

Abstract
Human amnion fibroblasts produce abundant prostaglandins toward the end of gestation, which is one of the major events leading to parturition. In marked contrast to its well-described antiinflammatory effect, glucocorticoids have been shown to up-regulate cyclooxygenase-2 (COX-2) expression in human amnion fibroblasts. The mechanisms underlying this paradoxical induction of COX-2 by glucocorticoids have not been resolved. Using cultured human amnion fibroblasts, we found that the induction of COX-2 mRNA expression by cortisol was a glucocorticoid receptor (GR)-dependent process requiring ongoing transcription. Upon transfection of a COX-2 promoter-driven reporter gene into the amnion fibroblasts, cortisol stimulated the COX-2 promoter activity. This was abolished by mutagenesis of a cAMP response element (CRE) at −53 to approximately −59bp as well as by cotransfection of a plasmid expressing dominant-negative CRE-binding protein (CREB). The phosphorylation level of CREB-1 was significantly increased by cortisol treatment of the amnion fibroblasts, whereas the effect was attenuated either by the protein kinase A inhibitor H89 or the p38 -MAPK inhibitor SB203580. The induction of the COX-2 promoter activity and the phosphorylation of CREB-1 were also blocked by the GR antagonist RU486. Chromatin immunoprecipitation (ChIP) assay revealed that the binding of CREB-1 to the CRE of the COX-2 promoter was increased by cortisol treatment of the amnion fibroblasts. In conclusion, cortisol, via binding to GR, stimulated COX-2 expression by increasing phosphorylated CREB-1 binding to the CRE of the COX-2 gene. Cortisol may phosphorylate CREB-1 by activating either protein kinase A or p38-MAPK in the amnion fibroblasts.
Cortisol induces COX-2 by increasing phosphorylated CREB via PKA or p38-MAPK and its binding to the CRE of the COX-2 gene in human amnion fibroblasts.
Preterm labor occurs in approximately 8–10% of all pregnancies (1, 2), which accounts for more than 75% of the perinatal mortality and morbidity rate (1, 2). Although the survival rates for very early preterm births have increased because of technological advances, the neurodevelopmental disabilities and recurrent health problems are common into early childhood or even persist into adolescence especially for cardiovascular and metabolic disorders (2). Despite progress in this field, the lack of identification of the mechanism of human parturition has limited the specific and effective diagnosis and treatment of preterm labor. In most mammalian species, there is an increase in glucocorticoid concentration in maternal and fetal circulations as well as amniotic fluid toward the end of gestation and at the onset of labor (3, 4). This surge of glucocorticoid is believed to be crucial to maturation of fetal organs as well as to be integral to the cascade of events in the initiation and maintenance of labor (5).
Prostaglandins (PGs), especially PGE2 and PGF2α, have been identified as key factors inducing cervical ripening, myometrial contraction, and fetal membrane rupture at term (6, 7). The increase in PG synthesis at term and parturition is believed to be associated with increased expression of the enzyme cyclooxygenase (COX), the enzyme that converts arachidonic acid to PGH2, the precursor of the two series prostanoids, which is a rate-limiting step in the formation of PG (8). At least three different COX enzyme isoforms exist, COX-1, COX-2, and COX-3. COX-1 is a constitutively expressed enzyme, being found in most mammalian cells, whereas COX-3 is an inactive alternatively spliced variant of COX-1. COX-2, however, is an inducible enzyme, being rapidly up-regulated in inflammatory cells and responsible for abundant PG production (9). Glucocorticoids are commonly used in the treatment of immune and inflammatory disorders. One of the major reported mechanisms of glucocorticoid modulation of the inflammatory response is to repress COX-2 expression and subsequent PG production (10, 11). However, in pregnancy, there is a paradoxical concurrent increase in COX-2 expression and subsequent PG production with the rise of glucocorticoid concentration in the fetal membranes at term (12, 13, 14, 15, 16), which is believed to be one of the major events leading to labor (6).
The human fetal membranes consist of two layers of tissues, the amnion and chorion, with the amnion as a major source of PG at term gestation (6, 7, 8, 12). Although both amnion epithelial cells and fibroblasts are capable of PG synthesis, amnion fibroblasts produced much more PGE2 per cell than amnion epithelial cells at term (13, 14). We and others have shown that glucocorticoids exert potent stimulation of PG output by inducing the expression of COX-2 in the amnion fibroblasts (13, 14), which is in marked contrast to the reported inhibitory action of glucocorticoids on COX-2 expression and PG production at inflammation (10, 11).
Sequence analysis of the 5′-flanking region of the COX-2 gene reveals several potential transcription regulatory sequences, including a C/EBP (CCAAT enhancer binding protein) motif, two AP-2 (activator protein-2) sites, three Sp1 sites, two NFκB (nuclear factor-κB) sites, a CRE (cAMP response element), and a TATA box (17, 18). Although glucocorticoids are known to down-regulate the induced COX-2 expression by proinflammatory cytokines by interfering with NFκB function at inflammation (10, 19), the mechanisms of glucocorticoid-induced COX-2 expression in human amnion fibroblasts remain largely unknown. Neither are we clear about the promoter response element mediating glucocorticoid-induced COX-2 expression in human amnion fibroblasts. In this study, we investigated the promoter elements whereby glucocorticoids stimulate COX-2 expression in human amnion fibroblasts and compared these effects to an immortalized cell line, human fetal lung fibroblasts (HFL-1).
Results
Differential effects of cortisol on COX-2 mRNA expression in human amnion fibroblasts and HFL-1 cells
Treatment of human amnion fibroblasts with cortisol (0.01–1 μm) induced COX-2 mRNA expression in a concentration-dependent manner (Fig. 1A). In accordance with the up-regulation of COX-2 mRNA expression, PGE2 output from cultured human amnion fibroblasts was also significantly increased by cortisol (1 μm) (Fig. 1B). The stimulatory effect of cortisol (1 μm) on COX-2 mRNA expression in human amnion fibroblasts was completely blocked by either the glucocorticoid receptor (GR) antagonist RU486 (1 μm) (Fig. 1C) or by the mRNA transcription inhibitor 5,6-dichlorobenzimidazole riboside (DRB, 75 μm) (Fig. 1D) or reversed by the protein synthesis inhibitor cycloheximide (CHX, 10 μm) (Fig. 1E), suggesting this stimulatory effect of cortisol on COX-2 expression is a GR-mediated induction of mRNA transcription via induction of de novo protein synthesis. In contrast, cortisol (1 μm) significantly inhibited COX-2 mRNA expression and PGE2 output in HFL-1 cells (Fig. 2).
Fig. 1.

A, Concentration-dependent induction of COX-2 mRNA expression by cortisol (F) treatment of human amnion fibroblasts for 24 h; B, stimulation of PGE2 production by cortisol (1 μm) treatment of human amnion fibroblasts for 24 h; C, inhibition of cortisol-induced (1 μm) COX-2 mRNA expression by cotreatment of human amnion fibroblasts with GR antagonist RU486 (RU) (1 μm) for 24 h; D, inhibition of cortisol-induced (1 μm) COX-2 mRNA expression by pretreatment for 0.5 h and cotreatment with mRNA transcription inhibitor DRB (75 μm) for 24 h in human amnion fibroblasts; E, reversal of cortisol-induced (1 μm) COX-2 mRNA expression by pretreatment for 0.5 h and cotreatment with protein synthesis inhibitor CHX (10 μm) for 24 h in human amnion fibroblasts. n = 3–4; *, P < 0.05; **, P < 0.01 vs. respective control (ctr); ##, P < 0.01 vs. cortisol.
Fig. 2.

Inhibition of COX-2 mRNA expression (A) and PGE2 production (B) by cortisol (F, 1 μm) treatment of human fetal lung fibroblasts (HFL-1) for 24 h. n = 3; **, P < 0.01 vs. control (ctr).
Sequence analysis of the COX-2 promoter cloned from human amnion fibroblasts
Sequencing of the COX-2 promoter (−886 to +31 bp) cloned from human amnion fibroblasts (Fig. 2) showed full alignment with the COX-2 promoter sequence reported in NCBI GenBank (GenBank no. U44805). Bioinformatic analysis of the cloned COX-2 promoter with Transcription Element Search System (TESS) revealed a TATA box at −26 to −31 bp, a CRE at −53 to −59 bp, two NFκB sites at −233 to −214 and −448 to −439 bp, respectively, and a putative glucocorticoid response element (GRE) at −725 to −718 bp (Fig. 3).
Fig. 3.

Sequence of the cloned COX-2 promoter spanning from −886 to +31 bp from human amnion fibroblasts. The arrows pointing right indicate the positions of 5′ primers used for progressive deletion of the promoter fragment with PCR, and the arrow pointing left indicates the position of the 3′ primer. The boxed bold letters indicate the putative transcription factor binding sites analyzed with TESS.
Differential effects of cortisol on the COX-2 promoter activity in human amnion fibroblasts and HFL-1 cells
Upon transfection of the constructed plasmid carrying −886 bp of the COX-2 promoter and luciferase reporter gene into human amnion fibroblasts, cortisol (0.01–1 μm) up-regulated the COX-2 promoter activity in a concentration-dependent manner (Fig. 4A). The effect of cortisol (1 μm) was fully abolished by the GR antagonist RU486 (1 μm) (Fig. 4B), suggesting that this stimulatory effect of cortisol on the COX-2 promoter activity was dependent on GR. Progressive deletion of the COX-2 promoter from the 5′ end to the 3′ end revealed that cortisol (1 μm) significantly stimulated all the constructed COX-2 promoter activities including −886, −694, −340, −201, −101, and −66 bp (Fig. 4C), suggesting the element responsible for the up-regulating effect of cortisol on the COX-2 promoter activity in human amnion fibroblasts resides downstream to −66 bp and upstream to the transcription start site.
Fig. 4.

A, Concentration-dependent induction of COX-2 promoter (−886 bp) activity by cortisol (F) treatment of human amnion fibroblasts for 24 h; B, inhibition of cortisol-induced (1 μm) increase of COX-2 promoter (−886 bp) activity by cotreatment of human amnion fibroblasts with GR antagonist RU486 (RU) (1 μm) for 24 h; C, cortisol (1 μm) treatment of human amnion fibroblasts for 24 h increased the COX-2 promoter activities of −886, −694, −340, −201, −101, and −66 bp (top panel). The primer sequences for subcloning the COX-2 promoter fragments are listed in the bottom panel. The lowercase letters indicate the added nucleotides for the digestion with restriction enzymes (sense, KpnI; antisense, BglII). n = 3–4; **, P < 0.01 vs. control (ctr) without cortisol treatment; ##, P < 0.01 vs. cortisol. Luc, Luciferase.
In contrast, cortisol (∼0.01–1 μm) down-regulated the COX-2 promoter activity (−886 bp) in a concentration-dependent manner in HFL-1 cells (Fig. 5A). This inhibitory effect of cortisol (1 μm) could also be fully abolished by the GR antagonist RU486 (1 μm) (Fig. 5B), suggesting that this inhibitory effects of cortisol on the COX-2 promoter activity was dependent on GR as well. In addition to the inhibition of the COX-2 promoter activity by cortisol itself, cortisol (1 μm) also significantly attenuated the induction of the COX-2 promoter activity by IL-1β (10 ng/ml) in HFL-1 cells (Fig. 5C). Progressive deletion of the COX-2 promoter from the 5′ end to the 3′ end revealed that the element responsible for the down-regulating effect of cortisol on the COX-2 promoter activity in HFL-1 cells localized between −453 and −340 bp, because cortisol (1 μm) significantly inhibited the COX-2 promoter activities of −886, −694, and −453 bp but not those of −340, −201, and −101 bp (Fig. 5D).
Fig. 5.

A, Concentration-dependent inhibition of COX-2 promoter (−886 bp) activity by cortisol (F) treatment of human fetal lung fibroblasts (HFL-1) for 24 h; B, blockade of cortisol-induced (1 μm) inhibition of COX-2 promoter (−886 bp) activity by cotreatment of HFL-1 cells with GR antagonist RU486 (RU) (1 μm) for 24 h; C, inhibition of IL-1β-induced (10 ng/ml) increase of COX-2 promoter (−886 bp) activity by cotreatment of HFL-1 cells with cortisol (1 μm) for 24 h; D, cortisol (1 μm) treatment of HFL-1 cells for 24 h decreased the COX-2 promoter activities of −886, −694, and −453 but not −340, −201, or −101 bp (top panel). The primer sequences for subcloning the COX-2 promoter fragments are listed in the bottom panel. The lowercase letters indicate the added nucleotides for the digestion with restriction enzymes (sense, KpnI; antisense, BglII). n = 3; *, P < 0.05; **, P < 0.01 vs. control (ctr) without cortisol treatment; ##, P < 0.01 vs. cortisol. Luc, Luciferase.
CRE and NFκB binding sites mediate the differential effects of cortisol on COX-2 promoter activity in human amnion fibroblasts and HFL-1 cells
Analysis of the promoter sequence downstream to −66 bp and upstream to the transcription start site with TESS revealed a TATA box at −26 to −31 bp and a CRE at −53 to −59 bp. Because the CRE is often located within the proximal region of transcriptional start site, providing the interaction of CRE-binding protein (CREB) with basal transcription factors at the site of the TATA box for initiation of transcription, we predicted that this CRE was very likely to mediate the stimulatory effect of cortisol on the COX-2 promoter in human amnion fibroblasts. Site-directed mutagenesis of this CRE indeed abolished the up-regulatory effect of cortisol (1 μm) and the stimulatory effect of forskolin (100 μm) as well on the COX-2 promoter activity in human amnion fibroblasts (Fig. 6A), indicating that cortisol and forskolin use the same CRE to increase the COX-2 promoter activity in human amnion fibroblasts. Consistent with the promoter activity, forskolin (100 μm) also significantly increased COX-2 mRNA level in cultured human amnion fibroblasts (Fig. 6B).
Fig. 6.
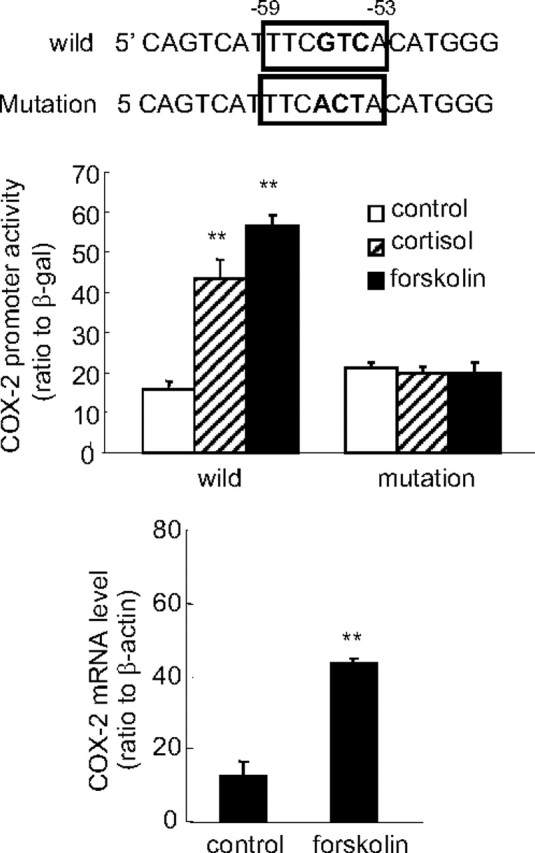
Treatment of human amnion fibroblasts with forskolin (100 μm), an activator of adenylyl cyclase, for 24 h increased COX-2 promoter (−66 bp) activity (top panel) and COX-2 mRNA level (bottom panel). Introduction of nucleotide mutations into the CRE at −53 to −59 bp diminished the stimulation of COX-2 promoter (−66 bp) activity by both forskolin (100 μm) and cortisol (1 μm) in human amnion fibroblasts. The boxed nucleotide sequence shown on top is the CRE of the COX-2 promoter, and the bold letters represent the mutated nucleotides. n = 3; **, P < 0.01 vs. control without forskolin or cortisol treatment.
Analysis of the promoter sequence downstream to −453 bp and upstream to −340 bp with TESS revealed a NFκB binding site at −448 to −439 bp. Because NFκB is one of the key transcription factors mediating the induction of COX-2 by proinflammatory cytokines and cortisol was demonstrated to be able to attenuate the induction of COX-2 by IL-1β in HFL-1 cells, we predicted that this NFκB binding site was very likely to mediate the inhibition of COX-2 expression by cortisol itself in HFL-1 cells. Introduction of nucleotide mutations into this NFκB binding site indeed fully abolished the inhibitory effect of cortisol on COX-2 promoter activity in HFL-1 cells (Fig. 7).
Fig. 7.

Introduction of nucleotide mutations into the NFκB binding site at −448 to −439 bp diminished the inhibition of COX-2 promoter (−453 bp) activity by cortisol (1 μm) in human fetal lung fibroblasts (HFL-1). The boxed nucleotide sequence shown on top is the NFκB binding site of the COX-2 promoter, and the bold letters represent the mutated nucleotides. n = 3; **, P < 0.01 vs. control without cortisol treatment.
Differential regulation of CREB-1 phosphorylation by cortisol in human amnion fibroblasts and HFL-1 cells
Both CREB-1 and phospho (p)-CREB-1 antibodies used in Western blotting analysis revealed two major protein bands around 43 and 37 kDa, which were believed to be CREB-1 and activating transcription factor-1 (ATF-1), respectively. The level of phosphorylated CREB-1 but not the total CREB-1 was increased by cortisol (1 μm) treatment of the human amnion fibroblasts (Fig. 8). The effect of cortisol (1 μm) in human amnion fibroblasts was not only blocked by GR antagonist RU486 (10 μm) but also blocked by the protein kinase A (PKA) inhibitor H89 (20 μm) or the p38-MAPK inhibitor SB203580 (10 μm) (Fig. 8), suggesting that activation of both PKA and p38-MAPK is involved in the phosphorylation of CREB-1 by cortisol, and the activation is mediated by GR. Consistently, inhibition of PKA with H89 (20 μm) and inhibition of p38-MAPK with SB203580 (10 μm) also significantly attenuated the induction of the COX-2 mRNA expression and the COX-2 promoter activity by cortisol (1 μm) in human amnion fibroblasts (Fig. 9), suggesting that CREB-1 phosphorylated by both PKA and p38 MAPK plays an important role in the induction of COX-2 expression by cortisol in human amnion fibroblasts. In marked contrast to human amnion fibroblasts, cortisol (1 μm) treatment of HFL-1 cells for 12 h significantly decreased p-CREB-1 level but not the total CREB-1 (Fig. 10A). A time-course study showed that the reduction in p-CREB-1 level required longer cortisol incubation time (6 and 12 h) and shorter incubation time (0.5 and 1 h) did not cause obvious changes in p-CREB-1 level in HFL-1 cells (Fig. 10B), suggesting that the reduction of p-CREB-1 level by cortisol requires ongoing protein synthesis. Analysis of the mRNA levels of the proteins possibly involved in the dephosphorylation of CREB-1 revealed that there were no obvious changes in the levels of the known CREB phosphatases such as protein phosphatase 1 (PPP1) and PPP2, upon cortisol treatment of the HFL-1 cells for 12 h (Fig. 10C). However, cortisol treatment of the HFL-1 cells for 12 h dramatically increased the level of MAPK phosphatase-1 (MKP-1), a known phosphatase that dephosphorylates MAPK (Fig. 10C). In consideration of the role of MAPK in the phosphorylation of CREB (20, 21), we speculate that the induction of MKP-1 by cortisol would cause dephosphorylation of the MAPK family, which could in turn result in the decreased level of p-CREB. In addition, activation of MKP-1 by glucocorticoids has indeed been reported to be associated with the reduction of COX-2 mRNA level in HeLa cells (22).
Fig. 8.

Cortisol (F, 1 μm) treatment of human amnion fibroblasts for 12 h increased the level of p-CREB-1 but not the total CREB-1 protein level. Cotreatment of the amnion fibroblasts with cortisol (1 μm) and GR antagonist RU486 (RU, 1 μm) or PKA inhibitor H89 (20 μm) or p38-MAPK inhibitor SB203580 (SB, 10 μm) abolished the induction of phosphorylation of CREB-1 by cortisol. n = 3; **, P < 0.01 vs. vehicle control (ctr).
Fig. 9.

Cotreatment of human amnion fibroblasts with cortisol (1 μm) and PKA inhibitor H89 (20 μm) or p38-MAPK inhibitor SB203580 (SB, 10 μm) for 24 h abolished the induction of COX-2 mRNA (A and B) and COX-2 promoter (−886 bp) activity (C and D) by cortisol (1 μm). n = 3–4; **, P < 0.01 vs. vehicle control; #, P < 0.05; ##, P < 0.01 vs. vehicle control (cortisol without H89 or SB203580).
Fig. 10.

A, Cortisol (F, 1 μm) treatment of HFL-1 cells for 12 h decreased the level of p-CREB-1 but not the total CREB-1 protein level; B, time course of the effect of cortisol (1 μm) treatment on the level of p-CREB-1 in HFL-1 cells; C, effect of cortisol (1 μm, 12 h) treatment on PPP1, PPP2, and MKP-1 mRNA levels in HFL-1 cells. n = 3–4; *, P < 0.05 vs. respective vehicle controls (ctr).
Stimulation of CREB-1 binding to the COX-2 promoter by cortisol and attenuation of cortisol-induced COX-2 expression by dominant-negative (dn)CREB in human amnion fibroblasts
ChIP assay demonstrated that cortisol (1 μm) increased the binding of CREB-1 to the COX-2 promoter fragment containing the putative CRE in human amnion fibroblasts (Fig. 11A). Transfection of human amnion fibroblasts with vector carrying dnCREB not only attenuated the induction of COX-2 mRNA expression by cortisol (1 μm) but also inhibited the induction of COX-2 promoter activity by cortisol (1 μm) in human amnion fibroblasts (Fig. 11, B and C).
Fig. 11.
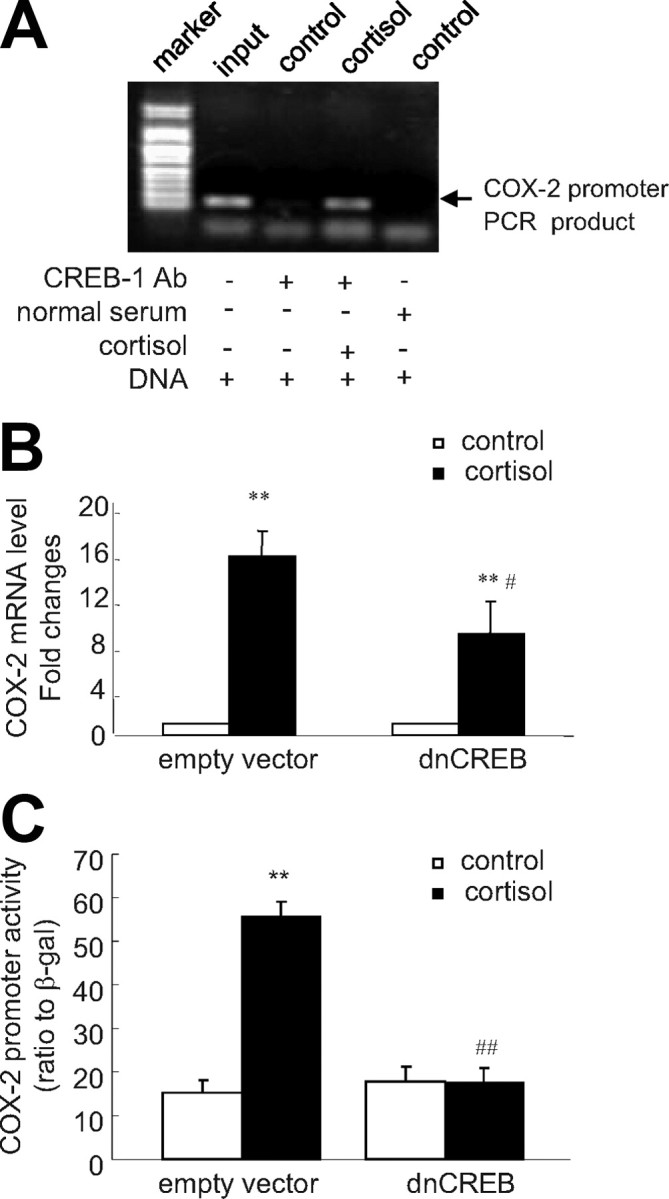
A, ChIP demonstrated that cortisol (1 μm) stimulated the binding of CREB-1 to the CRE in the COX-2 promoter. Top panel is a representative gel of the PCR products amplified from the DNA fragments immunoprecipitated by CREB-1 antibody. B and C, Transfection of the plasmid expressing dnCREB into human amnion fibroblasts blocked the induction of COX-2 mRNA and promoter (−886 bp) activity by cortisol (1 μm, 24 h). n = 3; **, P < 0.01 vs. vehicle control; #, P < 0.05; ##, P < 0.01 vs. empty vector with cortisol. Ab, Antibody.
Discussion
In this study, we found that cortisol paradoxically increased the COX-2 expression via phosphorylation of CREB and its binding to the CRE in the COX-2 promoter in human amnion fibroblasts. In contrast, cortisol classically inhibited the COX-2 expression via a NFκB binding site in the COX-2 promoter in HFL-1 cells, implying that the up-regulation of the COX-2 expression by cortisol could be a unique phenomenon in human amnion fibroblasts at the end of pregnancy. In consideration of the fact that the amnion fibroblasts are the major source of PG at the end of human pregnancy and the crucial roles of PG in parturition (4, 5), this paradoxical induction of COX-2 expression by cortisol in the amnion fibroblasts could be one of the indispensable events leading to parturition at the end of pregnancy.
As a well-recognized antiinflammatory hormone, glucocorticoids inhibit COX-2 expression and PG synthesis in virtually every known system (10, 11). In this study, we found that cortisol was not only capable of attenuating IL-1β-induced COX-2 expression but also capable of inhibiting COX-2 expression by itself in HFL-1 cells. Classically, the effects of glucocorticoids are mediated through intracellular GR. Upon binding of glucocorticoids to GR in the cytosol, heat-shock protein 90 (HSP90) is displaced from the receptor complex, which allows receptor dimerization and movement into the nucleus. Within the nucleus, upon binding of GR to the GRE within the promoter region of the respective gene, either a stimulatory or an inhibitory effect of glucocorticoids on gene expression is executed. In addition, the antiinflammatory effect of glucocorticoids can also be achieved via GR interference with other transcription factors, such as AP-1, NFκB, and CREB that are involved in inflammatory responses (23, 24, 25, 26). Sequence analysis of the COX-2 promoter revealed a putative GRE at −718 to −725 bp. However, it appears unlikely that this putative GRE mediates the inhibition of cortisol on COX-2 expression in HFL-1 cells because deletion of the fragment bearing this putative GRE did not affect the inhibitory effect of cortisol. Instead of GRE, the distal NFκB (nuclear factor κ-light-chain-enhancer of activated B cells) site appeared to be the crucial element mediating the inhibitory effect of cortisol on the COX-2 expression in HFL-1 cells. There are two NFκB sites in the COX-2 promoter. The proximal one is located at −233 to −214 bp, and the distal one is found at −448 to −439 bp. An earlier study reported that although both of the NFκB sites were functional for the basal transcriptional activity of the COX-2 promoter, mutation of the distal one rather than the proximal one affected the sensitivity of COX-2 gene to IL-1β stimulation and the inhibition of IL-1β stimulation by dexamethasone in WISH cells (19). Our study confirmed that only the distal NFκB site was functional in mediating the suppressing effect of cortisol on COX-2 expression in HFL-1 cells. The NFκB is a protein complex that acts as a key transcription factor in regulating the immune response to infection (24, 25). It is normally sequestered in the cytoplasm by IκB (25). Glucocorticoids normally inactivate NFκB via induction of IκB expression (24, 25). In addition, upon binding with glucocorticoid, GR can also have direct tethering interaction with NFκB to prevent its binding to the NFκB sites of the proinflammatory genes (26, 27). These NFκB inactivating mechanisms by glucocorticoids may thus operate in HFL-1 cells suppressing COX-2 expression.
In the sheep, the glucocorticoid surge at the end of pregnancy triggers parturition by stimulating P-450 17α-hydroxylase as well as COX-2 expression in intrauterine tissues (28), which leads to progesterone withdrawal and increased estrogen synthesis as well as PG production (28). Glucocorticoids are also among a variety of hormones possibly involved in human parturition. Despite a lack of P-450 17α-hydroxylase in human placenta, glucocorticoids nevertheless stimulate the production of PG in the intrauterine tissues, especially in the amnion tissue (28). This stimulatory effect of glucocorticoids on PG production in the human amnion is in marked contrast to the classical antiinflammatory effect of glucocorticoids. Although this paradoxical phenomenon in human amnion has been recognized for more than 20 yr (16), the mechanisms leading to the paradox remain largely unknown. We and others have demonstrated that glucocorticoids up-regulate PG production in the amnion via induction of cPLA2 and COX-2 expression, the two key enzymes in PG synthesis (13, 14, 15, 16). Gibb and Lavoie (29) argued that this paradoxical effect of glucocorticoids on PG production in the amnion might be determined by the proinflammatory cytokine levels. They believed that low proinflammatory cytokine levels constituted a preconditional environment for the paradoxical effect of glucocorticoids to take place. We demonstrated in a previous study that cortisol increased PG synthesis in human amnion fibroblasts despite concurrent potent inhibition of the proinflammatory cytokine expression (30), suggesting that the paradoxical induction of PG synthesis by glucocorticoids could operate along with the concurrent classical inhibition of proinflammatory cytokines. In this study, we demonstrated that use of a CRE in the COX-2 promoter by glucocorticoids to up-regulate COX-2 expression might be a mechanism coexisting with the inhibitory effect of glucocorticoids on proinflammatory cytokine expression in human amnion fibroblasts.
The CRE was first identified as being an inducible enhancer of genes that can be transcribed in response to an increased cAMP level (31). There are multiple CREB including CREB-1, CREB-2, ATF-1, ATF-2, and ATF-3, which share a C-terminal leucine zipper for dimerization and DNA binding (32). CREB-1 is the most abundant CREB expressed in most of the cells (33). The CREB binds both constitutively and inductively to the CRE in the promoter of the target gene activating transcription. Extracellular stimuli that activate PKA can lead to phosphorylation of CREB on Ser133, resulting in a further enhancement of transcriptional activation (27, 34). Previous studies demonstrated that activation of cAMP/PKA pathway could lead to the activation of COX-2 expression in both amnion and nonamnion cells (35, 36). In this study, we found that phosphorylation of the CREB-1 was increased by cortisol via activating PKA, which might account, in part, for the mechanism of the induction of COX-2 by cortisol. In addition to the phosphorylation of CREB-1 via PKA pathway, p38 MAPK has also been reported to play a role in the activation of CREB in different cell types (20, 21). This study also found that the phosphorylation of CREB and the induction of COX-2 expression by cortisol was blocked not only by the PKA inhibitor H89 but also by the p38-MAPK inhibitor SB203580, suggesting that this activation of p38-MAPK in human amnion fibroblasts might operate along with the activation of PKA in the induction of COX-2 by cortisol. However, this activation of p38-MAPK by cortisol in human amnion fibroblasts is in marked contrast to the inhibition of the MAPK signaling pathway by cortisol in most other cell types (22, 37). By inhibiting p38-MAPK, dexamethasone was reported to destabilize COX-2 mRNA rather than to induce COX-2 expression at inflammation (22). This inhibition of p38-MAPK by cortisol may also account for the inhibition of CREB-1 phosphorylation in HFL-1 cells. The mechanisms underlying these apparent contradictory findings in human amnion fibroblasts vs. in most other cell types with regard to the effect of cortisol on p38-MAPK and CREB-1 phosphorylation are obviously an issue of interest requiring further study. Consistent with the blocking effect of GR antagonist on the induction of COX-2 expression by cortisol, the phosphorylation of CREB-1 by cortisol could also be blocked by GR antagonist in human amnion fibroblasts, suggesting activation of either PKA or p38-MAPK by cortisol may require the participation of GR via induction of de novo protein synthesis because blocking protein synthesis with CHX reversed the stimulatory effect of cortisol on COX-2 expression in human amnion fibroblasts. However, the exact identity of the protein induced by cortisol leading to the activation of PKA and p38-MAPK that ultimately phosphorylate CREB is another issue requiring further study.
In conclusion, we demonstrated in this study that via binding to GR, cortisol stimulated COX-2 expression by increasing the phosphorylated CREB-1 binding to the CRE of the COX-2 gene. Cortisol may phosphorylate CREB-1 by activating either PKA or p38-MAPK in the amnion fibroblasts (Fig. 12).
Fig. 12.

Simplified model illustrating the hypothesized mechanisms underlying the induction of COX-2 expression by cortisol in human amnion fibroblasts.
Materials and Methods
Human amnion fibroblast and fetal lung fibroblast cell culture
To study the effects of glucocorticoids on COX-2 mRNA expression and promoter activity, we prepared amnion fibroblasts from human fetal membranes and compared these effects with an immortalized cell line, HFL-1. Human fetal membranes were collected at elective cesarean section from patients not in labor at term under a protocol approved by the ethics committee of the School of Life Sciences, Fudan University. Patients treated with steroids or other antiinflammatory agents or with clinical indication of inflammation were excluded from this study. Amnion was peeled off the chorion, washed thoroughly in cold PBS (pH 7.4), and digested with 0.125% trypsin (Sigma Chemical Co., St. Louis, MO) twice for 30 min at 37 C. The trypsin digestion medium was discarded, and the amnion tissue was washed vigorously with PBS to get rid of the residual epithelial cells. The remaining amnion tissue was further digested with 0.1% collagenase type 1 (Worthington, NJ) at 37 C for 1 h. The digestion medium was then collected and centrifuged at 2300 rpm for 15 min. Cell pellets were collected, and the resuspended cells were loaded onto prepared discontinuous Percoll (GE Healthcare, Uppsala, Sweden) gradients (5, 20, 40, and 60%, respectively). The gradients were centrifuged at 2500 rpm for 20 min. A single band of cells around 20–40% Percoll concentration was collected and cultured in complete DMEM containing 10% newborn bovine serum (NBS) (Invitrogen, Auckland, New Zealand) and antibiotic-antimycotic (Invitrogen, Grand Island, NY). The identity of cells has been previously verified, and more than 90% of the cells are fibroblasts (14). The human fetal lung fibroblast cell line was obtained from American Type Culture Collection (Rockville, MD) and was cultured in complete MEM-α (Invitrogen, Grand Island, NY) supplemented with 10% NBS and antibiotic-antimycotic. Cell culture was maintained at 37 C with a water-saturated atmosphere of 5% CO2 in air.
Treatment of human amnion fibroblasts and fetal lung fibroblasts and determination of COX-2, PPP1, PPP2, and MKP-1 mRNA and PGE2 levels
On the third day of primary amnion fibroblast culture or upon 90% confluence of HFL-1 cells, the cells were washed with PBS and the culture media were changed to NBS-free media. The cells were then treated with different concentrations of cortisol (0.01–1 μm; Sigma). To examine whether the effect of cortisol on COX-2 mRNA expression is dependent on ongoing transcription and requires de novo protein synthesis, the cells were pretreated with mRNA transcription inhibitor DRB, 75 μm; Sigma) or protein synthesis inhibitor CHX (10 μm; Sigma) for 0.5 h before cortisol treatment, and the DRB or CHX treatment was continued thereafter along with cortisol treatment for 24 h. To explore whether the effect of cortisol on COX-2 expression is dependent on GR, the cells were treated with cortisol (24 h) in the presence or absence of GR antagonist RU486 (1 μm; Sigma). To study the interaction of cortisol with IL-1β, PKA, and p38-MAPK in the regulation of COX-2 expression, the cells were treated with cortisol (1 μm, 24 h) in the presence and absence of IL-1β (10 ng/ml; Biosource, Camarillo, CA) and forskolin (100 μm, a stimulator of adenylyl cyclase; Sigma), H89 (20 μm, an inhibitor of PKA, Sigma), or SB203580 (10 μm, an inhibitor of p38-MAPK; Sigma). To explore the mechanism of CREB-1 dephosphorylation by cortisol treatment in HFL-1 cells, mRNA levels of PPP1 and PPP2 catalytic subunits and α-isoforms as well as MKP-1 were also measured in response to cortisol treatment (1 μm, 12 h). After removal of the culture medium, cells were scraped into the cell lysis buffer for subsequent extraction of total RNA for analysis with PCR using UNIQ-10 RNA extraction kit (Sangon Biotech, Shanghai, China) according to the protocol provided by the company. One microgram of RNA was reverse-transcribed to cDNA with oligo(dT)12–18 primers using Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI), and cDNA was used for subsequent measurement of COX-2, PPP1, PPP2, and MKP-1 mRNA levels with quantitative real-time PCR (qRT-PCR). Reaction solution of qRT-PCR consisted of 2.0 μl diluted cDNA, 0.2 μm of each paired primer, and power SYBR Green PCR master mix (Toyobo, Osaka, Japan). The annealing temperature was set at 61 C. The absolute mRNA levels in each sample were calculated according to a standard curve set up using serial dilutions of known amounts of specific templates against corresponding cycle threshold values. To control for sampling errors, qRT-PCR for the housekeeping gene β-actin was routinely performed on each sample. The ratio of the target genes over β-actin in each sample was obtained to normalize the expression of the target gene. The specificity of the primers was verified by examining the melting curve as well as the sequences of the PCR products. The primer sequences for amplifying human COX-2, PPP1, PPP2, MKP-1, and β-actin genes are illustrated in Table 1. The culture media removed from amnion fibroblast and HFL-1 cells treated with cortisol (1 μm) were assayed for PGE2 level using a Cayman Chemical’s ACE enzyme immunoassay kit (Ann Arbor, MI).
Table 1.
Primer sequences used for qRT-PCR
| Sequence (5′–3′) | |
|---|---|
| COX-2 | |
| Sense | TGTGCAACACTTGAGTGGCT |
| Antisense | ACTTTCTGTACTGCGGGTGG |
| PPP1 | |
| Sense | GCCTGCTGGAAGGTGACATA |
| Antisense | CATAGATGCGGTTGATGCTG |
| PPP2 | |
| Sense | CCAGCTAGTGATGGAGGGAT |
| Antisense | GAGTAACATGTGGCTCGCCT |
| MKP-1 | |
| Sense | TGGAGGACAACCACAAGG |
| Antisense | 5CAAAGGATGGCACAGGAT |
| β-Actin | |
| Sense | GGGAAATCGTGCGTGACATTAAG |
| Antisense | TGTGTTGGCGTACAGGTCTTTG |
Cloning of human COX-2 promoter from human amnion fibroblasts and construction into the pGL3-enhancer plasmid carrying luciferase reporter gene
Human amnion fibroblasts were cultured as described above. Genomic DNA was extracted and purified using TIANamp genomic DNA kit (Tiangen Biotech, Beijing, China). Sense and antisense oligonucleotide primers corresponding to −886 to −869 bp (CGCCGCTTCCTTTGTCCA) and +14 to +31bp (ACTTGCAGTGAGCGTCAG) of the 5′-flanking region of the COX-2 gene (GenBank no. U44805) was used to amplify COX-2 gene promoter with PCR using genomic DNA extracted from human amnion fibroblasts as template, and a 917-bp DNA fragment was generated. The sense and antisense primers carried KpnI and BglII restriction enzyme sites, respectively, as linkers to the polycloning sites of pGL3-enhancer plasmid carrying luciferase reporter gene (Promega). Upon successful PCR amplification, the PCR product was initially cloned using a TA cloning kit (Takara, Dalian, China) and the released PCR product was then subcloned into KpnI and BglII sites of pGL3-enhancer plasmid upstream to the luciferase reporter gene. The insert was verified via sequencing by Invitrogen (Shanghai, China).
The 5′ progressive deletion and site-directed mutagenesis of the COX-2 gene promoter and construction into the pGL3-enhancer plasmid
Progressive deletion of the cloned COX-2 gene promoter was carried out with PCR using a combination of a common antisense primer as described above and a series of 5′ sense primers (progressing toward the transcription start site) as paired primer sets. The sequences and annealing positions of these primers are shown in Figs. 3, 4C, and 5C. Restriction sites for KpnI and BglII were designed into the sense and antisense primers, respectively. In addition to −886 bp, a series of 5′-deleted COX-2 gene promoters was produced as follows: −694, −453, −340, −201, −101, and −66 bp upstream to the transcription start site. Based upon 5′ deletion studies, specific nucleotide mutation was introduced into the predicted responsive elements (CRE and NFκB) for the effect of cortisol by designing the mutation sites into the elements of the sense primers (Figs. 6 and 7, nucleotides in bold letters). The PCR products were cut with KpnI and BglII and ligated into the polycloning sites of pGL3-enhancer plasmid. All the above cloned promoter fragments were verified by sequencing.
Transient transfection of human amnion fibroblasts and HFL-1 cells with pGL3-enhancer plasmid carrying COX-2 promoter-driven luciferase reporter gene
Both human amnion fibroblasts and HFL-1 cells were grown to a density of about 70% confluence in complete media containing 10% NBS and antibiotic-antimycotic before the start of transfection. On the day of transfection, the cells were washed with PBS and the media were changed to antibiotic-free media containing 10% NBS. The cells were cotransfected with 0.5 μg/well of pGL3-enhancer plasmid carrying COX-2 promoter and 0.05 μg/well of pSV-β-galactosidase plasmid using Lipofectamine LTX in Opti-MEM (Invitrogen, Carlsbad, CA). The above plasmids were extracted from the transformed bacteria using endotoxin-free Nucleobond PC500 EF kit (Macherey- Nagel, Duren, Germany). The pSV-β-galactosidase plasmid was used for transfection efficiency control. Transfection was allowed for 12 h. The media were changed to complete media. After 24 h, the cells were treated with cortisol (0.01–1 μm) in the presence and absence of RU486 (1 μm), IL-1β (10 ng/ml), forskolin (100 μm), H89 (20 μm), and SB203580 (10 μm) in serum-free medium for another 24 h. The cells were then lysed for subsequent measurement of luciferase activity using a luciferase assay system (Promega) and β-galactosidase activity using β-galactosidase enzyme assay system (Promega). The ratio of COX-2 promoter-driven luciferase activity against β-galactosidase activity was obtained to correct differential transfection efficiency in each well and to express the promoter activity. To study the role of the putative transcription factor, CREB, in the induction of COX-2 promoter activity and mRNA expression by cortisol, some of the amnion fibroblasts were transfected with RSV-SG plasmid carrying a dominant-negative mutant of CREB (dnCREB, 2 μg/well) in which Ser133 is replaced with Ala (kindly provided by Dr. Montminy, The Salk Institute, La Jolla, CA).
Western blotting analysis of total and phosphorylated CREB-1 level
The human amnion fibroblasts were treated with cortisol (1 μm) in serum-free medium in the presence of RU486 (10 μm), H89 (20 μm), and SB203580 (10 μm) for 12 h. The HFL-1 cells were treated with cortisol (1 μm) for 0.5, 1, 6, and 12 h in serum-free medium. The cells were then washed and collected in PBS containing phosphatase inhibitor. After spinning down, total cellular protein was extracted using a kit from Active Motif (Carlsbad, CA). The CREB transcription factor family includes CREB-1, CREB-2, ATF-1, ATF-2, and ATF-3, which share a C-terminal leucine zipper for dimerization and DNA binding. The transcription-activating activity of CREB is induced upon phosphorylation on Ser133. The expression of total CREB-1 protein (43 kDa) and phosphorylated CREB-1 protein was examined using a standard Western blotting protocol. Briefly, 50 μg protein of each sample was electrophoresed in 9% SDS-polyacrylamide gel and transferred to the nitrocellulose blot. The blot was blocked with nonfat milk solution and incubated with 1:500 dilution of rabbit antibody against the C terminus of CREB-1 and phosphorylated CREB-1 (Ser133) (Santa Cruz Biotechnology, Santa Cruz, CA) overnight. Both of the antibodies cross-react with the 37-kDa ATF-1. After washing, the blot was incubated with the secondary antirabbit IgG antibody conjugated with horseradish peroxidase (Santa Cruz) for 1 h. The enhanced chemiluminescence detection system (Millipore, Billerica, MA) was used to detect the bands with peroxidase activity. After stripping, the same blot was reprobed for β-actin for loading control examination.
ChIP assay demonstrating the binding of CREB-1 to COX-2 promoter in human amnion fibroblasts
Primary human amnion fibroblasts were prepared as described above. The cells were treated with cortisol (1 μm) for 24 h. Upon termination of treatment, ChIP assay was conducted using a kit from Upstate Biotechnology (Temecula, CA) and a method modified from the manufacturer’s protocol. Briefly, the cells were fixed with 1% formaldehyde to cross-link the transcription factors to chromatin DNA. After washing with PBS, the cells were incubated with glycine and then scraped off the dish in PBS containing protease inhibitor cocktail. After spinning down, the cells were resuspended with lysis buffer supplemented with protease inhibitor cocktail and broken up using a Dounce homogenizer to aid nuclei release. After spinning down, the nuclei were resuspended in digestion buffer supplemented with protease inhibitor cocktail. The shearing of chromatin DNA was carried out by sonication to produce an optimized size of input DNA around 500 bp. The sheared DNA was collected for subsequent immunoprecipitation with CREB-1 antibody (Santa Cruz) or normal serum as negative control. The immunoprecipitate was then incubated with protein A agarose/salmon sperm DNA, and the antibody/protein/DNA/agarose complex was washed and collected for subsequent reverse cross-linking. The sheared DNA recovered from reverse cross-linking was extracted with a DNA extraction kit for further PCR analysis. The positions and sequences of the primers used for PCR are the same as used in cloning the −66-bp promoter fragment spanning the predicted CRE promoter region (Fig. 4C). Real-time PCR was performed on the sheared DNA fragments and was stopped before saturation according to the observation of the amplification curve. PCR products were analyzed with 2% agarose gel electrophoresis.
Statistical analysis
All data are reported as mean ± sem. Paired Student’s t test or one-way ANOVA test followed by the Student-Newman-Keuls test was used to assess significant differences where appropriate. Significance was set at P < 0.05.
NURSA Molecule Pages:
Ligands: Hydrocortisone | RU486;
Nuclear Receptors: GR.
Footnotes
This work was supported by the Natural Science Foundation of China (30570680 and 30870935 to K.S.) and Shanghai Leading Academic Discipline Project (B111).
Disclosure Summary: The authors have nothing to disclose.
First Published Online October 1, 2009
X.O.Z., Z.Y., and C.M.G. have made equal contribution to this work.
Abbreviations: ATF-1, Activating transcription factor-1; ChIP, chromatin immunoprecipitation; CHX, cycloheximide; COX, cyclooxygenase; CRE, cAMP response element; CREB, CRE-binding protein; dn, dominant-negative; DRB, 5,6-dichlorobenzimidazole riboside; GR, glucocorticoid receptor; GRE, glucocorticoid response element; MKP-1, MAPK phosphatase-1; NBS, newborn bovine serum; NFκB, nuclear factor-κB; p-, phospho-; PG, prostaglandin; PKA, protein kinase A; PPP1, protein phosphatase 1; qRT-PCR, quantitative real-time PCR; TESS, Transcription Element Search System.
References
- 1.Creasy RK1991. Preventing preterm birth. N Engl J Med 325:727–729 [DOI] [PubMed] [Google Scholar]
- 2.Saigal S, Doyle LW2008. An overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet 371:261–269 [DOI] [PubMed] [Google Scholar]
- 3.Goldkrand JW, Schulte RL, Messer RH1976. Maternal and fetal plasma cortisol levels at parturition. Obstet Gynecol 47:41–45 [PubMed] [Google Scholar]
- 4.Sippell WG, Müller-Holve W, Dörr HG, Bidlingmaier F, Knorr D1981. Concentrations of aldosterone, corticosterone, 11-deoxycorticosterone, progesterone, 17-hydroxyprogesterone, 11-deoxycortisol, cortisol, and cortisone determined simultaneously in human amniotic fluid throughout gestation. J Clin Endocrinol Metab 52:385–392 [DOI] [PubMed] [Google Scholar]
- 5.Whittle WL, Patel FA, Alfaidy N, Holloway AC, Fraser M, Gyomorey S, Lye SJ, Gibb W, Challis JR2001. Glucocorticoid regulation of human and ovine parturition: the relationship between fetal hypothalamic-pituitary-adrenal axis activation and intrauterine prostaglandin production. Biol Reprod 64:1019–1032 [DOI] [PubMed] [Google Scholar]
- 6.Challis JR, Lye SJ, Gibb W1997. Prostaglandins and parturition. Ann NY Acad Sci 828:254–267 [DOI] [PubMed] [Google Scholar]
- 7.Kniss DA1999. Cyclooxygenases in reproductive medicine and biology. J Soc Gynecol Investig 6:285–292 [DOI] [PubMed] [Google Scholar]
- 8.Duchesne MJ, Thaler-Dao H, de Paulet AC1978. Prostaglandin synthesis in human placenta and fetal membranes. Prostaglandins 15:19–42 [DOI] [PubMed] [Google Scholar]
- 9.Roos KL, Simmons DL2005. Cyclooxygenase variants: the role of alternative splicing. Biochem Biophys Res Commun 338:62–69 [DOI] [PubMed] [Google Scholar]
- 10.Rhen T, Cidlowski JA2005. Antiinflammatory action of glucocorticoids: new mechanisms for old drugs. N Engl J Med 353:17117–17123 [DOI] [PubMed] [Google Scholar]
- 11.Newton R, Seybold J, Kuitert LM, Bergmann M, Barnes PJ1998. Repression of cyclooxygenase-2 and prostaglandin E2 release by dexamethasone occurs by transcriptional and post-transcriptional mechanisms involving loss of polyadenylated mRNA. J Biol Chem 273:32312–32321 [DOI] [PubMed] [Google Scholar]
- 12.Challis JR, Sloboda DM, Alfaidy N, Lye SJ, Gibb W, Patel FA, Whittle WL, Newnham JP2002. Prostaglandins and mechanisms of preterm birth. Reproduction 124:1–17 [DOI] [PubMed] [Google Scholar]
- 13.Economopoulos P, Sun M, Purgina B, Gibb W1996. Glucocorticoids stimulate prostaglandin H synthase type-2 (PGHS-2) in the fibroblast cells in human amnion cultures. Mol Cell Endocrinol 117:141–147 [DOI] [PubMed] [Google Scholar]
- 14.Sun K, Ma R, Cui X, Campos B, Webster R, Brockman D, Myatt L2003. Glucocorticoids induce cytosolic phospholipase A2 and prostaglandin H synthase type 2 but not microsomal prostaglandin E synthase (PGES) and cytosolic PGES expression in cultured primary human amnion cells. J Clin Endocrinol Metab 88:5564–5571 [DOI] [PubMed] [Google Scholar]
- 15.Zakar T, Hirst JJ, Mijovic JE, Olson DM1995. Glucocorticoids stimulate the expression of prostaglandin endoperoxide H synthase-2 in amnion cells. Endocrinology 136:1610–1619 [DOI] [PubMed] [Google Scholar]
- 16.Casey ML, MacDonald PC, Mitchell MD1985. Despite a massive increase in cortisol secretion in women during parturition, there is an equally massive increase in prostaglandin synthesis. A paradox? J Clin Invest 75:1852–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tazawa R, Xu XM, Wu KK, Wang LH1994. Characterization of the genomic structure, chromosomal location and promoter of human prostaglandin H synthase-2 gene. Biochem Biophys Res Commun 203:190–199 [DOI] [PubMed] [Google Scholar]
- 18.Appleby SB, Ristimäki A, Neilson K, Narko K, Hla T1994. Structure of the human cyclo-oxygenase-2 gene. Biochem J 302 (Pt 3):723–727 [DOI] [PMC free article] [PubMed]
- 19.Wang Z, Tai HH1998. Interleukin-1β and dexamethasone regulate gene expression of prostaglandin H synthase-2 via the NF-κB pathway in human amnion derived WISH cells. Prostaglandins Leukot Essent Fatty Acids 59:63–69 [DOI] [PubMed] [Google Scholar]
- 20.Bhat NR, Zhang P, Mohanty SB2007. p38 MAP kinase regulation of oligodendrocyte differentiation with CREB as a potential target. Neurochem Res 32:293–302 [DOI] [PubMed] [Google Scholar]
- 21.Keren A, Keren-Politansky A, Bengal E2008. A p38 MAPK-CREB pathway functions to pattern mesoderm in Xenopus Dev Biol 322:86–94 [DOI] [PubMed] [Google Scholar]
- 22.Lasa M, Brook M, Saklatvala J, Clark AR2001. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol Cell Biol 21:771–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hayashi R, Wada H, Ito K, Adcock IM2004. Effects of glucocorticoids on gene transcription. Eur J Pharmacol 500:51–62 [DOI] [PubMed] [Google Scholar]
- 24.Smoak KA, Cidlowski JA2004. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev 125:697–706 [DOI] [PubMed] [Google Scholar]
- 25.Baldwin AS Jr1996. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol 14:649–683 [DOI] [PubMed] [Google Scholar]
- 26.Sheppard KA, Phelps KM, Williams AJ, Thanos D, Glass CK, Rosenfeld MG, Gerritsen ME, Collins T1998. Nuclear integration of glucocorticoid receptor and nuclear factor κB signaling by CREB-binding protein and steroid receptor coactivator-1. J Biol Chem 273:29291–29294 [DOI] [PubMed] [Google Scholar]
- 27.Kassel O, Herrlich P2007. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol 275:13–29 [DOI] [PubMed] [Google Scholar]
- 28.Challis JRG, Matthews SG, Gibb W, Lye SJ2000. Endocrine and paracrine regulation of birth at term and preterm. Endocr Rev 21:514–550 [DOI] [PubMed] [Google Scholar]
- 29.Gibb W, Lavoie JC1990. Effects of glucocorticoids on prostaglandin formation by human amnion. Can J Physiol Pharmacol 68:671–676 [DOI] [PubMed] [Google Scholar]
- 30.Guo C, Yang Z, Li W, Zhu P, Myatt L, Sun K2008. Paradox of glucocorticoid-induced cytosolic phospholipase A2 group IVA messenger RNA expression involves glucocorticoid receptor binding to the promoter in human amnion fibroblasts. Biol Reprod 78:193–197 [DOI] [PubMed] [Google Scholar]
- 31.Comb M, Birnberg NC, Seasholtz A, Herbert E, Goodman HM1986. A cyclic AMP- and phorbol ester-inducible DNA element. Nature 323:353–356 [DOI] [PubMed] [Google Scholar]
- 32.Hai T, Hartman MG2001. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene 273:1–11 [DOI] [PubMed] [Google Scholar]
- 33.Tamai KT, Monaco L, Nantel F, Zazopoulos E, Sassone-Corsi P1997. Coupling signalling pathways to transcriptional control: nuclear factors responsive to cAMP. Recent Prog Horm Res 52:121–139; discussion 139–140 [PubMed] [Google Scholar]
- 34.Quinn PG1993. Distinct activation domains within cAMP response element-binding protein (CREB) mediate basal and cAMP-stimulated transcription. J Biol Chem 268:16999–17009 [PubMed] [Google Scholar]
- 35.Debey S, Meyer-Kirchrath J, Schrör K2003. Regulation of cyclooxygenase-2 expression by iloprost in human vascular smooth muscle cells. Role of transcription factors CREB and ICER. Biochem Pharmacol 65:979–988 [DOI] [PubMed] [Google Scholar]
- 36.Collins PL, Zink E, Moore RM, Roberts JM, Maguire ME, Moore JJ1993. Ritodrine: a β-adrenergic receptor antagonist in human amnion. Am J Obstet Gynecol 168:143–151 [DOI] [PubMed] [Google Scholar]
- 37.Clark AR, Lasa M2003. Crosstalk between glucocorticoids and mitogen-activated protein kinase signalling pathways. Curr Opin Pharmacol 3:404–411 [DOI] [PubMed] [Google Scholar]