

Abstract
Most receptor tyrosine kinases and G protein-coupled receptors (GPCRs) operate via a limited number of MAPK cascades but still exert diverse functions, and therefore signal specificity remains an enigma. Also, most GPCR ligands utilize families of receptors for mediation of diverse biological actions; however, the mammalian type I GnRH receptor (GnRHR) seems to be the sole receptor mediating GnRH-induced gonadotropin synthesis and release. Signaling complexes associated with GPCRs may thus provide the means for signal specificity. Here we describe a signaling complex associated with the GnRHR, which is a unique GPCR lacking a C-terminal tail. Unlike other GPCRs, this signaling complex is preformed, and exposure of LβT2 gonadotropes to GnRH induces its dynamic rearrangement. The signaling complex includes c-Src, protein kinase Cδ, -ε, and -α, Ras, MAPK kinase 1/2, ERK1/2, tubulin, focal adhesion kinase (FAK), paxillin, vinculin, caveolin-1, kinase suppressor of Ras-1, and the GnRHR. Exposure to GnRH (5 min) causes MAPK kinase 1/2, ERK1/2, tubulin, vinculin, and the GnRHR to detach from c-Src, but they reassociate within 30 min. On the other hand, FAK, paxillin, the protein kinase Cs, and caveolin-1 stay bound to c-Src, whereas kinase suppressor of Ras-1 appears in the complex only 30 min after GnRH stimulation. GnRH was found to activate ERK1/2 in the complex in a c-Src-dependent manner, and the activated ERK1/2 subsequently phosphorylates FAK and paxillin. In parallel, caveolin-1, FAK, vinculin, and paxillin are phosphorylated on Tyr residues apparently by GnRH-activated c-Src. Receptor tyrosine kinases and GPCRs translocate ERK1/2 to the nucleus to phosphorylate and activate transcription factors. We therefore propose that the role of the multiprotein signaling complex is to sequester a cytosolic pool of activated ERK1/2 to phosphorylate FAK and paxillin at focal adhesions.
A pre-formed signaling complex associated with the GnRH receptor sequesters activated ERK1/2 to phosphorylate FAK and paxillin at focal adhesions and regulates cell migration.
GnRH, a hypothalamic neurohormone, is secreted in a pulsatile manner to pituitary gonadotropes. It is the prime regulator of the synthesis and release of the gonadotropins LH and FSH. These stimulate gonadal sex hormone production and gametogenesis (1, 2, 3, 4, 5, 6). Upon stimulation, the GnRH receptor (GnRHR), a G protein-coupled receptor (GPCR), triggers a cascade of signaling events including sequential activation of phospholipase Cβ, phospholipase D, and phospholipase A2, Ca2+ mobilization and influx, activation of protein kinase Cs (PKCs) and MAPK cascades [ERK, Jun N-terminal kinase (JNK), and p38] and formation of prostaglandins and leukotrienes, culminating in LH and FSH synthesis and release (1, 2, 3, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23).
MAPK cascades in mammals include ERK1/2 (p42 and p44), JNK1/3, p38 (α, β, γ, δ), and ERK5 and act by a sequential phosphorylation and activation of their kinase components (24, 25). For instance, ERK1/2 is sequentially activated by Ras/Rap1, MAP4K, Rafs (MAP3K), and MAPK kinase (MEK)1/2 (MAPKK). The hallmark of members of the MAPK family is their ability to translocate to the nucleus and activate a variety of transcription factors (24, 25). MAPKs are known to participate in GnRH-induced transcriptional control of gonadotropin subunits and GnRHR genes (5, 6, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36).
Given that both receptor tyrosine kinases and GPCRs activate MAPKs with different responses, how then is signal specificity maintained? The presence of scaffold molecules and signaling complexes that bring together different MAPK cascade members and targeting them to specific sites in a spatio/temporal fashion, may provide signal specificity (37, 38, 39, 40, 41, 42). In this respect, a signaling platform for ERK activation by GnRH was reported to include the GnRHR, c-Raf kinase, Ca2+-calmodulin, and ERK2 and was localized to low-density membrane microdomains (lipid rafts/caveolae) (43, 44, 45). Furthermore, activation of ERK in human embryonic kidney (HEK)293 cells stably expressing the GnRHR was found to be dependent on its protein-protein interaction with focal adhesion kinase (FAK) and c-Src at focal adhesion (FA) complexes (46). Microtubules and actin filaments are already known to restrict cAMP formation by regulating the localization and interaction of GPCR-Gs-adenylyl cyclase in lipid rafts/caveolae (47). Also, in addition to its role in GPCR desensitization, β-arrestin is able to serve as an escort for signaling molecules such as c-Src and various components of the MAPK cascades. This impacts the temporal and spatial properties of ERK activation (48, 49, 50). Therefore it is thought that signal propagation through the MAPK cascades is probably regulated by a large network of context-specific signaling complexes, controlling the spatial and temporal aspects, as well as the magnitude of MAPK activation. This presumably gives rise to signal specificity (50).
Here we demonstrate the presence of a preformed signaling complex that seems to reside in microtubules at the boundaries of caveolae and FAs. The interacting proteins are c-Src, paxillin, FAK, caveolin1, tubulin, PKCδ, -ε, and -α, Ras, MEK1/2, ERK1/2, KSR-1 (kinase suppressor of Ras), and GnRHR. GnRH induces temporal changes in the complex composition. We propose that the role of this complex is to sequester a pool of activated ERK1/2 in the cytosol for the phosphorylation of FAK and paxillin at FAs. This could then lead to GnRH-mediated cell migration and spreading, both of which were recently attributed to the downstream actions of the GnRHR (46, 51).
Results
GnRH activates c-Src in gonadotrope LβT2 cells
Activation of the nonreceptor tyrosine kinase c-Src is associated with autophosphorylation of Tyr418 and dephosphorylation of Tyr529. We first exposed the cells to GnRH (10 nm; 5–30 min), lysed the cells, immunoprecipitated c-Src with anti-c-Src A/G agarose beads and followed by Western blotting with antiphospho-c-Src-Tyr418 antisera. As shown in Fig. 1A, GnRH stimulation resulted in enhanced c-Src-Tyr418 phosphorylation, hence activation. Preincubation with the c-Src inhibitor, PP2 (10 μm), abolished c-Src activation by GnRH (Fig 1A), confirming the possible use of PP2 in further studies.
Fig. 1.
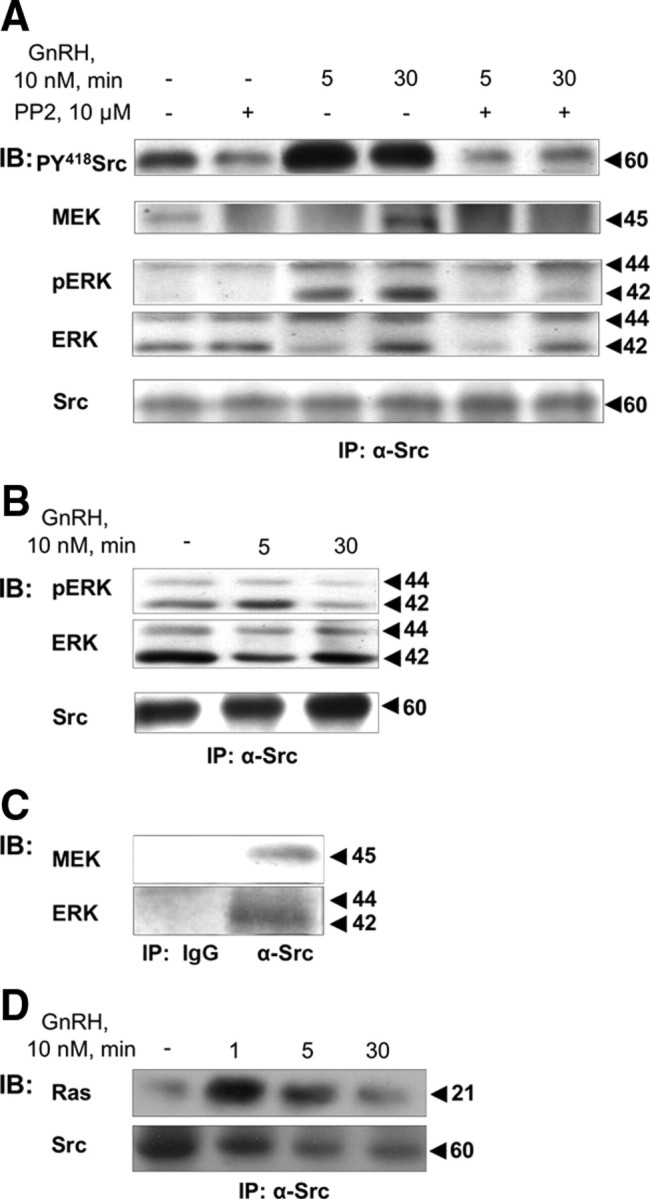
Activation of c-Src by GnRH and association of c-Src with Ras, MEK1/2, and ERK1/2. LβT2 cells were serum starved for 16 h before pretreatment with the c-Src inhibitor, PP2 (10 μm), for 30 min. Thereafter, GnRH (10 nm) was added for the indicated time. Cells were lysed, and proteins were subjected to SDS-PAGE after IP with α-Src antibodies. Samples were then immunoblotted (IB) with: A, antiphosphorylated Y418-Src antibodies (PY418 Src), anti-MEK antibodies, antiphospho-ERK (pERK), antitotal ERK (ERK), and anti-Src antibodies. B, Details as in panel A. Samples were IB with anti-phospho ERK (pERK), anti-total ERK (ERK), and anti-Src antibodies. C, Cells were lysed, and proteins were subjected to SDS-PAGE after IP with serum IgG, or α-Src antibodies. Samples were then IB with anti-MEK antibodies, or antitotal ERK (ERK). A representative blot is shown, and similar results were observed in two other experiments. D, LβT2 cells were serum starved for 16 h before treatment with GnRH (10 nm) for the indicated time. Cells were then lysed and subjected to SDS-PAGE after IP with α-Src antibodies and immunoblotting with specific anti-pan-Ras and anti-Src antibodies. Molecular mass (kDa) of the proteins are indicated on the right. A representative blot is shown, and similar results were observed in two other experiments.
Association between c-Src and MEK1/2
The modular SH3 and SH2 kinase domains of the Src family of tyrosine kinases serve as inducible linkers for a diverse array of signaling proteins. Because c-Src is activated by GnRH and is involved in MAPK (ERK, JNK, and p38) activation by GnRH (19, 32, 34, 52, 53), we decided to use it as a bait to identify binding partners forming a signaling complex for ERK1/2 activation. MEK1/2, the upstream activator of ERK1/2, is present in the c-Src immune complex even in control cells (Fig. 1A). Stimulation by GnRH (5 min) induced a transient detachment of MEK1/2 from the complex and its reassociation after 30 min post-GnRH treatment (n = 3, P < 0.05). Inhibition of c-Src by PP2 prevented the reappearance of MEK1/2 in the complex, indicating that activated c-Src is required for MEK1/2 reassociation. Similar results were observed with PP1, another c-Src inhibitor (data not shown). Moreover, the lack of association of MEK1/2 with the complex in PP2-treated control cells suggests that, in general, MEK1/2 is only associated with activated c-Src.
Association between c-Src and ERK1/2
With MEK1/2 identified in the c-Src immune complex, it was natural to look for ERK1/2 and its activation within the complex. We found c-Src activation to be required for GnRH-induced ERK1/2 activation because PP2 abolished ERK1/2 activation by GnRH in the c-Src immune complex (Fig. 1A). Total ERK1/2 (ERK), like MEK1/2, is present in the c-Src immune complex even in control cells (Fig. 1A). Notably, 5 min after GnRH stimulation, total ERK1/2 (ERK), like MEK1/2, appeared to detach from c-Src, in particular ERK1, returning to the complex after 30 min of GnRH treatment (n = 3; P < 0.05). This detachment could be most distinctly observed in the presence of PP2 after 5 min of GnRH stimulation (Fig. 1A). However, unlike MEK, inhibition of c-Src by PP2 did not prevent the reappearance of ERK in the complex after 30 min, indicating that activated c-Src is not required for ERK reassociation and that ERK reassociation occurs independent of MEK. The detachment of ERK after 5 min of incubation with GnRH is shown more clearly in Fig. 1B (n = 3; P < 0.05). Under the same conditions in which we could detect the detachment of ERK after 5 min of incubation with GnRH, the levels of c-Src in the immune complexes did not change (Fig. 1B). Also, during the detachment of ERK (5 min), there was an increase in pERK in the complex, suggesting that the remaining pool of ERK was phosphorylated in the complex (Fig. 1B). Using total lysates, we observed an approximately 20-fold induction of ERK1/2 activation by GnRH with kinetics similar to its activation within the complex. This activation was abolished with PP2 treatment (data not shown), suggesting that c-Src was involved in ERK1/2 activation in general. Control immunoprecipitation (IP) with serum IgG, followed by Western blotting for MEK1/2 and ERK1/2, gave no signal (Fig. 1C). Similarly, IP with anti-Src, followed by Western blotting with serum IgG gave no signal for MEK1/2, ERK1/2, and also for the interacting proteins described below (data not shown).
Association between c-Src and Ras
We next investigated the presence of Ras in the c-Src immune complex (Fig. 1D). Ras association with the complex could be detected in control cells, but unlike MEK and ERK, Ras did not detach from the complex after GnRH treatment. On the contrary, the association seemed to strengthen upon GnRH treatment (n = 3; P < 0.02 for 1 min and P < 0.05 for 5 and 30 min), suggesting enhanced interaction of the c-Src immune complex with Ras in the membrane (Fig. 1D).
The fate of ERK1/2 in GnRH-stimulated cells
It has been purported that several GPCR ligands induce rapid translocation of ERK1/2 to the nucleus to phosphorylate and activate transcription factors (24, 25). We wanted to determine whether this was the case with GnRH, and therefore treated the cells with GnRH (10 nm) to examine the fate of ERK1/2 by immunocytochemistry. 4′,6-diamidino-2-phenylindole (DAPI) staining was used to track the location of the nucleus. Under basal conditions, ERK1/2 was mainly observed in the cytoplasm (Fig. 2). Short treatment with GnRH (1 min) seems to redistribute some of the ERK1/2 molecules in FAs and in the nucleus. At the 30 min time point there was much more staining (∼35%) in the nucleus.
Fig. 2.

Localization of ERK upon GnRH stimulation. LβT2 cells were serum starved overnight and were stimulated with 10 nm GnRH for the indicated periods of time. The cells were then fixed and stained with antitotal ERK antibody and DAPI as described in Materials and Methods. Removal of the secondary antibody or preabsorption of the antibodies with the relevant peptide antigen resulted in disappearance or a marked reduction of the staining, respectively (data not shown). A representative image is shown, and similar results were observed in two other experiments. Bar, 30 μm.
The c-Src immune complex is selective for ERK1/2
Based on earlier findings by others suggesting the activation of MAPKs within scaffolds (37, 38, 39, 40, 41), we then attempted to determine whether the c-Src-based complex could act as a general scaffold for the activation of MAPKs (ERK, JNK, and p38). We therefore looked for JNK1/2 and p38 in the complex. GnRH was clearly able to activate both p38 and JNK in LβT2 cells, with a peak activation of p38 and JNK occurring after 15 and 30 min, respectively (Fig. 3A). However, total JNK and p38 were only found present in total lysates but not in c-Src immune complexes either in untreated or GnRH-treated cells (Fig. 3B). Thus, although c-Src mediates the activation of JNK and p38 by GnRH in αT3-1 and LβT2 cells (34, 52), it appears that the complex is selective for ERK1/2.
Fig. 3.
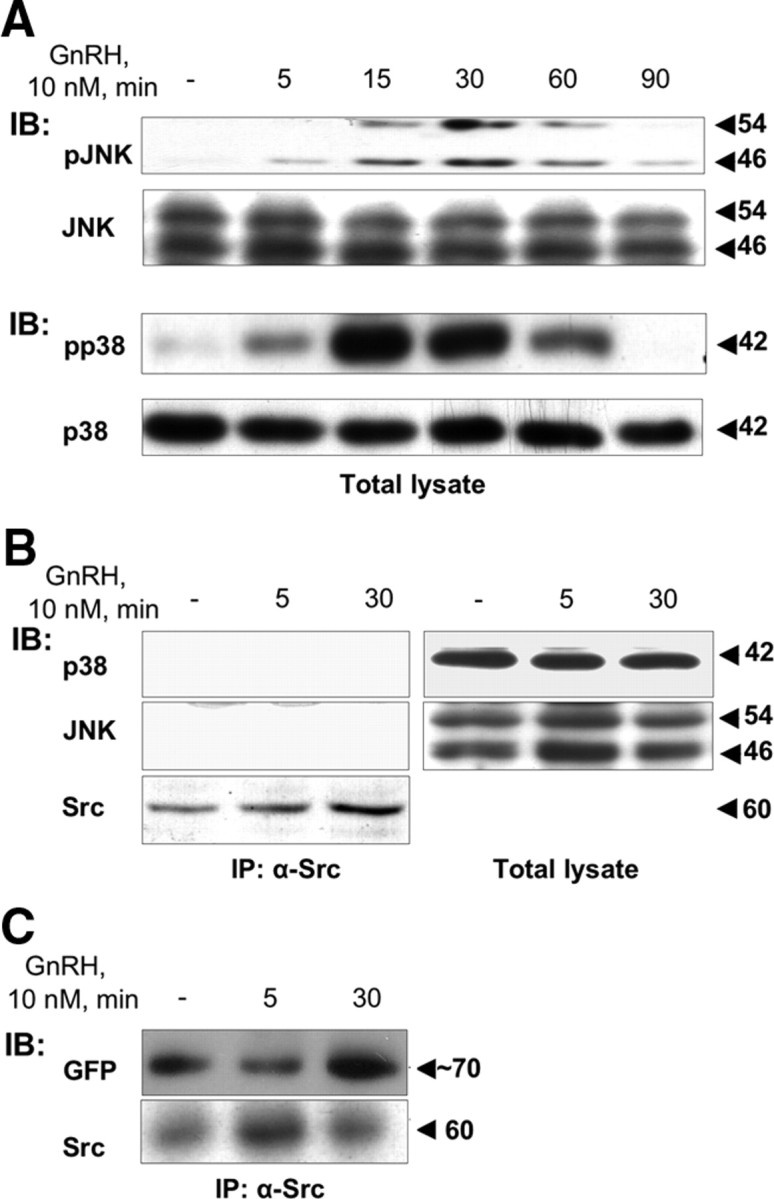
The c-Src immune complex is selective for ERK1/2. LβT2 cells were serum starved for 16 h before treatment with GnRH (10 nm) for the indicated time. Cells were lysed and subjected to SDS-PAGE directly (Total lysate) or after IP with α-Src antibodies (B and C). A, Activation pattern of JNK and p38MAPK as seen by Western blotting with antiphospho JNK (pJNK), or antiphospho p38 (pp38) antibodies. Total JNK and p38 were detected with polyclonal antibodies as a control for sample loading. B, p38 and JNK are in the total lystae but not in complex with Src. Total lysate or immunoprecipitated samples as above were probed with anti-JNK or anti-p38 antibodies. C, GnRHR associates with the complex. LβT2 cells were transfected with GnRHR-GFP as described in Materials and Methods, serum starved for 16 h before treatment with GnRH (10 nm) for the indicated time. Cells were lysed and subjected to SDS-PAGE after IP with α-Src antibodies. Immunoblotting (IB) was performed with anti-GFP antibodies. Molecular mass (kDa) of the proteins is indicated on the right. A representative blot is shown, and similar results were observed in two other experiments.
GnRHR is a binding partner in the c-Src immune complex
We then looked for the presence of the GnRHR in the c-Src-complex. The fate for the GnRHR was observed in GnRHR-green fluorescent protein (GFP) transfected cells after IP of c-Src and immunoblotting for GFP (Fig. 3C). The GnRHR-GFP could be detected in the complex in control cells. However, after GnRH treatment (5 min), this association was reduced and reappears after 30 min (n = 3; P < 0.05).
β-arrestin is not the scaffold in the GnRH-activated c-Src immune complex
Although we found Ras, MEK1/2, and ERK1/2 in the same complex as c-Src, given that there is no direct interaction between them (54), we searched for possible scaffold proteins that could assemble components of the ERK1/2 activation machinery (54). In addition to their role in GPCR desensitization, β-arrestin can also become an adaptor for signaling molecules such as c-Src, Ras, ERK1/2, JNK3, and MKK4, participating in MAPK activation (49, 55). Nevertheless, whereas arrestin 1 and 2 were found to be present in total lysates (Fig. 4A), they were not detected in the complex bearing c-Src, so that GnRH does not appear to recruit β-arrestin to the complex as a scaffold for MAPK activation.
Fig. 4.

α-Tubulin is present, but neither β-actin nor β-arrestin is found in the c-Src immune complex. LβT2 cells were serum starved for 16 h before treatment with GnRH (10 nm) for the indicated time. Cells were then lysed and subjected to SDS-PAGE directly (Total lysate), or after IP with α-Src antibodies. A, β-Arrestin is in the total lystae but not in the c-Src immune complex, as revealed by immunoblots (IB) of the c-Src immune complex with specific anti-β-arrestin antibodies. B, α-Tubulin is a binding partner in the GnRH-induced c-Src immune complex, as revealed by a proteomic analysis. SDS-PAGE of the c-Src immune complex was stained with Sypro Ruby, and a major GnRH-stimulated band was subjected to MALDI-TOF mass spectrometry. Peptide coverage map of the specific identified protein, α-tubulin, is shown. C, α-Tubulin is present in the c-Src immune complex, as revealed by IB of the c-Src immune complex with specific anti-α-tubulin antibodies. D, β-Actin is in the total lysate but not in the c-Src immune complex, as revealed by IB of the c-Src immune complex with specific anti-β-actin antibodies. Molecular mass (kDa) of the proteins is indicated on the right. A representative blot is shown, and similar results were observed in two other experiments.
α-Tubulin, but not β-actin, is a binding partner in GnRH-induced c-Src immune complex as revealed by proteomic analysis
We then took a proteomic approach by using c-Src immune complexes from control and GnRH-treated LβT2 cells and subjecting the samples to gel electrophoresis (Fig. 4B). We identified bands indicating GnRH-induced increases in association with c-Src. Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry then revealed α-tubulin as one of those proteins to be differentially associated with the complex under GnRH treatment (Fig. 4B). GnRH increased α-tubulin association with c-Src most markedly after 30 min of incubation (Fig. 4B). Once identified, we could now verify the results by immunoblotting the GnRH-stimulated c-Src immune complexes with anti-α-tubulin (Fig. 4C). As with Ras-MEK-ERK, α-tubulin was present in the immune complex even in untreated cells. Much like MEK-ERK, α-tubulin appeared to detach from the complex after 5 min exposure to GnRH but reassociated after 30 min. The data suggest the dynamic nature of the microtubules-based c-Src immune complex. We then tested the possibility that other structural proteins were resident in the complex, and for this, we checked for the presence of β-actin. We could detect β-actin in the total lysates, but not in the complex for both control and GnRH-treated cells (Fig. 4D).
Caveolin-1 is present in the c-Src immune complex
Previous studies have localized some GPCR-induced signaling complexes, including that of the GnRHR, to lipid rafts/caveolae (43, 44, 45, 47). We therefore searched for the presence of caveolin-1, a marker for caveolae, in the c-Src immune complex. Like the others, caveolin-1 was present in the complex even in untreated cells (Fig. 5A), except that, as in the case of Ras, caveolin-1 did not seem to detach from the complex after 5 min exposure to GnRH. Furthermore, caveolin-1 levels in the complex progressively increased with GnRH treatment. Also, exposure to GnRH resulted in enhanced phosphorylation of caveolin-1 on Tyr14 in the c-Src immune complex (n = 3; P < 0.02). Preincubation with the c-Src inhibitor PP2 resulted in a marked inhibition of the tyrosine phosphorylation of caveolin-1 (Fig. 5B). The data suggest an increase in association of the microtubule-based c-Src immune complex with caveolae after GnRH treatment.
Fig. 5.
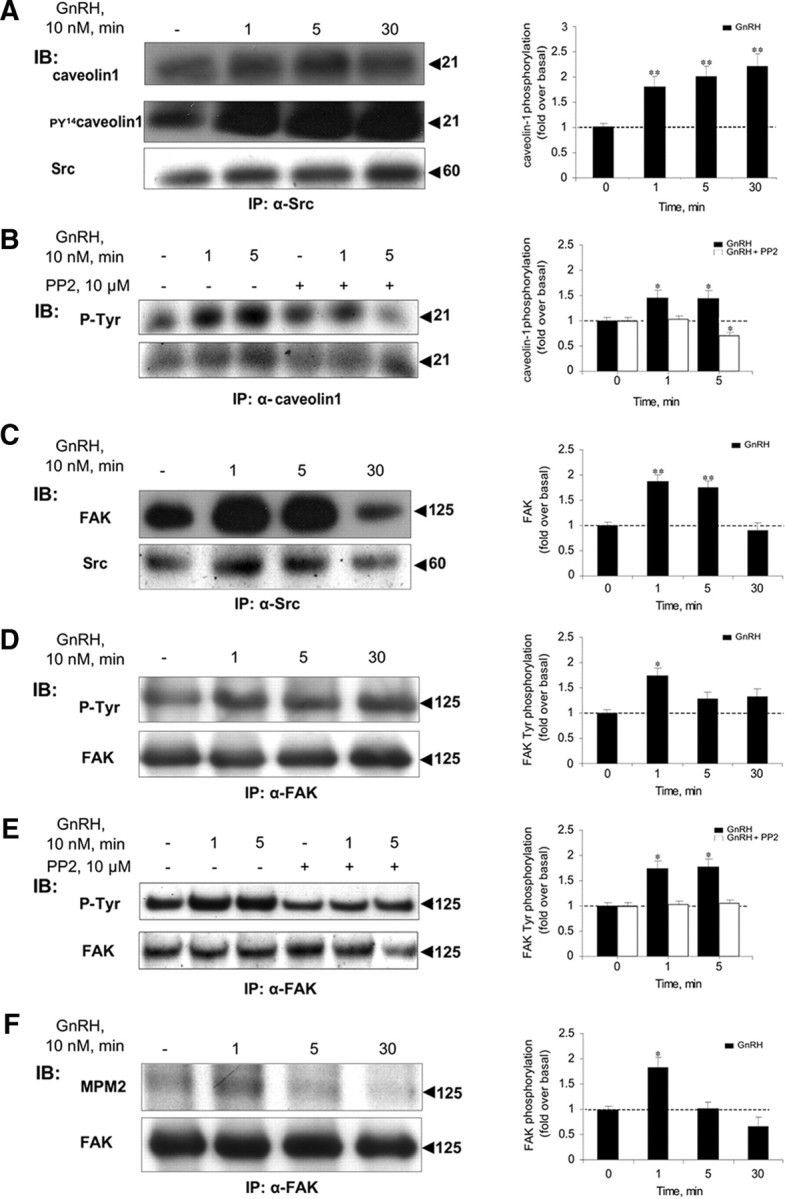
Caveolin-1 and FAK are present in the c-Src immune complex. LβT2 cells were serum starved for 16 h before treatment with GnRH (10 nm) for the indicated time. Another batch of the cells was preincubated with the c-Src inhibitor, PP2 (10 μm for 30 min), followed by GnRH stimulus for the indicated time. Cells were then lysed and subjected to SDS-PAGE after IP with the appropriate antibodies. A, Caveolin-1 is present in the c-Src immune complex, as revealed by immunoblots (IB) of the c-Src immune complex with specific anticaveolin-1 antibodies (first row). GnRH induces rapid phosphorylation of Tyr14 in caveolin-1 (n = 3; P < 0.02) in the c-Src immune complex, as revealed by IB of the c-Src immune complex with anti-PY14-caveolin-1 antibodies (second row). Blotting with α-Src antibodies is shown (third row). B, The c-Src inhibitor, PP2, markedly reduced the tyrosine phosphorylation of caveolin-1. Cells were treated as above, then lysed, and subjected to SDS-PAGE after IP with α-caveolin-1 antibodies, followed by IB with anti-p-Tyr antibodies. C, FAK is present in the c-Src immune complex, as revealed by IB of the c-Src immune complex with specific anti-FAK (first row) and anti-Src (second row) antibodies. D, GnRH induces rapid phosphorylation of FAK on tyrosine residues (n = 3; P < 0.05). Lysates from GnRH-treated cells were immunoprecipitated with anti-FAK antibodies, followed by IB with anti-P-Tyr (upper row), or anti-FAK (lower row) antibodies. E, The c-Src inhibitor, PP2, abolished the tyrosine phosphorylation of FAK. Cells were treated as above, then lysed, and subjected to SDS-PAGE after IP with α-FAK antibodies, followed by IB with anti-p-Tyr antibodies. F, GnRH induces rapid ERK-dependent phosphorylation of FAK on Ser/Thr residues (n = 3; P < 0.05). Lysates from GnRH-treated cells were immunoprecipitated with anti-FAK antibodies, followed by IB with anti-MPM2 (which recognizes phospho-ERK substrates) (upper row) or anti-FAK antibodies (lower row). The ratio between the phosphorylated form of the protein and the general amount was used for the quantification of the data. Representative blots are shown, and bars represent mean + sem from three experiments. *, P < 0.05; **, P < 0.02.
FAK is present in the c-Src immune complex
We then analyzed the c-Src immunoprecipitates from control and GnRH-treated LβT2 cells for the presence of the nonreceptor focal adhesion kinase (FAK). As with the rest, FAK was present in the immune complex even in untreated cells (Fig. 5C). Nevertheless, similar to caveolin-1, FAK did not seem to detach from c-Src after 5 min exposure to GnRH. Rather, FAK levels were found to increase with GnRH treatment. However, unlike caveolin-1, but like Ras, there was a sharp decrease in FAK levels after 30 min of incubation with GnRH. To investigate whether there might be modifications on FAK that could indicate its activation, we examined the phosphorylation status of FAK in GnRH-treated cells. Exposure to GnRH (1–30 min) enhanced phosphorylation of FAK at Tyr residues (Fig. 5D) (n = 3; P < 0.05). Preincubation with the c-Src inhibitor, PP2, abolished the tyrosine phosphorylation of FAK (Fig. 5E). Furthermore, using phospho-specific anti-ERK substrate antibodies (MPM2), we were able to demonstrate that FAK underwent additional phosphorylation at Ser/Thr residues by ERK, albeit in a more transient fashion (Fig. 5F) (n = 3; P < 0.05).
Vinculin is present in the c-Src immune complex
We further attempted to identify other binding partners known to reside in FAs. Vinculin was found to be present in the c-Src immune complex initially (Fig. 6A). However, like MEK, ERK, and α-tubulin, but unlike caveolin 1 and FAK, vinculin appeared to detach from the complex after 5 min exposure to GnRH (n = 3; P < 0.05), with no change in overall levels as adjudged from the analysis of total lysates. Nevertheless, vinculin reappeared in the complex after 30 min of GnRH stimulation (Fig. 6A) (n = 3; P < 0.05). Additionally, GnRH elevated levels of vinculin phosphorylation at Tyr residues (Fig. 6B), peaking at 1 min (n = 4; P < 0.05) and declining rapidly to below basal levels at 5–30 min. Preincubation with the c-Src inhibitor, PP2, abolished the tyrosine phosphorylation of vinculin (Fig. 6C).
Fig. 6.

Vinculin is present in the c-Src immune complex and undergoes phosphorylation by GnRH. LβT2 cells were serum starved for 16 h before treatment with GnRH (10 nm) for the indicated time. Another batch of the cells were preincubated with the c-Src inhibitor, PP2 (10 μm for 30 min), followed by GnRH stimulus for the indicated time. Cells were then lysed and subjected to SDS-PAGE after IP with the appropriate antibodies. A, Vinculin is present in the c-Src immune complex, as revealed by immunoblots (IB) of the c-Src immune complex with specific antivinculin (first row) and anti-Src (second row) antibodies. Molecular mass (kDa) of the proteins is indicated on the right. Vinculin appeares to detach from the complex after 5 min exposure to GnRH (n = 3; P < 0.05). A representative blot is shown, and similar results were found in three other experiments. B, GnRH-induces a rapid phosphorylation of vinculin on tyrosine residues. Lysates from GnRH-treated cells were immunoprecipitated with anti-P-Tyr antibodies, followed by IB with antivinculin antibodies. GnRH was found to elevate the levels of vinculin phosphorylation at Tyr residues peaking at 1 min (n = 4; P < 0.05). A representative blot is shown, and results (mean + sem) from three experiments are shown as bars. *, P < 0.05. C, The c-Src inhibitor, PP2, abolished the tyrosine phosphorylation of vinculin. Cells were treated as above, then lysed, and subjected to SDS-PAGE after IP with anti-P-Tyr antibodies, followed by IB with antivinculin antibodies.
Paxillin is present in the c-Src immune complex
Paxillin, like vinculin, belongs also to the FA protein family and is known to interact with vinculin and FAK (56). As seen in Fig. 7A, when analyzing the c-Src immunoprecipitates from control and GnRH-treated LβT2 cells, we could detect paxillin in control cells like the other binding partners. However, similar to caveolin-1 and FAK, paxillin does not detach from c-Src after 5 min exposure to GnRH, and its levels seem to increase in GnRH-treated cells during the 30-min treatment. GnRH stimulation resulted in a rapid (1 min) and dramatic rise in Tyr phosphorylation of paxillin (Fig. 7B) (n = 3; P < 0.02). Preincubation with the c-Src inhibitor, PP2, abolished the tyrosine phosphorylation of paxillin (Fig. 7C). Exposure to GnRH enhanced also ERK phosphorylation of paxillin with a peak at 1 min and declining thereafter (Fig. 7D) (n = 3; P < 0.05). Although ERK and paxillin are already present in the signaling complex at the start, they are not bound, and their interaction is detected only after 1 min of GnRH stimulation as detected by IP of paxillin and immunoblotting for ERK (Fig. 7E). The results are best explained by previous observations that once paxillin is phosphorylated by c-Src, it is capable of interacting with ERK (57). It seems therefore that vinculin and paxillin, both members of the FA protein family, direct the microtubule-based c-Src immune complex to FA in a dynamic and GnRH-dependent manner.
Fig. 7.

Paxillin and KSR-1, but not 14-3-3, are present in the c-Src immune complex. LβT2 cells were serum starved for 16 h before treatment with GnRH (10 nm) for the indicated time. Another batch of the cells were preincubated with the c-Src inhibitor, PP2 (10 μm for 30 min), followed by GnRH stimulus for the indicated time. Cells were then lysed and subjected to SDS-PAGE directly (Total lysate) or after IP with the appropriate antibodies as indicated. A, Paxillin is present in the c-Src immune complex, as revealed by immunoblots (IB) of the c-Src immune complex with antipaxillin (upper row) and anti-Src antibodies (lower row). B, GnRH induces rapid phosphorylation of paxillin on tyrosine residues. Lysates from GnRH-treated cells were immunoprecipitated with antipaxillin antibodies, followed by IB with anti-P-Tyr (upper lane), or antipaxillin (lower lane) antibodies. GnRH stimulation resulted in a rapid (1 min) and dramatic rise in Tyr phosphorylation of paxillin (n = 3; P < 0.02). C, The c-Src inhibitor, PP2, abolished the tyrosine phosphorylation of paxillin. Cells were treated as above, then lysed, and subjected to SDS-PAGE after IP with α-paxillin antibodies, followed by IB with anti-p-Tyr antibodies. D, GnRH induces rapid ERK-dependent phosphorylation of paxillin on Ser/Thr residues. Lysates from GnRH-treated cells were immunoprecipitated with antipaxillin antibodies, followed by IB with anti-MPM2 (which recognizes phospho-ERK substrates) (upper row), or antipaxillin antibodies (lower row). Exposure to GnRH enhanced ERK phosphorylation of paxillin with a peak at 1 min and declining thereafter (n = 3; P < 0.05). E, Activated paxillin interacts with ERK. Lysates from GnRH-treated cells were immunoprecipitated with antipaxillin antibodies, followed by IB with anti-ERK (upper row), or antipaxillin antibodies (lower row). Note that interaction occurs only after paxillin is activated as in panels B and D. F, KSR is present in the c-Src immune complex at later stages. Cell lysates from GnRH-treated cells were immunoprecipitated with anti-Src antibodies followed by IB with anti-KSR-1 antibodies (upper row), or anti-Src antibodies (lower row). G, 14-3-3 is present in the total lysate but not in the c-Src immune complex as revealed by IB with anti-14-3-3 antibodies (left three lanes), or by IP of c-Src, followed by IB with anti-14-3-3 antibodies (right three lanes). Molecular mass (kDa) of the proteins is indicated on the right. The ratio between the phosphorylated form of the protein and the general amount was used for the quantification of the data. A representative blot is shown, and results (mean + sem) from three experiments are shown as bars on the right (B and D). *, P < 0.05; **, P < 0.02.
KSR-1 is present in the c-Src immune complex at a later stage
KSR-1 is thought to act downstream of Ras and upstream, or parallel, to Raf-1 and serves as a scaffold for the ERK cascade (58, 59, 60). Unlike the other binding partners, KSR-1 was not detected in the complex only until after 30 min of incubation with GnRH (Fig. 7F). Because KSR-1 is known to interact with 14-3-3 protein at the membrane (61), and with 14-3-3 also implicated as a potential scaffold in the ERK pathway (62), we examined its presence in the c-Src immune complex. Although we could detect 14-3-3 in total lysates, we could not find it in the complex with c-Src either in control or GnRH-activated cells (Fig. 7G). Thus, KSR-1 appears to interact with the c-Src immune complex in a 14-3-3-independent manner at a later phase of complex formation.
To control the IP for caveolin-1, paxillin, and FAK shown in Figs. 5 and 7, we repeated the IP of the above proteins and in parallel used also serum IgG (Fig. 8). Cells were treated with or without GnRH, followed by IP of the above proteins, or the use of serum IgG and Western blotting for phosphotyrosine (p-Tyr) (Fig. 8A). As before, GnRH stimulation resulted in a rapid (1–5 min) rise in Tyr phosphorylation of caveolin-1, paxillin, and FAK, whereas IP with serum IgG gave no signal (Fig. 8A). Similarly, IP of the above proteins, or the use of serum IgG for IP and Western blotting for the above proteins, revealed the presence of the proteins in the IPs with no signal in the IgG IP (Fig. 8B).
Fig. 8.
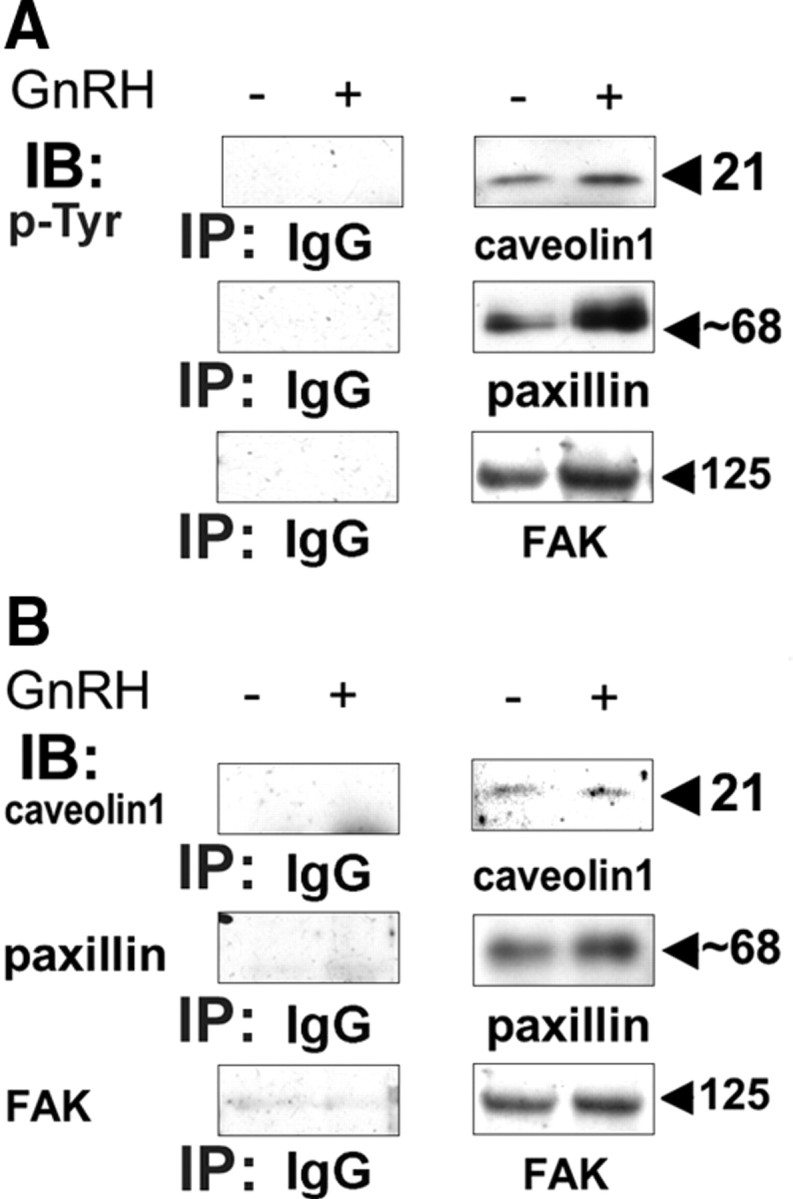
Specificity of the IPs. LβT2 cells were serum starved for 16 h before treatment with or without GnRH (10 nm) for 5 min. Cells were then lysed, and subjected to SDS-PAGE after IP with serum IgG, or antibodies to caveolin-1, or paxillin, or FAK. Samples were then immunoblotted (IB) with: anti-p-Tyr (A), or with antibodies to caveolin-1, or paxillin, or FAK (B). A representative blot is shown, and similar results were observed in two other experiments.
PKC is involved in ERK activation by GnRH in the c-Src immune complex
Because PKC is implicated in the signaling from the GnRHR to ERK, upstream of c-Src (53), we went on to investigate its presence and role in the c-Src immune complex. Preincubation with the pan-PKC inhibitor GF109203X markedly reduced ERK activation by GnRH, particularly at the 30-min time point, indicating that activation of ERK by GnRH in the c-Src immune complex is PKC dependent (Fig. 9A). Like before, ERK appeared to detach from the complex after 5 min of GnRH stimulation, but the PKC inhibitor blocked this exit (Fig. 9A), suggesting that the dynamic nature of ERK in the complex is PKC dependent.
Fig. 9.
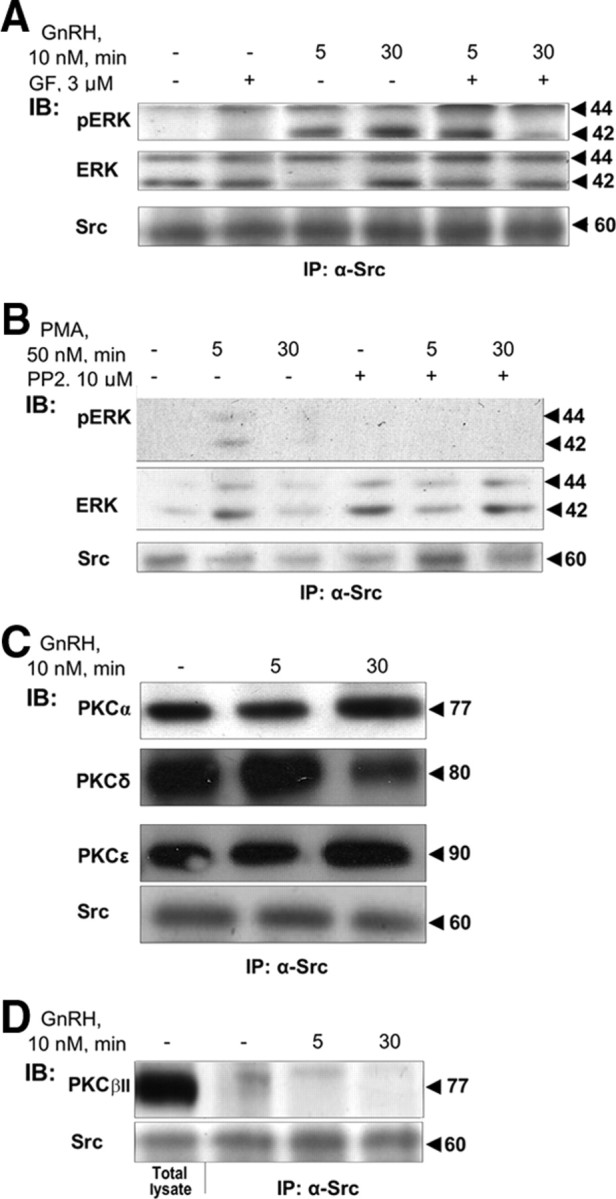
PKC is present and plays a role in GnRH-induced ERK activation in the c-Src immune complex. A, Activation of ERK by GnRH in the c-Src immune complex is PKC dependent. LβT2 cells were serum starved for 16 h before pretreatment with the pan-PKC inhibitor GF109203X (3 μm) (GF) for 30 min. Thereafter, GnRH (10 nm) was added for the indicated time. Cells were lysed, and proteins were subjected to SDS-PAGE after IP with α-Src antibodies. Samples were then immunoblotted (IB) with anti-phospho-ERK (pERK), anti-ERK (ERK), or anti-Src antibodies. B, PMA activation of ERK in the complex is c-Src dependent. LβT2 cells were serum starved for 16 h before pretreatment with the c-Src inhibitor, PP2 (10 μm), for 30 min. Thereafter, PMA (50 nm) was added for the indicated time. Cells were lysed, and proteins were subjected to SDS-PAGE after IP with α-Src antibodies. Samples were then IB with anti-phospho-ERK (pERK), anti-ERK (ERK), or anti-Src antibodies. C, PKCα, -δ, and -ε are present in the c-Src immune complex. LβT2 cells were serum starved for 16 h before treatment with GnRH (10 nm) for the indicated time. Cells were lysed, and proteins were subjected to SDS-PAGE after IP with α-Src antibodies. Samples were then IB with isoform-specific PKC antibodies. Molecular mass (kDa) of the proteins is indicated on the right. A representative blot is shown, and similar results were observed in two other experiments. D, PKCβII is absent from the c-Src immune complex. LβT2 cells were serum starved for 16 h before treatment with GnRH (10 nm) for the indicated time. Cells were lysed, and proteins were subjected to SDS-PAGE directly (Total lysate), or after IP with α-Src antibodies. Samples then received IB with anti-PKCβII antibodies. Molecular mass (kDa) of the proteins is indicated on the right. A representative blot is shown and similar results were observed in two other experiments.
To determine whether PKC-mediated GnRH activation of ERK in the c-Src immune complex required active c-Src, we treated cells with the PKC activator, phorbol 12-myristate 13-acetate (PMA), in the presence and absence of the c-Src inhibitor, PP2, shown above (Fig. 1) to be an effective inhibitor of c-Src activation by GnRH. PMA activation of pERK was best detected in the total lysate, but it was also found in the c-Src immune complex (Fig. 9B). Still, the effect was abolished by PP2, indicating that active c-Src is required for PKC activation of ERK in the c-Src immune complex. Still, whereas PP2 nearly abolished the GnRH effect at both 5 and 30 min (Fig. 1C), GF109203X was less effective (Fig. 9A), suggesting that c-Src contributes to the ERK response in a PKC-dependent and -independent manner.
PKCα, -δ, and -ε, but not -βII, are present in the c-Src immune complex
We further investigated the nature of the PKC isoforms present in the complex. Because GnRH activates PKCδ, -β, and -ε (63, 64), we tested their presence in the complex. We found PKCδ, -ε, and -α, but not PKCβII, to be present in the c-Src immune complex even under basal conditions. Moreover, they did not seem to detach from it upon GnRH treatment (Fig. 9, C and D, and Table 1).
Table 1.
Proteins associated with the c-Src immune complex during GnRHR action
| GnRH, min | − | 1 | 5 | 30 |
| IP-Src, IB | ||||
| MEK | + | nd | − | 2+ |
| ERK | 2++ | nd | − | 2++ |
| Tubulin | 2++ | 2+ | − | 2+ |
| Caveolin | + | + | 2+ | + |
| FAK | 2+ | 2++ | 2++ | + |
| Vinculin | 2+ | + | − | + |
| Paxillin | + | + | 2+ | + |
| KSR | − | nd | − | + |
| Ras | + | 2++ | 2+ | + |
| GnRHR | 2+ | 2+ | + | 2+ |
| PKCα | 2+ | nd | 2+ | 2++ |
| PKCδ | 2++ | nd | 2++ | + |
| PKCε | 2+ | nd | 2+ | 2++ |
| PKCβII | − | nd | − | − |
The table summarizes the list of the proteins found to be associated with c-Src and the dynamic nature of this association during a GnRH challenge for 1, 5, and 30 min. The relative strength of the association is also depicted. Data were collected from the figures. PKCβII represents a negative control. nd, Not determined.
Discussion
It is thought that receptor activation initiates the formation of signaling complex to gain receptor specificity (37, 38, 40). For the GnRHR, a signaling platform for ERK activation has been previously described that includes the GnRHR, c-Raf kinase, Ca2+-calmodulin, and ERK2, and is localized to low-density membrane microdomains (lipid rafts/caveolae) (43, 44, 45). Another signaling complex leading to ERK activation was described in HEK293 cells stably expressing the GnRHR and included FAK and c-Src at FA complexes (46). More recently Pyk2 was shown to act as a scaffold for a FA/cytoskeleton-dependent complex comprised of c-Src, Grb2, and mSos, which is involved in ERK translocation to the nucleus in GnRH-stimulated gonadotrope cells (65). Our results suggest a novel feature of these signaling complexes, namely, that they may already be preformed in the absence of ligand. The ligand, GnRH, then initiates a dynamic restructuring of the complex, which allows the retention of an active cytosolic pool of ERK, and subsequently directs it to phosphorylate FAK and paxillin at FA.
The preformed multiprotein signaling complex we have identified includes c-Src, FAK, paxillin, and caveolin-1 (all known to be scaffolding molecules), with additional members including Ras, MEK1/2, ERK1/2, α-tubulin, vinculin, PKC (α, δ, and ε), KSR-1, and the GnRHR. The nonreceptor tyrosine kinase c-Src has previously been reported to be associated with cellular structural elements, such as cytoskeletal stress fibers and FA complexes (66). Once GnRH is added, a restructuring process occurs, during which MEK, ERK, α-tubulin, and vinculin detach from the complex within 5 min and reassociate after 30 min. This detachment may be due to a decrease in microtubular stability known to occur after ERK activation (67). Once MEK, ERK, α-tubulin, and vinculin have been recruited to the complex after 30 min of GnRH stimulation, the levels of FAK decrease, apparently replaced by the scaffold molecule, KSR-1. The reappearance of MEK, ERK, α-tubulin, and vinculin in the complex may be linked to the activation of c-Src and PKC within the complex because, at least in the case of MEK and ERK, their reassociation with the complex required active c-Src and PKC, respectively.
It is thought that the interaction between MEK1/2 and ERK1/2 is crucial for ERK1/2 activation and its subcellular distribution (54). Initially, the interaction of ERK1/2 with MEK1/2 maintains its cytosolic localization (54, 68). Subsequent phosphorylation of ERK1/2 by MEK1/2 then induces a conformation change, releasing ERK1/2 from MEK1/2, and enabling ERK1/2 to translocate to the nucleus (54, 68). We therefore suggest that the role of the signaling complex might be to restrict an active pool of ERK from being translocated to the nucleus. This is supported by our findings that the signaling molecules involved in ERK activation, viz. PKC, c-Src, Ras, Raf, and MEK, are all present in the complex and their cross talk was described elsewhere (53). Nevertheless, the actual task of restricting ERK in the cytosol appears to be carried out here by paxillin instead, which is initially bound to c-Src but not to ERK. Once paxillin is phosphorylated by c-Src, it is then capable of interacting with ERK (Fig. 7D) (57). The activated ERK is subsequently redirected to FA to phosphorylate FAK and paxillin.
We searched for the presence of β-arrestin in the complex because binding of Raf-1 to β -arrestin has been reported to facilitate the assembly of MEK, and β-arrestin is also known to serve as an escort for signaling molecules such as c-Src. Together, there exists a β-arrestin-dependent modulation of both temporal and spatial properties of ERK activation (50). However, we failed to identify β-arrestin in the complex. Typically, β-arrestin would bind the phosphorylated c-tail of activated GPCRs during desensitization and later become a scaffold for ERK activation (55). It is possible that because the mammalian type I GnRHR lacks a c-carboxy tail, β-arrestin cannot bind it (49), and, as in our case, that GnRHR may undergo alternative pathways of signal termination (69).
A signaling platform including the GnRHR, c-Raf kinase, Ca2+-calmodulin, and ERK2 has been found to be localized to low-density membrane microdomains (lipid rafts/caveolae) (43, 44, 45). The presence here of the caveolae marker, caveolin-1, suggests a similar localization. Interestingly, caveolin-1 is known to inhibit ERK activity (70). Within the complex, caveolin-1 underwent GnRH-induced Tyr14 phosphorylation, apparently by the bound c-Src, which has been shown to phosphorylate this residue (71). It is therefore possible that the exit of MEK-ERK-tubulin-vinculin from c-Src was required to enable further ERK activation by removal of inhibition by caveolin-1. The presence of the signaling complex in both caveolae and FA is not contradictory because it has been shown that tyrosine-phosphorylated caveolin-1 is directed to FAs (72).
GnRH-induced ERK activation in the complex resulted in the phosphorylation of FAK and paxillin on Ser/Thr residues, as detected here using phosphorylation-specific antibodies (MPM2), which recognize the phosphorylation motif of ERK substrates (73). FAK has been shown to undergo phosphorylation on Ser910 in an ERK-dependent manner, whereas Ser126 and Ser130 of paxillin are phosphorylated in a Raf-dependent, ERK-regulated fashion. Hence, it is tempting to suggest that sequestering the c-Src immune complex to FA will enable active ERK to phosphorylate both paxillin and FAK.
FAK, caveolin-1, vinculin, and paxillin were also phosphorylated at Tyr residues in the GnRH-activated complex, apparently by c-Src, which is known to phosphorylate paxillin at Tyr118 and Tyr31 and FAK at Tyr 576, 577, 861, and 925 (56, 74). The tyrosine phosphorylation sites in vinculin have been mapped to residues 100 and 1065, whereas Tyr1065 located in the vinculin tail domain has been reported to be phosphorylated by c-Src in vitro (75). Indeed, preincubation with the c-Src inhibitor, PP2, resulted in a marked inhibition, or even abolished the tyrosine phosphorylation of FAK, caveolin-1, vinculin, and paxillin by GnRH, implicating c-Src as the tyrosine kinase participating in this process. It is known that vinculin can regulate survival and motility via ERK by controlling the accessibility of paxillin for FAK interaction (76). It is thought that Src phosphorylation of Tyr118 of paxillin creates an ERK binding site to allow ERK recruitment to FA. The activated paxillin then binds to Raf and MEK to activate ERK in the FAs. ERK phosphorylation of paxillin on Ser/Thr residues facilitates paxillin association with FAK (57, 74). Indeed, we could detect paxillin-ERK interaction only after paxillin became phosphorylated on Tyr in GnRH-treated cells. It is thought that the function of paxillin, like that of β-arrestin, is to prevent a pool of ERKs from being translocated to the nucleus and rather retain this pool in the cytosol (54) and our data supports this notion.
Paxillin is a FA-associated protein and a multidomain adapter protein (77). It is also known to bind α-tubulin (77), suggesting that paxillin can bridge the microtubule cytoskeleton and FA. This seems to be the present case. ERK’s association with the microtubule cytoskeleton is already documented, and it is thought that this association is also important for retaining some activated ERK in the cytoplasm to regulate cytoplasmic events (78). In our case, the complex seems to be associated with the microtubule cytoskeleton and FA, and ERK appears to be retained in the cytosol to phosphorylate FAK and paxillin, which may be important for FA turnover (78). Indeed, GnRH-mediated ERK activation is prevented by cytoskeletal disruption in HEK293 cells (46). Paxillin and FAK are mainly involved in cell migration, spreading, and morphogenesis (54). It has been shown that the activated GnRHR elicits changes in cell adhesion in HEK293 cells expressing the GnRHR (46). Furthermore, GnRH can induce cell migration of human prostatic carcinoma cell lines expressing the GnRHR (79). GnRH also induced dramatic changes in cellular morphology in the gonadotrope-derived αT3-1 cells (51). Induction of cellular movements by GnRH could also be detected in live murine pituitary slices and dissociated pituitary cells (51). Hence, ERK-mediated phosphorylation of FAK and paxillin in the complex may be required for GnRH induction of cell migration. Nevertheless, further studies are required to link the above complex with gonadotrope migration. Indeed, like RhoAGTPase, ERK is implicated in cell mobility by its activation at the leading edge of migrating cells, in pseudopodia growth and retraction during chemotaxis, and FA formation via paxillin phosphorylation (80, 81).
Further evidence of the dynamic nature of the complex was obtained when the scaffold molecule FAK appeared to be replaced by KSR-1 after 30 min of GnRH treatment. KSR binds ERKs via a D-domain-like motif in KSR and via the CRS/CD domain of ERK (58, 59, 60). It is likely that the spatial/temporal and the dynamic nature of the c-Src immune complex direct it to a locus that is accessible to KSR only after 30 min of incubation with GnRH. It should be noted that our immunocytochemistry results (Fig. 2) suggest that at this time part of ERK (∼35%) is the nucleus, but a cytosolic fraction is also detected. Thus, it seems that the function of KSR is to continue to maintain an active pool of ERK in the vicinity of the membrane for phosphorylation of cytosolic/membrane substrates.
It could be argued that the above data may suggest that c-Src can interact with each of the binding partners but possibly in independent complexes. However, we have also demonstrated direct interaction between paxillin and ERK (Fig. 7D) and phosphorylation of FAK and paxillin by ERK (Figs. 5 and 7), under conditions in which JNK, p38, and ERK5 were absent from the complex (Fig. 3 and data not shown). In addition, the interactions between the binding partners identified here are well documented. We therefore propose that this multiprotein signaling complex is established in the following manner: c-Src is bound separately to caveolin-1, FAK, paxillin, and the PKCs. Paxillin and FAK interact with each other (82). Paxillin interacts also with vinculin (82), with ERK (57, 82), and with α-tubulin (77). α-Tubulin can also bind to ERK (37, 78). ERK is bound to MEK, which binds Raf, which is itself bound to Ras in the membrane (24, 25). MEK binds paxillin and KSR, whereas KSR also binds Raf and ERK (58, 59, 60). These multiprotein interactions, some of which are phosphorylation dependent, are most likely involved in directing the signaling complex along the microtubule cytoskeleton to the network of FA to mediate cell adhesion, cytoskeletal remodeling, and cell migration (46, 51).
Materials and Methods
Materials
GnRH and PMA were obtained from Sigma (St. Louis, MO). PP2 and GF109203X were from A.G. Scientific, Inc. (San Diego, CA). Mouse monoclonal antiactive (antidiphospho-antibodies, doubly phosphorylated)-ERK, p38, and JNK antibodies, polyclonal antibodies to general MAPKs, actin, β-arrestin, and KSR-1 and mouse monoclonal antibodies to tubulin, actin, and PKCs were obtained from Sigma (Rehovot, Israel). Rabbit polyclonal antibodies to c-Src, caveolin-1, KSR-1, and FAK, mouse monoclonal anti-p-Tyr (PY99), anti-pan-Ras antibodies, and protein A/G Agarose beads were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Mouse monoclonal anti-v-Src (327) antibody was from Calbiochem (Darmstadt, Germany). Mouse monoclonal antiphospho-Ser/Thr-Pro (MPM2) antibody was from Upstate Biotechnology, Inc. (Lake Placid, NY). Mouse monoclonal antipaxillin antibody was from BD Transduction (BD Biosciences, San Jose, CA). Mouse monoclonal anti-14-3-3 antibody (8C3) was kindly provided by Dr. D. Klein, National Institutes of Health (Bethesda, MD). Secondary HRP-conjugated goat antimouse antibodies or goat antirabbit antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). All media and sera were obtained from Biological Industries (Beit-Haemek, Israel). Vectashield mounting medium was from Vector Laboratories, Inc. (Burlingame, CA). FuGENE6 was from Roche Applied Science (Indianapolis, IN), and TransIT-LT1 transfection reagent was obtained from Mirus BIO Corp. (Madison, WI). GnRHR-hemagglutinin and GnRHR-GFP constructs were kindly provided by Dr. Colin Clay (Colorado State University, Fort Collins, CO).
Cell culture and transfection
LβT-2 cells (a gift from Dr. P. Mellon, University of California, San Diego, CA) were grown in monolayer and cultured in DMEM supplemented with 10% fetal calf serum and antibiotics in humidified 5% CO2 at 37 C. Cells were starved overnight in 0.1% serum DMEM and then pretreated with the various inhibitors for 30 min. GnRH or PMA was then added for an appropriate time as indicated. LβT2 cells were transfected using FuGENE6 or its analog, TransIT-LT1 transfection reagent.
IP and immunoblotting
LβT2 cells were grown in 10-cm plates, serum starved (0.1% fetal calf serum) for 16 h, and later stimulated with the various ligands. The cells were then washed twice with ice-cold PBS and overlaid with 1 ml of hypotonic lysis buffer (50 mm HEPES, pH 7.4; 5 mm MgCl2; 1 mm Na3VO4; 50 mm β-glycerophosphate; 1 mm benzamidine; 10 μg/ml aprotinin; 10 μg/ml leupeptin; 1 mm phenylmethylsulfonylfluoride; and 0.5% Triton X-100). Cells were harvested, sonicated six times for 10 sec each time (Ultrasonic sonicator, XL2015; Heat Systems, Misonix, Inc., Farmingdale, NY). The cell debris was then pelleted by centrifugation (15,000 × g, 15 min, 4 C). The supernatants were collected for further experiment. For IP, protein A/G Agarose beads were mixed with an appropriate antibody (Ab) at room temperature for 1 h, after which the beads were washed twice with PBS. Cell lysates (300–500 μg) were added to the beads and rotated at 4 C for 3 h. Then immunocomplexes were washed twice with wash buffer (hypotonic lysis buffer with 150 mm NaCl, 0.1% Triton X-100) and heated to 100 C in Laemmli loading buffer. The supernatants were collected, and proteins were separated on 10% SDS-PAGE gel, followed by Western blotting analysis. Polyclonal antibodies to c-Src (Santa Cruz Biotechnology) were used for IP. After stripping, a mouse monoclonal anti-v-Src antibody (Calbiochem, Darmstadt, Germany) was used for c-Src immunoblotting. Part of the cell lysates were directly mixed with loading buffer for analysis of total lysates. For all experiments, control IPs were performed, where the antibody was omitted from A/G beads before addition of the cell lysate. We could not detect any nonspecific binding when the control sample was blotted with the appropriate antibody. The blots were detected using horseradish peroxidase-conjugated antimouse or antirabbit Fab antibodies (Jackson ImmunoResearch Laboratories, Inc.) followed by homemade enhanced chemiluminescence (83). The blots were autoradiographed on Fuji Super RX films, and the amount of phosphorylation was quantitated by densitometry (Bio-Rad 690 densitometer; Bio-Rad Laboratories, Inc., Hercules, CA). MALDI-TOF (Micromass, Manchester, UK) analysis was carried out by the Smoler Proteomic Center, Department of Biology, Technion (Haifa, Israel).
Immunofluorescent staining
LβT2 cells were grown on coverslips and were starved overnight. After GnRH stimulation, the cells were fixed with 3% paraformaldehyde followed by permeabilization with 0.2% Triton X-100. The fixed cells were incubated with anti-ERK antibody (1:100) followed by secondary antibody conjugated with rhodamine. DAPI staining was used to track the location of the nucleus. The stained cells were inspected under a deconvolution microscope (DeltaVision; Issaquah, WA).
Footnotes
This work was supported by The Israel Science Foundation (Grant 221/05), the German-Israeli Foundation for Research and Development (Grant I-751-168.2/2002), U.S.-Israel Binational Science Foundation (Grant 2007057), the Adams Super-Center for Brain Studies at Tel-Aviv University (to Z.N.) and from the EU Sixth Framework Program under the GROWTHSTOP (LSHC CT-2006-037731) to (R.S.). T.W. gratefully acknowledges funding by the Singapore Bioimaging Consortium (SBIC 003/2005). R.S. is the Incumbent of the Yale S. Lewine and Ella Miller Lewine Professorial chair for cancer research, and Z.N. is the incumbent of the Abraham E. Kazan Chair in Structural Biology.
Current address for S.L.: Institute of Molecular and Cell Biology, Cell Division and Cancer Laboratory, 61 Biopollis Drive, Proteos, Singapore 138673.
Disclosure Summary: The authors have nothing to disclose.
First Published Online July 23, 2009
Abbreviations: DAPI, 4′,6-Diamidino-2-phenylindole; FA, focal adhesion; FAK, focal adhesion kinase; GFP, green fluorescent protein; GnRHR, GnRH receptor; G-protein, guanine nucleotide-binding protein; GPCR, G-protein coupled receptor; HEK, human embryonic kidney; IP, immunoprecipitation; JNK, Jun N-terminal kinase; KSR, kinase suppressor of Ras; MALDI-TOF, matrix-assisted laser desorption ionization-time of flight mass spectrometry; MEK, MAPK kinase; PKC, protein kinase C; PKCs, PKC isoforms; PMA, phorbol 12-myristate 13-acetate; p-Tyr, phosphotyrosine.
References
- 1.Naor Z1990. Signal transduction mechanisms of Ca2+ mobilizing hormones: the case of gonadotropin-releasing hormone. Endocr Rev 11:326–353 [DOI] [PubMed] [Google Scholar]
- 2.Sealfon SC, Weinstein H, Millar RP1997. Molecular mechanisms of ligand interaction with the gonadotropin-releasing hormone receptor. Endocr Rev 18:180–205 [DOI] [PubMed] [Google Scholar]
- 3.Shacham S, Harris D, Ben-Shlomo H, Cohen I, Bonfil D, Przedecki F, Lewy H, Ashkenazi IE, Seger R, Naor Z2001. Mechanism of GnRH receptor signaling on gonadotropin release and gene expression in pituitary gonadotrophs. Vitam Horm 63:63–90 [DOI] [PubMed] [Google Scholar]
- 4.Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR2004. Gonadotropin-releasing hormone receptors. Endocr Rev 25:235–275 [DOI] [PubMed] [Google Scholar]
- 5.Dobkin-Bekman M, Naidich M, Pawson AJ, Millar RP, Seger R, Naor Z2006. Activation of Mitogen-activated protein kinase (MAPK) by GnRH is cell-context dependent. Mol Cell Endocrinol 252:184–190 [DOI] [PubMed] [Google Scholar]
- 6.Naor Z2009. Signaling by G-protein-coupled receptor (GPCR): studies on the GnRH receptor. Front Neuroendocrinol 30:10–29 [DOI] [PubMed] [Google Scholar]
- 7.Naor Z, Catt KJ1981. Mechanism of action of gonadotropin-releasing hormone. Involvement of phospholipid turnover in luteinizing hormone release. J Biol Chem 256:2226–2229 [PubMed] [Google Scholar]
- 8.Naor Z, Zer J, Zakut H, Hermon J1985. Characterization of pituitary calcium-activated, phospholipid-dependent protein kinase: redistribution by gonadotropin-releasing hormone. Proc Natl Acad Sci USA 82:8203–8207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naor Z1986. Phosphoinositide turnover, Ca2+ mobilization, protein kinase C activation and leukotriene action in pituitary signal transduction: effect of gonadotropin releasing hormone. Adv Prostaglandin Thromboxane Leukot Res 16:225–234 [PubMed] [Google Scholar]
- 10.Netiv E, Liscovitch M, Naor Z1991. Delayed activation of phospholipase D by gonadotropin-releasing hormone in a clonal pituitary gonadotrope cell line (αT3-1). FEBS Lett 295:107–109 [DOI] [PubMed] [Google Scholar]
- 11.Stojilkovic SS, Reinhart J, Catt KJ1994. Gonadotropin-releasing hormone receptors: structure and signal transduction pathways. Endocr Rev 15:462–499 [DOI] [PubMed] [Google Scholar]
- 12.Roberson MS, Misra-Press A, Laurance ME, Stork PJ, Maurer RA1995. A role for mitogen-activated protein kinase in mediating activation of the glycoprotein hormone α-subunit promoter by gonadotropin-releasing hormone. Mol Cell Biol 15:3531–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sundaresan S, Colin IM, Pestell RG, Jameson JL1996. Stimulation of mitogen-activated protein kinase by gonadotropin-releasing hormone: evidence for the involvement of protein kinase C. Endocrinology 137:304–311 [DOI] [PubMed] [Google Scholar]
- 14.Naor Z1997. GnRH receptor signaling: cross-talk of Ca2+ and protein kinase C. Eur J Endocrinol 136:123–127 [DOI] [PubMed] [Google Scholar]
- 15.Reiss N, Llevi LN, Shacham S, Harris D, Seger R, Naor Z1997. Mechanism of mitogen-activated protein kinase activation by gonadotropin-releasing hormone in the pituitary of αT3-1 cell line: differential roles of calcium and protein kinase C. Endocrinology 138:1673–1682 [DOI] [PubMed] [Google Scholar]
- 16.Mulvaney JM, Zhang T, Fewtrell C, Roberson MS1999. Calcium influx through L-type channels is required for selective activation of extracellular signal-regulated kinase by gonadotropin-releasing hormone. J Biol Chem 274:29796–29804 [DOI] [PubMed] [Google Scholar]
- 17.Mulvaney JM, Roberson MS2000. Divergent signaling pathways requiring discrete calcium signals mediate concurrent activation of two mitogen-activated protein kinases by gonadotropin-releasing hormone. J Biol Chem 275:14182–14189 [DOI] [PubMed] [Google Scholar]
- 18.Grosse R, Schmid A, Schöneberg T, Herrlich A, Muhn P, Schultz G, Gudermann T2000. Gonadotropin-releasing hormone receptor initiates multiple signaling pathways by exclusively coupling to G(q/11) proteins. J Biol Chem 275:9193–9200 [DOI] [PubMed] [Google Scholar]
- 19.Naor Z, Benard O, Seger R2000. Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends Endocrinol Metab 11:91–99 [DOI] [PubMed] [Google Scholar]
- 20.Kraus S, Naor Z, Seger R2001. Intracellular signaling pathways mediated by the gonadotropin-releasing hormone (GnRH) receptor. Arch Med Res 32:499–509 [DOI] [PubMed] [Google Scholar]
- 21.Ruf F, Fink MY, Sealfon SC2003. Structure of the GnRH receptor-stimulated signaling network: insights from genomics. Front Neuroendocrinol 24:181–199 [DOI] [PubMed] [Google Scholar]
- 22.Ferris HA, Shupnik MA2006. Mechanisms for pulsatile regulation of the gonadotropin subunit genes by GNRH1. Biol Reprod 74:993–998 [DOI] [PubMed] [Google Scholar]
- 23.Naor Z, Jabbour HN, Naidich M, Pawson AJ, Morgan K, Battersby S, Millar MR, Brown P, Millar RP2007. Reciprocal cross talk between gonadotropin-releasing hormone (GnRH) and prostaglandin receptors regulates GnRH receptor expression and differential gonadotropin secretion. Mol Endocrinol 21:524–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murphy LO, Blenis J2006. MAPK signal specificity: the right place at the right time. Trends Biochem Sci 31:268–275 [DOI] [PubMed] [Google Scholar]
- 25.Yoon S, Seger R2006. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24:21–44 [DOI] [PubMed] [Google Scholar]
- 26.Saunders BD, Sabbagh E, Chin WW, Kaiser UB1998. Differential use of signal transduction pathways in the gonadotropin-releasing hormone-mediated regulation of gonadotropin subunit gene expression. Endocrinology 139:1835–1843 [DOI] [PubMed] [Google Scholar]
- 27.Weck J, Fallest PC, Pitt LK, Shupnik MA1998. Differential gonadotropin-releasing hormone stimulation of rat luteinizing hormone subunit gene transcription by calcium influx and mitogen-activated protein kinase-signaling pathways. Mol Endocrinol 12:451–457 [DOI] [PubMed] [Google Scholar]
- 28.Call GB, Wolfe MW1999. Gonadotropin-releasing hormone activates the equine luteinizing hormone β promoter through a protein kinase C/mitogen-activated protein kinase pathway. Biol Reprod 61:715–723 [DOI] [PubMed] [Google Scholar]
- 29.Roberson MS, Zhang T, Li HL, Mulvaney JM1999. Activation of the p38 mitogen-activated protein kinase pathway by gonadotropin-releasing hormone. Endocrinology 140:1310–1318 [DOI] [PubMed] [Google Scholar]
- 30.Yokoi T, Ohmichi M, Tasaka K, Kimura A, Kanda Y, Hayakawa J, Tahara M, Hisamoto K, Kurachi H, Murata Y2000. Activation of the luteinizing hormone beta promoter by gonadotropin-releasing hormone requires c-Jun NH2-terminal protein kinase. J Biol Chem 275:21639–21647 [DOI] [PubMed] [Google Scholar]
- 31.Fowkes RC, King P, Burrin JM2002. Regulation of human glycoprotein hormone α-subunit gene transcription in LβT2 gonadotropes by protein kinase C and extracellular signal-regulated kinase 1/2. Biol Reprod 67:725–734 [DOI] [PubMed] [Google Scholar]
- 32.Harris D, Bonfil D, Chuderland D, Kraus S, Seger R, Naor Z2002. Activation of MAPK cascades by GnRH: ERK and Jun N-terminal kinase are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHβ-subunit promoter. Endocrinology 143:1018–1025 [DOI] [PubMed] [Google Scholar]
- 33.Harris D, Chuderland D, Bonfil D, Kraus S, Seger R, Naor Z2003. Extracellular signal-regulated kinase and c-Src, but not Jun N-terminal kinase, are involved in basal and gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone α-subunit promoter. Endocrinology 144:612–622 [DOI] [PubMed] [Google Scholar]
- 34.Bonfil D, Chuderland D, Kraus S, Shahbazian D, Friedberg I, Seger R, Naor Z2004. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone β-subunit promoter. Endocrinology 145:2228–2244 [DOI] [PubMed] [Google Scholar]
- 35.Kanasaki H, Bedecarrats GY, Kam KY, Xu S, Kaiser UB2005. Gonadotropin-releasing hormone pulse frequency-dependent activation of extracellular signal-regulated kinase pathways in perifused LβT2 cells. Endocrinology 146:5503–5513 [DOI] [PubMed] [Google Scholar]
- 36.Xie J, Bliss SP, Nett TM, Ebersole BJ, Sealfon SC, Roberson MS2005. Transcript profiling of immediate early genes reveals a unique role for activating transcription factor 3 in mediating activation of the glycoprotein hormone α-subunit promoter by gonadotropin-releasing hormone. Mol Endocrinol 19:2624–2638 [DOI] [PubMed] [Google Scholar]
- 37.Reszka AA, Seger R, Diltz CD, Krebs EG, Fischer EH1995. Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc Natl Acad Sci USA 92:8881–8885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ1999. β-Arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science 283:655–661 [DOI] [PubMed] [Google Scholar]
- 39.Burack WR, Shaw AS2000. Signal transduction: hanging on a scaffold. Curr Opin Cell Biol 12:211–216 [DOI] [PubMed] [Google Scholar]
- 40.Pierce KL, Lefkowitz RJ2001. Classical and new roles of β-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci 2:727–733 [DOI] [PubMed] [Google Scholar]
- 41.Morrison DK, Davis RJ2003. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol 19:91–118 [DOI] [PubMed] [Google Scholar]
- 42.Shaul YD, Seger R2007. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta 1773:1213–1226 [DOI] [PubMed] [Google Scholar]
- 43.Navratil AM, Bliss SP, Berghorn KA, Haughian JM, Farmerie TA, Graham JK, Clay CM, Roberson MS2003. Constitutive localization of the gonadotropin-releasing hormone (GnRH) receptor to low density membrane microdomains is necessary for GnRH signaling to ERK. J Biol Chem 278:31593–31602 [DOI] [PubMed] [Google Scholar]
- 44.Roberson MS, Bliss SP, Xie J, Navratil AM, Farmerie TA, Wolfe MW, Clay CM2005. Gonadotropin-releasing hormone induction of extracellular-signal regulated kinase is blocked by inhibition of calmodulin. Mol Endocrinol 19:2412–2423 [DOI] [PubMed] [Google Scholar]
- 45.Bliss SP, Navratil AM, Breed M, Skinner DC, Clay CM, Roberson MS2007. Signaling complexes associated with the type I GnRH receptor: colocalization of ERK2 and GnRH receptor within membrane rafts. Mol Endocrinol 21:538–549 [DOI] [PubMed] [Google Scholar]
- 46.Davidson L, Pawson AJ, Millar RP, Maudsley S2004. Cytoskeletal reorganization dependence of signaling by the gonadotropin-releasing hormone receptor. J Biol Chem 279:1980–1993 [DOI] [PubMed] [Google Scholar]
- 47.Head BP, Patel HH, Roth DM, Murray F, Swaney JS, Niesman IR, Farquhar MG, Insel PA2006. Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. J Biol Chem 281:26391–26399 [DOI] [PubMed] [Google Scholar]
- 48.Caunt CJ, Finch AR, Sedgley KR, McArdle CA2006. Seven-transmembrane receptor signalling and ERK compartmentalization. Trends Endocrinol Metab 17:276–283 [DOI] [PubMed] [Google Scholar]
- 49.Caunt CJ, Finch AR, Sedgley KR, McArdle CA2006. GnRH receptor signalling to ERK: kinetics and compartmentalization. Trends Endocrinol Metab 17:308–313 [DOI] [PubMed] [Google Scholar]
- 50.Wang Q, Lu R, Zhao J, Limbird LE2006. Arrestin serves as a molecular switch, linking endogenous alpha2-adrenergic receptor to SRC-dependent, but not SRC-independent, ERK activation. J Biol Chem 281:25948–25955 [DOI] [PubMed] [Google Scholar]
- 51.Navratil AM, Knoll JG, Whitesell JD, Tobet SA, Clay CM2007. Neuroendocrine plasticity in the anterior pituitary: gonadotropin-releasing hormone-mediated movement in vitro and in vivo Endocrinology 148:1736–1744 [DOI] [PubMed] [Google Scholar]
- 52.Levi NL, Hanoch T, Benard O, Rozenblat M, Harris D, Reiss N, Naor Z, Seger R1998. Stimulation of Jun N-terminal kinase (JNK) by gonadotropin-releasing hormone in pituitary α T3-1 cell line is mediated by protein kinase C, c-Src, and CDC42. Mol Endocrinol 12:815–824 [DOI] [PubMed] [Google Scholar]
- 53.Benard O, Naor Z, Seger R2001. Role of dynamin, Src, and Ras in the protein kinase C-mediated activation of ERK by gonadotropin-releasing hormone. J Biol Chem 276:4554–4563 [DOI] [PubMed] [Google Scholar]
- 54.Chuderland D, Seger R2005. Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Mol Biotechnol 29:57–74 [DOI] [PubMed] [Google Scholar]
- 55.Lefkowitz RJ, Shenoy SK2005. Transduction of receptor signals by β-arrestins. Science 308:512–517 [DOI] [PubMed] [Google Scholar]
- 56.Schaller MD, Otey CA, Hildebrand JD, Parsons JT1995. Focal adhesion kinase and paxillin bind to peptides mimicking β integrin cytoplasmic domains. J Cell Biol 130:1181–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishibe S, Joly D, Zhu X, Cantley LG2003. Phosphorylation-dependent paxillin-ERK association mediates hepatocyte growth factor-stimulated epithelial morphogenesis. Mol Cell 12:1275–1285 [DOI] [PubMed] [Google Scholar]
- 58.Denouel-Galy A, Douville EM, Warne PH, Papin C, Laugier D, Calothy G, Downward J, Eychéne A1998. Murine Ksr interacts with MEK and inhibits Ras-induced transformation. Curr Biol 8:46–55 [DOI] [PubMed] [Google Scholar]
- 59.Yu W, Fantl WJ, Harrowe G, Williams LT1998. Regulation of the MAP kinase pathway by mammalian Ksr through direct interaction with MEK and ERK. Curr Biol 8:56–64 [DOI] [PubMed] [Google Scholar]
- 60.Stewart S, Sundaram M, Zhang Y, Lee J, Han M, Guan KL1999. Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol Cell Biol 19:5523–5534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Müller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK2001. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell 8:983–993 [DOI] [PubMed] [Google Scholar]
- 62.Freed E, Symons M, Macdonald SG, McCormick F, Ruggieri R1994. Binding of 14-3-3 proteins to the protein kinase Raf and effects on its activation. Science 265:1713–1716 [DOI] [PubMed] [Google Scholar]
- 63.Harris D, Reiss N, Naor Z1997. Differential activation of protein kinase C δ and ε gene expression by gonadotropin-releasing hormone in αT3-1 cells. Autoregulation by protein kinase C. J Biol Chem 272:13534–13540 [DOI] [PubMed] [Google Scholar]
- 64.Shraga-Levine Z, Ben-Menahem D, Naor Z1994. Activation of protein kinase C β gene expression by gonadotropin-releasing hormone in α T3-1 cell line. Role of Ca2+ and autoregulation by protein kinase C. J Biol Chem 269:31028–31033 [PubMed] [Google Scholar]
- 65.Maudsley S, Naor Z, Bonfil D, Davidson L, Karali D, Pawson AJ, Larder R, Pope C, Nelson N, Millar RP, Brown P2007. Proline-rich tyrosine kinase 2 mediates gonadotropin-releasing hormone signaling to a specific extracellularly regulated kinase-sensitive transcriptional locus in the luteinizing hormone β-subunit gene. Mol Endocrinol 21:1216–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parsons JT, Parsons SJ1997. Src family protein tyrosine kinases: cooperating with growth factor and adhesion signaling pathways. Curr Opin Cell Biol 9:187–192 [DOI] [PubMed] [Google Scholar]
- 67.Gundersen GG, Cook TA1999. Microtubules and signal transduction. Curr Opin Cell Biol 11:81–94 [DOI] [PubMed] [Google Scholar]
- 68.Rubinfeld H, Hanoch T, Seger R1999. Identification of a cytoplasmic-retention sequence in ERK2. J Biol Chem 274:30349–30352 [DOI] [PubMed] [Google Scholar]
- 69.Shacham S, Cheifetz MN, Fridkin M, Pawson AJ, Millar RP, Naor Z2005. Identification of Ser153 in ICL2 of the gonadotropin-releasing hormone (GnRH) receptor as a phosphorylation-independent site for inhibition of Gq coupling. J Biol Chem 280:28981–28988 [DOI] [PubMed] [Google Scholar]
- 70.Engelman JA, Chu C, Lin A, Jo H, Ikezu T, Okamoto T, Kohtz DS, Lisanti MP1998. Caveolin-mediated regulation of signaling along the p42/44 MAP kinase cascade in vivo. A role for the caveolin-scaffolding domain. FEBS Lett 428:205–211 [DOI] [PubMed] [Google Scholar]
- 71.Sanguinetti AR, Cao H, Corley Mastick C2003. Fyn is required for oxidative- and hyperosmotic-stress-induced tyrosine phosphorylation of caveolin-1. Biochem J 376:159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee H, Volonte D, Galbiati F, Iyengar P, Lublin DM, Bregman DB, Wilson MT, Campos-Gonzalez R, Bouzahzah B, Pestell RG, Scherer PE, Lisanti MP2000. Constitutive and growth factor-regulated phosphorylation of caveolin-1 occurs at the same site (Tyr-14) in vivo: identification of a c-Src/Cav-1/Grb7 signaling cassette. Mol Endocrinol 14:1750–1775 [DOI] [PubMed] [Google Scholar]
- 73.Westendorf JM, Rao PN, Gerace L1994. Cloning of cDNAs for M-phase phosphoproteins recognized by the MPM2 monoclonal antibody and determination of the phosphorylated epitope. Proc Natl Acad Sci USA 91:714–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brown MC, Turner CE2004. Paxillin: adapting to change. Physiol Rev 84:1315–1339 [DOI] [PubMed] [Google Scholar]
- 75.Zhang Z, Izaguirre G, Lin SY, Lee HY, Schaefer E, Haimovich B2004. The phosphorylation of vinculin on tyrosine residues 100 and 1065, mediated by SRC kinases, affects cell spreading. Mol Biol Cell 15:4234–4247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Subauste MC, Pertz O, Adamson ED, Turner CE, Junger S, Hahn KM2004. Vinculin modulation of paxillin-FAK interactions regulates ERK to control survival and motility. J Cell Biol 165:371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Herreros L, Rodríguez-Fernandez JL, Brown MC, Alonso-Lebrero JL, Cabañas C, Sánchez-Madrid F, Longo N, Turner CE, Sánchez-Mateos P2000. Paxillin localizes to the lymphocyte microtubule organizing center and associates with the microtubule cytoskeleton. J Biol Chem 275:26436–26440 [DOI] [PubMed] [Google Scholar]
- 78.Gundersen GG, Cook TA1999. Microtubules and signal transduction. Curr Opin Cell Biol 11:81–94 [DOI] [PubMed] [Google Scholar]
- 79.Enomoto M, Utsumi M, Park MK2006. Gonadotropin-releasing hormone induces actin cytoskeleton remodeling and affects cell migration in a cell-type-specific manner in TSU-Pr1 and DU145 cells. Endocrinology 147:530–542 [DOI] [PubMed] [Google Scholar]
- 80.Brahmbhatt AA, Klemke RL2003. ERK and RhoA differentially regulate pseudopodia growth and retraction during chemotaxis. J Biol Chem 278:13016–13025 [DOI] [PubMed] [Google Scholar]
- 81.Ishibe S, Joly D, Liu ZX, Cantley LG2004. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol Cell 16:257–267 [DOI] [PubMed] [Google Scholar]
- 82.Brown MC, Perrotta JA, Turner CE1996. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J Cell Biol 135:1109–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang JY, Widmann C2001. Antiapoptotic signaling generated by caspase-induced cleavage of RasGAP. Mol Cell Biol 21:5346–5358 [DOI] [PMC free article] [PubMed] [Google Scholar]