

Abstract
The majority of the biological effects of estrogens in the reproductive tract are mediated by estrogen receptor (ER)α, which regulates transcription by several mechanisms. Because the tissue-specific effects of some ERα ligands may be caused by tissue-specific transcriptional mechanisms of ERα, we aimed to identify the contribution of DNA recognition to these mechanisms in two clinically important target organs, namely uterus and liver. We used a genetic mouse model that dissects DNA binding-dependent vs. independent transcriptional regulation elicited by ERα. The EAAE mutant harbors amino acid exchanges at four positions of the DNA-binding domain (DBD) of ERα. This construct was knocked in the ERα gene locus to produce ERα(EAAE/EAAE) mice devoid of a functional ERα DBD. The phenotype of the ERα(EAAE/EAAE) mice resembles the general loss-of-function phenotype of αER knockout mutant mice with hypoplastic uteri, hemorrhagic ovaries, and impaired mammary gland development. In agreement with this phenotype, the expression pattern of the ERα(EAAE/EAAE) mutant mice in liver obtained by genome-wide gene expression profiling supports the observation of a near-complete loss of estrogen-dependent gene regulation in comparison with the wild type. Further gene expression analyses to validate the results of the microarray data were performed by quantitative RT-PCR. The analyses indicate that both gene activation and repression by estrogen-bound ERα rely on an intact DBD in vivo.
Gene expression analysis in a mutant ER knock-in mouse model shows that estrogen response requires a functional DNA binding domain.
Estrogens not only regulate processes essential for female reproductive functions, such as uterine growth, mammary epithelial cell proliferation, and hypothalamo-pituitary feedback regulation of the ovaries, but they are also involved in male reproductive development and physiology, in bone homeostasis, in blood vessel physiology, metabolism, and in various functions in the central nervous system.
The variety of physiological responses to estrogens is initiated by the binding of steroidal estrogens such as 17β-estradiol (E2) to their cognate receptors, estrogen receptors α and β (ERα and ERβ). These receptors, members of the nuclear receptor superfamily of ligand-activated transcription factors, can either induce specific gene transcription upon binding to estrogen-responsive elements (EREs) in target promoters or regulate genes by interference with other transcription factors bound to DNA, or influence cytoplasmic signaling pathways (1). c-Myc, CyclinD1, p21 (cyclin-dependent kinase inhibitor), and igf-1, for example, have been described to be regulated by estrogen via an interaction of the ER with other transcription factors such as nuclear factor κB (NF-κB) and activator protein-1 (AP-1) (2, 3, 4). On such promoters, AP-1 or NF-κB-responsive elements rather than EREs mediate transcriptional regulation by estrogens (5, 6). Cross talk of transcription factors also seems to play a role in hormone-mediated gene repression, because several estrogen-repressed genes such as fibroblast growth factor-inducible kinase (fnk), JAK-binding protein (JAB), lipopolysaccharide-induced c-x-c chemokine (LIX), bcl-3, and short heterodimer partner (SHP) were suggested to be regulated by an interaction of NF-κB and ERα (7).
The set of target genes regulated by estrogens differs from tissue to tissue, e.g. from liver to uterus, as described elsewhere (8). A recent study showed that also in cancerous tissue, the set of estrogen-regulated genes differs depending on the tissue origin (9). This indicates that different ER transcriptional mechanisms may prevail in different tissues, depending on the tissue-specific distribution of cofactors or other ER-interacting proteins.
A mechanistic understanding of the tissue specificity of estrogen signaling would have therapeutic implications: selective ER modulators (SERMs) like tamoxifen and raloxifene are synthetic molecules that show different degrees of estrogen agonism or antagonism in a tissue-specific manner. It is still incompletely understood, why a compound, such as tamoxifen, is able to work in an estrogen-agonistic manner in one tissue and as an antagonist in another. Whereas several studies suggest that differential tissue-specific expression of cofactors in different cell types governs the degree of tamoxifen agonism (2), other studies suggest that the differential activity of SERMs depends on the type of response element (10).
A close understanding of the significance of different transcriptional pathways can therefore guide the identification of new ligands with tissue-selected profiles. Clinically relevant examples of tissue-selective estrogen action are the uterus and the liver. Estrogen-induced uterine growth constitutes an important classical estrogen effect that is part of the efficacy of oral contraceptives and cyclical hormone treatments for the control of uterine bleeding. On the other hand, estrogen-induced uterine growth is a critical issue during postmenopausal hormonal therapy, where it needs to be counteracted by a progestin (11). In the liver, however, estrogen action has several facets: the effects of estrogen on lipid and lipoprotein metabolism may well contribute to beneficial metabolic effects of estrogens (12), whereas the up-regulation of some serum proteins, such as thrombin and fibrinogen, is suspected to contribute to an increased risk of venous thromboembolism after estrogen and SERM treatment (13, 14).
The goal of the present study is to investigate, with the help of a genetic mouse model, the contribution of DNA binding-dependent ERα transcriptional regulation on tissue-specific gene expression in liver and uterus in vivo.
The EAAE mouse model was generated to analyze whether classical, DNA binding-dependent ERα actions differentially contribute to estrogen action in liver and uterus. By targeting murine ERα with a construct that harbors a four-amino acid exchange in the DNA recognition helix (EAAE), the resulting ERα protein is incapable of binding to an ERE. In this mouse model, gene expression evoked by oral ethinyl estradiol (EE) application, thus closely mimicking the classical route of application of synthetic ER modulators was measured. These experiments showed that estrogen-regulated gene transcription is dependent on a functional DNA-binding domain (DBD) of ERα in both liver and uterus.
Results
The EAAE mutation in ERα abolishes ERE interaction and leads to infertility
To dissect classical ERE-dependent from ERE-independent actions of murine ERα, a mutant was generated harboring four amino acid exchanges in the DNA recognition helix, i.e. Y201E, K210A, K214A, R215E, hence named EAAE (Fig. 1A). In transfected HeLa cells, the EAAE mutant ERα is no longer able to activate an ERE-containing reporter gene but can still repress an NF-κB-responsive promoter in the presence of RelA (Fig. 1B).
Fig. 1.

Generation and initial characterization of the ERαEAAE mutant. The location of the four mutated amino acids in the first zinc finger of the ERα (Y201 was changed to E, K210 to A, K214 to A, R215 to E), hence the name EAAE, is indicated (A). In Hela cells, the mutated ERα is unable to activate a luciferase reporter with 2 ERE neither with nor without 10−8 m E2, but the mutated ERα can still repress a NF-κB-responsive promoter (ICAM-tk-Luc with three NF-κB sites) in an E2-dependent manner (B). Female homozygous ERαEAAE mice develop hypoplastic uteri (indicated by an arrow) and have blunted mammary gland development (C). In nuclear protein extracts from the livers of wild-type and EAAE mice, the ERα protein is present, but not in liver-specific ERα knockout mice (31 ) (D). Ctrl, Control.
In addition, chromatin immunoprecipitation (ChIP) on the ERE-containing pS2 promotor in HeLa cells shows detectable [2.1 (±0.4)-fold enrichment] binding of the wild-type ERα but no binding [1.3 (±1.0) fold enrichment] of the ERα(EAAE/EAAE) mutant. On the indirectly regulated Cyclin D1 promotor harboring AP-1 and SP-1 sites (3) ChIP shows detectable (yet attenuated compared with wild type) binding of the ERα(EAAE/EAAE) [2.2 (±0.8)-fold enrichment] (see Table 1). Of note, the ERα(EAAE/EAAE) has been shown to still interact in a ligand-dependent manner with coactivators, such as Baf-57 (15).
Table 1.
ChIP shows lack of recruitment of the ERα(EAAE/EAAE) to classical ERE promoter in response to E2
| pS2 promoter | CyclinD1 promoter | ||
|---|---|---|---|
| ERα | DMSO | 1 | 1 |
| E2 | 2.1 (±0.4) | 14.1 (±7.6) | |
| ERα(EAAE/EAAE) | DMSO | 1 | 1 |
| E2 | 1.3 (±1.0) | 2.2 (±0.8) |
To demonstrate the modified DNA interaction of ERα(EAAE/EAAE) in response to E2 a ChIP was performed on the pS2 and CyclinD1 promoter. Hela cells were transfected with ERα or ERα(EAAE/EAAE)DNA separately, treated with 10 nm E2 (n = 3) or 0.1% DMSO (n = 2) and the DNA-protein complexes were pulled out with the HC-20 ERα antibody. The bound DNA was quantified by quantitative PCR. DMSO- treated samples were set at 1.0. DMSO, Dimethylsulfoxide.
By gene targeting in embryonic stem cells, the Estra locus coding for the mouse ERα gene was modified such that ERα expresses the EAAE mutations. A mouse line was derived from embryonic stem cells that harbor the ERα EAAE allele. Heterozygous mice were viable and fertile and did not show any physiological abnormalities (Fig. 1C). Homozygous ERα(EAAE/EAAE) mice were born in normal Mendelian ratio. Immunoblot analysis of liver nuclear extracts showed that ERα protein is present in ERα(EAAE/EAAE) mice (Fig. 1D). Note that the antibody used (MC-20) detects the C-terminal ligand-binding domain (LBD) of murine ERα, indicating that the ERα-EAAE mutant is expressed in full length.
In contrast to littermate controls, mice homozygous for the EAAE mutation are infertile and show a severely hypoplastic uterus (indicated by an arrow), hemorrhagic ovaries, and blunted ductal mammary gland development (Fig. 1C). The phenotype of the reproductive organs is akin to the phenotype described for mice completely devoid of ERα (16, 17). These results indicate that a functional DBD of ERα is essential for female fertility and that ERα protein activities outside the DNA recognition helix cannot compensate for a lack of DNA binding by the mutant receptor in maintaining the function of the female reproductive tract. A similar conclusion holds true for male EAAE mice, which are also infertile and show a phenotype similar to male ERα knockout (αERKO) mice (data not shown). To investigate whether DNA binding-independent functions of ERα may play a role in estrogen action outside the reproductive tract, we finally focused our attention on the liver.
The DBD of ERα is required for estrogen-induced gene expression in the uterus
The severely hypoplastic uteri of homozygous ERα(EAAE/EAAE) mice are reminiscent of the phenotype of global αERKO knockout mice (16). To verify that this phenotype is caused by an inability of the mutant receptor to activate gene expression, we analyzed the expression of several known ERα target genes and their induction by estrogen treatment in the uterus of mutant and wild-type animals. Genes such as Lifr (leukemia-inhibitory factor receptor), p21, Tgm2 (transglutaminase 2), Wnt4 (wingless-related murine mammary tumor virus integration site 4) und Pgr (progesterone receptor) are known to be induced by oral EE treatment (8, 18). We could show that in addition to known estrogen target genes such as Wnt4 (Fig. 2C), Pgr (Fig. 2E) and p21 (Fig. 2F), genes like Gdf15 (growth and differentiation factor 15) (Fig. 2A), Il17ra (IL 17 receptor a) (Fig. 2B), and Mmd2 (monocyte to macrophage differentiation-associated 2) (Fig. 2D) are induced 4 h and much lower 24 h after EE treatment in the wild-type uterus as measured by quantitative RT-PCR (qRT-PCR).
Fig. 2.

Gene expression analysis in the uterus of ERα(EAAE/EAAE) and wild-type mice. The expression of G(Gdf15) (A), Il17ra (B), Wnt4 (C), Mmd 2 (D), Pgr (E), and p21(F) was examined in the uterus of ERα(EAAE/EAAE) and wild-type mice (A–F) by qRT-PCR (relative expression level normalized to cyclophilin) V, Vehicle.
In contrast, in the uterus of homozygous ERα(EAAE/EAAE) mice, Gdf15, Il17ra, Wnt4, Mmd2, Pgr, and p21 are not EE regulated (Fig. 2 and Table 2).
Table 2.
TaqMan probes used for classical PCR or on Micro Fluidic Cards
| Gene name | Assay | Gene ID |
|---|---|---|
| Cyclophilin | Mm00478295_m1 | NM_011149.2 |
| Leukemia-inhibitory factor receptor (Lifr) | Mm00442940_m1 | NM_013584.1 |
| IL-7 receptor a (Il17ra) | Mm00434214_m1 | NM_008359.1 |
| Cyclin-dependent inhibitory factor (p21) | Mm00432448_m1 | NM_001111098.1 |
| Monocyte to macrophage differentiation-associated 2 (Mmd2) | Mm00558356_m1 | NM_175217.6 |
| Transglutaminase 2 (Tgm2) | Mm00436980_m1 | NM_009373.3 |
| Growth differentiation factor 15 (Gdf15) | Mm00442228_m1 | NM_011819.1 |
| Coagulation factor III (F3) | Mm00438853_m1 | NM_010171.2 |
| Presenilin 2 (Psen2) | Mm00448405_m1 | NM_011183.1 |
| FGF-inducible kinase (fnk) | Mm01187219_g1 | NM_013807.2 |
| Wingless-related MMTV integration site 4 (Wnt4) | Mm00437341_m1 | NM_009523.1 |
| Progesterone receptor (Pgr) | Mm00435625_m1 | NM_008829.2 |
| IGF-1 (Igf-1) | Mm00439561_m1 | NM_001111274.1 |
| NM_001111275.1 | ||
| NM_001111276.1 |
FGF, Fibroblast growth factor; ID, identification; MMTV, murine mammary tumor virus.
The latter gene, p21 (Fig. 2F), has been described to be regulated in an ERE-independent manner (4). Similar to p21, insulin-like growth factor-1 (igf-1) has been described to be an ERE-independently regulated, uterus-specific ERα target gene (2). Therefore, we investigated the expression of igf-1 in wild-type and ERα(EAAE/EAAE) mutant mice in both uterus and liver (see below). igf-1 and p21 are induced both 4 h and 24 h after EE application in the wild-type uterus, as shown by qRT-PCR, but not in the mutant (Fig. 3A and Fig. 2F). In the liver igf-1 is only minimally repressed by EE in the wild-type but not in the ERα(EAAE/EAAE) mutant, as shown both by qRT-PCR and Illumina profiling (Fig. 3B). p21, on the other hand, is induced by EE in the wild-type in both liver and uterus. Similar to igf-1, p21 is also not induced in the liver of the ERα(EAAE/EAAE) mutant (Fig. 4D). Thus, tissue-specific induction of igf-1 by estrogens [regulated by EE in the wild-type uterus and only slightly in the wild-type liver (Fig. 3, A and B)] relies on a functional ERα DBD in vivo, indicating that a functional ERα DBD is essential for the transcriptional response to estrogen in the uterus, and that a mutation of the DBD is equivalent to a loss of receptor.
Fig. 3.

Igf-1, a uterus-specific ERα target gene, is regulated by direct DNA interaction. The expression of igf-1 was investigated in uterus (A) and liver (B) of wild-type (wt) and ERα(EAAE/EAAE) mutant (EAAE) mice. The mice were treated with vehicle (V) or EE for 4 or 24 h, and the expression levels of igf-1 determined by qRT-PCR are shown by gray bars described by the left axis (relative expression level normalized to cyclophilin) (A and B). The expression analysis done by GEP is illustrated by black filled squares and described by the right axis (normalized signal intensity) (B).
Fig. 4.
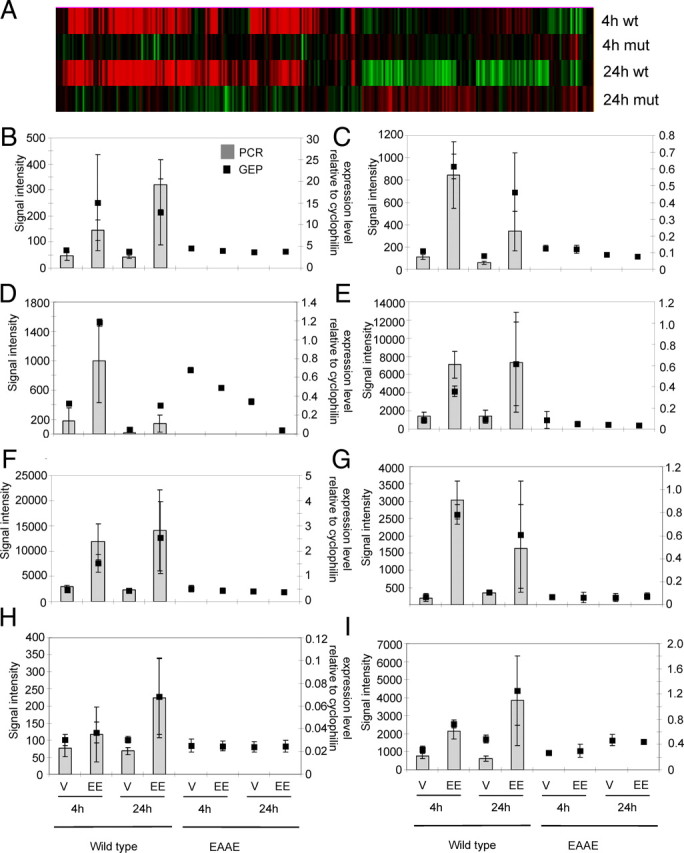
Genome-wide gene expression profiling of RNA isolated from the liver of ERα(EAAE/EAAE) and wild-type mice and selected single gene RT-PCR analysis. RNA was isolated from the liver of homozygous EAAE mutants and wild-type mice treated for 4 h or 24 h with EE (100 μg/kg) or vehicle, and the cRNA was hybridized to Illumina Mouse SentrixWG-6 version 1.1 Bead Chips. The heat map (hierarchical clustering, Genedata Expressionist) presents the gene expression ratio of EE vs. vehicle (P < 0.001; n = 4–5 per group). The red bars indicate genes that are induced and the green bars indicate genes that are repressed by EE treatment (A). Six genes that, according to GEP with Illumina BeadChips, are induced by EE in the liver of wild-type but not in the EAAE mutant mice (indicated by black squares, left axis) were validated by qRT-PCR (gray bars; right axis): Lifr (B), Il17ra (C), p21 (D), Mmd 2 (E), Tgm2 (F), Gdf15 (G), F3 (H), and Psen2 (I). Mut, Mutant; V, vehicle; wt, wild type.
Orally applied, EE regulates a variety of genes in the livers of wild-type but not ERα(EAAE/EAAE) mice.
EE regulates a variety of genes in the liver of wild-type but not ERα (EAAE/EAAE) mice
To understand the contribution of direct vs. indirect DNA binding of estrogen-activated ERα or ERα(EAAE/EAAE) on gene expression in the murine liver, RNA was isolated from the liver of wild-type and ERα(EAAE/EAAE) mice treated perorally with 100 μg/kg EE or vehicle for 4 or 24 h. A genome-wide gene expression study on Illumina Sentrix Mouse WG-6v1.1 arrays interrogated about 47.000 murine transcripts. In the liver of the wild-type animals, 78 genes (fold change >1.5; P < 0.001) are regulated by EE after both application periods (Table 3 and supplemental Table S1).
Table 3.
Number of genes regulated by EE compared with vehicle in the liver of wild-type and ERα(EAAE/EAAE) mutants (P < 0.001; fold change >1.5)
| Up-regulated | Down-regulated | |||
|---|---|---|---|---|
| 4 h | wt | 57 | 1 | |
| mut | 0 | 0 | ||
| 24 h | wt | 4 | 16 | |
| mut | 0 | 1 | ||
mut, Mutant; wt, wild type.
However, there are only very weak gene expression changes in the liver of the mutant mice after EE treatment after both 4 and 24 h. Only one gene ID, 4932417H02Rik, a riken clone, is up-regulated and one gene, Lrrc59 (leucin-rich repeat containing 59), is down-regulated after 24 h treatment in the mutant (P = 0.001; fold change >1.5), but no differential expression was detected 4 h after EE treatment in the mutant mice (P = 0.001; fold change >1.5) (Table 3 and supplemental Table S1).
The induction of representative estrogen-regulated genes, i.e. Lifr, Il17ra, p21, Mmd2, Tgm2, Gdf15, F3(coagulation factor III), and Psen2 (Presenelin 2), was validated by quantitative RT-PCR (Fig. 4, B-I and Table 2). The induction of Lifr, Tgm2, and p21 by EE is in line with observations described by Boverhof et al. (8) and Hewitt et al. (19, 20). These genes and Mmd2, Gdf15, F3, and Psen2 were detected by our genome-wide gene expression profiling as induced by EE in the liver (Fig. 4, B–I and Table 4).
Table 4.
Fold change of genes regulated by EE vs. vehicle detected by genome-wide gene expression analysis and validated by qRT-PCR
| Gene name | TaqMan probe | Gene ID | 4 h wt | 4 h mut | 24 h wt | 24 h mut |
|---|---|---|---|---|---|---|
| Leukemia-inhibitory factor receptor (Lifr) | Mm00442940_m1 | NM_013584.1 | 5.2 | 1.0 | 3.2 | 1.0 |
| (5.9 × 10−5) | (0.909) | (0.068) | (0.769) | |||
| IL-17 receptora (Il17ra) | Mm00434214_m1 | NM_008359.1 | 5.9 | 1.0 | 4.6 | 0.8 |
| (2.11 × 10−7) | (0.742) | (0.073) | (0.130) | |||
| Monocyte to macrophage differentiation-associated 2 (Mmd2) | Mm00558356_m1 | NM_175217.6 | 3.6 (4.27 × 10−4) | 0.7 (0.535) | 7.1 (0.181) | 0.9 |
| Transglutaminase 2 (Tgm2) | Mm00436980_m1 | NM_009373.3 | 2.9 | 0.9 | 5.9 | 1.0 |
| (8.25 × 10−5) | (0.368) | (0.079) | (0.499) | |||
| Growth differentiation factor 15 (Gdf15) | Mm00442228_m1 | NM_011819.1 | 10.8 | 0.5 | 4.5 | 1.3 |
| (7.14 × 10−5) | (0.432) | (0.260) | (0.683) | |||
| Coagulation factor III (F3) | Mm00438853_m1 | NM_010171.2 | 1.2 | 0.9 | 2.5 | 1.0 |
| (0.202) | (0.650) | (0.126) | (0.862) | |||
| Presenilin 2 (Psen2) | Mm00448405_m1 | NM_011183.1 | 2.1 | 1.2 | 2.3 | 1.1 |
| (5.21 × 10−5) | (0.733) | (0.056) | (0.566) | |||
| Wingless-related MMTV integration site 4 (Wnt4) | Mm00437341_m1 | NM_009523.1 | 1.0 | 1.1 | 1.1 | 1.1 |
| (0.970) | (0.475) | (0.406) | (0.153) | |||
| Progesterone receptor (Pgr) | Mm00435625_m1 | NM_008829.2 | 1.0 | 1.0 | 0.9 | 1.0 |
| (0.648) | (0.702) | (0.888) | (0.554) | |||
| IGF-1 (Igf-1) | Mm00439561_m1 | NM_001111274.1 | 1.0 | 1.0 | 0.7 | 0.9 |
| NM_001111275.1 | (0.658) | (0.887) | (0.060) | (0.177) | ||
| NM_001111276.1 |
ID, Identification; MMTV, murine mammary tumor virus; mut, mutant; wt, wild type. P values are in parentheses.
The Illumina microarray data and the qRT-PCR for these genes are consistent with regard to direction and magnitude of fold change after EE treatment (Fig. 4, B–I, and Table 4), showing increased expression in the liver of wild-type mice after 4 h and 24 h of EE treatment but no change in the liver of mutant mice. This suggests that the EE-dependent increased transcription of these genes requires direct DNA binding by ERα along with 78 genes activated by EE in the liver of wild-type mice vs. one gene in the mutant.
Next, we analyzed genes repressed by EE. As Fig. 4A shows, some genes, which are not regulated by EE after 4 h, are actually repressed after 24 h. This set of genes is not regulated in the liver of ERα(EAAE/EAAE) mice. Examples for these genes are Gsta4 (glutathione S-transferase, α 4), Arrdc3 (arrestin domain containing 3), Nsbp1 (nucleosome binding protein 1), and Ugp2 (UDP-glucose pyrophosphorylase 2). The expression data and results of Welch two-sample t tests are shown in Fig. 5, A–D. EE does not repress the four genes either 4 h (data not shown) or 24 h (Fig. 5) after application in the liver of ERα(EAAE/EAAE) mice, because the Welch two-sample t test“ data of ERα(EAAE/EAAE) mice are higher than 0.001 and their gene expression differences are not significant. These results indicate that not only the genes induced by EE treatment but also the genes repressed by EE are regulated by the ERα only after direct binding of the receptor to DNA.
Fig. 5.

Genes repressed by EE in murine liver. Some of the genes analyzed by the genome-wide expression study are repressed by a 24-h EE treatment. The diagrams show results of the Illumina BeadChip analysis: Gsta4 (A), Arrdc3 (B), Nsbp1 (C), and Ugp2 (D); P values of Welch two-sample t tests describe the significance of gene repression by EE vs. vehicle; V, vehicle.
IL-1β-mediated repression of fnk and LIX requires direct binding of ERα to DNA
For the analysis of gene repression in the liver mediated via the IL-1β pathway, which is well known to repress fnk and LIX expression, ovariectomized ERα wild-type and ERα(EAAE/EAAE) mice were treated with EE (100 μg/kg) or vehicle for 5 d, followed by a single application of IL-1β (20 μg/kg) to activate NF-κB (7) (Fig. 6). As expected, in the liver of wild-type mice EE represses the NF-κB-activated genes fnk and LIX. In the liver of ERα(EAAE/EAAE) animals, however, the expression of fnk and LIX is not repressed after EE application. This observation shows that in vivo gene repression of NF-κB-induced genes by ERα requires a functional DBD.
Fig. 6.

Repression of fnk and LIX in the liver of wild-type, but not ERα(EAAE/EAAE) mice. To investigate the role of the mutated ERα in repression of gene expression, wild-type and ERα(EAAE/EAAE) ovariectomized mice were treated sc for 5 d with EE (100 μg/kg) or vehicle (V). On d 5, 1 h after compound treatment, IL-1β (20 μg/kg) was applied by ip injection, which activates NF-κB. The expression level of fnk and LIX was analyzed by qRT-PCR.
Discussion
The focus of this work was to analyze DNA binding-dependent vs. independent function of the EE-liganded ERα in a murine genetic model. The major finding of this study is that in vivo in both liver and uterus, gene expression changes evoked by oral EE application, both gene induction and gene repression, rely on direct binding of ERα to DNA. We could show that the majority of estrogen-regulated genes in the liver, as well as representative genes regulated in the uterus including igf-1, are not estrogen responsive any longer in ERα(EAAE/EAAE) mice. The heat plot of liver gene expression profiles of ERα wild-type and EAAE mutant mice 4 and 24 h after EE treatment (Fig. 4A) and especially the expression of igf-1 in these animals as uterus-specific target gene illustrate that signaling mechanisms independent of the ERα DBD do not significantly contribute to the transcriptional response to oral estrogens in the liver or to the induction of well-known estrogen target genes in the uterus (Figs. 2 and 4A). A comparison of this study with the microarray study published by Hewitt et al. (2003) with murine uteri of αERKO mice shows that only very few genes that remain estrogen regulated in both mutants can be detected. The wild-type gene expression of the liver we describe in our experiments is in line with investigations performed by Boverhof et al. (8). These authors published igf-1 also to be repressed very weakly in the wild-type liver after 24 h, and they also observed a regulation of Lifr, Jund1, Ets2, Mad2l1, Stat5a, and Tgm2 in the liver, as in this study (see supplemental data; compare Ref. 8).
In addition, the hypoplastic uteri, the hemorrhagic ovaries, and the blunted mammary gland development seen in ERα(EAAE/EAAE) mice are similar to the phenotype of mice lacking ERα (16, 17), indicating that a mutation of four amino acids in the DNA recognition helix in the ERα(EAAE/EAAE) is equivalent to a loss-of-function mutant of the ERα.
A number of studies have been published addressing alternative ER signaling with the help of genetic mouse models. O′Brien et al. (4, 18) studied a mouse model with an ERα mutated at the DNA recognition helix called NERKI (nonclassical ER knock-in) mouse. In contrast to the EAAE model used here, two amino acids are substituted, namely E207 vs. A and G208 vs. A, in the DNA recognition sequence of the first zinc finger eliminating ERα binding to EREs (18). This mutation is distinct from the one used here because E207 and G208 remain intact in the EAAE mutation. The most striking difference in the phenotypes of these two models is that the heterozygous NERKI females (AA/+) are described to be infertile and to have grossly enlarged uteri with cystic hyperplasia, precluding the analysis of NERKI homozygous mice (4, 18). This has not been observed with either heterozygous ERα(+/EAAE) or ERα(+/−) mice, both of which are fertile and phenotypically normal. The αERKO and the ERα(EAAE/EAAE) mice show hypoplastic uteri and hemorrhagic ovaries only as homozygous mutants. Thus, in genetic terms, the NERKI is a gain-of-function allele in that it displays a heterozygous phenotype that neither the complete αERKO nor the partial ERα(EAAE/EAAE) loss-of-function mutant of the same gene shows when present in the heterozygous state. The reason for these discrepancies may be that the NERKI mutation generates a different interaction site in the ERα DBD or favors certain protein-protein interactions, whereas the EAAE mutant should be interpreted as a distinct loss-of-function allele of the ERα in which all protein domains are present but where the DBD is nonfunctional.
Recently, another function-selective ERα mutant has been published that resembles the EAAE mutant both genetically (loss-of-function) and phenotypically (21). The ENERKI (estrogen-nonresponsive estrogen receptor-α knock-in) mutant was designed to probe for ligand-independent activation of ERα. Heterozygous ENERKI mice were fertile (no phenotype described), and homozygous ENERKI mice could be bred and resulted in a αERKO-like phenotype, indicating ligand-dependent receptor activation. The similar phenotypes of these two mutants, one addressing the upstream activation of the receptor, the other the downstream transcriptional mechanisms, nicely complement each other: Whereas the ERα(EAAE/EAAE) mouse described here underscores that the DBD is necessary for estrogen action and the LBD is not sufficient. The ENERKI mouse shows that an intact estrogen-binding domain is necessary (and the DBD and AF-1 only are insufficient) for physiological response to estrogens (21).
Of note, a functional DBD is also important for both physiological and pharmacological gene repression by estrogens. We observed only one gene repressed by EE in the EAAE mutant compared with 17 in wild-type mice (P 0.001). Furthermore, repression of IL-1β-induced NF-κB activity in the liver (as described by Ref. 7) was absent in ERα(EAAE/EAAE) mice similar to αERKO mice (22). The NF-κB-induced genes fnk and LIX were repressed by EE in wild-type mice (7) (Fig. 6), but the expression levels were indistinguishable in ERα(EAAE/EAAE) mice regardless of whether the mice were treated with EE or vehicle, as described for αERKO mice (7). We conclude that the EE-liganded, EAAE-mutated ERα is not able to repress gene expression because of the mutated DNA-binding site, which contrasts to expectation from in vitro experiments showing repression of NF-κB on a synthetic promoter (Fig. 1b) as well as detectable, yet attenuated, recruitment to AP-1/Sp1 elements in a native chromatin context (Table 1). The discrepancy between the in vivo and in vitro data may be explained by a stronger requirement of ERα-DBD for repression of endogenous genes in their native chromatin environment in vivo that is dispensable on transfected promoters in transfected cells. This assumption is supported by the attenuated recruitment of the ERα(EAAE/EAAE) compared with the wild-type receptor to AP-1/Sp-1 elements observed via ChIP.
The requirement for an intact DBD for induction or repression of gene transcription by ERα in vivo is distinct from what has been identified for another nuclear receptor, the glucocorticoid receptor (23). A recently published study aimed at identifying ERα binding loci in mouse liver by ChIP (24) showed that the majority of ERα binding sites contain at least an ERE half-site, lending further support to the notion that a functional ERα-ERE interaction is essential for ERα-mediated transcriptional responses in mouse liver. We conclude that different transcriptional mechanisms cannot fully explain tissue specificity of EE and SERM signaling in uterus and liver. Tissue-specific expression of coactivators may also contribute to tissue specificity of EE and SERM signaling or differential epigenetic regulation of ERα target genes, e.g. via chromatin modification. The latter mechanism has been suggested to contribute to ERα-mediated tissue-specific gene regulation by a recent study of ERα cistromes in different cell types (25).
In summary, we show that both physiological development of the female reproductive tract as well as the transcriptional response to orally applied estrogens in both liver and uterus are contingent upon a functional ERα DBD. Of note, the mere presence of several other estrogen-binding proteins, such as the ERβ or other putative receptors, do not seem to contribute significantly to the action of oral estrogens on two relevant target organs, liver and uterus, in the absence of a functional ERα DBD.
Materials and Methods
Generation and initial characterization of EAAE knock-in mice
To selectively modify the mouse Estra locus coding for ERα, a gene targeting construct was generated based on a 9-kb BamHI fragment encompassing exon 3 of the mouse Estra gene (26). This fragment, obtained from the RPCI21 mouse genomic library was modified using homologous recombination in Escherichia coli to carry the required codon exchanges (changing Y201E, K210A, K214A, R215E, inserted by PCR) and a PGKtkneo cassette flanked by loxP sites (27, 28). Embryonic d 14 stem cells were transfected with the linearized targeting construct and selected for construct integration (29). G418-resistant clones were characterized by Southern blot using external genomic probes from the Estra locus (data not shown and Ref. 26). Clones that had undergone homologous recombination were transiently transfected with the expression plasmid pOG-Cre (23) and selected with 1 mm gancyclovir to isolate subclones that have lost the selection cassette after loxP recombination. This was verified by Southern blot analysis (data not shown). From the resulting embryonic stem cell clones, chimeric mice were generated by blastocyst injection and uterine transfer. By breeding these chimeras to C57BL/6 mice, the ERαflox mouse line was established. To generate homozygous ERα(EAAE/EAAE) mice, heterozygous mice on a C57BL/6 background were intercrossed. For initial pathological investigations, mice were dissected at 9 wk of age, ovaries and uterus were photographed, and mammary gland whole mounts were prepared as described elsewhere (30).
For detection of ER in liver nuclear extracts, snap-frozen liver samples were homogenized in a Dounce homogenizer in 10 mm HEPES (pH 7.9), 1.5 mm MgCl2, 10 mm KCl, 0.5 mm dithiothreitol, Mini complete protease inhibitors (Roche Molecular Biochemicals; Basel; Switzerland), 0.1% Nonidet P-40, incubated 5 min on ice and centrifuged 15 min at 14,000 rpm in a tabletop centrifuge at 4 C. Pelleted nuclei were resuspended in lysis-buffer (20 mm HEPES, pH 7.9; 420 mm NaCl; 1.5 mm MgCl2; 0.2 mm EDTA; 0.5 dithiothreitol; Mini complete protease inhibitors; 10% glycerol) for 15 min on ice. Debris was removed by an additional 15-min 14,000 rpm centrifugation step. Protein content was determined using a BCA Assay (Pierce Chemical Co., Woburn, MA), and 20 μg total protein was loaded on a 10% SDS-PAGE. Proteins were immunoblotted on a nylon membrane using a wet blot cell, the membrane was blocked with 5% skimmed milk in PBS-Tween 0.5%, and ER was detected with MC-20 antibody (sc-542; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) in blocking solution. Detection was performed with a horseradish peroxidase-labeled goat-antirabbit antibody and enhanced chemiluminescence detection system (Amersham, Buckinghamshire; UK).
Cell culture and transient transfections
HeLa cells were cultured in DMEM plus 10% fetal bovine serum (Life Technologies, Inc., Gaithersburg, MD). Cells were plated, 24 h before transfection, in phenol red-free medium with 5% dextran charcoal-stripped fetal calf serum. Cells were transfected using a modified calcium, phosphate coprecipitation method with either ICAM-tk-Luc reporter plasmid containing three NF-κB sites, pSG5-RelA and pSG5-ERα, or EAAE-ERα (plasmids described in Refs. 15 and 31) or another reporter plasmid, 2×ERE-pS2-pGL3, was transfected together with the ER expression plasmids pSG5-ERα or EAAE-ERα. After incubation for 16 h, cells were washed and incubated with 10−8 m 17β-estradiol for 24 h. Cell extracts were assayed for luciferase using a dual reporter assay as described elsewhere (15, 31).
Animals
Animals were housed in approved facilities, and all experiments were performed to the highest institutional standards in accordance with local regulations. For the global genome-wide gene expression profile, 10-wk-old wild-type mice (C57BL/6) and EAAE mice were ovariectomized. After 14 d of recovery, mice were treated perorally with either 100 μl vehicle (benzylbenzoat rizinus oil 1/4) or 100 μg/kg EE in vehicle (five mice per treatment group). The mice were dissected 4 or 24 h later, and uterus and liver were frozen in liquid nitrogen.
For the gene repression analysis with IL-1β, 10-wk-old mice were also ovariectomized and 14 d later, five mice per treatment group were treated by daily sc injections of vehicle or EE (100 μg/kg). On the fifth day of treatment, 1 h after receiving the sc injection, the mice received an ip injection of PBS containing 20 μg/kg IL-1β (GTX29723; Gene Tex, San Antonio, TX). The mice were dissected 1 h later and the livers were frozen in liquid nitrogen.
Total RNA extraction
Liver tissues were homogenized using liquid nitrogen and mortar and pistil. The uterine tissues were homogenized by a Precellys24 Lysis and Homogenizer (Bertin Technologies; Saint-Quentin-en-Yveline Cedex, France) with 2.8-mm ceramic beads (6000 rpm two times for 20 sec; 20 sec break). Total RNA was prepared using a RNeasy Mini Kit with a deoxyribonuclease I digestion performed on the column (QIAGEN; Hilden, Germany). The quality of the RNA was analyzed on an Agilent Bioanalyzer (Agilent Technologies; Santa Clara, CA). The RNA integrity values ranged from 6.8–8.8. The concentration of the RNA was determined with a Peqlab Nanodrop (Peqlab Biotechnologies GmbH; Erlangen, Germany).
Production of labeled cRNA for Illumina BeadChip analysis
Using the Illumina Total Prep RNA Amplification Kit (Ambion; Cambridgeshire; UK; IL-1791) 100 ng of deoxyribonuclease I-digested total RNA was subjected to first- and second-strand cDNA synthesis. Purification of cDNA was performed with cDNA filter cartridges included in the kit. The in vitro transcription was performed according to the manufacturer’s protocol. The biotinylated cRNA was purified with a cRNA filter cartridge. cRNA (1.5 μg) was hybridized to Illumina Mouse Sentrix WG-6v1.1 BeadChips interrogating more than 47,000 murine transcripts based on the MEEBO, RefSq, and RIKEN Famtom2 content. The BeadChips were stained and washed according to the manufacturer’s instructions. The BeadChips were scanned on an Illumina Beadstation 500× (Illumina; San Diego, CA). RNA isolated from four to five mice per treatment group was hybridized to Illumina BeadChips.
BeadChip data quality control
BeadChip raw data output contains the average signal intensity and the detection P value for each probe (one or several different 50 mer detector oligonucleotides per interrogated transcript with approximately 30 replicate beads per probe type). Signals were logarithmized, and a quality control was carried out using box plots of signal intensity, unsupervised hierarchical clustering, and Principal Components Analysis for both unnormalized and LOWESS-normalized data (Illumina BeadStudio version 3.1 and R). The analysis revealed even signal distribution within the set of experiments and highly correlated expression data with the exception of one outlier experiment that was removed in subsequent analyses. Analysis of signal to GC-content relation showed a maximum of signal intensity for a GC content of 20–30 of 50 nucleotides indicating optimal hybridization conditions.
Statistical analysis
Signals were logarithmized (log2) and LOWESS-normalized. N-way ANOVA was used to investigate the influence of the factors time, animal, and treatment. A three-way ANOVA of all three factors was used to compare the relative contributions of these factors to overall expression changes. Most variability could be explained by the factor time, mutant status showed medium effects, and treatment showed only minor effects. The three-way ANOVA with effect treatment identified genes differentially expressed by compound treatment irrespective of time and animal, thereby revealing potential adverse effects. We also constructed a two-level factor hypothetical effect summarizing animal and treatment. One level of this factor corresponds to compound-treated wild-type animals, the only group that is expected to show strong effects by the compound; the other level corresponds to the remaining groups, i.e. vehicle-treated animals and mutants. A two-way ANOVA of the factors, time and hypothetical effect, identified genes that correlate with the expected response, irrespective of time. Finally, four pair-wise t tests were used to identify differential expression caused by compound treatment for each time point and each animal type (wild type or mutant). Genes with P < 0.001 and a fold change larger than 1.5 in at least one of the four pair-wise comparisons are summarized in supplemental Table S1.
Clustering
A heat map was generated by hierarchical clustering of probes based on logarithmized fold changes from the four comparisons described above. Only probes with P < 0.001 in at least one of the four pair-wise comparisons entered the clustering process (302 of 46,643). Eucledian distance with average linkage was used. P values were not corrected for multiple testing.
Quantitative real-time PCR
Selected regulated genes identified by genome-wide gene expression profiling (GEP) were analyzed by qRT-PCR using an ABI PRISM 7700 Sequence Detection System and 7900 HT Fast Real-Time PCR System according to the manufacturer‘s protocol (Applied Biosystems; Foster City, CA). For the qRT-PCR 1 μg total RNA was reverse transcribed with the SuperScript III First-Strand Synthesis System for RT-PCR from Invitrogen (Carlsbad, CA) using random primers. For reactions with Micro Fluidic Cards, 500 ng RNA was reverse transcribed with High Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems). TaqMan probes and Micro Fluidic Cards (Applied Biosystems TaqMan Low Density Array) were obtained from Applied Biosystems (Table 1). The Platinum qPCR SuperMix UDG from Invitrogen was used for qRT-PCR. Taq Man Universal PCR Master Mix (Applied Biosystems) was used for analysis with Micro Fluidic Cards. The data were analyzed using Sequence Detector version 1.7 and 2.3 and SDS RQ Manager 1.2 software (PE Applied Biosystems, Foster City, CA) and normalized to cyclophilin.
Amplification was carried out as follows: 1) 50 C, 2 min (for uracil-N-glycosylase incubation); 2) 94 C, 10 min (denaturation); 3) 97 C, 30 sec; 4) 59.7 C, 1 min (denaturation/ amplification) using the 7900 HT Fast Real-Time PCR System. ABI PRISM 7700 Sequence Detection System was used as follows: 1) 50 C, 2 min 2) 95 C, 10 min (denaturation); 3) 95 C, 15 sec; 4) 60 C, 1 min (denaturation/ amplification). In both systems the reactions took 40 cycles. All the listed genes with the exception of fnk (Fig. 6) and igf-1 (Fig. 3) were analyzed in duplicates using Micro Fluidic Cards. Each sample was normalized to cyclophilin. Finally the mean of the normalized values of one treatment group was calculated. To validate the Illumina microarray analysis, four to five animals per treatment group were used. Igf-1 (Fig. 3) was validated in triplicates by qRT-PCR.
The expression study of fnk (Fig. 6) was performed in two replicates with three to five animals per group.
ChIP
HeLa cells were cultivated in DMEM (GIBCO) supplemented with 10% fetal calf serum, at 37 C under 7.5% CO2. ChIP experiments were conducted with 5 × 106 cells synchronized after 3 d of culture in 2% dextran-charcoal-treated fetal calf serum and transfected with 10 μg hERα full-length (Hego expression plasmid) DNA and 10 μg EAAE construct (hER(EAAE)pSG5). These DNAs and also a Renilla luciferase construct as transfection control (phRL-TK 1.5μg per plate) were transfected with FuGene 6-reagent (Roche). The ChIP started with a treatment with 2.5 μm α-amanitin for 2 h, followed by exposure to 10−8 m E2 or dimethylsulfoxide (0.1%). Chromatin was cross linked using 1.5% formaldehyde for 5 min and 125 mm glycine for 5 min at 37 C. The cells were collected after two washings with PBS in collection buffer (10 mm Tris-HCl, pH 8; and 150 mm NaCl; 1 mm EDTA) and were slightly centrifuged (1000 × g) at 4 C. The cell pellet was lysed in 300 μl lysis buffer [1% sodium dodecyl sulfate (SDS), 10 mm EDTA, 50 mm Tris-HCl (pH 8), 10 mm β-glycerophosphate, 0.5% empigen BB (Sigma)] and sonicated two times for 8 min at maximum and pulsed (30 sec on/30 sec off) settings (Bioruptor; Diagenode, Liège, Belgium).
The DNA fragments under the range of 200-1000 kb. After centrifugation 60 μl was used as input, and the remainder was diluted with 3 ml IP buffer [0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl (pH 8), 167 mm NaCl, 10 mm β-glycerophosphate]. The dilution was precleared with 60 μl of salmon sperm DNA/Protein A agarose-50% slurry (Millipore Corp., Bedford, MA) for 2 h at 4 C. After splitting each sample in two, one of the dilutions was incubated with 4 μg ERα antibody (HC-20; sc-543) (Santa Cruz) and the other with 20 μl preimmune IgG (Sigma) overnight at 4 C. Complexes were recovered by a 2-h incubation at 4 C with 70 μl salmon sperm DNA/Protein A agarose-50% slurry. Precipitates were washed with 500 μl washing buffer I [2 mm EDTA, 20 mm Tris-HCl (pH 8), 0.1% SDS, 1% Triton X-100, 150 mm NaCl], washing buffer II [2 mm EDTA, 20 mm Tris-HCl (pH 8), 0.1% SDS, 1% Triton X-100, 500 mm NaCl], washing buffer III [1 mm EDTA, 10 mm Tris-HCl (pH 8), 1% Nonidet P-40, 10 mm deoxycholate, 0.25 m LiCl] and then twice with 1 mm EDTA, 10 mm Tris-HCl (pH 8). Precipitated chromatin complexes were removed from the beads via a 30-min incubation with 50 μl of 1% SDS, 0.1 m NaHCO3, with vortexing each 5 min. This step was repeated with 10-min incubation times. All buffers were supplemented with 1× protease inhibitor cocktail (complete minus EDTA, Roche). Cross-linking was reversed by an overnight incubation at 65 C. DNA was purified with QIAquick columns (QIAGEN), as indicated by the manufacturer. Quantitative PCRs were done with 13 μl SybrGreen (Invitrogen), 2 μl DNA, 8.5 μl H2O, and 1.5 μl pS2 primer [−409fwd-5′-ATG GGC TTC ATG AGC TCC-3′; −266rev-5′-AGG GTA AAT ACT GTA CTC AC-3′] (20 pmol/μl), cyclophilin primer [hCyclo_Taq_fwd-5′-GAA GTT GGC CGC ATG AAG A-3′; hCyclo_Taq_rev-5′-GCC TAA AGT TCT CGG CCG T-3′] (20 pmol/μl) and cyclin D1 primer [cyclinD1 Fwd (−204)-5′-GGC GAT TTG CAT TTC TAT GA-3′: cyclin D1 Rv (+32)-5′-CAA AAC TCC CCT GTA GTC CGT-3′] (20 pmol/μl).
The data were analyzed using Sequence Detector version 1.6.3 (PE Applied Biosystems). ABI PRISM 7700 Sequence Detection System was used as follows: 1) 50 C, 2 min; 2) 95 C, 10 min (denaturation); 3) 95 C, 15 sec; 4) 60 C, 1 min (denaturation/amplification).
Acknowledgments
We thank John Schwabe for helpful advice on the ERα DBD mutation; Heidrun Kern, Heike Alter, and Sandra Fehsenfeld [Deutsches Krebsforschungszentrum (DKFZ)] for expert technical assistance in generating the gene-targeted mouse model; Anke Schultze for animal work; Martina Sperling and Daniela Launhardt for Illumina array experiments; and Frank Kleinjung (MicroDiscovery, Berlin) and Jörg Müller for help with analysis of Illumina data.
NURSA Molecule Pages:
Ligands: 17α-ethinylestradiol | 17β-estradiol;
Nuclear Receptors: ER-α.
Footnotes
B.D.S. was supported by a Human Frontier Fellowship. Work in the laboratory of G.S. was supported by the Deutsche Forschungsgemeinschaft through Collaborative Research Centre 636 and by the European Union through Grant LSHM-CT-2005-018652 (CRESCENDO).
Present address for B.D.S.: Phenex-Pharmaceuticals AG, c/o Werksgelände BASF, Tor 5, D-67056 Ludwigshafen, Germany.
Disclosure Summary: D. A.-D., G. L., J. S., S. T., H. S., A. S., and T. W. are employees of Bayer Schering Pharma AG; G. L., A. S., and T. W. hold shares in Bayer AG; B.D.S. has a patent that was received for the mice; M.G.P. and G.S. have nothing to declare.
First Published Online July 2, 2009
D.L.A.-D. and B.D.S. contributed equally to this study.
Abbreviations: AP-1, Activator protein-1; ChIP, chromatin immunoprecipitation; DBD, DNA-binding domain; E2, 17β-estradiol; EE, ethinylestradiol; ER, estrogen receptor; ERE, estrogen-responsive element; αERKO, αER knock-out; ENERKI, estrogen-nonresponsive ERα knock-in; GEP, genome-wide gene expression profiling; LBD, ligand-binding domain; NERKI, nonresponsive ERα knock-in; NF-κB, nuclear factor κB; qRT-PCR, quantitative RT-PCR; SDS, sodium dodecyl sulfate; SERM, selective estrogen receptor modulator.
References
- 1.Hall JM, Couse JF, Korach KS2001. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem 276:36869–36872 [DOI] [PubMed] [Google Scholar]
- 2.Shang Y, Brown M2002. Molecular determinants for the tissue specificity of SERMs. Science 295:2465–2468 [DOI] [PubMed] [Google Scholar]
- 3.Castro-Rivera E, Samudio I, Safe S2001. Estrogen regulation of cyclin D1 gene expression in ZR-75 breast cancer cells involves multiple enhancer elements. J Biol Chem 276:30853–30861 [DOI] [PubMed] [Google Scholar]
- 4.O'Brien JE, Peterson TJ, Tong MH, Lee EJ, Pfaff LE, Hewitt SC, Korach KS, Weiss J, Jameson JL2006. Estrogen-induced proliferation of uterine epithelial cells is independent of estrogen receptor α binding to classical estrogen response elements. J Biol Chem 281:26683–26692 [DOI] [PubMed] [Google Scholar]
- 5.Dubik D, Shiu RP1992. Mechanism of estrogen activation of c-myc oncogene expression. Oncogene 7:1587–1594 [PubMed] [Google Scholar]
- 6.Umayahara Y, Kawamori R, Watada H, Imano E, Iwama N, Morishima T, Yamasaki Y, Kajimoto Y, Kamada T1994. Estrogen regulation of the insulin-like growth factor I gene transcription involves an AP-1 enhancer. J Biol Chem 269:16433–16442 [PubMed] [Google Scholar]
- 7.Evans MJ, Eckert A, Lai K, Adelman SJ, Harnish DC2001. Reciprocal antagonism between estrogen receptor and NF-κB activity in vivo. Circ Res 89:823–830 [DOI] [PubMed] [Google Scholar]
- 8.Boverhof DR, Fertuck KC, Burgoon LD, Eckel JE, Gennings C, Zacharewski TR2004. Temporal- and dose-dependent hepatic gene expression changes in immature ovariectomized mice following exposure to ethynyl estradiol. Carcinogenesis 25:1277–1291 [DOI] [PubMed] [Google Scholar]
- 9.Schaner ME, Ross DT, Ciaravino G, Sorlie T, Troyanskaya O, Diehn M, Wang YC, Duran GE, Sikic TL, Caldeira S, Skomedal H, Tu IP, Hernandez-Boussard T, Johnson SW, O'Dwyer PJ, Fero MJ, Kristensen GB, Borresen-Dale AL, Hastie T, Tibshirani R, van de Rijn M, Teng NN, Longacre TA, Botstein D, Brown PO, Sikic BI2003. Gene expression patterns in ovarian carcinomas. Mol Biol Cell 14:4376–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS1997. Differential ligand activation of estrogen receptors ERα and ERβ at AP1 sites. Science 277:1508–1510 [DOI] [PubMed] [Google Scholar]
- 11.Turgeon JL, McDonnell DP, Martin KA, Wise PM2004. Hormone therapy: physiological complexity belies therapeutic simplicity. Science 304:1269–1273 [DOI] [PubMed] [Google Scholar]
- 12.Blum A, Cannon 3rd RO1998. Effects of oestrogens and selective oestrogen receptor modulators on serum lipoproteins and vascular function. Curr Opin Lipidol 9:575–586 [DOI] [PubMed] [Google Scholar]
- 13.Battaglioli T, Martinelli I2007. Hormone therapy and thromboembolic disease. Curr Opin Hematol 14:488–493 [DOI] [PubMed] [Google Scholar]
- 14.Douketis JD, Gordon M, Johnston M, Julian JA, Adachi JR, Ginsberg JS2000. The effects of hormone replacement therapy on thrombin generation, fibrinolysis inhibition, and resistance to activated protein C: prospective cohort study and review of literature. Thromb Res 99:25–34 [DOI] [PubMed] [Google Scholar]
- 15.García-Pedrero JM, Kiskinis E, Parker MG, Belandia B2006. The SWI/SNF chromatin remodeling subunit BAF57 is a critical regulator of estrogen receptor function in breast cancer cells. J Biol Chem 281:22656–22664 [DOI] [PubMed] [Google Scholar]
- 16.Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O1993. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA 90:11162–11166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M2000. Effect of single and compound knockouts of estrogen receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development 127:4277–4291 [DOI] [PubMed] [Google Scholar]
- 18.Jakacka M, Ito M, Martinson F, Ishikawa T, Lee EJ, Jameson JL2002. An estrogen receptor (ER)α deoxyribonucleic acid-binding domain knock-in mutation provides evidence for nonclassical ER pathway signaling in vivo.Mol Endocrinol 16:2188–2201 [DOI] [PubMed] [Google Scholar]
- 19.Hewitt SC, Collins J, Grissom S, Deroo B, Korach KS2005. Global uterine genomics in vivo: microarray evaluation of the estrogen receptor α-growth factor cross talk mechanism. Mol Endocrinol 19:657–668 [DOI] [PubMed] [Google Scholar]
- 20.Hewitt SC, Deroo BJ, Hansen K, Collins J, Grissom S, Afshari CA, Korach KS2003. Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Mol Endocrinol 17:2070–2083 [DOI] [PubMed] [Google Scholar]
- 21.Sinkevicius KW, Burdette JE, Woloszyn K, Hewitt SC, Hamilton K, Sugg SL, Temple KA, Wondisford FE, Korach KS, Woodruff TK, Greene GL2008. An estrogen receptor-α knock-in mutation provides evidence of ligand-independent signaling and allows modulation of ligand-induced pathways in vivo Endocrinology 149:2970–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans MJ, Lai K, Shaw LJ, Harnish DC, Chadwick CC2002. Estrogen receptor α inhibits IL-1β induction of gene expression in the mouse liver. Endocrinology 143:2559–2570 [DOI] [PubMed] [Google Scholar]
- 23.Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schütz G1998. DNA binding of the glucocorticoid receptor is not essential for survival. Cell 93:531–541 [DOI] [PubMed] [Google Scholar]
- 24.Gao H, Fält S, Sandelin A, Gustafsson JA, Dahlman-Wright K2008. Genome-wide identification of estrogen receptor α-binding sites in mouse liver. Mol Endocrinol 22:10–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krum SA, Miranda-Carboni GA, Lupien M, Eeckhoute J, Carroll JS, Brown M2008. Unique ERα cistromes control cell type-specific gene regulation. Mol Endocrinol 22:2393–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wintermantel TM, Campbell RE, Porteous R, Bock D, Gröne HJ, Todman MG, Korach KS, Greiner E, Pérez CA, Schütz G, Herbison AE2006. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron 52:271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vente A, Korn B, Zehetner G, Poustka A, Lehrach H1999. Distribution and early development of microarray technology in Europe. Nat Genet 22:22. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Buchholz F, Muyrers JP, Stewart AF1998. A new logic for DNA engineering using recombination in Escherichia coli Nat Genet 20:123–128 [DOI] [PubMed] [Google Scholar]
- 29.Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G1999. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23:99–103 [DOI] [PubMed] [Google Scholar]
- 30.Wintermantel TM, Bock D, Fleig V, Greiner EF, Schütz G2005. The epithelial glucocorticoid receptor is required for the normal timing of cell proliferation during mammary lobuloalveolar development but is dispensable for milk production. Mol Endocrinol 19:340–349 [DOI] [PubMed] [Google Scholar]
- 31.Valentine JE, Kalkhoven E, White R, Hoare S, Parker MG2000. Mutations in the estrogen receptor ligand binding domain discriminate between hormone-dependent transactivation and transrepression. J Biol Chem 275:25322–25329 [DOI] [PubMed] [Google Scholar]
- 32.Wintermantel TM, Elzer J, Herbison AE, Fritzemeier KH, Schutz G2006. Genetic dissection of estrogen receptor signaling in vivo. Ernst Schering Found Symp Proc:25–44 [DOI] [PubMed]