

Abstract
Human adipose tissue secretes a number of proinflammatory mediators that may contribute to the pathophysiology of obesity-related disorders. Understanding the regulatory pathways that control their production is paramount to developing effective therapeutics to treat these diseases. Using primary human adipose-derived stem cells as a source of preadipocytes and in vitro differentiated adipocytes, we found IL-8 and monocyte chemoattractant protein-1 (MCP-1) are constitutively secreted by both cell types and induced in response to serum deprivation. MicroRNA profiling revealed the rapid induction of microRNA 132 (miR-132) in these cells when switched to serum-free medium. Furthermore, miR-132 overexpression was sufficient to induce nuclear factor-κB translocation, acetylation of p65, and production of IL-8 and MCP-1. Inhibitors of miR-132 decreased acetylated p65 and partially inhibited the production of IL-8 and MCP-1 induced by serum deprivation. MiR-132 was shown to inhibit silent information regulator 1 (SirT1) expression through a miR-132 binding site in the 3′-untranslated region of SirT1. Thus, in response to nutritional availability, induction of miR-132 decreases SirT1-mediated deacetylation of p65 leading to activation of nuclear factor-κB and transcription of IL-8 and MCP-1 in primary human preadipocytes and in vitro differentiated adipocytes.
miR-132 regulates chemokine production in human adipocytes through repression of SirT1 mediated deacetylation of NFκB.
Historically, adipose tissue was thought to serve only as the storage site of excess energy; however, numerous studies have now identified a variety of proteins secreted from adipose, including both hormones and cytokines, which suggest a significant role for adipose in regulating whole-body energy metabolism and inflammation (1, 2, 3, 4, 5, 6, 7, 8, 9). These studies have led to the now generally accepted concept that adipose is a secretory organ with both endocrine and immunological activities (10). Obesity is a growing worldwide epidemic with type 2 diabetes and cardiovascular disease as common comorbidities. The development and progression of insulin resistance and atherosclerosis may result from a state of chronic inflammation within the adipose (11, 12). The pathogenesis of the inflammation is a matter of debate. Regardless of the cellular source of proinflammatory mediators within adipose, inhibition of their production may be therapeutically advantageous for the treatment of type 2 diabetes as well as cardiovascular disease. As a key transcription factor in inflammatory cascades in immune cells (13) and human adipocytes (14), nuclear factor-κB (NFκB) plays an important role in both innate and adaptive immunity. Chronic activation of NFκB in vivo has been shown to emulate the chronic inflammatory state observed with obesity and produces an insulin-resistant phenotype in IKKβ transgenic mice (15). However, the role of persistent activation of NFκB has not been directly assessed in human adipose. Furthermore, the mechanisms regulating the activation of NFκB and the production of proinflammatory mediators remain poorly characterized in human adipocytes.
MicroRNAs are small oligonucleotides that posttranscriptionally regulate gene expression by binding to the 3′-untranslated region (3′-UTR) of target gene sequences resulting in inhibition of transcription or translation (16, 17). Although over 700 microRNAs have been identified and various target predictions have been made, the function of these are just now being elucidated. Although the regulation of cell differentiation (18, 19, 20) by microRNAs and their dysregulation in various cancers (21, 22) is well documented, their role in signal transduction is less well understood.
To investigate the role of microRNAs in the production and secretion of proinflammatory mediators in human adipocytes, we profiled the expression of over 250 microRNAs in primary human preadipocytes and in vitro differentiated adipocytes under conditions of serum deprivation. We identified the induction of microRNA 132 (miR-132) and found it plays a key role in the response to serum deprivation in both preadipocytes and in vitro differentiated adipocytes. Furthermore, we demonstrate for the first time, the repression of silent information regulator 1 (SirT1) protein levels by miR-132 and propose a pathway where miR-132 represses the SirT1-mediated deacetylation of p65 resulting in NFκB activation and IL-8 and monocyte chemoattractant protein-1 (MCP-1) production.
Results
Serum deprivation induces IL-8 and MCP-1
To assess the production of proinflammatory mediators by primary human preadipocytes and in vitro differentiated adipocytes, cells were cultured in the presence or absence of serum, and at various times, the medium was analyzed for the presence of interferon-γ (IFNγ), IL-1β, IL-10, IL-12p70, IL-13, IL-2, IL-4, IL-5, IL-8, TNFα, and MCP-1. Serum deprivation induced a significant increase in the chemokines IL-8 and MCP-1 in both cell types compared with cells grown in serum-containing medium (Fig. 1). No other cytokine tested was significantly produced from either cell type under these conditions (data not shown). These data show both cell types constitutively express IL-8 and MCP-1 when grown in serum-containing medium. However, the in vitro differentiated adipocytes secrete higher basal levels of IL-8 and MCP-1 and, in terms of chemokine production, are more sensitive to the stress of serum deprivation compared with the preadipocytes. These data suggest the regulation of chemokine production by nutrient availability.
Fig. 1.

Serum deprivation induces IL-8 and MCP-1. Human preadipocytes and in vitro differentiated adipocytes were incubated in serum-containing medium (▪) or serum-free medium (♦) for the indicated times. After incubation, medium was collected, and the quantity of IL-8 and MCP-1 was determined using MSD assay kits. Proteins were quantitated by electrochemiluminescence detection and a MSD SECTOR Imager 6000. Data represent the mean of eight replicates ± sem. These data are representative of three independent experiments.
Serum deprivation induces miR-132
To identify pathways involved in the induction of chemokines by serum deprivation, we investigated the effect of serum deprivation on the expression of microRNAs in human preadipocytes and in vitro differentiated human adipocytes. Initially, we measured the expression of 255 microRNAs using real-time PCR (data not shown). We identified an increase in miR-132 expression and, in subsequent studies, confirmed this observation in both cell types in response to serum deprivation (Fig. 2). Serum deprivation rapidly induced the biphasic expression of miR-132 with peaks at 1 and 24 h in both cell types. The similarities between the miR-132 expression profile and chemokine production suggest a possible role for miR-132 in the regulation of chemokines in these cells.
Fig. 2.

Serum deprivation induces miR-132 expression. In vitro differentiated human adipocytes and human preadipocytes were treated with serum-containing medium (▪) or serum-free medium (♦) for the indicated times. Cells were lysed and total RNA extracted as described in Materials and Methods. Expression of miR-132 was determined using real-time PCR. Data represent the mean of four replicates and are representative of three independent experiments. Data are expressed as fold change relative to 0 h and were normalized to input of total RNA into the reverse transcription reaction.
miR-132 induces IL-8 and MCP-1
To gain insight into the possible relationship between induction of miR-132 and IL-8 and MCP-1 production, we explored the role of miR-132 by overexpressing miR-132 in preadipocytes and in vitro differentiated adipocytes. Transfection of pre-miR-132 in preadipocytes for 48 h induced a concentration-dependent increase of IL-8 and MCP-1 (Fig. 3). No other cytokine tested (IFNγ, IL-1β, IL-10, IL-12p70, IL-13, IL-2, IL-4, IL-5, or TNFα) was significantly produced from either cell type under these conditions (data not shown). The induction of both chemokines appeared biphasic with concentration of transfected pre-miR 132 with an initial plateau at 12.5–25 nm and subsequently at 200–250 nm. However, miR-132 was more efficacious at stimulating IL-8 production. We next investigated whether the ability of miR-132 to induce IL-8 and MCP-1 was unique to the preadipocytes. Transfection of miR-132 for 48 h was similarly effective at inducing both chemokines in the in vitro differentiated adipocytes (data not shown). These data demonstrate that miR-132 overexpression is sufficient to induce IL-8 and MCP-1.
Fig. 3.

MiR-132 induces IL-8 and MCP-1. Human preadipocytes were plated (3 × 104 cells per well) and transfected with varying concentrations of pre-miR-132 or a scrambled oligonucleotide as a negative control (250 nm). After 48 h incubation, medium was collected and the quantity of IL-8 (white bars) and MCP-1 (black bars) was determined using MSD assay kits. Proteins were quantitated by electrochemiluminescence detection and a MSD SECTOR Imager 6000. Data are expressed as light units of cytokine and represent the mean of eight replicates ± sem. These data are representative of three independent experiments.
Inhibitors of miR-132 partially inhibit induction of IL-8 and MCP-1 by serum deprivation
To determine whether blocking endogenous miR-132 affects the induction of IL-8 and MCP-1 by serum deprivation, human preadipocytes were transfected with inhibitors of miR-132. Inhibitors to miR-132 decreased the induction of IL-8 in a concentration-dependent manner (Fig. 4A). At 50 nm transfected inhibitor, there was an approximate decrease of 33% (P = 0.0002). MiR-132 inhibitor was maximally effective at 250 nm (2-fold, P = 0.000002) compared with serum withdrawal alone (Fig. 4A). Likewise, miR-132 inhibitors partially blocked the induction of MCP-1 by serum deprivation (Fig. 4B). The maximally effective concentration of miR-132 inhibitor was 100 nm with a decrease of 33% compared with cells grown in serum-free medium alone. This brought the level of MCP-1 down to about 2-fold greater then the constitutive production seen in cells grown in serum-containing medium. These changes were specific to the inhibitor used because the negative control had no significant effect on the production of IL-8 and MCP-1.
Fig. 4.

MiR-132 inhibitor partially blocks IL-8 and MCP-1 production. Human preadipocytes were transfected for 24 h in DMEM/F12 containing 10% FBS with varying concentrations of miR-132 inhibitor or a scrambled oligonucleotide negative control (500 nm). Cells were then incubated with fresh serum-containing medium or with serum-free medium for 24 h as indicated. After incubation, medium was collected, and the quantity of IL-8 (A) and MCP-1 (B) was determined using MSD assay kits. Proteins were quantitated by electrochemiluminescence detection and a MSD SECTOR Imager 6000. Data are expressed as light units of IL-8 and MCP-1 and represent the mean of eight replicates ± sem. These data are representative of three independent experiments.
Serum deprivation and miR-132 induces acetylation of p65 NFκB
NFκB plays a central role in the production of proinflammatory mediators, and IL-8 and MCP-1 are known transcriptional targets. Therefore, we initially assessed the role of NFκB by using 2-[(aminocarbonyl)amino]-5-(4-flurophenyl)-3-thiophenecarboxamide (TPCA1), a potent and selective inhibitor of IκB kinase 2 (IKK-2). Pretreatment of either preadipocytes or in vitro differentiated adipocytes with 50 nm TPCA1 blocked the induction of IL-8 and MCP-1 by serum deprivation, suggesting NFκB activation is necessary for chemokine production (data not shown). We also measured translocation of the transcription factor from the cytosol to the nucleus in human preadipocytes. After 1 h incubation in serum-free medium, the translocation of NFκB from the cytosol into the nucleus was seen (data not shown). Likewise, after 24 h transfection, pre-miR-132 induced a similar response. To investigate the mechanism of NFκB activation, we measured the acetylation of lysine 310 of the p65 component of the complex. After 1 h in serum-free medium, an increase in endogenous acetylated lysine 310 p65 was seen in the preadipocytes (Fig. 5A) and in vitro differentiated adipocytes (data not shown) that was not seen in cells grown in serum-containing medium. Transfection of a miR-132 inhibitor decreased the acetylation of lysine 310 p65, whereas a negative control oligonucleotide had no effect. Likewise, overexpression of miR-132 was sufficient to induce acetylation of lysine 310 p65 after 24 h transfection, whereas the transfection of a scrambled oligonucleotide control had no effect on acetylation of lysine 310 of p65 in cells grown in serum-free medium (Fig. 5B). Taken together, these data suggest the regulation of the acetylation/deacetylation pathway of NFκB by serum deprivation and miR-132.
Fig. 5.
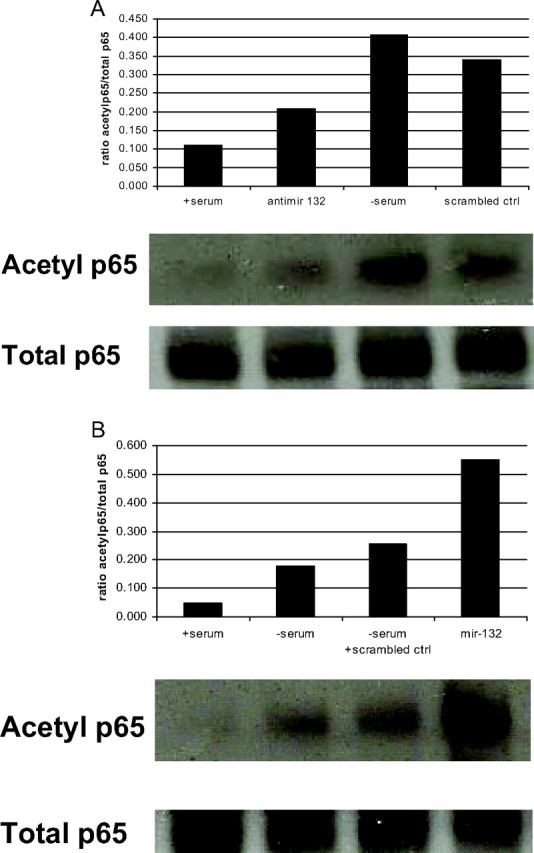
Serum deprivation and miR-132 overexpression induces acetylation of lysine 310 p65 and miR-132 inhibitors decrease acetylation. Human preadipocytes (1 × 106 cells) were plated in 25-cm2 flasks. Cells were transfected for 24 h with a miR-132 inhibitor or scrambled oligonucleotide (A) or pre-miR-132 or a scrambled oligonucleotide control (B) before addition of serum-containing medium or serum-free medium for 1 h. After incubation, cells were lysed and protein was immunoprecipitated overnight at 4 C using anti-total p65. Proteins were resolved by SDS-PAGE and blotted with anti-acetyl lysine 310 antibody. Enhanced chemiluminescence detection was used. These data are representative of two independent experiments.
Serum deprivation and miR-132 regulate SirT1 expression
Because miR-132 appears to regulate the acetylation of p65, we searched for protein targets that are known to be involved in acetylation/deacetylation pathways. The nicotinamide adenine dinucleotide-dependent deacetylase SirT1 was found as a predicted target of miR-132 in MiRBase (23, 24, 25) (http://microrna.sanger.ac.uk/). Thus, we assessed the protein levels of SirT1 in human preadipocytes and in vitro differentiated adipocytes in response to serum deprivation and miR-132 overexpression. A significant decrease in SirT1 protein levels were observed at 1 and 3 h in cells incubated in serum-free medium compared with cells grown in serum-containing medium (Fig. 6A). Likewise, overexpression of miR-132 for 24 h decreased SirT1 protein levels relative to cells transfected with a scrambled oligonucleotide control (Fig. 6B) while transfection of antimirs to miR-132 blocked the decrease in SirT1 seen with serum deprivation (data not shown). The decrease in SirT1 protein with miR-132 overexpression suggests the direct translational repression of SirT1 by miR-132 and the possible regulation of SirT1-mediated deacetylation of p65 by miR-132.
Fig. 6.
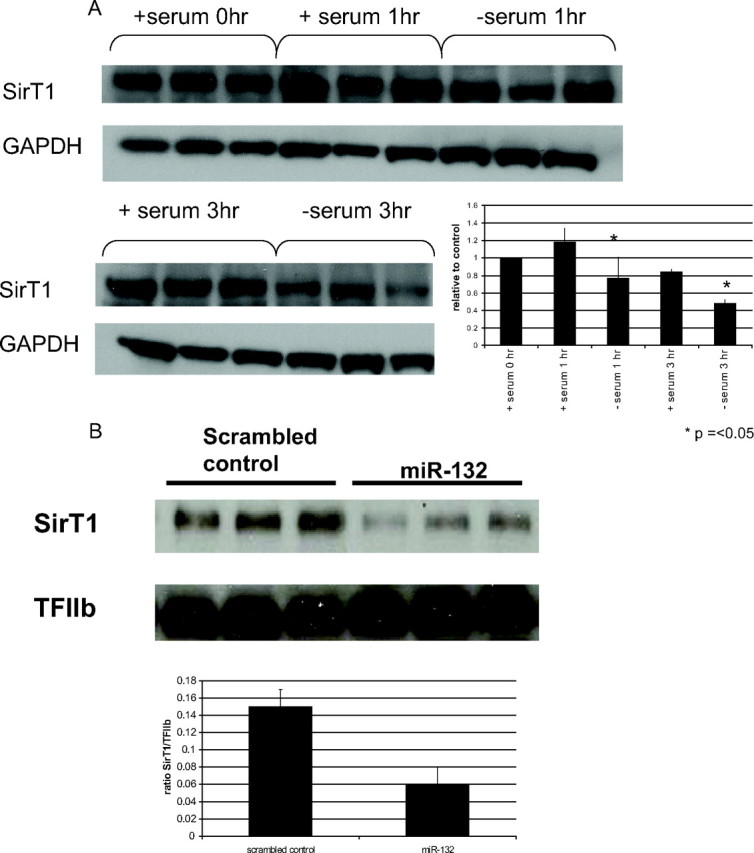
Serum deprivation and miR-132 overexpression decreases SirT1 protein levels. Human preadipocytes were plated in 12-well plates. Cells were incubated in serum-containing medium or serum-free medium for various times (A) or transfected with pre-miR-132 for 24 h (B). Cells were lysed, collected, and analyzed as described in Materials and Methods. The insets show the quantitation of SirT1 protein by densitometry. These data are representative of three independent experiments.
MiR-132 represses SirT1 expression through binding the 3′-UTR in the SirT1 gene
We first assessed whether serum deprivation or miR-132 overexpression affected SirT1 mRNA levels. Through gene expression analysis of SirT1 mRNA using real-time PCR, we found there was no change in SirT1 mRNA under any of these treatment conditions (data not shown). Thus, a luciferase reporter construct containing the 3′-UTR of the SirT1 gene and the predicted miR-132 binding site was prepared to assess whether the binding of miR-132 to the SirT1 3′-UTR mediates translational repression. After cotransfection of the reporter construct and pre-miR-132 or a scrambled oligonucleotide control in human preadipocytes, there was an approximately 2-fold decrease in luciferase activity of the reporter construct in cells cotransfected with pre-miR-132 compared with the scrambled oligonucleotide control (Fig. 7). These data suggest miR-132 represses SirT1 translation through directly binding the response element in the SirT1 3′-UTR.
Fig. 7.

miR-132 mediates translational repression of SirT1 through a binding site in SirT1 3′-UTR. Human preadipocytes (1 × 105 cells) were plated in 96-well plates and cotransfected with 200 ng pMIR-Report luciferase vector including the 3′-UTR of SirT1 containing the predicted miR-132 binding site and pre-miR-132 or scrambled oligonucleotide using an electroporator. Luciferase assays were performed using the dual-luciferase assay system 6–8 h after transfection. Data represent the mean of four biological replicates. The data are representative of two independent experiments.
Discussion
Studies of microRNAs have yielded unique insights into the regulation of important biological processes. Yamakuchi et al. (26) recently demonstrated the translational repression of SirT1 by miR-34a resulting in the activation of p53 and apoptosis, thus identifying miR-34a as a tumor suppressor. Our study is the first report to demonstrate the regulation of SirT1 by miR-132 and subsequent chemokine production through activation of NFκB. Previously, miR-132 was shown to be a transcriptional target of cAMP response element binding protein (CREB), which regulates neuronal morphogenesis through translational repression of p250GAP (27). Additionally, it’s been shown to regulate the expression of methyl-CpG-binding protein (MeCP2) (28) and the angiotensin II type 1 receptor (AT1R) (29). MiR-132 was also reported to be induced by lipopolysaccharide in THP-1 cells (30), but a function was not ascribed. The biphasic expression pattern of miR-132 appears to parallel the biphasic activity profile of NFκB (31) and suggests miR-132 expression may be regulated by NFκB and represent a positive feedback loop. However, in silico promoter analysis failed to identify NFκB binding sites in the region of miR-132. As mentioned above, CREB has been shown to regulate miR-132 expression in neuronal cells (27). Additionally, CREB is regulated by high glucose in THP-1 cells where it has been shown to induce cyclooxygenase 2 (COX2), an enzyme that metabolizes arachidonic acid to prostaglandins (32). In our studies, CREB may regulate expression of miR-132 in response to the treatment conditions used where serum was absent and the concentration of glucose was relatively high (17.5 mm). Additional studies are needed to determine the exact mechanism of miR-132 transcription in human adipocytes.
SirT1 is highly regulated by nutrient availability. SirT1 protein levels in vivo are increased with starvation, fasting, and calorie restriction (33), whereas SirT1 protein decreases with age and senescence (33). Most interestingly, glucose appears to regulate SirT1 protein levels. Incubation of PC12 and HEK293 cells in the absence of both serum and glucose induces SirT1 protein expression through either an increase in transcription (34) or posttranscriptional regulation (35). In contrast, Nedachi et al. (36) showed low serum and high glucose represses SirT1 protein in C2C12 cells. Likewise, Rodgers et al. (37) found high glucose mediated the posttranscriptional repression of SirT1 protein in mouse hepatocytes in conditions of low serum. Corroborating these findings, our study shows SirT1 protein levels decrease when human preadipocytes and in vitro differentiated adipocytes are incubated in serum-free conditions and high glucose (17.5 mm) and extends these studies by proposing the posttranscriptional regulation of SirT1 by miR-132. Inhibition of SirT1 in response to high glucose in adipocytes should also result in activation of peroxisome proliferator-activated receptor-γ (PPARγ), which allows the cells to store glucose during times of excess energy (38). However, our data suggest nutrient availability may result in persistent activation of NFκB and chemokine production.
SirT1 has recently been implicated in the regulation of inflammation. Recently, Zhu et al. (39) showed resveratrol, a SirT1 activator, decreased TNFα-induced MCP-1 secretion in 3T3-L1 adipocytes. Pfluger et al. (40) showed overexpression of SirT1 in mice resulted in a lower recovery of IL-6 and TNFα in serum of transgenic mice fed a high-fat diet and an attenuated response to TNFα-induced NFκB activation in transgenic mouse embryonic fibroblasts. Thus, increased SirT1 activity appears to be antiinflammatory in mice. In contrast, when inhibited, SirT1 appears to be proinflammatory. Kwon et al. (41) showed HIV Tat binds SirT1 and inhibits the SirT1-mediated deacetylation of p65 NFκB resulting in hyperactivated NFκB, a greater transactivation efficiency and an exaggerated immune response. Likewise, Yang et al. (42) recently showed that cigarette smoke extract caused a dose- and time-dependent decrease in SirT1 activity and protein and a concomitant increase in NFκB-dependent proinflammatory mediator release in MonoMac 6 cells. More recently, studies using small interfering RNA (siRNA) to knock down SirT1 reported an increase in TNFα-induced MCP-1 and other proinflammatory genes in 3T3-L1 adipocytes (43). The authors went on to show that knockdown of SirT1 led to an increase in acetylation of lysine 310 of p65. Taken together with the findings reported here, these data demonstrate a decrease in SirT1 activity increases activation of NFκB and transcription of proinflammatory mediators.
Reversible acetylation of signaling proteins has emerged as a key posttranslational modification regulating activity of p53 and Forkhead proteins. Several reports have shown the reversible acetylation of NFκB (44, 45, 46, 47). Furthermore, Yeung et al. (44) demonstrated the attenuation of NFκB activity by SirT1 and histone deacetylases leading to the enhancement of TNFα-induced apoptosis. Acetylation of lysine 310 of the p65 component of the NFκB complex has been shown to be necessary for the full activation of NFκB (45) and affect the duration and the selectivity of gene transcription (46). Ito (47) reported the acetylated lysine 310 p65 complex preferentially bound the NFκB binding site on the promoter region of IL-8 but not granulocyte-macrophage colony-stimulating factor. We found miR-132 inhibitors blocked the accumulation of endogenous acetylated lysine 310 p65 and partially inhibited the induction of IL-8 and MCP-1 in serum-deprived cells. These data confirm the importance of acetylated lysine 310 p65 on the transcriptional activation of NFκB and extends it to conditions of nutrient availability in human preadipocytes and adipocytes. MiR-132 indirectly regulates acetylation of NFκB through repression of SirT1. SirT1 also binds to and regulates p300 (48), the acetyltransferase involved in NFκB activation. The decrease in SirT1 protein may have a possible dual effect on acetylation of NFκB through both inhibition of deacetylation and activation of acetylation.
Previously, IL-8 and MCP-1 were shown to be constitutively secreted by both human preadipocytes and isolated adipocytes implying a normal biological function for these chemokines (49, 50, 51, 52). Excessive production through dysregulated NFκB is thought to play a pathogenic role in metabolic diseases. Indeed, circulating levels of IL-8 and MCP-1 are higher in obese then lean individuals and are associated with parameters of insulin resistance, atherosclerosis, and cardiovascular disease (53, 54, 55, 56, 57, 58, 59). Our data support the central role of NFκB in the inflammatory process in adipose and implies the miR-132 repression of SirT1-mediated deacetylation of p65 could be a mechanism of NFκB dysregulation contributing to the chronic inflammatory state in adipose tissue. Preventing the decrease in SirT1 protein should block the activation of NFκB and may be a therapeutic alternative to activating SirT1.
Materials and Methods
Cell culture and transfection
Primary human adipose-derived stem cells were collected from the abdominal sc adipose of a 37-yr-old female with a BMI of 23.3 (Zen-Bio Biological Systems and Services, Research Triangle Park, NC) and used as a source for preadipocytes and differentiated adipocytes. Cells (passages 4–8) were cultured in sc preadipocyte medium (Zen-Bio) until treatment. To differentiate into adipocytes, the sc preadipocytes were plated in preadipocyte medium and then after 24 h were differentiated in DMEM/F12 plus 10% fetal bovine serum (FBS) medium (Invitrogen, Carlsbad, CA) containing 200 μm isobutylmethylxanthine, 20 nm dexamethasone, 20 nm GW347845X, a PPARγ agonist and 20 nm human insulin. After incubation for 7 d, the medium was changed to DMEM/F12 plus 10% FBS containing 20 nm insulin. All experiments on mature in vitro differentiated adipocytes were begun after d 14. Differentiation of cells was more than 90% as assessed by quantitating triglyceride accumulation using the Trinder assay (Sigma Chemical Co., St. Louis, MO) and real-time PCR analysis of fatty acid-binding protein 4 (FABP4 or ap2), PPARγ, and fatty acid synthase (FAS) mRNA, as markers of adipocyte differentiation. These results showed less than 5% interwell variability among differentiated cultures of adipocytes. Serum deprivation was carried out by incubating cells in DMEM/F12 plus 1% BSA-fatty acid free. Experiments done to overexpress or inhibit miR-132 were done using Dharmafect 2 siRNA transfection reagent and miRIDIAN microRNA 132 mimic (pre-miR-132) UAACAGUCUACAGCCAUGGUCG and mimic negative control UCACAACCUCCUAGAAAGAGUAGA (Dharmacon, LaFayette, CO) or miRCURY LNA knockdown probes CGACCATGGCTGTAGACTGTTA and scramble-miR control GTGTAACACGTCTATACGCCCA (Exiqon, Inc., Woburn, MA). The negative control oligonucleotides were tested at the highest concentration of the pre-miR-132 or inhibitor used in the experiment. Thus, we did not control for each concentration of microRNA used or transfect constant amounts of total microRNA. Uptake of microRNAs into cells was assessed using Cy-3-labeled pre-miR negative control and Cy-3-labeled anti-miR negative control (Ambion, Austin, TX) and Dharmafect 2 siRNA transfection reagent. Unless otherwise noted, all transfections were done in serum-containing medium.
mRNA gene expression analysis
Total RNA was isolated from cells using Promega total RNA isolation kit (Promega, Madison, WI). RNA was reverse-transcribed using the ABI high-capacity cDNA archive kit (Applied Biosystems, Foster City, CA). Reactions containing 10 ng cDNA, SirT1 primers and probes (Assays on Demand; Applied Biosystems), and Universal PCR Master Mix (Applied Biosystems) were analyzed using a 7900HT sequence detector system (Applied Biosystems) as recommended by the vendor. The relative abundance of SirT1 mRNA was normalized to β-actin (Actb) mRNA.
MicroRNA gene expression analysis
Total RNA was isolated by removing the treatment medium, washing the cells with PBS, and adding 1 ml TRIzol reagent per well. A phenol-chloroform extraction was performed, and 1.25 vol 100% ethanol was added to the aqueous phase. The microRNAs were isolated according to the manufacturer’s protocol using a mirVana microRNA isolation kit from Ambion (Austin, TX). The microRNAs were then converted to cDNA, and gene expression analysis was done using the TaqMan microRNA assays (Applied Biosystems).
NFκB translocation
Translocation of NFκB was assessed in human preadipocytes using the NFκB activation kit (ThermoScientific) and the Cellomics ArrayScan II high-content imaging system. Briefly, human preadipocytes (3 × 104 cells per well) were plated into 96-well plates. After incubation overnight, cells were treated by incubating in serum-free medium or transfected with pre-miR-132 (100 nm) (Dharmacon) for 24 h. After treatment for various times, cells were fixed and stained with anti-NFκB antibody followed by an AlexaFluor 488-conjugated secondary antibody. The ratio of fluorescence in the cytosol and nucleus was calculated as a quantitative measure of NFκB translocation.
Western blot analysis
Protein lysates were prepared using M-PER cell lysis buffer (Pierce, Rockford, IL) containing a protease cocktail inhibitor mix (Boehringer Mannheim, Mannheim, Germany). Protein lysates (10 μg total protein) were resolved by electrophoresis on 10% or 4–12% SDS-polyacrylamide gels (Invitrogen, Carlsbad, CA) and transferred to nitrocellulose (Invitrogen). Proteins were detected by immunoblotting using anti-acetyl lysine 310 p65, anti-NFκB p65, anti-SirT1(C14H4), anti-GAPDH, antirabbit IgG horseradish peroxidase-linked antibody, anti-TFIIB (Cell Signaling Technologies, Beverly, MA). All primary antibodies were used at 1:1000 dilution and incubated overnight at 4C. Antirabbit IgG HRP was used as the secondary antibody at 1:2000 dilution and detected using ECL reagents (Amersham, Piscataway, NJ).
Plasmid construction
A 1.7-kb DNA fragment containing the SirT1 3′-UTR and the predicted miR-132 binding site was PCR amplified from human liver cDNA (QUICK-Clone cDNA; Clontech, Mountain View, CA). Primers were designed based on Genbank NM_012238 and included a 5′ Sac1 site and a 3′ Mlu1 site to accommodate subcloning into the pMIR-REPORT plasmid (Ambion, Austin, TX).
Luciferase assays
Primary cultures of human preadipocytes (1 × 105 cells per well) were plated in 96-well plates. After 24 h, 200 ng pMIR-Report luciferase vector including the 3′-UTR of SirT1, miR-132, or scrambled oligonucleotide were transfected using a Nucleofector (Amaxa, Walkersville, MD). Luciferase assays were performed using the dual-luciferase assay system (Promega) 6–8 h after transfection.
Cytokine measurements
Media were collected at various times from preadipocytes and differentiated adipocytes to analyze secreted cytokines. MesoScale Discovery (MSD) assay kits were used according to the manufacturer’s recommendations to quantitate IFNγ, IL-1β, IL-10, IL-12p70, IL-13, IL-2, IL-4, IL-5, IL-8, TNFα, and MCP-1 (MesoScale Discovery, Gaithersburg, MD) protein levels using electrochemiluminescence detection and a MSD SECTOR Imager 6000.
Footnotes
Disclosure Summary: All authors are employees of GlaxoSmithKline (GSK). J.C.S., K.M.W., and J.F. own GSK stock options.
First Published Online October 9, 2009
Abbreviations: CREB, cAMP response element binding protein; FBS, fetal bovine serum; IFNγ, interferon-γ; MCP-1, monocyte chemoattractant protein-1; miR-132, microRNA 132; MSD, MesoScale Discovery; NFκB, nuclear factor-κB; PPARγ, peroxisome proliferator-activated receptor-γ; siRNA, small interfering RNA; SirT1, silent information regulator 1; 3′-UTR, 3′-untranslated region.
References
- 1.Schäffler A, Schölmerich J, Salzberger B2007. Adipose tissue as an immunological organ: Toll-like receptors, C1q/TNFs and CTRPs. Trends Immunol 28:393–399 [DOI] [PubMed] [Google Scholar]
- 2.Matarese G, La Cava A2004. The intricate interface between immune system and metabolism. Trends Immunol 25:193–200 [DOI] [PubMed] [Google Scholar]
- 3.Juge-Aubry CE, Henrichot E, Meier CA2005. Adipose tissue: a regulator of inflammation. Best Pract Res Clin Endocrinol Metab 19:547–566 [DOI] [PubMed] [Google Scholar]
- 4.Tilg H, Moschen AR2006. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 6:772–783 [DOI] [PubMed] [Google Scholar]
- 5.Schaffler A, Muller-Ladner U, Scholmerich J, Buchler C2006. Role of adipose tissue as an inflammatory organ in human diseases. Endocrinol Rev 27:449–467 [DOI] [PubMed] [Google Scholar]
- 6.Warne JP2003. Tumor necrosis factor α: a key regulator of adipose tissue mass. J Endocrinol 177:351–355 [DOI] [PubMed] [Google Scholar]
- 7.Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW2004. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology 145:2273–2282 [DOI] [PubMed] [Google Scholar]
- 8.Hoch M, Eberle AN, Peterli R, Peters T, Seboek D, Keller U, Muller B, Linscheid P2008. LPS induces interleukin-6 and interleukin-8 but not tumor necrosis factor-α in human adipocytes. Cytokine 41:29–37 [DOI] [PubMed] [Google Scholar]
- 9.Chung S, Lapoint K, Martinez K, Kennedy A, Sandberg MB, McIntosh MK2006. Preadipocytes mediate lipopolysaccaride-induced inflammation and insulin resistance in primary cultures of newly differentiated human adipocytes. Endocrinology 147:5340–5351 [DOI] [PubMed] [Google Scholar]
- 10.Caspar-Bauguil S, Cousin B, Galinier A, Segafredo C, Nibbelink M, André M, Casteilla L, Pénicaud L2005. Adipose tissues as an ancestral immune organ: site-specific change in obesity. FEBS Lett 579:3487–3492 [DOI] [PubMed] [Google Scholar]
- 11.Wellen KE, Hotamisligil GS2005. Inflammation, stress and diabetes. J Clin Invest 115:1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone Jr MA, Luster AD, Luscinskas FW, Rosenzweig A1999. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 398:718–723 [DOI] [PubMed] [Google Scholar]
- 13.Caamaño J, Hunter CA2002. NF-κB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev 15:414–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Creely SJ, McTernan PG, Kusminski CM, Fisher M, Da Silva NF, Khanolkar M, Evans M, Harte AL, Kumar S2007. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab 292:740–747 [DOI] [PubMed] [Google Scholar]
- 15.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE2005. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med 11:183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ambros V2004. The function of animal microRNAs. Nature 431:350–355 [DOI] [PubMed] [Google Scholar]
- 17.He L, Hannon GJ2004. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5:522–531 [DOI] [PubMed] [Google Scholar]
- 18.Chen CZ, Li L, Lodish HF, Bartel DP2004. MicroRNAs modulate hematopoietic lineage differentiation. Science 303:83–86 [DOI] [PubMed] [Google Scholar]
- 19.Monticelli S, Ansel KM, Xiao C, Socci ND, Krichevsky AM, Thai TH, Rajewsky N, Marks DS, Sander C, Rajewsky K, Rao A, Kosik KS2005. MicroRNA profiling of the murine hematopoietic system. Genome Biol 6:R71 [DOI] [PMC free article] [PubMed]
- 20.Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, Dean NM, Freier SM, Bennett CF, Lollo B, Griffey R2004. MicroRNA-143 regulates adipocyte differentiation. J Biol Chem 279:52361–52365 [DOI] [PubMed] [Google Scholar]
- 21.Fabbri M, Garzon R, Andreeff M, Kantarjian HM, Garcia-Manero G, Calin GA2008. MicroRNAs and noncoding RNAs in hematological malignancies: molecular, clinical and therapeutic implications. Leukemia 22:1095–1105 [DOI] [PubMed] [Google Scholar]
- 22.Dalmay T2008. MicroRNAs and cancer. J Intern Med 263:366–375 [DOI] [PubMed] [Google Scholar]
- 23.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ2008. miRBase: tools for microRNA genomics. Nucleic Acid Res 36(Database issue):D154–D158 [DOI] [PMC free article] [PubMed]
- 24.Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ2006. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acid Res 34(Database issue):D140–D144 [DOI] [PMC free article] [PubMed]
- 25.Griffiths-Jones S2004. The microRNA Registry. Nucleic Acid Res 32(Database issue):D109–D111 [DOI] [PMC free article] [PubMed]
- 26.Yamakuchi M, Ferlito M, Lowenstein CJ2008. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA 105:13421–13426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, Impey S2005. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci USA 102:16426–16431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH2007. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci 10:1513–1514 [DOI] [PubMed] [Google Scholar]
- 29.Elton TS2008. MiR-132 regulates angiotensin II type 1 receptor expression through a protein coding region binding site. Circulation 118:S513
- 30.Taganov KD, Boldin MP, Chang KJ, Baltimore D2006. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci USA 103:12481–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ladner KJ, Caligiuri MA, Guttridge DC2003. Tumor necrosis factor-regulated biphasic activation of NFκB is required for cytokine-induced loss of skeletal muscle gene products. J Biol Chem 278:2294–2303 [DOI] [PubMed] [Google Scholar]
- 32.Shanmugam N, Gaw Gonzalo IT, Natarajan R2004. Molecular mechanisms of high glucose-induced cyclooxygenase-2 expression in monocytes. Diabetes 53:795–802 [DOI] [PubMed] [Google Scholar]
- 33.Kwon HS, Ott M2008. The ups and downs of SIRT1. Trends Biol Sci 609:1–9 [DOI] [PubMed] [Google Scholar]
- 34.Nemoto S, Fergusson MM, Finkel T2004. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 306:2105–2108 [DOI] [PubMed] [Google Scholar]
- 35.Kanfi Y, Peshti V, Gozlan YM, Rathaus M, Gil R, Cohen HY2008. Regulation of SIRT1 protein levels by nutrient availability. FEBS Lett 582:2417–2423 [DOI] [PubMed] [Google Scholar]
- 36.Nedachi T, Kadotani A, Ariga M, Katagiri H, Kanzaki M2008. Ambient glucose levels qualify the potency of insulin myogenic actions by regulating SIRT1 and FoxO3a in C2C12 myocytes. Am J Physiol Endocrinol Metab 294:E668–E678 [DOI] [PubMed]
- 37.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P2005. Nutrient control of glucose homeostasis through a complex of PGC1-α and SIRT1. Nature 434:113–118 [DOI] [PubMed] [Google Scholar]
- 38.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L2004. Sirt1 promotes fat mobilization in white adipocytes by repressing PPARγ. Nature 429:771–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu J, Yong W, Wu X, Yu Y, Lv J, Liu C, Mao X, Zhu Y, Xu K, Han X, Liu C2008. Anti-inflammatory effect of resveratrol on TNF-α-induced MCP-1 expression in adipocytes. Biochem Biophys Res Commun 369:471–477 [DOI] [PubMed] [Google Scholar]
- 40.Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschöp MH2008. Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci USA 105:9793–9798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwon HS, Brent MM, Getachew R, Jayakumar P, Chen LF, Schnolzer M, McBurney MW, Marmorstein R, Greene WC, Ott M2008. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe 3:158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I2007. Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/NF-κB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol 292:L567–L576 [DOI] [PubMed]
- 43.Yoshizaki T, Milne JC, Imamura T, Schenk S, Sonoda N, Babendure JL, Lu JC, Smith JJ, Jirousek MR, Olefsky JM2009. SIRT1 exerts anti-inflammatory effect and improves insulin sensitivity in adipocytes. Mol Cell Biol 29:1363–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW2004. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23:2369–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen LF, Fischle W, Verdin E, Greene WC2001. Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293:1653–1657 [DOI] [PubMed] [Google Scholar]
- 46.Chen LF, Mu Y, Greene WC2002. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J 21:6539–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ito K2007. Impact of post-translational modifications of proteins on the inflammatory process. Biochem Soc Trans 35:281–283 [DOI] [PubMed] [Google Scholar]
- 48.Bouras T, Fu M, Sauve AA, Wang F, Quong AA, Perkins ND, Hay RT, Gu W, Pestell RG2005. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem 280:10264–10276 [DOI] [PubMed] [Google Scholar]
- 49.Bruun JM, Pedersen SB, Richelsen B2001. Regulation of interleukin 8 production and gene expression in human adipose tissue in vitro J Clin Endocrinol Metab 86:1267–1273 [DOI] [PubMed] [Google Scholar]
- 50.Gerhardt CC, Romero IA, Cancello R, Camoin L, Strosberg AD2001. Chemokines control fat accumulation and leptin secretion by cultured human adipocytes. Mol Cell Endocrinol 175:81–92 [DOI] [PubMed] [Google Scholar]
- 51.Bruun JM, Lihn AS, Madan AK, Pedersen SB, Schiøtt KM, Fain JN, Richelsen B2004. Higher production of IL-8 in visceral vs. subcutaneous adipose tissue. Implication of nonadipose cells in adipose tissue. Am J Physiol Endocrinol Metab 286:E8–E13 [DOI] [PubMed]
- 52.Christiansen T, Richelsen B, Bruun JM2005. Monocyte chemoattractant protein 1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int J Obes 29:146–150 [DOI] [PubMed] [Google Scholar]
- 53.Wang B, Jenkins JR, Trayhurn P2005. Expression and secretion of inflammation-related adipokines by human adipocytes differentiated in culture: integrated response to TNF-α. Am J Physiol Endocrinol Metab 288:E731–E740 [DOI] [PubMed]
- 54.Charo IF, Taubman MB2004. Chemokines in the pathogenesis of vascular disease. Circ Res 95:858–866 [DOI] [PubMed] [Google Scholar]
- 55.Sartipy P, Loskutoff DJ2003. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci USA 100:7265–7270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bruun JM, Lihn AS, Pedersen SB, Richelsen B2005. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): implication of macrophages resident in the AT. J Clin Endocrinol Metab 90:2282–2289 [DOI] [PubMed] [Google Scholar]
- 57.Moreau M, Brocheriou I, Petit L, Ninio E, Chapman MJ, Rouis M1999. Interleukin-8 mediates downregulation of tissue inhibitor of metalloproteinase-1 expression in cholesterol-loaded human macrophages: relevance to stability of atherosclerotic plaque. Circulation 99:420–426 [DOI] [PubMed] [Google Scholar]
- 58.Straczkowski M, Dzienis-Straczkowska S, Stêpieñ A, Kowalska I, Szelachowska M, Kinalska I2002. Plasma interleukin-8 concentrations are increased in obese subjects and related to fat mass and tumor necrosis factor-α system. J Clin Endocrinol Metab 87:4602–4606 [DOI] [PubMed] [Google Scholar]
- 59.Zozuliñska D, Majchrzak A, Sobieska M, Wiktorowicz K, Wierusz-Wysocka B1999. Serum interleukin-8 level is increased in diabetic patients. Diabetologia 42:117–118 [DOI] [PubMed] [Google Scholar]