogenic compounds in small femoral arteries of male and female estrogen receptor-β knockout mice. Am J Physiol Heart Circ Physiol 290:H823–H829 [DOI] [PubMed]
- 24.Helguero LA, Faulds MH, Gustafsson JA, Haldosén LA2005. Estrogen receptors α (ERα) and β (ERβ) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene 24:6605–6616 [DOI] [PubMed] [Google Scholar]
- 25.Fehmann HC, Noll B, Goke R, Goke B, Trautmann ME, Arnold R1990. Atrial natriuretic factor has a weak insulinotropic action in the isolated perfused rat pancreas. Res Exp Med (Berl) 190:253–258 [DOI] [PubMed] [Google Scholar]
- 26.Uehlinger DE, Weidmann P, Gnädinger MP, Hasler L, Bachmann C, Shaw S, Hellmüller B, Lang RE1986. Increase in circulating insulin induced by atrial natriuretic peptide in normal humans. J Cardiovasc Pharmacol 8:1122–1129 [DOI] [PubMed] [Google Scholar]
- 27.Ashcroft FM2005. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest 115:2047–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ropero AB, Soria B, Nadal A2002. A nonclassical estrogen membrane receptor triggers rapid differential actions in the endocrine pancreas. Mol Endocrinol 16:497–505 [DOI] [PubMed] [Google Scholar]
- 29.Pedram A, Razandi M, Levin ER2006. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol 20:1996–2009 [DOI] [PubMed] [Google Scholar]
- 30.Wong JK, Le HH, Zsarnovszky A, Belcher SM2003. Estrogens and ICI182,780 (Faslodex) modulate mitosis and cell death in immature cerebellar neurons via rapid activation of p44/p42 mitogen-activated protein kinase. J Neurosci 23:4984–4995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ackermann S, Hiller S, Osswald H, Lösle M, Grenz A, Hambrock A2009. 17β-Estradiol modulates apoptosis in pancreatic β-cells by specific involvement of the sulfonylurea receptor (SUR) isoform SUR1. J Biol Chem 284:4905–4913 [DOI] [PubMed] [Google Scholar]
- 32.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER2005. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307:1625–1630 [DOI] [PubMed] [Google Scholar]
- 33.Thomas P, Pang Y, Filardo EJ, Dong J2005. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146:624–632 [DOI] [PubMed] [Google Scholar]
- 34.Mårtensson UE, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Gräde PO, Owman C, Rosen CJ, Adamo ML, Lundquist I, Rorsman P, Nilsson BO, Ohlsson C, Olde B, Leeb-Lundberg LM2009. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology 150:687–698 [DOI] [PubMed] [Google Scholar]
- 35.Sarkar SN, Huang RQ, Logan SM, Yi KD, Dillon GH, Simpkins JW2008. Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc Natl Acad Sci USA 105:15148–15153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levin ER2001. Cell localization, physiology, and nongenomic actions of estrogen receptors. J Appl Physiol 91:1860–1867 [DOI] [PubMed] [Google Scholar]
- 37.Nadal A, Diaz M, Valverde MA2001. The estrogen trinity: membrane, cytosolic, and nuclear effects. Physiology 16:251–255 [DOI] [PubMed] [Google Scholar]
- 38.Abrahám IM, Han SK, Todman MG, Korach KS, Herbison AE2003. Estrogen receptor β mediates rapid estrogen actions on gonadotropin-releasing hormone neurons in vivo. J Neurosci 23:5771–5777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hammes SR, Levin ER2007. Extranuclear steroid receptors: nature and actions. Endocr Rev 28:726–741 [DOI] [PubMed] [Google Scholar]
- 40.Auricchio F, Migliaccio A, Castoria G2008. Sex-steroid hormones and EGF signalling in breast and prostate cancer cells: targeting the association of Src with steroid receptors. Steroids 73:880–884 [DOI] [PubMed] [Google Scholar]
- 41.Fu XD, Simoncini T2008. Extra-nuclear signaling of estrogen receptors. IUBMB Life 60:502–510 [DOI] [PubMed] [Google Scholar]
- 42.Garcia-Segura LM, Diz-Chaves Y, Perez-Martin M, Darnaudery M2007. Estradiol, insulin-like growth factor-I and brain aging. Psychoneuroendocrinology 32 (Suppl 1):S57–S61 [DOI] [PubMed]
- 43.Levin ER2008. Rapid signaling by steroid receptors. Am J Physiol Regul Integr Comp Physiol 295:R1425–R1430 [DOI] [PMC free article] [PubMed]
- 44.Ordóñez-Morán P, Muñoz A2009. Nuclear receptors: genomic and non-genomic effects converge. Cell Cycle 8:1675–1680 [DOI] [PubMed] [Google Scholar]
- 45.Morales A, Díaz M, Guelmes P, Marín R, Alonso R2005. Rapid modulatory effect of estradiol on acetylcholine-induced Ca2+ signal is mediated through cyclic-GMP cascade in LHRH-releasing GT1–7 cells. Eur J Neurosci 22:2207–2215 [DOI] [PubMed] [Google Scholar]
- 46.El-Mowafy AM, Alkhalaf M, Jaffal SM2007. Nongenomic activation of the GC-A enzyme by resveratrol and estradiol downstream from membrane estrogen receptors in human coronary arterial cells. Nutr Metab Cardiovasc Dis 17:508–516 [DOI] [PubMed] [Google Scholar]
- 47.Pedram A, Razandi M, Lubahn D, Liu J, Vannan M, Levin ER2008. Estrogen inhibits cardiac hypertrophy: role of estrogen receptor-β to inhibit calcineurin. Endocrinology 149:3361–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verspohl EJ, Ammon HP1989. Atrial natriuretic peptide (ANP) acts via specific binding sites on cGMP system of rat pancreatic islets without affecting insulin release. Naunyn Schmiedebergs Arch Pharmacol 339:348–353 [DOI] [PubMed] [Google Scholar]
- 49.Sorenson RL, Brelje TC2009. Prolactin receptors are critical to the adaptation of islets to pregnancy. Endocrinology 150:1566–1569 [DOI] [PubMed] [Google Scholar]
- 50.Cleary MP, Grossmann ME2009. Minireview: obesity and breast cancer: the estrogen connection. Endocrinology 150:2537–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lukanova A, Lundin E, Zeleniuch-Jacquotte A, Muti P, Mure A, Rinaldi S, Dossus L, Micheli A, Arslan A, Lenner P, Shore RE, Krogh V, Koenig KL, Riboli E, Berrino F, Hallmans G, Stattin P, Toniolo P, Kaaks R2004. Body mass index, circulating levels of sex-steroid hormones, IGF-I and IGF-binding protein-3: a cross-sectional study in healthy women. Eur J Endocrinol 150:161–171 [DOI] [PubMed] [Google Scholar]
- 52.Simpson ER, Ackerman GE, Smith ME, Mendelson CR1981. Estrogen formation in stromal cells of adipose tissue of women: induction by glucocorticosteroids. Proc Natl Acad Sci USA 78:5690–5694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O1998. Generation and reproductive phenotypes of mice lacking estrogen receptor β. Proc Natl Acad Sci USA 95:15677–15682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopez MJ, Wong SK, Kishimoto I, Dubois S, Mach V, Friesen J, Garbers DL, Beuve A1995. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature 378:65–68 [DOI] [PubMed] [Google Scholar]
- 55.Valdeolmillos M, Nadal A, Contreras D, Soria B1992. The relationship between glucose-induced K+ATP channel closure and the rise in [Ca2+]i in single mouse pancreatic β-cells. J Physiol 455:173–186 [DOI] [PMC free article] [PubMed] [Google Scholar]