

Abstract
In the present study, we evaluated the regulation of G protein-coupled receptor (GPR)30 expression in estrogen receptor (ER)-positive endometrial, ovarian, and estrogen-sensitive, as well as tamoxifen-resistant breast cancer cells. We demonstrate that epidermal growth factor (EGF) and TGFα transactivate the GPR30 promoter and accordingly up-regulate GPR30 mRNA and protein levels only in endometrial and tamoxifen-resistant breast cancer cells. These effects exerted by EGF and TGFα were dependent on EGF receptor (EGFR) expression and activation and involved phosphorylation of the Tyr1045 and Tyr1173 EGFR sites. Using gene-silencing experiments and specific pharmacological inhibitors, we have ascertained that EGF and TGFα induce GPR30 expression through the EGFR/ERK transduction pathway, and the recruitment of c-fos to the activator protein-1 site located within GPR30 promoter sequence. Interestingly, we show that functional cross talk of GPR30 with both activated EGFR and ERα relies on a physical interaction among these receptors, further extending the potential of estrogen to trigger a complex stimulatory signaling network in hormone-sensitive tumors. Given that EGFR/HER2 overexpression is associated with tamoxifen resistance, our data may suggest that ligand-activated EGFR could contribute to the failure of tamoxifen therapy also by up-regulating GPR30, which in turn could facilitates the action of estrogen. In addition, important for resistance is the ability of tamoxifen to bind to and activate GPR30, the expression of which is up-regulated by EGFR activation. Our results emphasize the need for new endocrine agents able to block widespread actions of estrogen without exerting any stimulatory activity on transduction pathways shared by the steroid and growth factor-signaling networks.
EGF and TGFα generate a stimulatory network in estrogen-sensitive cancer cells up-regulating GPR30, which interacts with activated EGFR and ERα.
Estrogens regulate the growth and differentiation of many normal and neoplastic tissues including breast, endometrial, and ovarian tumors (1, 2). The effects of 17β-estradiol (E2) are mainly mediated by binding to estrogen receptor (ER) α and ERβ and transcriptional stimulation of estrogen-responsive genes (3, 4). However, cell membrane-initiated events induced via growth factor receptors and/or G protein-coupled receptors have largely been associated with rapid estrogen effects (5). Recently, several reports, including our own (6, 7, 8, 9, 10, 11, 12), have described estrogen/antiestrogen binding and activation properties of the G protein-coupled receptor (GPR)30, which has therefore been proposed as a candidate for triggering a broad range of biological activities initiated at the level of the plasma membrane by the aforementioned agents.
Prolonged exposure to estrogens is a major risk factor for the progression of breast cancer (13), which expresses elevated levels of ERα in approximately 70% of cases (14). Consequently, ERα antagonists such as tamoxifen, ICI 182,780 (ICI), and raloxifene have been used in the pharmacological management of ERα-positive breast cancer to inhibit the mitogenic actions of estrogen (15, 16, 17). Although there is general concordance between ERα expression and responsiveness to ER-targeted agents, as indicated by a greater 5-yr disease-free survival for ERα-positive patients receiving tamoxifen (18), approximately one in four patients does not respond to tamoxifen therapy from the onset, and in most patients tamoxifen treatment produces agonist effects after a few years (19). In this setting, the existence of an alternative receptor for estrogen, such as GPR30, which is activated by ER antagonists, may represent an additional mechanism potentially involved in the resistance to tamoxifen treatment.
Cross talk between estrogen and the epidermal growth factor (EGF) receptor (EGFR) transduction pathways has been shown to occur in the development and progression of breast cancer (20, 21). The expression of EGFR is generally found to be inversely correlated with the levels of ERα; however, both receptors are involved in the progression of breast cancer, regardless of which are the dominant pathway present (22). Interestingly, long-term blockade of ERα function with tamoxifen leads to the overexpression of EGFR or HER2, which is a member of the EGFR family of membrane receptors (23). Of note, GPR30 is involved in EGFR transactivation and MAPK phosphorylation by estrogen, even in cancer cells lacking ER (6, 24, 25). In addition, we recently have demonstrated that EGF-activated EGFR signaling up-regulates GPR30 expression, which in turn mediates estrogen action in ER-negative breast cancer cells (12).
It has been reported that EGFR activation by its cognate ligand EGF, as well as by TGFα, triggers different biological responses, such as cell proliferation, differentiation, migration, and survival (26). TGFα is structurally related to EGF and widely expressed in developing embryos and normal adult tissues including the colon, skin, mammary gland, liver, and kidney (27). Given that TGFα is also produced by activated macrophages and eosinophils (28, 29), its potential role in immunological processes has been also suggested (30). Moreover, TGFα was shown to act as a potent angiogenetic factor up-regulating the expression of vascular endothelial growth factor (VEGF) (31) and was found inappropriately expressed in neoplasia (27). Indeed, high levels of TGFα were associated with an increased expression of EGFR, indicating autocrine, paracrine, and juxtacrine functions exerted during tumorigenesis (27, 30).
The signal transduction cascade that translates EGFR activation to ERK phosphorylation has been investigated at length (32, 33). The binding of the cognate ligand stimulates the tyrosine kinase activity of the intracellular portion of the receptor, which results in increased phosphorylation of several tyrosine residues within the receptor dimer (34). These phosphorylated tyrosines serve as recruitment/docking sites for several adaptor proteins, which lead to receptor stabilization/degradation and thereafter ERK activation (35, 36, 37, 38, 39). The intensity and duration of EGFR transactivation and subsequent ERK stimulation are key points in response to a variety of physiological as well as nonphysiological stimuli, which promote growth effects in cancer cells (32, 35).
In the present study, we have evaluated the regulation of GPR30 expression in ER-positive endometrial, ovarian, and breast estrogen-sensitive as well as tamoxifen-resistant (TamR) tumor cells. We demonstrate that the up-regulation GPR30 by EGF and TGFα was dependent on activation of EGFR/ERK signaling in distinct cellular contexts. Moreover, we provide new insights regarding the cross talk between GPR30 and both activated EGFR and ERα, which relies on a direct physical interaction among these receptors leading to stimulatory effects in estrogen-sensitive cancer cells.
Results
EGF and TGFα transactivate GPR30 promoter constructs through an activator protein-1 (AP-1) site in ER-positive endometrial and TamR breast cancer cells
We began our study evaluating whether 24-h treatments with EGF, E2, ICI, 1-[4-(-6-bromobenzol [1, 3] diodo-5-yl)-3a,4,5,9b-tetrahidro3H-cyclopenta[c]quinolin-8yl]-ethanone (G-1), TGFα, and IGF-I could transactivate the GPR30 promoter sequence in endometrial (Ishikawa), ovarian (BG-1), and breast (wild-type MCF7 or TamR MCF7-TR1) tumor cells (Fig. 1, A–D), which all express ERα but different levels of EGFR (Fig. 1E). Interestingly, in Ishikawa and MCF7-TR1 cells, only EGF and TGFα induced transactivation of the GPR30 promoter construct (Fig. 1, A–D), which was abrogated in presence of the EGFR inhibitor AG1478 (AG) or the MAPK kinase (MEK) inhibitor PD98059 (PD) (Fig. 2A-B). Considering the aforementioned results and the very low levels of EGFR expression observed in BG-1 and wild-type MCF7 cells (Fig. 1E), we engineered these cells to express EGFR to evaluate whether the promoter of GPR30 could be transactivated by EGF and TGFα. As shown in supplemental Fig. 1, published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org), expression of EGFR allowed for GPR30 promoter responsiveness to EGF and TGFα treatments, confirming that EGFR is indeed required to mediate the transactivation of the GPR30 promoter construct. To determine the sequence(s) involved in the GPR30 promoter’s responsiveness to these ligands, we transfected Ishikawa and MCF7-TR1 cells with the aforementioned expression vector mutated in the potential AP-1 (GPR30AP1mut), or the stimulatory protein-1 (SP-1) (GPR30SP1mut) sites (12). Only the AP-1 mutation site abolished GPR30 transactivation by EGF and TGFα treatments for 24 h (Fig. 2, C and D), suggesting that this sequence is implicated in the transcriptional activation of the GPR30 promoter by these ligands (see below).
Fig. 1.
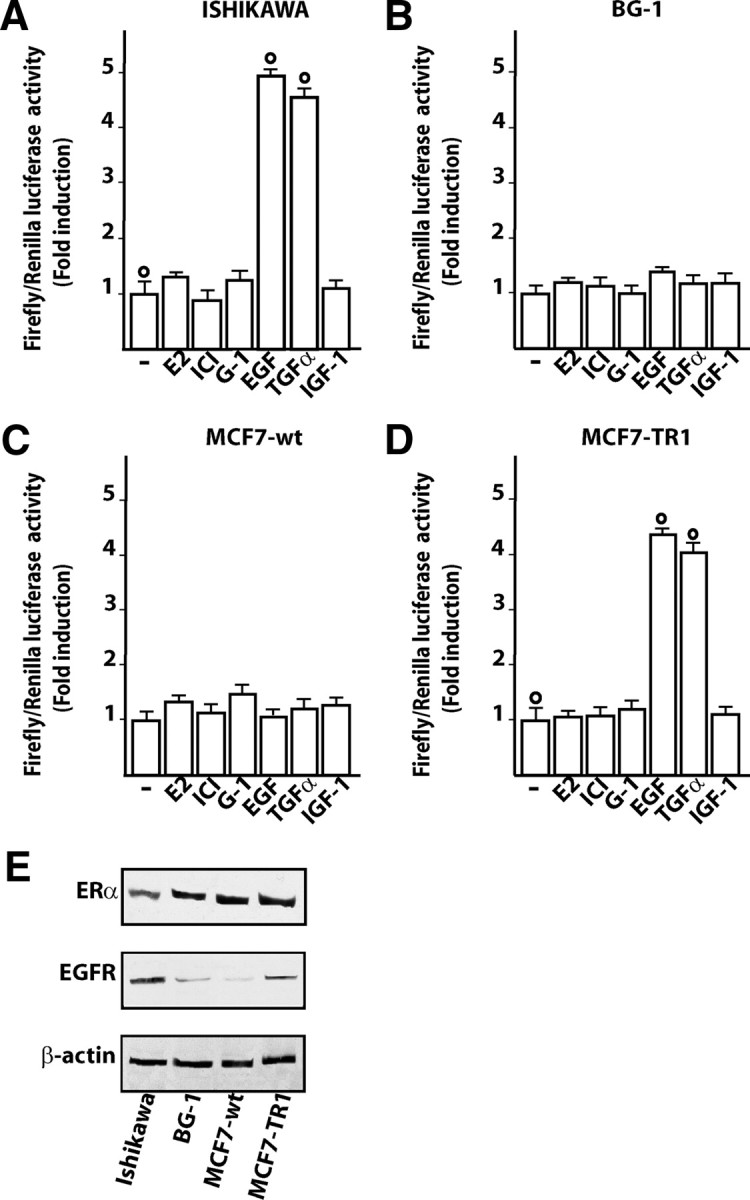
EGF and TGFα transactivate the promoter of GPR30 in Ishikawa and MCF7-TR1 cells but not in BG-1 and MCF wild type (MCF7-wt) cells. Ishikawa (A), BG-1 (B), MCF7-wt (C), and MCF7-TR1 (D) cells were transfected with a reporter plasmid encoding the GPR30 promoter and treated for 24 h with 100 nm E2, 1 μm ICI, 1 μm G-1, 100 ng/ml EGF, 100 ng/ml TGFα, or 100 ng/ml IGF-I. The luciferase activities were normalized to the internal transfection control, and values of cells receiving vehicle (−) were set as 1-fold induction upon which the activity induced by treatments was calculated. Each column represents the mean ± sd of three independent experiments performed in triplicate. ○, P < 0.05, for cells receiving vehicle (−) vs. treatment. E, ERα and EGFR protein expression in the indicated cells. β-Actin served as a loading control.
Fig. 2.

An AP-1 site within the GPR30 promoter sequence is required for the transactivation by EGF and TGFα in Ishikawa and MCF7-TR1 cells. A and B, cells were transfected with the plasmid encoding the GPR30 promoter and stimulated for 24 h with 100 ng/ml EGF or 100 ng/ml TGFα and 10 μm of the EGFR inhibitor tyrphostin AG or the MEK inhibitor PD, as indicated. C and D, cells were transfected with the reporter plasmids containing AP-1 (GPR30AP1mut) and SP-1 (GPR30SP1mut) mutations generated within the GPR30 promoter and treated for 24 h with 100 ng/ml EGF or 100 ng/ml TGFα. The luciferase activities were normalized to the internal transfection control, and values of cells receiving vehicle (−) were set as 1-fold induction upon which the activity induced by treatments was calculated. Each column represents the mean ± sd of three independent experiments performed in triplicate. ○ and •, P < 0.05, for cells receiving vehicle (−) vs. treatment.
EGF and TGFα up-regulate GPR30 expression
Having established that the GPR30 promoter is transactivated by EGF and TGFα, we next asked whether these ligands were able to stimulate GPR30 expression. A time course study performed by real time quantitative RT-PCR revealed that the mRNA levels of GPR30 were up-regulated with EGF and TGFα treatments in Ishikawa and TamR MCF7-TR1 cells (Fig. 3, A and B). In both cell types, EGF and TGFα also induced GPR30 protein levels, which were blocked in the presence of AG and PD (Fig. 4, A and B), or with knockdown of EGFR expression using a short hairpin RNA (shRNA) (Fig. 4, C and D). We did not observe GPR30 stimulation at either the mRNA or protein levels in BG-1 and MCF7 wild-type cells (data not shown). Cumulatively, these data indicate that the GPR30 response to EGF and TGFα depends on the cell context and, in particular, on EGFR expression and function in the cell.
Fig. 3.

Time course of GPR30 mRNA expression. Evaluation of GPR30 mRNA expression by real-time PCR in Ishikawa (A) and MCF7-TR1 (B) cells treated with 100 ng/ml EGF and TGFα as indicated. Data shown are the mean ± sd of three independent experiments.
Fig. 4.

EGF and TGFα up-regulate GPR30 protein levels in Ishikawa and MCF7-TR1 cells. A and B, immunoblots of GPR30 from cells treated for 8 h with vehicle (−), 100 ng/ml EGF, or 100 ng/ml TGFα alone and in combination with 10 μm of the EGFR inhibitor tyrphostin AG, the MEK inhibitor PD, or by silencing EGFR expression (C and D). The efficacy of EGFR silencing was evaluated by immunoblots as indicated. β-Actin served as a loading control. Data shown are representative of three independent experiments.
EGF and TGFα induce EGFR Tyr1045 and Tyr1173 phosphorylation and Erk1/2 activation
To provide further insight into the mechanisms by which EGF and TGFα trigger GPR30 expression, we evaluated the ability of both ligands to phosphorylate different tyrosine sites in the EGFR. In Ishikawa and MCF7-TR1 cells, short treatments (5 min) with EGF or TGFα induced EGFR phosphorylation at its Tyr1045 and Tyr1173 sites (Fig. 5, A and B), whereas no phosphorylation was observed at the Tyr992 or Tyr1068 residues (data not shown). In BG-1 and MCF7 wild-type cells, neither EGF nor TGFα stimulated EGFR phosphorylation in any of these sites (data not shown), indicating that specific tyrosine phosphorylation in response to ligand stimulation is dependent on the cell context. On the other hand, EGFR expression was observed in MCF7-TR1 and to a higher extent in Ishikawa cells (Fig. 5, A and B), whereas none was detected in BG-1 or MCF7 wild-type cells (data not shown), allowing a diverse cell responsiveness to EGFR ligands. Next, EGFR activation in Ishikawa and MCF7-TR1 cells by EGF and TGFα was paralleled by rapid ERK1/2 phosphorylation (Fig. 5C), confirming that the EGFR/ERK signaling could drive GPR30 expression (Fig. 2, A and B, and Fig. 4, A–D).
Fig. 5.

EGF and TGFα induce EGFR phosphorylation and Erk1/2 activation in Ishikawa and MCF7-TR1 cells. Phosphorylation of EGFR Tyr1045 and Tyr1173 sites (A and B) and ERK1/2 (C) after a rapid treatment (5 min) with 100 ng/ml EGF or TGFα in Ishikawa and MCF7-TR1 cells. Total EGFR and ERK2 levels were used as a loading control. Data shown are representative of three independent experiments.
EGF and TGFα stimulate c-fos expression and its recruitment to an AP-1 site located within the GPR30 promoter sequence
In our previous studies (6, 9, 10, 11, 12) we have demonstrated using different hormone-sensitive tumor cells that activation of the EGFR/ERK transduction pathway induced the expression of the immediate early gene c-fos, which stimulates normal cell growth and cellular transformation, mainly by forming a AP-1 transcription complex with members of the c-jun family (40). Treatment of Ishikawa and MCF7-TR1 cells with EGF and TGFα for 2 h led to a strong induction of c-fos protein, which was abolished in the presence of either AG and PD (Fig. 6, A and B), or with silencing of EGFR expression (Fig. 6, C and D), suggesting that EGFR/ERK signaling mediates c-fos expression upon exposure to these ligands. To evaluate whether the up-regulation of c-fos induced by EGF and TGFα could be involved in inducing GPR30 expression, we next performed chromatin immunoprecipitation (ChIP) analysis using immunoprecipitation of cell chromatin with an anti-c-fos GPR30 promoter. Treatment for 2 h with either EGF or TGFα strongly recruited c-fos to the AP-1 site in Ishikawa (Fig. 7) and MCF7-TR1 cells (data not shown). We did not visualize any ethidium bromide staining using primer pairs to amplify a control DNA sequence that does not contain the AP-1 site as a negative control (Fig. 7).
Fig. 6.

EGF and TGFα up-regulate c-fos protein levels in Ishikawa and MCF7-TR1 cells. Immunoblots of c-fos from cells treated for 2 h with vehicle (−), 100 ng/ml EGF, or 100 ng/ml TGFα alone and in combination with 10 μm of the EGFR inhibitor tyrphostin AG, the MEK inhibitor PD (A and B), or by silencing EGFR expression (C and D). The efficacy of EGFR silencing was evaluated by immunoblots as indicated. β-Actin served as a loading control. Data shown are representative of three independent experiments.
Fig. 7.

EGF and TGFα induce the recruitment of c-fos at the AP-1 site in the GPR30 promoter sequence. c-fos is recruited to the AP-1 site located within the GPR30 promoter sequence in Ishikawa cells treated for 2 h with 100 ng/ml EGF or TGFα. The amplification of a region lacking the AP-1 site (control) shows no recruitment in the above mentioned experimental conditions. In control samples, nonspecific IgG was used instead of the primary antibody.
EGFR and GPR30 interact directly
On the basis of the present and previous data (41) indicating that a functional cross talk exists between GPR30 and EGFR in tumor cells, we next evaluated their potential direct interaction using co-immunoprecipitation assays in a time course study. In particular, in Ishikawa cells EGF stimulated the immunoprecipitation of GPR30 with activated EGFR, as revealed using the antiphospho-EGFR Tyr1173 antibody (Fig. 8, A and B). Results similar to those obtained within the first 24 h of treatment were observed on d 5 of treatment (supplemental Fig. 2) following the same experimental conditions of the growth studies (see below). Next, the immunoprecipitation of GPR30 with the anti-EGFR antibody was lowered in the presence of both AG and PD (Fig. 8C), indicating that the interaction between GPR30 and EGFR requires EGFR activation and an active ERK signaling. Thereafter, we also determined that the expression of EGFR is not increased by EGF treatment in Ishikawa cells (Fig. 8D), hence the enhanced interaction between GPR30 and EGFR upon EGF exposure does not rely on EGFR up-regulation by its cognate ligand. Finally, the coimmunoprecipitation of GPR30 with EGFR using the anti-EGFR antibody was no longer evident silencing EGFR expression by shRNA, hence demonstrating the specificity of the interaction between the two proteins (Fig. 8E).
Fig. 8.
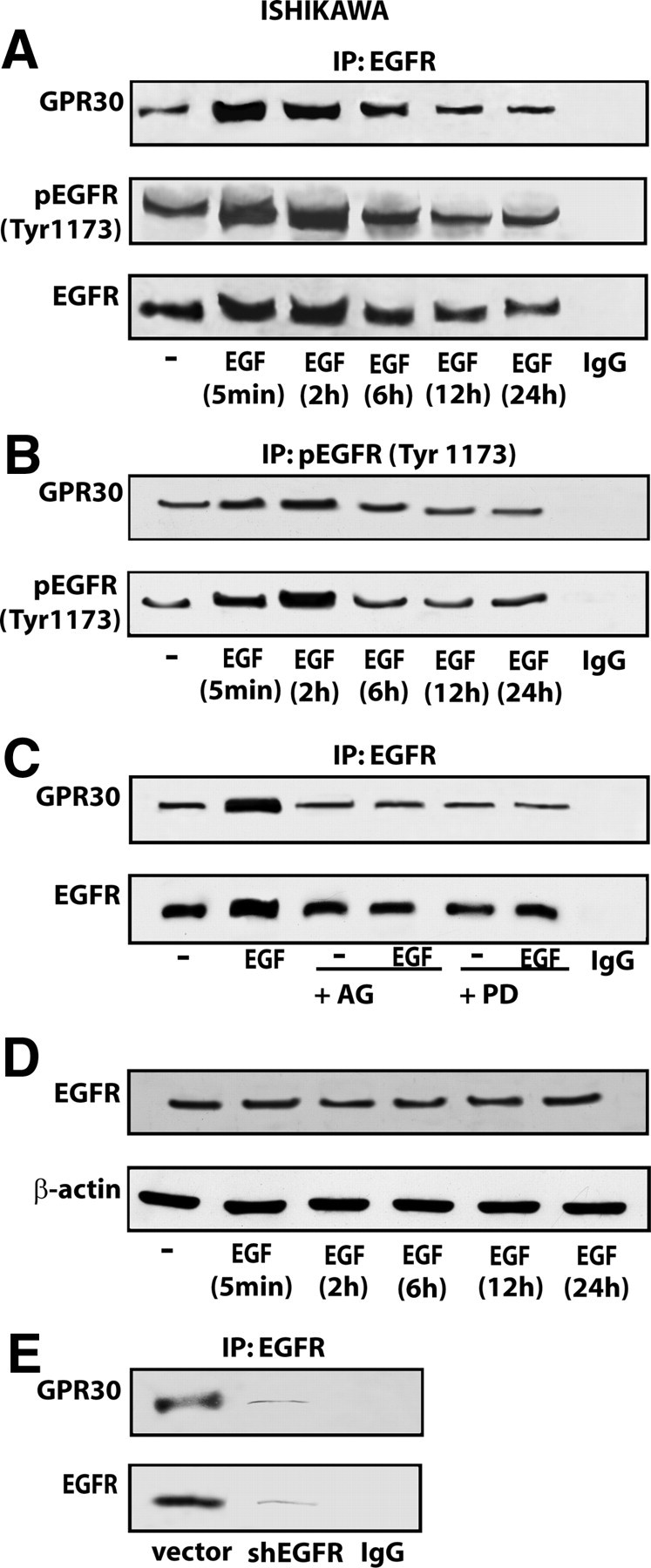
EGFR coimmunoprecipitates with GPR30 in Ishikawa cells. A and B, time course studies of GPR30 coimmunoprecipitation with EGFR and EGFR Tyr1173 in Ishikawa cells treated with 100 ng/ml EGF. C, The enhanced association of GPR30 with EGFR in Ishikawa cells treated for 2 h with 100 ng/ml EGF is prevented in presence of 10 μm of AG and PD, EGFR, and MEK inhibitors, respectively. D, Time course evaluation of EGFR protein expression in Ishikawa cells treated with 100 ng/ml EGF. E, The coimmunoprecipitation of GPR30 with EGFR is no longer evident knocking down EGFR expression. The coimmunoprecipitation assays were performed using antibodies against EGFR or EGFR Tyr1173 followed by immunoblotting for GPR30 as indicated. In control samples, nonspecific IgG was used instead of the primary antibody. Data shown are representative of three independent experiments. IP, Immunoprecipitation.
The proliferative effects induced by E2 in Ishikawa cells require both GPR30 and ERα
As a biological counterpart of the aforementioned results, we assessed cell proliferation upon exposure to E2, EGF, and TGFα alone or in combination, and by silencing GPR30, EGFR, or ERα expression. Because the MCF-7-TR1 cell line is cultured in medium containing tamoxifen which acts as an ER antagonist but also as a GPR30 activator, only Ishikawa cells were used in proliferation assays as well as in coimmunoprecipitation studies. First, we ascertained that the growth stimulation induced by each ligand is further increased treating cells with EGF or TGFα in the presence of E2 (Fig. 9A). Thereafter, we evaluated the proliferation of Ishikawa cells using knockdown of GPR30 because E2 is known to bind and activate GPR30 (7, 8), and the expression of which is up-regulated by EGF and TGFα treatment as we have demonstrated in the present and our previous studies (12). Interestingly, cell growth induced by E2 alone, and the additional stimulation observed in the presence of either EGF or TGFα was abrogated with silencing of GPR30 (Fig. 9B). The proliferative effects stimulated by E2 were still observed when EGFR expression was silenced (Fig. 9C) but were abrogated when ERα protein levels were reduced (Fig. 9D). Taken together, these findings suggest that estrogen-dependent proliferation of Ishikawa cells requires both GPR30 and ERα. Hence, to provide further insight into the mechanisms by which these receptors cooperate in mediating estrogen action, we assessed their direct association using coimmunoprecipitation assays. Of note, GPR30 and ERα showed a physical interaction that was further increased by E2 (Fig. 10A). These findings were similar to those obtained on d 5 of treatment (supplemental Fig. 3) following the same experimental conditions of the growth studies. Next, the enhanced GPR30 coimmunoprecipitation with ERα in the presence of E2 was prevented using the ER-antagonist ICI (Fig. 10B), suggesting that a ligand-dependent interaction may occur between the two receptors. However, further studies are needed to determine the exact mechanism(s) by which E2 stimulates the coprecipitation of GPR30 with ERα. It is worthwhile to note that EGF was able to up-regulate the expression of ERα (Fig. 10C), whereas the enhanced coprecipitation of GPR30 with ERα by E2 was still evident silencing EGFR expression (Fig. 10D). When either GPR30 or ERα was knocked down, only the single expression of the targeted receptor was abrogated (Fig. 10E).
Fig. 9.

ERα and GPR30 are required for Ishikawa cell proliferation induced by E2. A, Cell growth stimulated by EGF and TGFα is further increased in the presence of E2. Cells were treated with vehicle or 100 nm E2 and/or 100 ng/ml EGF and TGFα in medium containing 2.5% charcoal-stripped FBS (medium was refreshed and treatments were renewed every 2 d). B–D, The growth effects induced by E2 alone or in combination with EGF and TGFα in Ishikawa cells were abolished silencing GPR30 or ERα expression. Cells were transfected with an empty vector, shGPR30, or shERα and the next day were treated with vehicle (−), 100 nm E2, and/or 100 ng/ml EGF and TGFα in medium containing 2.5% charcoal-stripped FBS. Medium, transfections, and treatments were renewed every 2 d. C, Ishikawa cell proliferation induced by EGF and TGFα was abrogated using a shEGFR, whereas the growth effects stimulated by E2 were still maintained. Cells were transfected with an empty vector or a shEGFR and the next day were treated with vehicle (−), 100 nm E2, and/or 100 ng/ml EGF and TGFα. Medium, transfections, and treatments were renewed every 2 d. The efficacy of ERα, EGFR, and GPR30 silencing was evaluated by immunoblots, as indicated. Each column represents the mean ± sd of three independent experiments performed in triplicate. ○, •, □, ▪, and ♦, P < 0.05, for cells receiving vehicle (−) vs. treatment.
Fig. 10.
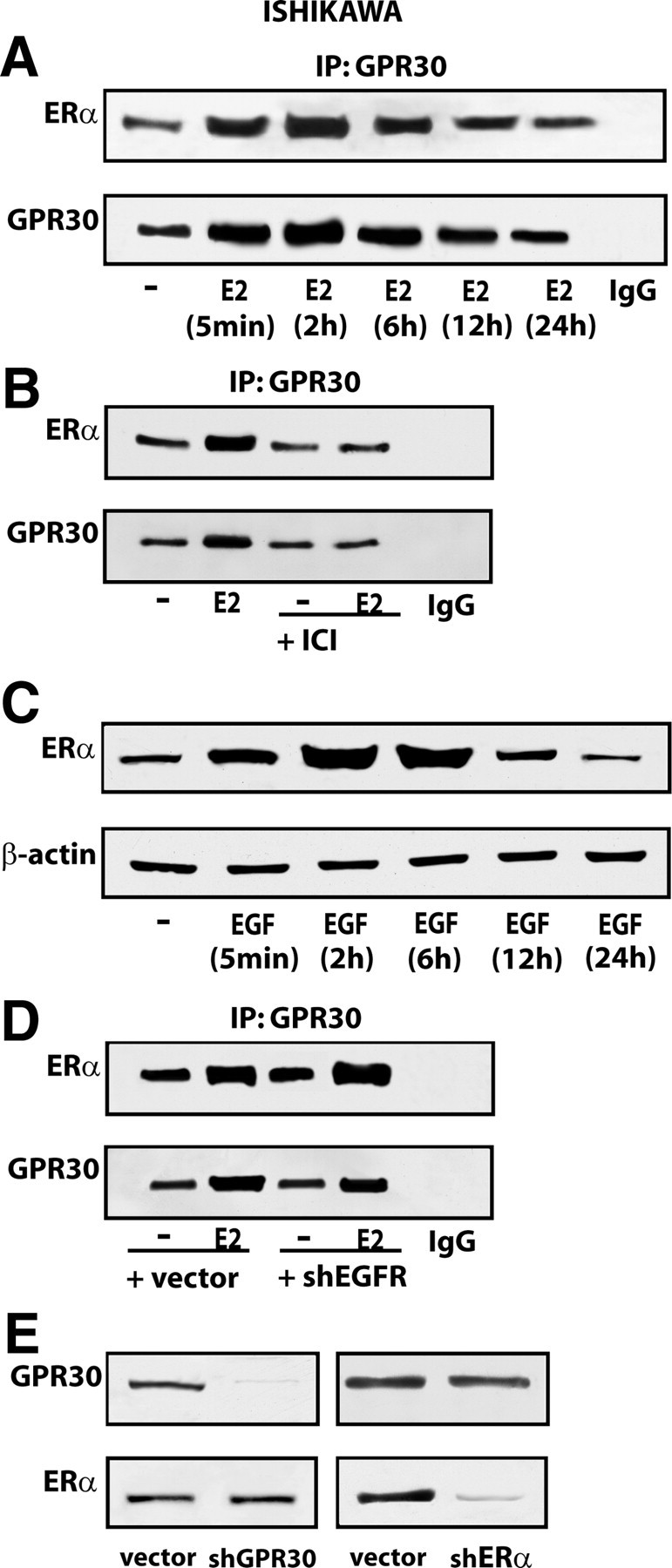
GPR30 coimmunoprecipitates with ERα in Ishikawa cells. A, Time course study of ERα coimmunoprecipitation with GPR30 in Ishikawa cells treated with 100 nm E2. B, The enhanced association of ERα with GPR30 in Ishikawa cells treated for 2 h with 100 nm E2 is prevented by 10 μm ICI. C, Time course evaluation of ERα protein expression in Ishikawa cells treated with 100 ng/ml EGF. D, ERα coimmunoprecipitates with GPR30 even silencing EGFR in Ishikawa cells treated with 100 nm E2 for 2 h; the efficacy of EGFR knockdown is shown in Fig. 9C. E, GPR30 silencing does not alter ERα expression as well as ERα knockdown does not influence GPR30 protein levels. In control samples, nonspecific IgG was used instead of the primary antibody. Data shown are representative of three independent experiments. IP, Immunoprecipitation.
Discussion
In the present study, we have provided new insights regarding the regulation and function of GPR30 in ER-positive cancer cells. We have demonstrated that EGF- and TGFα-dependent activation of the EGFR signaling pathway leads to the transactivation of the GPR30 promoter and up-regulation of GPR30 expression at both the mRNA and protein levels. The response to both ligands was evident in the Ishikawa endometrial and the TamR MCF7-TR1 breast cancer cells, but not in BG-1 ovarian or wild-type MCF7 breast tumor cells. Response was dependent on sufficient EGFR expression and activation at the Tyr1045 and Tyr1173 EGFR phosphorylation sites. By knocking down EGFR expression and using specific EGFR and MEK inhibitors, we have also explored the molecular mechanisms by which the EGFR/ERK transduction pathway mediates GPR30 expression, including the expression of c-fos and its recruitment to the AP-1 site located within the GPR30 promoter region. As a biological counterpart, the proliferative effects induced by EGF and TGFα in Ishikawa cells through the EGFR/ERK transduction pathway were enhanced by estrogen treatment in a GPR30- and ERα-dependent manner. The latter findings were nicely supported by our novel evidence showing that GPR30 physically interacts with activated EGFR and ERα, generating a functional protein complex assembly.
EGFR (also called ErbB1/HER1) is the prototypical member of a family of four structurally related receptor tyrosine kinases. The other members include: ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4, and together they have been shown to play an integral role in the development and growth of the mammary gland and uterus (42, 43). Individually, EGFR recognizes members of a family of small polypeptide ligands that are homologous to EGF, including amphiregulin, betacellulin, heparin-bound EGF, neuregulins, and TGFα. In particular, TGFα has been implicated in a variety of physiological processes, as well as in tumor growth and progression, via an autocrine/paracrine loop triggering EGFR binding and activation (27). Moreover, a potential role for TGFα has been suggested in drug-resistant HER2-overexpressing tumors (44), as well as in TamR breast cancers characterized by increased EGFR/HER2 expression and/or activation of downstream transduction pathways like the Erk1/2 signaling network (45).
The EGFR transduction pathway has been implicated in estrogen action (46). Intrauterine estrogen administration increased EGF concentrations (47) and EGFR autophosphorylation (48), whereas neutralizing antibodies against EGF inhibited estrogen-induced uterine growth (49). In addition, in vitro experiments demonstrated that estrogen stimulates various EGFR-associated signaling cascades, including MAPK activation, which is dependent on the rapid release of heparin-bound EGF, and activation of matrix metalloproteinase-2 and -9 (21, 24, 25, 50, 51).
Recently, GPR30 has been identified as a candidate for promoting rapid estrogen action that can occur in an ER-independent manner through the EGFR transduction pathway (6, 9, 10, 11, 12, 24, 25, 50, 51). However, our previous investigations have demonstrated that a cross talk between GPR30 and ERα, including EGFR signaling, mediates the effects of estrogen, as well as those exerted by the selective GPR30 ligand G-1 (11, 52). In this context, the present study provides novel insights regarding the physical association between GPR30 and activated EGFR and ERα, leading to the generation of a complex involved in relevant biological responses, such as gene expression and cell proliferation. Considering the ability of EGF and TGFα to up-regulate GPR30 expression, which in turn potentiates the action of estrogen, we could assume that a positive feedback loop does occur within the ligand EGFR-, GPR30-, and ER-activated signaling network leading to the stimulation of estrogen-sensitive tumors. Taken together, the present data broaden our knowledge regarding the cross talk between EGF and estrogen signaling, which can also take place in an ER-independent manner through EGFR activation, as we have demonstrated for GPR30 regulation and function (12). It should noted, however, that either GPR30 or ERα are required to mediate estrogen-induced activity in cell contexts harboring both receptors.
Treatment with antiestrogenic compounds such as tamoxifen has considerably reduced the risk of recurrence and mortality in ERα-positive breast cancer (18); however, both de novo and acquired resistance remain major obstacles in tamoxifen-treated patients (53, 54, 55, 56, 57). Several studies have suggested that overexpression of EGFR or HER2 contributes to tamoxifen resistance (54, 58, 59, 60, 61, 62, 63, 64), even though the mechanisms involved in the failure of tamoxifen therapy are probably multifactorial and remain to be fully understood (Ref. 65 and references therein). Increased levels of EGFR and/or HER2 were found in breast cancer patients with poor response to tamoxifen treatment (66, 67) and in some sublines of MCF7 cells exhibiting acquired tamoxifen resistance (68, 69, 70). Moreover, EGFR/HER2 tyrosine kinase inhibitors effectively reversed resistance in these cell types (65, 71), strongly suggesting the involvement of this transduction pathway in sensitivity to tamoxifen therapy. Considering that the model system provided by TamR MCF7-TR1 breast cancer cells does not allow the evaluation of the entire findings observed in Ishikawa uterine cells, the role exerted by GPR30 in the resistance to tamoxifen in breast cancer cells remains to be fully investigated. However, on the basis of our data we may argue that ligand-activated EGFR signaling could contribute to tamoxifen resistance, at least in part, up-regulating GPR30, which in turn facilitates estrogen action. Given that tamoxifen binds to and activates GPR30 (7, 8, 9, 10, 72), an EGFR-dependent up-regulation of GPR30 could be further involved in the failure of treatment with tamoxifen due to its stimulatory activity exerted through GPR30.
GPR30 overexpression was recently associated with lower survival rates in endometrial cancer patients (73) and higher risk of developing metastatic disease in patients with breast cancer (74). Therefore, GPR30 levels may not only characterize estrogen sensitivity and the potential for a response to endocrine pharmacological interventions in these tumors but could also be predictive of biologically aggressive, resistant phenotypes consistent with adverse outcome and survival. Overall, the potential for GPR30-mediated signaling should be taken into account in estrogen-sensitive tumors to discover novel antiestrogens. Therefore, our data emphasize the need for new endocrine agents able to selectively block estrogen action without exerting any stimulatory effects through transduction pathways shared by the steroid- and growth factor-signaling networks. Finally it is noteworthy that the physical and functional interaction between GPR30, EGFR, and ERα discovered in this study may represent a potential target for novel pharmacotherapeutic approaches in estrogen-sensitive tumors.
Materials and Methods
Reagents
E2, EGF, IGF-I, PD, and TGFα were purchased from Sigma-Aldrich Corp. (Milan, Italy). G-1 was bought from Merck KGaA (Frankfurt, Germany). AG and ICI 182,780 were obtained from Biomol Research Laboratories, Inc. (DBA, Milan, Italy) and Tocris Chemicals (Bristol, UK), respectively. All compounds were solubilized in dimethylsulfoxide, except E2 and PD, which were dissolved in ethanol.
Cell culture
Ishikawa endometrial cancer cells and BG-1 ovarian cancer cells were maintained in DMEM without phenol red supplemented with 10% fetal bovine serum (FBS). MCF7 wild-type (MCF7-wt) and MCF7 tamoxifen resistant (MCF7-TR1) breast cancer cells were cultured in DMEM and MEM, respectively, with phenol red supplemented with 10% FBS. MCF7-TR1 cells were generated in the laboratory of Dr. Fuqua as previously described (75) by maintaining cells in MEM with 10% FBS, 6 ng/ml insulin, penicillin (100 U/ml), streptomycin (100 μg/ml), and adding 4-hydroxytamoxifen in 10-fold increasing concentrations every 4 wk (from 10−9 m to 10−6 m final). Cells were thereafter routinely maintained with 10−6 m 4-hydroxytamoxifen. The day before experiments for immunoblots, real-time PCR, ChIP, and coimmunoprecipitation assay cells were switched to medium without serum; thereafter cells were treated as indicated. Cells used for coimmunoprecipitation assays after 5 d of treatment were cultured as described for the growth evaluation (see below).
Plasmids
The luciferase expression vector for the GPR30 promoter (GPR30), a 641-bp fragment within the 5′-flanking region of the GPR30 gene and its mutants in AP-1 (GPR30AP1mut) and SP-1 (GPR30SP1mut) were obtained as previously described (12). The EGFR expression vector (pEGFR) was bought from Addgene, Inc. (Cambridge, MA). The SureSilencing (sh)EGFR, shERα, and negative control plasmid (shRNA) were purchased from SA Bioscience Corp. (Frederick, MD) and were used according to the manufacturer’s recommendations. Short hairpin constructs against human GPR30 (shGPR30) were generated and used as previously described (12).
Transfection and luciferase assays
Cells (1 × 105) were plated into 24-well dishes with 500 μl of regular growth medium per well the day before transfection. The medium was replaced with that lacking serum on the day of transfection performed using Fugene 6 reagent as recommended by the manufacturer (Roche Diagnostics, Milan, Italy), with a mixture containing 500 ng of GPR30 promoter plasmid and 2 ng of pRL-TK. After 5 h, the serum-free medium containing the indicated treatments was renewed after which cells were incubated for an additional 18 h. Luciferase activity was measured with the Dual Luciferase kit (Promega, Milan, Italy) according to the manufacturer’s recommendations. Firefly luciferase values were normalized to the internal transfection control provided by the Renilla luciferase activity. The normalized relative light unit values obtained from cells treated with vehicle were set as 1-fold induction upon which the activity induced by treatments was calculated.
Reverse transcription and real-time PCR
Ishikawa and MCF7-TR1 cells were grown in 10-cm dishes in regular growth medium and then switched to medium lacking serum for 24 h. Thereafter, treatments were added for the times indicated, and cells were processed for mRNA extraction using Trizol reagent (Invitrogen, Milan, Italy) according to the manufacturer’s protocol. RNA was quantified spectrophotometrically, and its quality was checked by electrophoresis through agarose gel stained with ethidium bromide. Only samples that were not degraded and showed clear 18S and 28S bands under ultraviolet light were used for real-time PCR. Total cDNA was synthesized from RNA by reverse transcription using the murine leukemia virus reverse transcriptase (Invitrogen) following the protocol provided by the manufacturer. The expression of selected genes was quantified by real-time PCR using Step One sequence detection system (Applied Biosystems, Inc, Milano, Italy), following the manufacturer’s instructions. Gene-specific primers were designed using Primer Express version 2.0 software (Applied Biosystems. Inc.). Assays were performed in triplicate, and the mean values were used to calculate expression levels, using the relative standard curve method. For GPR30 and the ribosomal protein 18S, which was used as a control gene to obtain normalized values, the primers were: 5′-ACACACCTGGGTGGACACAA-3′ (GPR30 forward); 5′-GGAGCCAGAAGCCACATCTG-3′ (GPR30 reverse); 5′-GGCGTCCCCCAACTTCTTA-3′ (18S forward) and 5′-GGGCATCACAGACCTGTTATT-3′ (18S reverse).
Western blotting.
Cells were grown in 10-cm dishes, exposed to ligands for 2 h, and then lysed in 500 μl of RIPA buffer (20 mm Tris-HCl, pH 7.5; 100 mm NaCl; 0.5% Nonidet P-40; 0.5 mm EDTA; 0.5 mm phenylmethylsulfonylfluoride) a mixture of protease inhibitors containing 1,7 mg/ml aprotinin, 1 mg/ml leupeptin, 200 mmol/liter phenylmethylsulfonylfluoride, 200 mmol/liter sodium orthovanadate, and 100 mmol/liter sodium fluoride. Protein concentration was determined using Bradford reagent according to the manufacturer’s recommendations (Sigma-Aldrich). Equal amounts of whole-protein extract were resolved on a 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel, transferred to a nitrocellulose membrane (Amersham Biosciences, Milan, Italy), probed overnight at 4 C with antibodies against GPR30 (LS-A4271) (MBL-Eppendorf, Milan, Italy), c-fos (H-125), EGFR (1005), ERα (F-10), β-actin (C-2), phosphorylated ERK1/2 (E-4), ERK2 (C-14), and pEGFR Tyr 1173 (sc-12351) (all purchased from Santa Cruz Biotechnology, Inc., DBA), pEGFRs (Tyr1045, Tyr 1068, Tyr992) (Cell Signaling Technologies, Inc., Celbio, Milan, Italy) and then revealed using the ECL Western Blotting Analysis System (Amersham Biosciences).
ChIP
Cells grown in 10-cm plates were shifted for 24 h to medium lacking serum and then treated for 2 h with vehicle or 100 ng/ml EGF and TGFα. ChIP assay was performed as we have previously described (76). The immunocleared chromatin was precipitated with anti-c-fos antibody. A 4 μl volume of each sample was used as template to amplify by PCR two fragments located into the GPR30 promoter region: one fragment of 261 bp containing the AP-1 site and the second fragment of 364 bp (from −937 to −1301) not containing the AP-1 site. The primer pairs used to amplify the first fragment were: 5′-CGTGCCCATACCTTCATTGCTTCC-3′ (forward) and 5′-CCTGGCCGGGTGTCTGTAG-3′ (reverse) whereas the primer pairs used to amplify the second fragment were: 5′-CCGTGGCCCGCTGCATAGAGAAC-3′ (forward) and 5′-GAGAGGGAGAAGTGGGCTGTC-3′ (reverse). The PCR conditions were 45 sec at 94 C, 40 sec at 58 C, and 90 sec at 72 C. The amplification products obtained in 25 cycles were analyzed in a 2% agarose gel and visualized by ethidium bromide staining. Of the initial preparations of soluble chromatin, 3 μl was amplified to control input DNA before precipitation.
Coimmunoprecipitation
After stimulation with 100 ng/ml EGF or 100 nm E2 as indicated, Ishikawa cells were washed with PBS and lysed using 500 μl RIPA buffer with a mixture of protease inhibitors containing 1.7 mg/ml aprotinin, 1 mg/ml leupeptin, 200 mmol/liter phenylmethylsulfonyl fluoride, 200 mmol/liter sodium orthovanadate, and 100 mmol/liter sodium fluoride. Samples were then centrifuged at 13,000 rpm for 10 min, and protein concentrations were determined using Bradford reagent. Protein (400 μg) was then incubated overnight with: 900 μl of immunoprecipitation buffer with inhibitors, 2 μg of EGFR or ERα antibody (1:1000), 20 μl of Proteina A/G agarose immunoprecipitation reagent (Santa Cruz Biotechnology, Inc.). Samples were centrifuged at 13,000 rpm for 5 min at 4 C to pellet beads. Pellets were washed four times with 500 μl of PBS and centrifuged at 13,000 rpm for 5 min at 4 C. Supernatants were collected, resuspended in 20 μl RIPA buffer with protease inhibitors, 2× SDS sample buffer (40 mm Tris-HCl; 4% glycerol; 2% SDS) and β-mercaptoethanol and heated to 95 C for 5 min. Samples were then run on 10% SDS-PAGE, transferred to nitrocellulose, and probed with rabbit anti-GPR30 or anti-EGFR antibody. Western blot analysis and ECL detection were performed as described above. For EGFR, GPR30, and ERα silencing experiments, cells were plated in 10-cm dishes and transfected with 5 μg of the corresponding shRNA in medium without serum for 48 h, using Fugene 6 reagent as recommended by the manufacturer (Roche Diagnostics).
Proliferation assay
For quantitative proliferation assay, 1 × 105 cells were seeded in 24-well plates in regular growth medium. Cells were washed once they had attached and then incubated in medium containing 2.5% charcoal-stripped FBS with the indicated treatments; medium was renewed every 2 d (with treatments) before dimethylthiazoldiphenyltetra-zoliumbromide assays. A concentration of 200 ng/liter of the indicated shRNA was transfected using Fugene 6 Reagent as recommended by the manufacturer the day before treatments and then renewed every 2 d before counting.
Statistical analysis
Statistical analysis was performed using ANOVA followed by Newman-Keuls’ testing to determine differences in means. P < 0.05 was considered as statistically significant.
NURSA Molecule Pages:
Ligands: 17β-estradiol | Fulvestrant;
Nuclear Receptors: ER-α.
Footnotes
This work was supported by grants from Associazione Italiana per la Ricerca sul Cancro, Ministero dell’Università e Ricerca Scientifica e Tecnologica, Regione Calabria, and Provincia di Cosenza.
Disclosure Summary: The authors have nothing to disclose.
First Published Online September 11, 2009
Abbreviations: AG, AG1478; AP-1, activator protein-1; ChIP, chromatin immunoprecipitation; E2, 17β-estradiol; EGF, epidermal growth factor; EGFR, EGF receptor; ER, estrogen receptor; FBS, fetal bovine serum; G-1,1-[4-(-6-bromobenzol [1,3] diodo-5-yl)-3a, 4, 5, 9b-tetrahidro-3H-cyclopenta[c]quinolin-8yl] ethanone; GPR30, G protein-coupled receptor 30; ICI, ICI 182,780; MEK, MAPK kinase; PD, PD98059; SDS, sodium dodecyl sulfate; sh, short hairpin; SP-1, stimulatory protein-1; TamR, tamoxifen-resistant.
References
- 1.Pike MC, Pearce CL, Wu AH2004. Prevention of cancers of the breast, endometrium and ovary. Oncogene 23:6379–6391 [DOI] [PubMed] [Google Scholar]
- 2.Hacker NF2005. Uterine cancer. In: Berek JS, Hacker NF, eds. Practical gynecologic oncology. Philadephia: Lippincott Williams & Wilkins; 397–442
- 3.Tsai SY, Tsai MJ, O’Malley BW1989. Cooperative binding of steroid hormone receptors contributes to transcriptional synergism at target enhancer elements. Cell 57:443–448 [DOI] [PubMed] [Google Scholar]
- 4.Kumar V, Chambon P1988. The estrogen receptor binds to its responsive element as a ligand-induced homodimer. Cell 55:145–156 [DOI] [PubMed] [Google Scholar]
- 5.Kampa M, Pelekanou V, Castanas E2008. Membrane-initiated steroid action in breast and prostate cancer. Steroids 73:953–960 [DOI] [PubMed] [Google Scholar]
- 6.Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, Montanaro D, Musti AM, Picard D, Andò S2004. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17β-estradiol and phytoestrogens in breast cancer cells. J Biol Chem 279:27008–27016 [DOI] [PubMed] [Google Scholar]
- 7.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER2005. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 307:1625–1630 [DOI] [PubMed] [Google Scholar]
- 8.Thomas P, Pang Y, Filardo EJ, Dong J2005. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146:624–632 [DOI] [PubMed] [Google Scholar]
- 9.Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Andò S, Maggiolini M2006. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17β-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol 20:631–646 [DOI] [PubMed] [Google Scholar]
- 10.Vivacqua A, Bonofiglio D, Albanito L, Madeo A, Rago V, Carpino A, Musti AM, Picard D, Andò S, Maggiolini M2006. 17β-Estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the G protein coupled-receptor GPR30. Mol Pharmacol 70:1414–1423 [DOI] [PubMed] [Google Scholar]
- 11.Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Andò S, Maggiolini M2007. G-protein couplet receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res 67:1859–1866 [DOI] [PubMed] [Google Scholar]
- 12.Albanito L, Sisci D, Aquila S, Brunelli E, Vivacqua A, Madeo A, Lappano R, Pandey DP, Picard D, Mauro L, Andò S and Maggiolini M2008. Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells. Endocrinology 149:3799–3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dupont WD, Page DL1991. Menopausal estrogen replacement therapy and breast cancer. Arch Intern Med 151:67–72 [PubMed] [Google Scholar]
- 14.Scott JA, McGuire WL1991. New molecular markers of prognosis in breast cancer. New York: Raven Press; 179–196
- 15.Ponzone R, Biglia N, Jacomuzzi ME, Mariani L, Dominguez A, Sismondi P2006. Antihormones in prevention and treatment of breast cancer. Ann NY Acad Sci 1089:143–158 [DOI] [PubMed] [Google Scholar]
- 16.Jordan VC, Fritz NF, Tormey DC1987. Endocrine effects of adjuvant chemotherapy and long-term tamoxifen administration on node-positive patients with breast cancer. Cancer Res 47:624–630 [PubMed] [Google Scholar]
- 17.Howell A, DeFriend D, Robertson J, Blamey R, Walton P1995. Response to a specific antioestrogen (ICI 182780) in tamoxifen-resistant breast cancer. Lancet 345:29–30 [DOI] [PubMed] [Google Scholar]
- 18.Early Breast Cancer Trialist’ Collaborative Group2005. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-years survival: an overview of the randomized trials. Lancet 365:1687–1717 [DOI] [PubMed] [Google Scholar]
- 19.Herynk MH, Fuqua SA2007. Estrogen receptors in resistance to hormone therapy. Adv Exp Med Biol 608:130–143 [DOI] [PubMed] [Google Scholar]
- 20.Nicholson RI, Hutcheson IR, Harper ME, Knowlden JM, Barrow D, McClelland RA, Jones HE, Wakeling AE, Gee JM2002. Modulation of epidermal growth factor receptor in endocrine-resistant, estrogen-positive breast cancer. Ann NY Acad Sci 963:104–115 [DOI] [PubMed] [Google Scholar]
- 21.Razandi M, Pedram A, Park ST, Levin ER2003. Proximal events in signalling by plasma membrane estrogen receptors. J Biol Chem 278:2701–2712 [DOI] [PubMed] [Google Scholar]
- 22.Song RX, Zhang Z, Santen RJ2005. Estrogen rapid action via protein complex formation involving ERα and Src. Trends Endocrinol Metab 16:347–353 [DOI] [PubMed] [Google Scholar]
- 23.Chung YL, Sheu ML, Yang SC, Lin CH, Yen SH2002. Resistance to tamoxifen-induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. Int J Cancer 97:306–312 [DOI] [PubMed] [Google Scholar]
- 24.Filardo EJ, Quinn JA, Bland KI, Frackelton Jr AR2000. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30 and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14:1649–1660 [DOI] [PubMed] [Google Scholar]
- 25.Filardo EJ, Quinn JA, Frackelton Jr AR, Bland KI2002. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol 16:70–84 [DOI] [PubMed] [Google Scholar]
- 26.Hsieh M, Conti M2005. G-protein-coupled receptor signalling and the EGF network in endocrine system. Trends Endocrinol Metab 16:320–326 [DOI] [PubMed] [Google Scholar]
- 27.Booth BW, Smith GH2007. Roles of transforming growth factor-α in mammary development and disease. Growth Factors 25:227–235 [DOI] [PubMed] [Google Scholar]
- 28.Madtes DK, Raines EW, Sakariassen KS, Assoian RK, Sporn MB, Bell GI, Ross R1988. Induction of transforming growth factor α in activated human alveolar macrophages. Cell 53:285–293 [DOI] [PubMed] [Google Scholar]
- 29.Wong DT, Weller PF, Galli SJ, Elovic A, Rand TH, Gallagher GT, Chiang T, Chou MY, Matossian K, McBride J, Todd R1990. Human eosinophils express transforming growth factor α. J Exp Med 172:673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar V, Bustin SA, McKay IA1995. Transforming growth factor α. Cell Biol Int 19:373–388 [DOI] [PubMed] [Google Scholar]
- 31.Swendeman S, Mendelson K, Weskamp G2008. VEGF-A stimulates ADAM17-dependent shedding of VEGFR2 and crosstalk between VEGFR2 and ERK signalling. Circ Res 24:916–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis TS, Shapiro PS, Ahn NG1998. Signal transduction through MAP kinase cascades. Adv Cancer Res 74:49–139 [DOI] [PubMed] [Google Scholar]
- 33.Schaeffer HJ, Weber MJ1999. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol 19:2435–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo L, Kozlosky CJ, Ericsson LH, Daniel TO, Cerretti DP, Johnson RS2003. Studies of ligand-induced site-specific phosphorylation of epidermal growth factor receptor. J Am Soc Mass Spectrom 14:1022–1031 [DOI] [PubMed] [Google Scholar]
- 35.Zwick E, Hackel PO, Prenzel N, Ullrich A1999. The EGF receptor as central transducer of heterologous signalling systems. Trends Pharmacol Sci 20:408–412 [DOI] [PubMed] [Google Scholar]
- 36.Iordanov MS, Choi RJ, Ryabinina OP, Dinh TH, Bright RK, Magun BE2002. The UV (ribotoxic) stress response of human keratinocytes involves the unexpected uncoupling of the Ras-extracellular signal-regulated kinase signaling cascade from the activated epidermal growth factor receptor. Mol Cell Biol 22:5380–5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, Lipkowitz S, Yarden Y1999. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell 4:1029–1040 [DOI] [PubMed] [Google Scholar]
- 38.Ettenberg SA, Keane MM, Nau MM, Frankel M, Wang LM, Pierce JH, Lipkowitz S1999. cbl-b inhibits epidermal growth factor receptor signaling. Oncogene 18:1855–1866 [DOI] [PubMed] [Google Scholar]
- 39.Pennock S, Wang Z2008. A Tale of Two Cbls: Interplay of c-Cbl and Cbl-b in epidermal growth factor receptor downregulation. Mol Cell Biol 28:3020–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curran T, Franza Jr BR1988. Fos and Jun: the AP1 connection. Cell 55:395–397 [DOI] [PubMed] [Google Scholar]
- 41.Filardo EJ, Quinn JA, Sabo E2008. Association of the membrane estrogen receptor, GPR30, with breast tumor metastasis and transactivation of the epidermal growth factor receptor. Steroids 73:870–873 [DOI] [PubMed] [Google Scholar]
- 42.Stern DF2000. Tyrosine kinase signaling in breast cancer: ErbB family receptor tyrosine kinases. Breast Cancer Res 2:176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ebert AD, Wechselberger C, Martinez-Lacaci I, Bianco C, Weitzel HK, Salomon DS2000. Expression and function of EGF-related peptides and their receptors in gynecological cancer—from basic science to therapy. J Recept Signal Transduct Res 20:1–46 [DOI] [PubMed] [Google Scholar]
- 44.Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, Arteaga CL2007. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res 13:4909–4919 [DOI] [PubMed] [Google Scholar]
- 45.Gee JM, Robertson JF, Gutteridge E, Ellis IO, Pinder SE, Rubini M, Nicholson RI2005. Epidermal growth factor receptor/HER2/insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocr Relat Cancer 12(Suppl 1):S99–S111 [DOI] [PubMed]
- 46.Cunha GR, Wiesen JF, Werb Z, Young P, Hom YK, Cooke PS, Lubahn DB2000. Paracrine mechanisms of mouse mammary ductal growth. Adv Exp Med Biol 480:93–97 [DOI] [PubMed] [Google Scholar]
- 47.DiAugustine RP, Petrusz P, Bell GI, Brown CF, Korach KS, McLachlan JA, Teng CT1988. Influence of estrogens on mouse uterine epidermal growth factor precursor protein and messenger ribonucleic acid. Endocrinology 122:2355–2363 [DOI] [PubMed] [Google Scholar]
- 48.Mukku VR, Stancel GM1985. Regulation of epidermal growth factor receptor by estrogen. J Biol Chem 260:9820–9824 [PubMed] [Google Scholar]
- 49.Nelson KG, Takahashi T, Bossert NL, Walmer DK, McLachlan JA1991. Epidermal growth factor replaces estrogen in the stimulation of female genital tract growth and differentiation. Proc Natl Acad Sci USA 88:21–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Filardo EJ2002. Epidermal Growth Factor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signalling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol 80:231–238 [DOI] [PubMed] [Google Scholar]
- 51.Filardo EJ, Thomas P2005. GPR30: a seven-transmembrane-spanning estrogen receptor that triggers EGF release. Trends Endocrinol Metab 16:362–367 [DOI] [PubMed] [Google Scholar]
- 52.Albanito L, Lappano R, Madeo A, Chimento A, Prossnitz ER, Cappello AR, Dolce V, Abonante S, Pezzi V, Maggiolini M2008. G-protein-coupled receptor 30 and estrogen receptor-α are involved in the proliferative effects induced by atrazine in ovarian cancer cells. Environ Health Perspect 116:1648–1655 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Dowsett M, Houghton J, Iden C, Salter J, Farndon J, A’Hern R, Sainsbury R, Baum M2006. Benefit from adjuvant tamoxifen therapy in primary breast cancer patients according oestrogen receptor, progesterone receptor, EGF receptor and HER2 status. Ann Oncol 17:818–826 [DOI] [PubMed] [Google Scholar]
- 54.Arpino G, Green SJ, Allred DC, Lew D, Martino S, Osborne CK, Elledge RM2004. HER-2 amplification, HER-1 expression, and tamoxifen rensponse in estrogen receptor-positive metastatic breast cancer: A southwest oncology group study. Clin Cancer Res 10:5670–5676 [DOI] [PubMed] [Google Scholar]
- 55.Arpino G, Weiss H, Lee AV, Schiff R, De Placido S, Osborne CK, Elledge RM2005. Estrogen receptor-positive, progesterone receptor-negative breast cancer. Association with growth factor receptor expression and tamoxifen resistance. J Natl Cancer Inst 97:1254–1261 [DOI] [PubMed] [Google Scholar]
- 56.Linke SP, Bremer TM, Herold CD, Sauter G, Diamond C2006. A multimarker model to predict outcome in tamoxifen-treated breast cancer patients. Clin Cancer Res 12:1175–1183 [DOI] [PubMed] [Google Scholar]
- 57.Osborne CK, Shou J, Massarweh S, Schiff R2005. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res 11:865s–870s [PubMed] [Google Scholar]
- 58.Berry DA, Muss HB, Thor AD, Dressler L, Liu ET, Broadwater G, Budman DR, Henderson IC, Barcos M, Hayes D, Norton L2000. HER-2/neu and p53 expression versus tamoxifen resistance in estrogen receptor-positive, node-positive breast cancer. J Clin Oncol 18:3471–3479 [DOI] [PubMed] [Google Scholar]
- 59.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R2004. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst 96:926–935 [DOI] [PubMed] [Google Scholar]
- 60.Pérez-Tenorio G, Stål O2002. Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br J Cancer 86:540–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gee JM, Robertson JF, Ellis IO, Nicholson RI2001. Phosphorylation of ERK1/2 mitogen-activated protein kinase is associated with poor response to anti-hormonal therapy and decreased patient survival in clinical breast cancer. Int J Cancer 95:247–254 [DOI] [PubMed] [Google Scholar]
- 62.Nicholson S, Halcrow P, Sainsbury JR, Angus B, Chambers P, Farndon JR, Harris AL1988. Epidermal growth factor receptor (EGFR) status associated with failure of primary endocrine therapy in elderly postmenopausal patients with breast cancer. Br J Cancer 58:810–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giltnane JM, Rydén L, Cregger M, Bendahl PO, Jirström K, Rimm DL2007. Quantitative measurement of epidermal growth factor receptor is a negative predictive factor for tamoxifen response in hormone receptor positive premenopausal breast cancer. J Clin Oncol 25:3007–3014 [DOI] [PubMed] [Google Scholar]
- 64.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R2008. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res 68:826–833 [DOI] [PubMed] [Google Scholar]
- 65.Giordano C, Cui Y, Barone I, Andò S, Mancini MA, Berno V, Fuqua SA2009. Growth factor-induced resistance to tamoxifen is associated with a mutation of estrogen receptor α and its phosphorylation at serine 305. Breast Cancer Res Treat 10.1007/s10549-009-0334-0 [DOI] [PMC free article] [PubMed]
- 66.De Laurentiis M, Arpino G, Massarelli E, Ruggiero A, Carlomagno C, Ciardiello F, Tortora G, D’Agostino D, Caputo F, Cancello G, Montagna E, Malorni L, Zinno L, Lauria R, Bianco AR, De Placido S2005. A meta-analysis on the interaction between HER-2 expression and response to endocrine treatment in advanced breast cancer. Clin Cancer Res 11:4741–4748 [DOI] [PubMed] [Google Scholar]
- 67.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R2003. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst 95:353–361 [DOI] [PubMed] [Google Scholar]
- 68.Benz CC, Scott GK, Sarup JC, Johnson RM, Tripathy D, Coronado E, Shepard HM, Osborne CK1992. Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res Treat 24:85–95 [DOI] [PubMed] [Google Scholar]
- 69.Nicholson RI, Gee JM, Knowlden J, McClelland R, Madden TA, Barrow D, Hutcheson I2003. The biology of antihormone failure in breast cancer. Breast Cancer Res Treat 80 (Suppl 1):S29–S34; discussion, S35 [DOI] [PubMed]
- 70.Hutcheson IR, Knowlden JM, Madden TA, Barrow D, Gee JM, Wakeling AE, Nicholson RI2003. Oestrogen receptor-mediated modulation of the EGFR/MAPK pathway in tamoxifen-resistant MCF-7 cells. Breast Cancer Res Treat 81:81–93 [DOI] [PubMed] [Google Scholar]
- 71.Kurokawa H, Lenferink AE, Simpson JF, Pisacane PI, Sliwkowski MX, Forbes JT, Arteaga CL2000. Inhibition of HER2/neu (erbB-2) and mitogen-activated protein kinases enhances tamoxifen action against HER2-overexpressing, tamoxifen-resistant breast cancer cells. Cancer Res 60:5887–5894 [PubMed] [Google Scholar]
- 72.Prakash Pandey D, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D2009. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J 28:523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith HO, Leslie KK, Singh M, Qualls CR, Revankar CM, Joste NE, Prossnitz ER2007. GPR30: a novel indicator of poor survival for endometrial carcinoma. Am J Obstet Gynecol 196:386.e1–9; discussion 386.e9–e11 [DOI] [PubMed] [Google Scholar]
- 74.Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, Steinhoff MM, Sabo E2006. Distribution of GPR30, a seven membrane spanning estrogen receptor, in primary breast cancer and its association with clinico pathologic determinants of tumor progression. Clin Cancer Res 12:6359–6366 [DOI] [PubMed] [Google Scholar]
- 75.Herman ME, Katzenellenbogen BS1996. Response-specific antiestrogen resistance in a newly characterized MCF-7 human breast cancer cell line resulting from long-term exposure to trans-hydroxytamoxifen. J Steroid Biochem Mol Biol 59:121–134 [DOI] [PubMed] [Google Scholar]
- 76.Morelli C, Garofalo C, Sisci D, del Rincon S, Cascio S, Tu X, Vecchione A, Sauter ER, Miller Jr WH, Surmacz E2004. Nuclear insulin receptor substrate 1 interacts with estrogen receptor α at ERE promoters. Oncogene 23:7517–7526 [DOI] [PubMed] [Google Scholar]