

Abstract
Grb14 belongs to the Grb7 family of molecular adapters and was identified as an inhibitor of insulin signaling. Grb14 binds to activated insulin receptors (IR) and inhibits their catalytic activity. To gain more insight into the Grb14 molecular mechanism of action, we generated various mutants and studied the Grb14-IR interaction using coimmunoprecipitation and bioluminescence resonance energy transfer (BRET) experiments. Biological activity was further analyzed using the Xenopus oocyte model and a functional complementation assay measuring cellular proliferation rate in Grb14 knockout mouse embryonic fibroblasts. These studies identified two important interaction sites, Grb14 L404-IR L1038 and Grb14 R385-IR K1168, involving the IR αC-helix and activation loop, respectively. Interestingly, the former involves residues that are likely to be crucial for the specificity of IR binding with regard to other members of the Grb7 family. In addition, mutation of the Grb14-S370 residue suggested that its phosphorylation status controlled the biological activity of the protein. We further demonstrated that insulin-induced Grb14-PDK1 interaction is required in addition to Grb14-IR binding to mediate maximal inhibition of insulin signaling. This study provides important insights into the molecular determinants of Grb14 action by demonstrating that Grb14 regulates insulin action at two levels, through IR binding and by interfering with downstream pathways. Indeed, a precise knowledge of the molecular mechanism of insulin signaling inhibition by Grb14 is a prerequisite for the development of insulin-sensitizing molecules to treat pathophysiological states such as obesity or type 2 diabetes.
Grb14 regulates insulin signaling and action affecting both insulin receptor binding and interfering with downstream pathways, including PDK1.
Alterations in insulin signaling and action lead to pathophysiological states such as obesity and type 2 diabetes, which are worldwide expanding diseases. The mechanisms causing insulin resistance are not completely understood but are likely to involve postreceptor defects. Insulin initiates its actions by binding to its transmembrane receptor, thereby inducing conformational changes and activation of the tyrosine kinase domain of the receptor. Once autophosphorylated on critical tyrosine residues and activated, the insulin receptor (IR) phosphorylates intracellular substrates [IR substrates (IRS) and Shc], that are the first effectors of the insulin signaling pathways (1). After insulin stimulation, two main signaling cascades are activated, the phosphatidylinositol 3-kinase-Akt and the Ras-MAPK pathways, respectively, involved in the metabolic and the mitogenic actions of insulin (1). The duration and magnitude of insulin signaling are regulated by control mechanisms that are set into motion immediately after insulin binding to its receptor. They involve the action of tyrosine or lipid phosphatases (2, 3), the recruitment of adaptor proteins like the Grb7 family of proteins and suppressors of cytokine signaling (SOCS) proteins (4, 5), and the serine/threonine phosphorylation of IRS proteins (6). In the perspective of developing new therapeutic strategies to improve insulin sensitivity, it is thus important to elucidate the molecular mechanism of action of the negative regulators of insulin signaling.
The Grb7 family of proteins, which includes Grb7, Grb14, and Grb10, interacts with growth factor receptors and regulates their signaling pathways. They have mainly been studied for their implications in cancer and insulin signaling (4, 7). The Grb14 isoform seems to be more specifically involved in the regulation of insulin action. Indeed, Grb14 expression level in white adipose tissue is negatively correlated with insulin sensitivity (8), and Grb14-deficient mice exhibit improved glucose homeostasis and enhanced insulin signaling (9). In addition, we recently showed that Grb14 is physiologically recruited to the IR in vivo in rat liver and that this interaction hinders IR catalytic activity (10). Members of the Grb7 family of proteins are characterized by a common multidomain structure, including a conserved polyproline motif in the N terminus, a Ras-associating domain, a PH domain, and a C-terminal SH2 domain. They also contain a so-called BPS (between PH and SH2 domains) region, also known as PIR (phosphorylated insulin receptor interacting region) that is unique to this family of proteins. The BPS region is responsible for the interaction between Grb14 and the activated IR catalytic domain (11, 12). Crystallographic studies revealed that Grb14 acts as a pseudosubstrate inhibitor bound in the peptide-binding groove of the kinase, thus indicating how Grb14 functions as a selective protein inhibitor of IR activity (13). However, the pseudosubstrate motif is conserved in Grb7 and Grb10, suggesting that the selectivity of the Grb14 effect might be linked to other interaction sites between Grb14 and the IR. Furthermore, in addition to its inhibitory action on IR catalytic activity, Grb14 acts also at more distal steps in insulin signaling pathways, such as insulin-induced phosphatidylinositol-dependent kinase 1 (PDK1) plasma membrane translocation and sterol response element binding protein-1c maturation (14, 15). To progress in the elucidation of the molecular mechanism of action of Grb14, two main points remained then to be investigated: 1) the identification of additional interaction sites between Grb14 and the IR and 2) the evaluation of the relative impact of Grb14 binding to the IR vs. downstream effects of Grb14 on insulin signaling. Indeed, a clear understanding of the various Grb14 interferences with insulin signaling and action is a prerequisite for the development of specific inhibitors of Grb14 that could be used to improve insulin sensitivity in pathophysiological situations characterized by an insulin-resistant state, such as obesity or type 2 diabetes.
The aim of the present work was to analyze at a molecular level the determinants of Grb14 interactions to obtain new information on the multifaceted functions of this adapter in the insulin signaling cascade. We thus used a site-directed mutagenesis strategy to target several residues on Grb14 and on the IR to address their role in protein-protein interaction and in the final action of insulin. The activity of the designed mutant proteins was analyzed by coimmunoprecipitation (co-IP) and bioluminescence resonance energy transfer (BRET) experiments to test their binding ability. The consequence of the mutations on the biological effects of insulin was further investigated using the Xenopus oocyte model (16) and functional complementation experiments in mouse embryonic fibroblasts (MEF) obtained from Grb14 knockout (KO) mice.
Results
Identification of IR domains involved in the interaction with Grb14
From the crystallographic studies of the IR tyrosine kinase domain (IRK), it appeared that the reorientation of the activation loop upon receptor autophosphorylation induced residues of the αC-helix to become solvent exposed and therefore available for protein-protein interactions (17), leading to the proposition that they could interact with the BPS region (18). We mutated three residues located in the αC-helix, L1038, I1042, and L1045 into alanine and investigated their ability to bind to Grb14. COS-7 cells were transiently transfected with wild-type (WT) Grb14 and the various IR constructs, and after 10 min insulin stimulation, proteins were immunoprecipitated with anti-IR antibodies. Under insulin stimulation, WT Grb14 was recruited to the IR, as previously observed (12, 13). The three substitutions chosen in the αC-helix differentially modulated the effect of insulin on Grb14 binding to the IR; mutation of the L1038 residue into an alanine drastically reduced insulin-induced Grb14 recruitment (−55%, P < 0.05), whereas the L1045A mutation enhanced it by 2.5-fold (P < 0.05), and the I1042A mutation had no effect (Fig. 1A). The increase in the formation of the IR-Grb14 L1045A complex could also be detected in the basal state (P < 0.05). Because mutations of the αC-helix residues did not impair insulin-induced autophosphorylation of the receptors, alteration in mutant receptor activation did not account for the differences observed (Fig. 1A). These data thus give clear evidence that the IR αC-helix is involved in Grb14 binding.
Fig. 1.

Effects of mutations of the insulin receptor on its interaction with Grb14. Grb14 and IR (WT or mutants) were transiently transfected in COS cells, and after treatment with insulin (100 nm) for 10 min, immunoprecipitations were performed with an anti-IR antibody. Antibodies used for Western blotting are shown on the right side of the blots. The blots shown are representative of three independent experiments. The autoradiograms were quantified by scanning densitometry, and the means ± sd are represented as histograms. *, P < 0.05. AU, Arbitrary units. A, Effect of mutation of residues L1038, I1042, and L1045A of the IR αC-helix on Grb14-IR interaction; B, effect of mutation of the K1168 residue situated in the IR catalytic loop on Grb14-IR interaction.
The crystal structure of the BPS bound to the activated IRK also revealed that residues F381, R387, and E394 of Grb14 were all in close proximity to the IR K1168 residue, suggesting that this lysine could be involved in the formation of the complex (13). K1168 is situated in the receptor activation loop, which is subjected to a major conformational change upon autophosphorylation (17). As shown in Fig. 1B, the IR K1168A mutant displayed a reduced ability (−50%, P < 0.05) to bind to Grb14 in the presence of a conserved insulin-induced autophosphorylation, providing evidence for its role in the IR interaction with Grb14.
All together, these results demonstrate that in addition to the substrate peptide binding groove of the kinase, various IRK domains may also be important for the interaction with Grb14.
Role of the R385 residue in the inhibitory effect of Grb14 on insulin signaling
In addition to the interaction of the pseudosubstrate region, the crystallographic study of the BPS/IRK complex identified R387 in Grb14 (R387 in the human sequence, corresponding to R385 in the rat sequence) as a key residue stabilizing the conformation of this region (13). As mentioned above, R387 is also in close proximity to the K1168 IR residue. To analyze its involvement in IR binding and insulin signaling, we substituted an asparagine for arginine 387. This mutant was designed as R385N in our study because we used the rat sequence. The interaction between this R385N mutant of Grb14 and the IR was first analyzed in co-IP experiments. As shown in Fig. 2A, the R385N mutation almost suppressed the effect of insulin on Grb14/IR interaction. To monitor the dynamics of this interaction in living cells, we used the BRET technique. In the absence of insulin, only a very low BRET signal was detected between IR-luc and WT Grb14-YFP, but insulin rapidly increased this signal, as previously described (19). The R385N mutation induced a 40% decrease in the insulin-induced BRET signal (Fig. 2B), thus providing additional evidence for a role of this residue in the formation of the Grb14/IR complex. To gain insight into the apparent discrepancy between the absence of IR interaction of the R385 Grb14 mutant as tested in co-IP experiments and the decreased interaction of this mutant in the BRET assay in living cells, we performed in vitro BRET experiments. Lysates prepared from IR-luc transfected cells stimulated or not with insulin and from Grb14-EYFP transfected cells were incubated together for 30 min. After addition of coelenterazine, we detected a BRET signal when WT Grb14 lysates were incubated with lysates from insulin-stimulated IR-luc-expressing cells and not from unstimulated cells (supplemental Fig. S1, published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). In contrast, no BRET signal was detected when stimulated IR-luc was incubated with the R385N Grb14 mutant. All together, these data suggest that the R385N mutation drastically reduces the affinity of Grb14 for the IR.
Fig. 2.

Role of the R385 residue of Grb14 on Grb14-IR interaction and on insulin action. A, Grb14 (WT or R385N mutant) and IR were transiently transfected in COS cells and co-IP as described in Fig. 1. The blot shown is representative of three independent experiments. The autoradiograms were quantified by scanning densitometry, and the means ± sd are represented as histograms. ***, P < 0.001. B, Dynamics of insulin-induced interaction between Grb14-EYFP (WT or R385N mutant) and IR-Rluc in HEK293T cells measured by BRET. A representative experiment is shown in the upper panel; lower panel, quantification of insulin-stimulated BRET at time 20 min. Data are expressed as percentage of BRET induced in the presence of WT Grb14. Results are means ± sd of three independent experiments. ***, P < 0.001. C, Effect of Grb14 (WT or R385N mutant) on insulin-induced X. laevis oocyte maturation. Oocytes were injected with increasing amounts of purified GST-Grb14 fusion proteins 1 h before insulin stimulation and analyzed 20 h later for the appearance of GVBD. Results are means ± sd of three independent animals, each concerning 20 oocytes.
To determine whether the alteration of Grb14 binding to the IR was accompanied by modifications in the action of insulin, we took advantage of the Xenopus oocyte model. Xenopus oocytes are physiologically arrested at the G2 stage of the first meiotic prophase, and their entry into the M phase, leading to maturation [germinal vesicle breakdown (GVBD)], can be induced by insulin (20). This model presents several advantages: 1) it is a physiological in vivo system, 2) the amount of microinjected protein can be carefully controlled in all cells, 3) it is an exquisitely sensitive system due to the all-or-none response of oocyte maturation (21). This model has previously been used to investigate Grb14 inhibitory action on insulin signaling (16, 22). Purified GST-Grb14 or GST-Grb14-R385N proteins were microinjected into oocytes 1 h before stimulation with insulin (10−6 m). Microinjection of increasing amounts of GST-Grb14 showed that whereas 15 ng had no effect, 20 ng Grb14 totally blocked insulin-induced oocyte maturation. In contrast, 90 ng of the R385N mutant was needed to inhibit the effect of insulin on the GVBD (Fig. 2C). This result suggests that the impairment in the formation of the Grb14-IR complex due to the R385N mutation prevented Grb14 inhibition of insulin action.
The S370 residue modulates the Grb14 inhibitory effect on insulin signaling
We previously demonstrated that Grb14 was phosphorylated by protein kinase Cζ (PKCζ) and that this phosphorylation improved its inhibitory effect on insulin signaling (22). In addition, the crystal structure of the BPS/IRK complex showed that S372 (S372 in the human sequence corresponding to S370 in the rat one) could interact with basic residues nearby in IRK (Hubbard, S., personal communication), and that the −3 residue is an arginine (R369), thus making a quasi-PKC consensus site. To investigate whether phosphorylation of S370 could modulate the effect of Grb14 on IR binding and insulin action, we mutated this residue into an alanine or an aspartic acid residue that, respectively, precluded phosphorylation (S370A) or mimicked a constitutive phosphorylation (S370D). As shown in Fig. 3A, in co-IP experiments, the Grb14 S370A mutant displayed an impaired capability to bind to the insulin-activated IR (−35%, P < 0.01), whereas the interaction with the S370D mutant was about 1.5-fold increased (P < 0.001). Insulin-induced Grb14 recruitment to the IR was also monitored using the BRET technique. Intriguingly, no difference was observed in the induction by insulin of Grb14-IR binding, either in the kinetics of the recruitment or in the maximal effect (Fig. 3B). Similar data were obtained using an in vitro BRET assay, in which the interaction between IR and Grb14 was assessed by incubating cell extracts containing Grb14-EYFP with cell extracts containing tyrosine-phosphorylated IR-luc (supplemental Fig. S2). This suggests that the discrepancy between the co-IP and BRET data are unlikely to be due to more stringent in vitro conditions in co-IP experiments. To further investigate the role of this site in the interaction between Grb14 and the IR, we mutated the IR R1092 residue, which was close to S370 in the crystal structure of the complex (13). The IR R1092A mutant was unable to bind to Grb14 in co-IP experiments (data not shown). However, this mutation also suppressed the effect of insulin on receptor autophosphorylation, thus precluding unambiguous interpretation of the consequence of the mutation on the formation of the complex. Then, to investigate the inhibitory effect of the Grb14 S370 mutants on IR tyrosine kinase activity, we transfected COS cells with expression vectors for IR, IRS-1, and WT or mutant Grb14. As shown in Fig. 3C, in the presence of either S370A or S370D mutant IRS-1, tyrosine phosphorylation was totally blocked, as it was with WT Grb14. This suggests that the S370 substitution did not reduce Grb14 inhibitory action on IR tyrosine kinase activity.
Fig. 3.
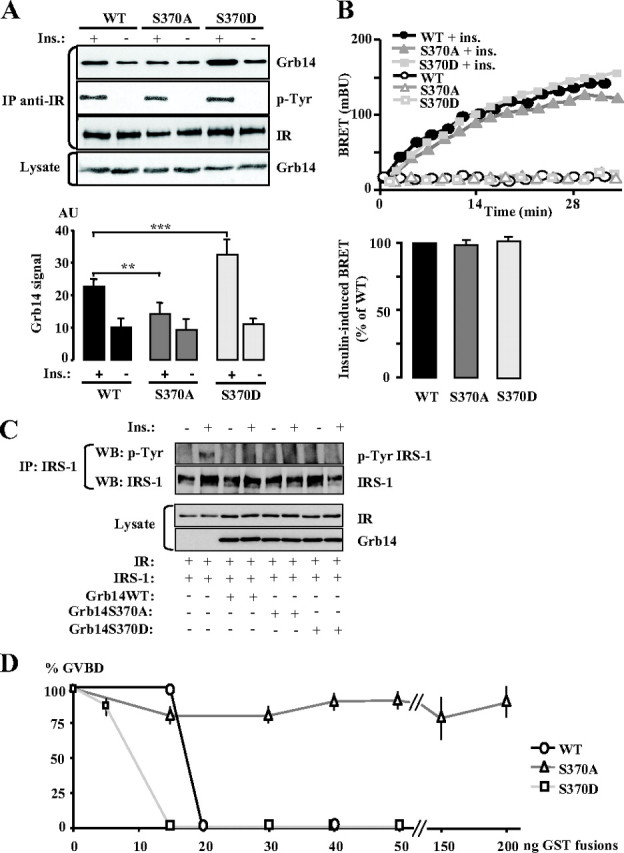
Consequences of the phosphorylation status of Grb14 S370 residue on Grb14-IR interaction and on insulin action. A, Grb14 (WT, S370A, or S370D mutants) and IR were transiently transfected in COS cells, and co-IP as described in Fig. 1. The blot shown is representative of five independent experiments. The autoradiograms were quantified by scanning densitometry, and the means ± sd are represented as histograms. ***, P < 0.001. B, Dynamics of insulin-induced interaction between Grb14-EYFP (WT or S370 mutants) and IR-Rluc in HEK293T cells measured by BRET. A representative experiment is shown in the upper panel; lower panel, insulin-stimulated BRET at time 20 min. Data are expressed as percentage of BRET induced in the presence of WT Grb14. Results are means ± sd of four independent experiments. C, COS cells were transiently transfected with the indicated expression vectors and stimulated or not with 100 nm insulin for 10 min. The tyrosine phosphorylation state of IRS-1 was analyzed by Western blotting after immunoprecipitation of the protein. D, Effect of Grb14 (WT or S370 mutants) on insulin-induced X. laevis oocyte maturation. Insulin-induced maturation of oocytes injected with increasing amounts of purified GST-Grb14 fusion proteins was performed as described in Fig. 2C. Results are means ± sd of three independent animals, each concerning 20 oocytes.
The biological activity of Grb14 S370 mutants was then investigated in the Xenopus oocyte system. As shown in Fig. 3D, Grb14 inhibition of insulin-induced oocyte maturation was almost abolished by the nonphosphorylatable S370A mutant. Inversely, the S370D mutant more efficiently inhibited the effect of insulin on the GVBD, because oocyte maturation was totally suppressed when 15 ng of this mutant was injected, whereas 20 ng was needed with the WT protein. These data suggest that the phosphorylation state of the S370 residue can modulate the inhibitory effect of Grb14 on insulin signaling.
Role of the Grb14-PDK1 interaction in the inhibitory action of Grb14 on insulin signaling
Grb14 is an adapter protein supposed to interact with other partners in addition to the IR. Among them, it was reported that PDK1 interacted constitutively with the E193 residue of Grb14 and that this interaction was required for a correct activation of Akt in response to insulin (14). It was thus interesting to investigate in our models the consequences of the E193A mutation on Grb14 activity. We first verified Grb14’s ability to bind to PDK1 and the implication of the E193 residue in this interaction in Xenopus oocytes. GST-Grb14 WT and E193A mutant were microinjected in Xenopus oocytes, which were or not stimulated with insulin. Grb14 was immunoprecipitated, and the association with endogenous PDK1 was analyzed by Western blot. In the absence of insulin, PDK1 did not bind to WT or mutant Grb14. However, 5 min after the addition of insulin, the formation of a Grb14-PDK1 complex could be detected, and substitution of the E193 residue of Grb14 abolished this recruitment (Fig. 4A). This result thus confirms that PDK1 interacts with Grb14 and further demonstrates that this interaction is induced by insulin in the oocytes.
Fig. 4.
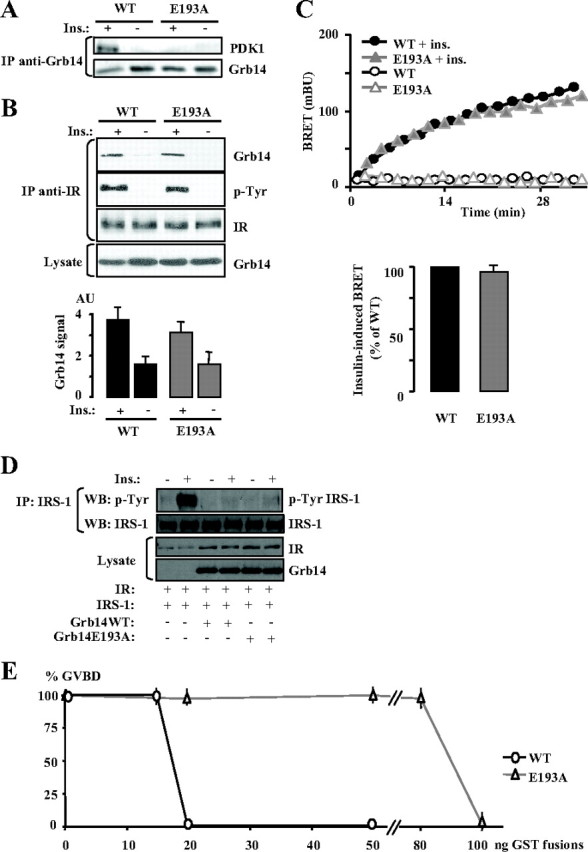
Functional consequences of the mutation of the Grb14 interaction site with PDK1. A, Insulin stimulation of the Grb14-PDK1 interaction. X. laevis oocytes were microinjected with GST-Grb14 WT or E193A mutant and stimulated with insulin for 5 min. Lysate from 20 oocytes was immunoprecipitated using an anti-Grb14 antibody and immunodetected with the indicated antibodies. The blot presented is representative of three independent experiments. B, Grb14 (WT or E193A mutant) and IR were transiently transfected in COS cells, and co-IP as described in Fig. 1. The blot shown is representative of five independent experiments. The autoradiograms were quantified by scanning densitometry, and the means ± sd are represented as histograms. C, Dynamics of insulin-induced interaction between Grb14-EYFP (WT or E193A mutant) and IR-Rluc in HEK293T cells measured by BRET. A representative experiment is shown in the upper panel; lower panel, insulin-stimulated BRET at time 20 min. Data are expressed as percentage of BRET induced in the presence of WT Grb14. Results are means ± sd of three independent experiments. D, COS cells were transiently transfected with the indicated expression vectors and stimulated or not with 100 nm insulin for 10 min. The tyrosine phosphorylation state of IRS-1 was analyzed by Western blotting after immunoprecipitation of the protein. E, Effect of Grb14 (WT or E193A mutant) on insulin-induced X. laevis oocyte maturation. Insulin-induced maturation of oocytes injected with increasing amounts of purified GST-Grb14 fusion proteins was performed as described in Fig. 2C. Results are means ± sd of three independent animals, each concerning 20 oocytes.
Immunoprecipitation experiments performed in transiently transfected cells, as well as BRET assays, showed that insulin-induced Grb14-IR interaction was not altered by the E193A mutation (Fig. 4, B and C). This result was anticipated because the PDK1 binding motif is situated in the N-terminal domain of Grb14, outside of the BPS and SH2 domains responsible for IR binding. The inhibitory effect of the Grb14 E193A mutant on IR tyrosine kinase activity was further investigated in COS cells transfected with expression vectors for IR, IRS-1, and WT or mutant Grb14. As shown in Fig. 4D, IRS-1 tyrosine phosphorylation was similarly inhibited in the presence of WT Grb14 or the E193A mutant. This indicates that the E193A mutant, which binds correctly to the activated IR, also exerts its pseudosubstrate inhibitory action on the IR kinase activity. The consequences of the suppression of PDK1 recruitment on Grb14 were then investigated in Xenopus oocytes. Interestingly, the Grb14 E193A mutant displayed an impaired ability to inhibit insulin-induced GVBD because a 5-fold greater amount was required to block the GVBD as compared with the WT protein (Fig. 4E). This suggests that the interaction with PDK1 was required for the inhibitory action of Grb14. This is the first evidence that inhibition by Grb14 of IR tyrosine kinase is not sufficient to inhibit insulin signaling.
Functional consequences of Grb14 mutations
A useful approach to gain more insights into the biological consequences of the different Grb14 mutations is to perform functional complementation by measuring proliferation rate in Grb14-deficient cells transfected with the different Grb14 mutants. MEF were obtained from Grb14 WT and KO mice, and the proliferation rate of these cells was measured in the presence of 100 nm insulin or 10% fetal bovine serum (FBS). In both conditions, the proliferation rate of KO MEF was significantly increased when compared with WT MEF (+33 and +80% in insulin and FBS, respectively) (Fig. 5, A and B, left). However, when cultured in the presence of insulin alone, the MEF proliferation rate was much lower than in the presence of FBS (−38 and −55% for WT and KO cells, respectively), and cells stopped growing after 48 h (data not shown). The functional complementation assays were therefore performed in KO MEF cultured in the presence of 10% FBS, the results reflecting the effect of Grb14 on signaling by growth factors present in the serum, including insulin. To establish that the increase in cell proliferation rate of KO MEF was due to the absence of Grb14, KO cells were transfected with a plasmid allowing the expression of a Grb14-EYFP fusion or of EYFP as a negative control. Transfected cells were sorted by flow cytometry to be plated at the same cell density, and proliferation was measured 48 h after plating. As shown in Fig. 5B (right panel), in KO MEF, the increase in cell number measured after 2 d culture was not altered by the expression of EYFP, whereas it was inhibited by 80% by the expression of Grb14-EYFP. As a result, cell proliferation was similar in WT cells and in KO cells rescued by the expression of Grb14. These data thus demonstrate that Grb14 exerts a strong inhibitory action on MEF cell proliferation.
Fig. 5.

Effects of Grb14 mutants on MEF proliferation. MEF proliferation rate was measured using a cell proliferation assay as described in Materials and Methods. Absorbance (OD) at 570 nm is proportional to the number of cells. Black bars, Cell number at d 0; gray bars, cell number after 2 d culture. Results are means ± sd of three independent experiments. **, P < 0.01; ***, P < 0.001 when compared with WT. A, Cell proliferation of WT MEF or MEF KO for Grb14 cultured in the presence of insulin (100 nm). B, Cell proliferation of MEF cultured in the presence of 10% FBS; left, cell proliferation of WT or KO MEF; right, cell proliferation of KO MEF transiently transfected with Grb14-EYFP or pEYFP control vectors (Ctrl). C, Cell proliferation of MEF KO for Grb14 cultured in the presence of 10% FBS and transiently transfected with plasmids coding for Grb14-EYFP (WT or mutants) or with empty vector (Ctrl) as indicated.
To evaluate the biological activity of the various Grb14 mutants, we then performed complementation studies in KO MEF using Grb14-EYFP mutant constructs (Fig. 5C). After 2 d culture, the cell number was increased by 110% in KO MEF expressing the Grb14 R385N mutant, as compared with the 35% increase (P < 0.001) in KO MEF rescued with WT Grb14, showing that the R385N mutation significantly blunted the inhibitory effect of Grb14 on cell proliferation. However, cells expressing this mutant still proliferated more slowly than KO cells (162% increase for KO vs. 110% for R385N, P < 0.01), indicating that Grb14 inhibitory effect was not completely abolished by this mutation.
MEF KO cells expressing the S370D Grb14 mutant displayed a very low proliferation rate (13%, vs. 35% with Grb14 WT, P < 0.01), demonstrating that mimicking constitutive phosphorylation of this residue significantly enhanced the inhibitory action of Grb14 on cell division. On the other hand, MEF KO cells expressing the S370A mutant had an enhanced proliferation rate when compared with WT Grb14 (53%, P < 0.01), although it remained much lower than in control KO cells (P < 0.001). These data further suggest that the phosphorylation status of the S370 residue may affect the inhibitory action of Grb14 on cell growth.
Finally, we investigated the consequences of the mutation of the PDK1 binding motif on the efficiency of Grb14 to inhibit cell proliferation. As shown in Fig. 5C, KO cells expressing the Grb14 E193A mutant displayed a proliferation rate significantly higher than KO MEF rescued by WT Grb14 (54 vs. 35%, P < 0.001), showing that PDK1 recruitment is an important component of the inhibitory mechanism of Grb14. However, control KO cells still divided much more rapidly (162 vs. 54%, P < 0.001), providing further evidence that additional molecular mechanisms contribute to the control of cell division by Grb14.
Discussion
Among the Grb7 family of adapters, Grb14 is likely to be particularly involved in the regulation of insulin signaling, as assessed by its tissue expression (8, 11), by its inhibitory effect on IR catalytic activity (12), by its co-internalization with the IR in insulin-stimulated rat liver (10), and by the phenotype of mice deficient for the Grb14 gene (9). Although the elucidation of the crystal structure of the complex between the tyrosine kinase domain of the IR and the BPS region of Grb14 demonstrated that Grb14 acts as a pseudosubstrate inhibitor of the kinase (13), information is still lacking regarding the molecular mechanisms by which full-length Grb14 inhibits insulin action. To progress in the elucidation of these molecular mechanisms, based on information available from the crystal structure of the BPS/IRK complex and on Grb14 activity, we designed site-directed mutations on IR and Grb14 and investigated the biological consequences of these mutations on the formation of the IR-Grb14 complex and on insulin action. This study allowed us to identify additional interaction sites between Grb14 and the IR and to evaluate the relative impact of IR binding vs. downstream effects in the Grb14 mechanism of action on insulin signaling. In addition, it provided evidence for a role of Grb14 in cellular proliferation.
Upon autophosphorylation and activation of the IRK, two domains are subjected to major conformational changes: the activation loop of the kinase, which bears the three phosphorylated tyrosine residues Y1158, Y1162, and Y1163, and the αC-helix (amino acids 1038–1054) whose residues become more solvent exposed (17). The structure of the complex showed that the α1 helix of the Grb14 BPS interacts with residues of the IR αC-helix and, more particularly, that residues L404 and W406 of Grb14 make Van der Waals contacts with L1038 and I1042, respectively (13). In this study, we showed that mutation of the L1038 residue induced a striking decrease in insulin-induced Grb14 binding, thus confirming the involvement of the αC-helix in the formation of the Grb14-IR complex. In addition, this result is in agreement with the decreased complex formation induced by the L404Q substitution reported by Depetris et al. (13). The role of this IR L1038-Grb14 L404 interaction is likely to be particularly relevant for the selectivity of the IR-Grb14 complex formation, because these two residues are not conserved in the related proteins IGF-I receptor and Grb7/10. However, additional data including the substitution of IR L1038 with the corresponding IGF-I receptor residue are necessary to substantiate this hypothesis. The IR I1042 mutant was not altered in its ability to recruit Grb14 under insulin stimulation. These data apparently contrast with the reduced binding ability of the W406A mutant (13). However, the W406 residue also binds to the phosphorylated Y1163 residue, suggesting that its interaction with the IR activation loop could prevail over its interaction with the αC-helix. The increased complex formation observed with the L1045 mutant, which occurred both in the basal state and after insulin stimulation, suggests that this substitution enhanced Grb14 affinity for the IR. The crystal structure of the BPS/IRK complex does not afford sufficient information to explain this intriguing modification. Indeed, L1045 also makes Van der Waals contacts with Y984, a residue very close to the N-terminal end of the crystallized molecule. No structural information is yet available to correctly interpret the interactions occurring between the N terminus of the IRK domain and the C terminus of the BPS domain that are cocrystallized. All together, these results give evidence that the IR αC-helix directly interacts with the C-terminal end of the crystallized BPS region (amino acids 361–419). To further investigate whether the short noncrystallized region situated between the BPS and the SH2 domain could play a role in the binding to the IR, we mutated the P420 residue, because proline mutations cause major structural changes. As tested by co-IP and BRET experiments, the Grb14 P420A mutant was not affected in its IR binding ability, suggesting that C-terminal noncrystallized region of the PIR-BPS region is unlikely to significantly contribute to the interaction between Grb14 and the IR (data not shown).
From the crystal structure of the IR-Grb14 complex, it appeared that the R385 residue was likely to be implicated in the formation or the stability of the Grb14-IR complex, because 1) it is salt bridged to the Grb14 D380 and E394 residue, 2) together with D380 and E394 it is in close proximity to the IR K1168 residue, and 3) E394 is salt bridged to K1168 (13). Mutation of R385 could thus either destabilize the BPS structure by suppressing the salt bridge with D380 or decrease the interaction with the IR. In this study, we showed that both the Grb14 R385N mutant and the IR K1168A mutant displayed impaired ability to interact in response to insulin. The decreased IR-Grb14 complex formation due to these mutations thus highlights another important interaction point between the two proteins, implicating the activation loop of the receptor and a residue situated outside of the pseudosubstrate motif of Grb14. In BRET experiments performed in intact cells, the interaction between IR and the Grb14 R385N mutant was also reduced, albeit to a lower extent than in co-IP experiments. In living cells, the recruitment of Grb14 at the plasma membrane immediately after insulin stimulation is likely to mobilize a multiprotein complex containing Grb14 and also other proteins including cytoskeleton proteins, allowing us to target Grb14 in close proximity to the IR. The substitution of the R385 residue, by reducing the affinity of Grb14 for the IR, could destabilize the IR-Grb14 complex without completely disrupting it. The biological activity of the Grb14 R385N mutant was markedly impaired when compared with WT Grb14, because a 4.5-fold higher amount was required to inhibit Xenopus oocyte maturation under insulin induction. As measured in the MEF KO proliferation assay, the R385N mutation strikingly decreased the inhibitory effect of Grb14, because the increase in cell number in the proliferation assay was 3.2-fold higher in KO cells transfected with this mutant compared with WT Grb14. On the other hand, when compared with control KO cells, expression of the R385N mutant significantly decreased cell proliferation by 30%, suggesting that it still can exert an inhibitory action in this cellular model. Several hypotheses can be raised to explain this residual activity: 1) as shown by BRET experiments, the R385N mutant still binds, albeit with lower efficiency, to the activated IR and can thus inhibit IR kinase activity; 2) Grb14 also exerts its action by interfering with signaling steps distal from the IR that are unlikely to be altered by the mutation (14, 15); and 3) the MEF proliferation assay was performed in the presence of 10% fetal calf serum, and we cannot exclude that Grb14 also interferes with other growth factor receptor signaling pathways that could be differently altered by the R385N mutation (23).
We previously reported that Grb14 was a target for PKCζ phosphorylation and that the phosphorylated form displayed an increased inhibitory action on insulin signaling (22). We mutated the S370 residue, because it was proposed to be a good candidate for phosphorylation (13). Interestingly, compared with WT Grb14, the S370D mutant displayed an increased inhibitory effect on insulin-induced oocyte maturation as well as an increased inhibition of KO MEF proliferation, whereas conversely the nonphosphorylatable S370A mutant was less potent than WT Grb14 to decrease cell proliferation and was unable to alter oocyte maturation. These data argue in favor of a regulatory role of S370 phosphorylation status on Grb14 biological activity. The consequences of the S370 substitution on IR binding are less clear, because co-IP experiments suggest that phosphorylation of this residue improves the formation of the IR-Grb14 complex, but this was not supported by the BRET experiments. We verified that this discrepancy was not due to the presence of the YFP fusion, because the mutated Grb14-YFP fusion proteins displayed similar modifications in their IR binding ability in co-IP experiments. On the other hand, S370 substitution did not alter insulin-induced IR catalytic activity, as assessed by IRS-1 tyrosine phosphorylation level, suggesting that Grb14 S370 mutants bound similarly to the activated IR. Deciphering the role of the S370 residue in IR binding would therefore require additional experiments, avoiding overexpression that could favor the detection of a protein complex with reduced affinity. Thus, although we cannot clearly distinguish whether this phosphorylation modifies IR binding or the binding to another protein acting after the formation of the IR-Grb14 complex, our data suggest that phosphorylation of the Grb14 S370 residue may play a regulatory role on the inhibitory action of the protein.
An important aspect for understanding the Grb14 inhibitory effect on insulin signaling is the appraisal of the role of the interaction with the IR. This prompted us to analyze the biological function of a Grb14 mutant that normally bound the IR. We chose to substitute the E193 residue, which was reported to mediate PDK1 binding (14). We confirmed that the mutation of this residue suppressed PDK1 interaction and also provided new evidence that the formation of the Grb14-PDK1 complex is induced by insulin stimulation. This contrasts with a previous study reporting a constitutive interaction between the two proteins (14). This discrepancy could be due to the overexpression of both Grb14 and PDK1 in the first study, which may artificially force their interaction, whereas in the present work, we studied Grb14 binding to endogenous PDK1. In Grb14-injected oocytes, PDK1 was undetectable in anti-Grb14 immunoprecipitates performed in the absence of insulin, but after only 5 min exposure to insulin, the complex was formed. This suggests that in the basal state, the binding sites are not accessible and that insulin signaling triggers a conformational change of the proteins that enables their interaction. Surprisingly, the Grb14 E193A mutant, which was normally recruited by the activated IR, had a much lower potency to inhibit insulin signaling in Xenopus oocytes than WT Grb14. Thus, PDK1 recruitment is likely to be an important component of the Grb14 inhibitory mechanism. This was confirmed by the complementation experiments performed in KO MEF, showing that the Grb14 E193A mutant was less effective than WT Grb14 in reducing cell proliferation. Interestingly, the comparison between the proliferation rate of KO MEF reconstituted with WT Grb14 and KO MEF transfected with the E193A mutant allows us to estimate that about 20% of the inhibitory action of Grb14 is dependent on its interaction with PDK1.
In conclusion, the present study provides new important information on the molecular mechanism of action of Grb14 on insulin signaling. We have identified additional interaction sites in the Grb14-IR complex, including the IR αC-helix. Among them, the Grb14 L404-IR L1038 interaction site appears to be particularly interesting in the perspective of developing specific inhibitors of Grb14 action on insulin signaling, because it involves residues that are not conserved in related proteins. In addition, we have provided evidence that although IR binding represents a major mechanism implicated in the Grb14 inhibitory effect, interference with downstream effectors, including PDK1, is also an important aspect of the Grb14 molecular mechanism of action.
Materials and Methods
Reagents and antibodies
All chemicals were from Sigma-Aldrich (St. Louis, MO). Enzymes were from New England Biolabs (Beverly, MA). Oligonucleotides were from Invitrogen Corp. (Carlsbad, CA). Human recombinant insulin was from Novo-Nordisk (Stockholm, Sweden) (Actrapid, 100 IU/ml). Rabbit polyclonal anti-Grb14 was described previously (11). Polyclonal anti-IRβ antibody used for Western blotting and monoclonal anti-IRβ antibody used for immunoprecipitation experiments were from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-pTyr (phospho-tyrosine) was from Amersham (Piscataway, NJ), anti-IRS-1 was from Upstate (Lake Placid, NY), and anti-PDK1 was from Cell Signaling Technology (Beverly, MA).
Vector constructions
Grb14 cDNA was extracted by enzymatic digestion from the pECE-Grb14 vector (11) and introduced in the pGex-3X vector at the BamHI site and in the pEYFP-N1 vector between the XhoI and BamHI sites. pECE-Grb14 was used for transfection in COS-7 cells, pGex3X-Grb14 for GST fusion protein production, and pEYFP-N1-Grb14 for BRET and proliferation experiments. The peT vector (24) was used for IR transfection in COS-7 cells and the pcDNA3-IR-luc (19) for BRET experiments. All constructs were verified by DNA sequencing. The mutated codons are underlined.
Site-directed mutagenesis
The QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) was used following the manufacturer’s instructions. For Grb14 mutations, the following primers were used: R385N (5′-C TTC TCA GGT CAG AAG ACC AAC GTC ATA GAC AAC CCC-3′), S370A (5′-CCC ATG AGA AGC GTA GCA GAG AAT TCC CTA GTA GC-3′), S370D (5′-CCC ATG AGA AGC GTA GAC GAG AAT TCC CTA GTA GC-3′), and E193A (5′-AAT TAT GCC AAA TAT GCA TTT TTT AAG AAC CCA ATG-3′). Mutants of the insulin receptor were constructed with the following primers: L1038A (5′-C GAG TCA GCC AGT GCC CGA GAG CGG ATT GA-3′), I1042A (5′-C AGT CTC CGA GAG CGG GCT GAG TTC CTC AAT GAG-3′), L1045A (5′-GAG CGG ATT GAG TTC GCC AAT GAG GCC TCG GT-3′), and K1168A (5′-C CGG AAA GGG GGC GCG GGT CTG CTC CC-3′). All mutations were verified by DNA sequencing. The mutated codons are underlined.
Immunoprecipitation and Western blotting
COS-7 cells were cultured in DMEM with 1 g/liter d-glucose (GIBCO BRL, Carlsbad, CA), 10% fetal calf serum (GIBCO), 100 U/ml penicillin, and 50 μg/ml streptomycin. At 70% confluence, COS-7 cells were transfected with Lipofectamine (Invitrogen) following the manufacturer’s instructions. Two days after transfection, cells were serum deprived for 24 h and stimulated or not by 100 nm insulin for 10 min. Cells were then scraped in lysis buffer (20 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 30 mm sodium pyrophosphate, 50 mm NaF, 1% Triton X-100, 0.1% BSA, 1 μg/ml pepstatin A, 2 μg/ml leupeptin, 5 μg/ml aprotinin, 1 mm phenylmethylsulfonyl fluoride, and 1 mm orthovanadate), solubilized for 30 min at 4 C in lysis buffer, and centrifuged at 15,000 × g for 15 min at 4 C. Supernatant proteins were quantified, and 500 μg proteins were incubated overnight at 4 C with 1 μg monoclonal anti-IRβ antibody in the presence of protein G-agarose beads. The beads were washed three times in lysis buffer, and the resulting immunoprecipitates were subjected to SDS-PAGE analysis and immunodetected with the indicated antibodies. Immunoreactive bands were revealed using the ECL detection kit (Pierce, Rockford, IL). Autoradiograms were scanned and quantified using an image processor program (ImageJ). Quantifications of Western blots represent the immunoprecipitated Grb14 signal adjusted to the immunoprecipitated IRβ signal and to the total Grb14 signal.
BRET experiments
HEK293T cells were transfected in 12-well plates using FuGENE 6 (Roche, Indianapolis, IN) according to the manufacturer’s instructions. We transfected cells with pcDNA3-IR-luc (400 ng) together with WT or mutated pEYFP-N1-Grb14 (80 ng). The day after transfection, cells were transferred into 96-well microplates (PerkinElmer, Norwalk, CT). Twenty-four hours later, cells were serum starved in PBS for 15 min at 37 C, followed by addition of 2.5 μm of the substrate of Renilla luciferase (Coelenterazine h; Molecular Probes, Eugene, OR). After 15 min incubation at room temperature, 5 nm insulin was added, and light emission acquisition at 480 and 530 nm was immediately started using the Fusion Universal Microplate Analyzer (PerkinElmer). The recruitment of Grb14 to the activated insulin receptor was monitored for 36 min after the addition of insulin. The BRET signal was expressed in milli-BRET units, corresponding to the BRET ratio multiplied by 1000. The BRET ratio has been previously defined as the emission ratio 530/480 nm obtained when the two partners are coexpressed, corrected by the ratio 530/480 nm obtained for the luciferase-fused protein expressed alone (25, 26).
Xenopus oocyte studies
BL21-DE3 bacteria were transformed with the pGex-3X-Grb14 WT and mutated vectors, and GST fusion proteins were produced as described previously (27). Xenopus oocytes were handled and microinjected as described previously (16). GVBD in response to insulin was determined by the appearance of a white spot at the center of the animal pole of the oocytes. Results are expressed as percentages of GVBD 20 h after insulin stimulation. For Grb14 immunoprecipitations, oocytes were injected with 15 ng GST-Grb14 WT or 50 ng GST-Grb14 E193A purified proteins and were stimulated with insulin (10−6 m) for 5 min. Twenty treated oocytes were lysed in 200 μl buffer [25 mm MOPS (pH adjusted to 7.2), 60 mm β-glycerophosphate, 15 mm paranitrophenyl phosphate, 15 mm EDTA, 15 mm MgCl2, 2 mm dithiothreitol, 1 mm orthosodium vanadate, 1 mm NaF, 1 mm phenylphosphate, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor, and 10 μm benzamidine] added with 0.5% Triton X-100. Supernatants were precleared with protein A-agarose (Sigma) for 1 h at 4 C. Immunoprecipitations were performed by adding anti-Grb14 antibodies for 3 h and protein A-agarose for 1 h at 4 C. Immune complexes were analyzed by Western blotting (28).
Proliferation experiments
MEF were obtained from Grb14 KO mice embryos (9). Post-crisis MEF were cultured in DMEM with 4.5 g/liter d-glucose (GIBCO), 10% fetal calf serum (GIBCO), 100 U/ml penicillin, and 50 μg/ml streptomycin. MEF were transfected in 100-mm dishes using the polyethylenimine-adenofection technique described previously (29): 6 μg pEYFP-Grb14 vectors, 180 plaque-forming units per cell of Ad-RSV-nlsLacZ (Rous sarcoma virus promoter driving the nlsLacZ gene), and 75 μm polyethylenimine incubated with the cells for 4 h. Two days later, MEF expressing Grb14-EYFP were sorted by flow cytometry using the Epics ALTRA sorter (Beckman Coulter, Fullerton, CA). Similar levels of fluorescence were obtained after cell sorting, indicating that equivalent amounts of the various Grb14 mutant proteins were expressed. Cells were then plated at 1000 cells per well in 96-well plates and assayed using the CellTiter 96 nonradioactive cell proliferation assay (Promega, Madison, WI) following the manufacturer’s instructions.
Statistical analysis
Results are expressed as means ± sd. Variance equality was determined with a Fisher test, and statistical significance was determined using a Student’s t test.
Acknowledgments
We thank Dr. Marthe Rizk-Rabin for her help in proliferation experiments and the technical facilities of the Institut Cochin for flow cytometry analysis and DNA sequencing.
Footnotes
This work was supported by grants from the Agence Nationale de la Recherche (ANR-06-PHYSIO-014-01), the Association pour la Recherche sur le Cancer (ARC 3487) and “La Ligue Contre Le Cancer,” Comité du Nord, 2006, 2008. D.G. is the recipient of a doctoral fellowship from the Ministère de l’Enseignement Supérieur et de la Recherche. R.J.D. is the recipient of research grants from the National Health and Medical Research Council of Australia and Diabetes Australia Research Trust.
Disclosure Summary: The authors have nothing to disclose.
First Published Online April 9, 2009
Abbreviations: BPS, Between PH and SH2 domains; BRET, bioluminescence resonance energy transfer; co-IP, coimmunoprecipitation; FBS, fetal bovine serum; GVBD, germinal vesicle breakdown; IR, insulin receptor; IRK, IR tyrosine kinase domain; IRS, IR substrate; KO, knockout; MEF, mouse embryonic fibroblast; PDK1, phosphatidylinositol-dependent kinase 1; PIR, phosphorylated insulin receptor interacting region; PKCζ, protein kinase Cζ; WT, wild type.
References
- 1.Virkamäki A, Ueki K, Kahn CR1999. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest 103:931–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asante-Appiah E, Kennedy BP2003. Protein tyrosine phosphatases: the quest for negative regulators of insulin action. Am J Physiol Endocrinol Metab 284:E663–E670 [DOI] [PubMed]
- 3.Sleeman MW, Wortley KE, Lai KM, Gowen LC, Kintner J, Kline WO, Garcia K, Stitt TN, Yancopoulos GD, Wiegand SJ, Glass DJ2005. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat Med 11:199–205 [DOI] [PubMed] [Google Scholar]
- 4.Holt LJ, Siddle K2005. Grb10 and Grb14: enigmatic regulators of insulin action—and more? Biochem J 388:393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Howard JK, Flier JS2006. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab 17:365–371 [DOI] [PubMed] [Google Scholar]
- 6.Gual P, Le Marchand-Brustel Y, Tanti JF2005. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 87:99–109 [DOI] [PubMed] [Google Scholar]
- 7.Shen TL, Guan JL2004. Grb7 in intracellular signaling and its role in cell regulation. Front Biosci 9:192–200 [DOI] [PubMed] [Google Scholar]
- 8.Cariou B, Capitaine N, Le Marcis V, Vega N, Béréziat V, Kergoat M, Laville M, Girard J, Vidal H, Burnol AF2004. Increased adipose tissue expression of Grb14 in several models of insulin resistance. FASEB J 18:965–967 [DOI] [PubMed] [Google Scholar]
- 9.Cooney GJ, Lyons RJ, Crew AJ, Jensen TE, Molero JC, Mitchell CJ, Biden TJ, Ormandy CJ, James DE, Daly RJ2004. Improved glucose homeostasis and enhanced insulin signalling in Grb14-deficient mice. EMBO J 23:582–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desbuquois B, Béréziat V, Authier F, Girard J, Burnol AF2008. Compartmentalization and in vivo insulin-induced translocation of the insulin-signaling inhibitor Grb14 in rat liver. FEBS J 275:4363–4377 [DOI] [PubMed] [Google Scholar]
- 11.Kasus-Jacobi A, Perdereau D, Auzan C, Clauser E, Van Obberghen E, Mauvais-Jarvis F, Girard J, Burnol AF1998. Identification of the rat adapter Grb14 as an inhibitor of insulin actions. J Biol Chem 273:26026–26035 [DOI] [PubMed] [Google Scholar]
- 12.Béréziat V, Kasus-Jacobi A, Perdereau D, Cariou B, Girard J, Burnol AF2002. Inhibition of insulin receptor catalytic activity by the molecular adapter Grb14. J Biol Chem 277:4845–4852 [DOI] [PubMed] [Google Scholar]
- 13.Depetris RS, Hu J, Gimpelevich I, Holt LJ, Daly RJ, Hubbard SR2005. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Mol Cell 20:325–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.King CC, Newton AC2004. The adaptor protein Grb14 regulates the localization of 3-phosphoinositide-dependent kinase-1. J Biol Chem 279:37518–37527 [DOI] [PubMed] [Google Scholar]
- 15.Carré N, Caüzac M, Girard J, Burnol AF2008. Dual effect of the adapter Grb14 on insulin action in primary hepatocytes. Endocrinology 149:3109–3117 [DOI] [PubMed] [Google Scholar]
- 16.Cailliau K, Le Marcis V, Béréziat V, Perdereau D, Cariou B, Vilain JP, Burnol AF, Browaeys-Poly E2003. Inhibition of FGF receptor signalling in Xenopus oocytes: differential effect of Grb7, Grb10 and Grb14. FEBS Lett 548:43–48 [DOI] [PubMed] [Google Scholar]
- 17.Hubbard SR1997. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J 16:5572–5581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moncoq K, Broutin I, Larue V, Perdereau D, Cailliau K, Browaeys-Poly E, Burnol AF, Ducruix A2003. The PIR domain of Grb14 is an intrinsically unstructured protein: implication in insulin signaling. FEBS Lett 554:240–246 [DOI] [PubMed] [Google Scholar]
- 19.Nouaille S, Blanquart C, Zilberfarb V, Boute N, Perdereau D, Roix J, Burnol AF, Issad T2006. Interaction with Grb14 results in site-specific regulation of tyrosine phosphorylation of the insulin receptor. EMBO Rep 7:512–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El-Etr M, Schorderet-Slatkine S, Baulieu EE1979. Meiotic maturation in Xenopus laevis oocytes initiated by insulin. Science 205:1397–1399 [DOI] [PubMed] [Google Scholar]
- 21.Ferrell Jr JE, Machleder EM1998. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science 280:895–898 [DOI] [PubMed] [Google Scholar]
- 22.Cariou B, Perdereau D, Cailliau K, Browaeys-Poly E, Béréziat V, Vasseur-Cognet M, Girard J, Burnol AF2002. The adapter protein ZIP binds Grb14 and regulates its inhibitory action on insulin signaling by recruiting protein kinase Cζ. Mol Cell Biol 22:6959–6970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kairouz R, Parmar J, Lyons RJ, Swarbrick A, Musgrove EA, Daly RJ2005. Hormonal regulation of the Grb14 signal modulator and its role in cell cycle progression of MCF-7 human breast cancer cells. J Cell Physiol 203:85–93 [DOI] [PubMed] [Google Scholar]
- 24.Ellis L, Clauser E, Morgan DO, Edery M, Roth RA, Rutter WJ 1986 Replacement of insulin receptor tyrosine residues 1162 and 1163. compromises insulin-stimulated kinase activity and uptake of 2-deoxyglucose. Cell 45:721–732 [DOI] [PubMed] [Google Scholar]
- 25.Angers S, Salahpour A, Joly E, Hilairet S, Chelsky D, Dennis M, Bouvier M2000. Detection of β2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc Natl Acad Sci USA 97:3684–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boute N, Pernet K, Issad T2001. Monitoring the activation state of the insulin receptor using bioluminescence resonance energy transfer. Mol Pharmacol 60:640–645 [PubMed] [Google Scholar]
- 27.Kasus-Jacobi A, Perdereau D, Tartare-Deckert S, Van Obberghen E, Girard J, Burnol AF1997. Evidence for a direct interaction between insulin receptor substrate-1 and Shc. J Biol Chem 272:17166–17170 [DOI] [PubMed] [Google Scholar]
- 28.Cailliau K, Perdereau D, Lescuyer A, Chen H, Garbay C, Vilain JP, Burnol AF, Browaeys-Poly E2005. FGF receptor phosphotyrosine 766 is a target for Grb14 to inhibit MDA-MB-231 human breast cancer cell signaling. Anticancer Res 25:3877–3882 [PubMed] [Google Scholar]
- 29.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP1995. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA 92:7297–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]