

Abstract
Laminin-5-rich extracellular matrix derived from 804G cells (804G-ECM) induces spreading, improves glucose-stimulated insulin secretion, and increases survival and proliferation of rat pancreatic β-cells. The aim of the study was to determine growth signaling pathways activated by ECM with a particular focus on Ca2+-dependent transcription factors. 804G-ECM increased rat β-cell proliferation, and this stimulation was glucose and Ca2+ dependent. NF-κB nuclear translocation as well as IκBα gene expression were also Ca2+ dependent. Inhibition of NF-κB almost completely blocked 804G-ECM-stimulated β-cell proliferation as did the soluble IL-1 receptor antagonist IL-1Ra. 804G-ECM-induced proliferation was also blocked by cyclosporin A and the VIVIT peptide, suggesting involvement of nuclear factor of activated T cells (NFAT)/calcineurin. Use of selective inhibitors further implicated other pathways in this process. Inhibition of phosphatidylinositol 3-kinase and protein kinase A both prevented β-cell replication stimulated by 804G-ECM. Conversely, inhibition of MAPK, c-Jun N-terminal kinase, p38, and glycogen synthase kinase-3β increased β-cell proliferation on 804G-ECM. Our results suggest that Ca2+ entry, which is necessary for increased β-cell proliferation on 804G-ECM, is also involved in 804G-ECM-induced NF-κB activity. It is proposed that increased cytosolic Ca2+ leads to activation of the transcription factors NFAT and NF-κB that in turn increase β-cell proliferation. Activation of phosphatidylinositol 3-kinase by 804G-ECM also increases proliferation possibly by synergistic coactivation of NFAT via inhibition of glycogen synthase kinase-3β, whereas IL-1β may amplify the process by feed-forward activation of NF-κB. Conversely, inhibition of the MAPK pathway increased β-cell proliferation, indicating a counterregulatory restraining role for this signaling pathway.
Extracellular matrix increases proliferation of rat pancreatic beta cells in a glucose-dependent fashion by raising cytosolic Ca2+ and activating the transcription factors NFAT and NF-κB.
The extracellular matrix (ECM) is a complex structural entity surrounding cells within mammalian tissues that is able to regulate many essential cellular functions, including gene expression (1, 2), survival, development (3, 4), migration, proliferation, shape, and secretion (5).
Our group has reported that the laminin-5-rich ECM secreted by 804G cells (804G-ECM) has a beneficial effect on rat pancreatic β-cell survival and proliferation and that it activates intracellular signaling pathways involving the signaling proteins focal adhesion kinase (FAK), Akt/protein kinase B (PKB), and ERK (6, 7). Furthermore, it was found that plating rat β-cells on 804G-ECM induces transient nuclear translocation of nuclear factor (NF)-κB and its transcriptional activity, which is followed by overexpression of IκBα and NF-κB mRNAs (8). 804G-ECM also triggers the low-grade expression of cytokines, notably IL-1β, which is released and acts in an autocrine fashion via its receptor IL-1R to positively reinforce the activation of NF-κB and possibly its own expression in β-cells plated on 804G-ECM (9, 10).
It is well established that in β-cells glucose metabolism induces an increase in intracellular Ca2+ and we have shown that such an increase is also required for spreading of rat islet β-cells on extracellular matrix (11, 12). Furthermore, Ca2+ is an essential regulator of the cell cycle and the amplitude and duration of the Ca2+ response control gene expression in various cell types (12).
Recently, nuclear factor of activated T cells (NFAT) had been shown to regulate pancreatic β-cell growth and function (13). A sustained increase in cytosolic Ca2+ is necessary to dephosphorylate NFAT by calcineurin, a Ca2+/calmodulin-dependent phosphatase, and induce its translocation into the nucleus (14). Glycogen synthase kinase (GSK)-3β is a negative regulator of this signaling pathway because it phosphorylates NFAT and induces its export from the nucleus. Inactivation of GSK3β by phosphorylation on Ser9 by Akt/PKB is necessary to assure NFAT-dependent transcription (14).
We have now investigated the role of Ca2+-dependent signaling pathways in the stimulation of β-cell growth by ECM, with a particular focus on NF-κB and NFAT.
Results
ECM increases expression of cell cycle proteins
In confirmation of previous findings, after 2 d culture, 804G-ECM significantly increased proliferation of rat β-cells vs. poly-l-lysine (PLL) (control) using standard culture conditions [DMEM, 10% fetal calf serum (FCS), 11.2 mm glucose]: 1.8 ± 0.4 vs. 4.6 ± 0.5% of BrdU-positive β-cells on PLL vs. 804G-ECM, respectively (P = 0.006). 804G-ECM also significantly modified expression of proteins of the cell cycle. Indeed, expression of proliferating cell nuclear antigen (PCNA) and cyclin D1 were significantly increased in cells plated on 804G-ECM compared with cells on PLL (Fig. 1, A and B), whereas expression of cyclin-dependent kinase 4 (CDK4) and cyclin D2 was similar between cells attached on 804G-ECM or on PLL (Fig. 1, B and C).
Fig. 1.

Levels of PCNA and cyclin D1 protein are increased in β-cells cultured 48 h on 804G-ECM vs. PLL. A, Upper panel, representative Western blot for PCNA and actin (confirming equivalent protein loading); lower panel, quantification of band intensities (PCNA/actin); n = 3. *, P = 0.03 vs. PLL. B, Upper panel, representative Western blot for cyclin D1, CDK4, and actin (confirming equivalent protein loading); lower panel, quantification of band intensities (cyclin D1/actin and CDK4/actin); n = 4. *, P = 0.04 vs. PLL. C, Upper panel, representative Western blot for cyclin D2 and actin (confirming equivalent protein loading); lower panel, quantification of band intensities (cyclin D2/actin); n = 3.
ECM increased rat β-cell proliferation is glucose and calcium dependent
Spreading of β-cells on 804G-ECM is glucose dependent (11). We have now investigated β-cell proliferation at low (2.8 mm) or high (11.2 mm, standard) glucose for 24 h. At 2.8 mm glucose, rat β-cell proliferation was similar for cells on PLL and 804G-ECM (Fig. 2A). Increasing the glucose concentration (11.2 mm) significantly enhanced β-cell proliferation both on PLL and on 804G-ECM when compared with cells exposed to 2.8 mm glucose. However, a 6.8-fold increase in β-cell proliferation was observed with glucose on 804G-ECM against a 3.3-fold increase on PLL (Fig. 2A), indicating a synergistic or permissive effect of glucose on the increased proliferation of β-cells on ECM. As expected, the accumulation of insulin released to the medium during the 24-h culture period was significantly increased when cells were cultured with 11.2 mm glucose when compared with 2.8 mm (Fig. 2B). However, no significant difference was observed between PLL and 804G-ECM even at high glucose concentration. These results suggest that the beneficial effect of matrix on β-cell proliferation seen at 11.2 mm glucose is not simply due to an increase in insulin release of cells attached on 804G-ECM.
Fig. 2.
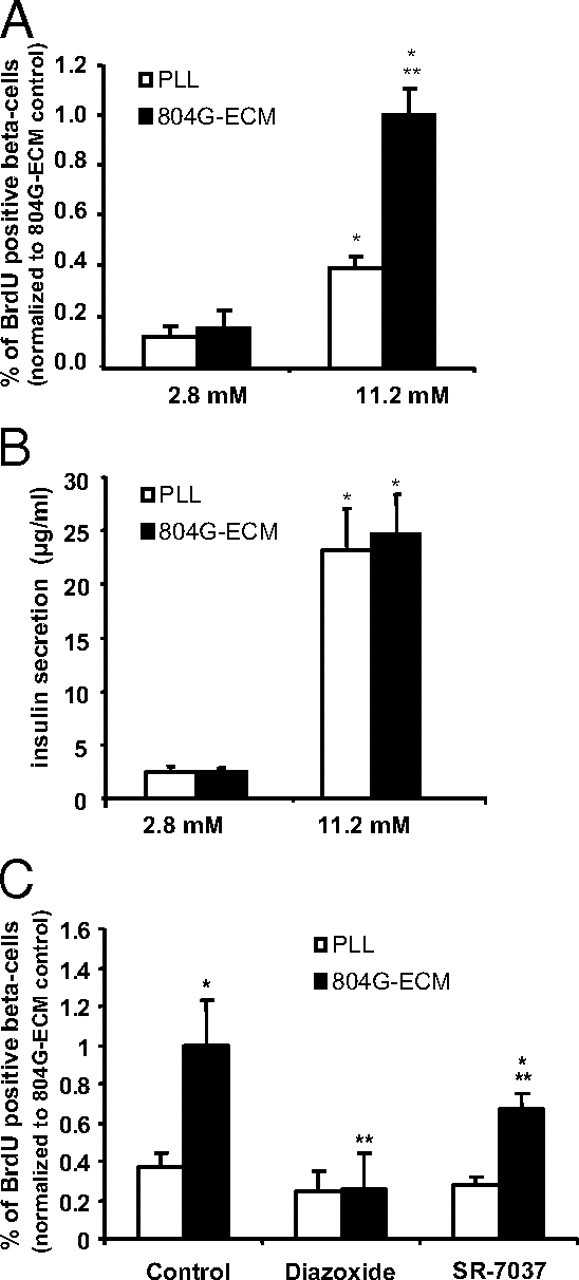
Increased proliferation of rat β-cells on 804G-ECM is glucose and calcium dependent. A, Rat β-cells were attached either on PLL or 804G-ECM and cultured in DMEM, 10% FCS with 2.8 or 11.2 mm glucose as indicated. After 24 h culture, cells were incubated for another 24 h in the presence of BrdU (10 μm). Immunofluorescence for BrdU (proliferation) and insulin (β-cells) was performed and the number of BrdU plus insulin double-positive β-cells expressed as a percentage of the total number of (insulin-positive) β-cells and then normalized to 804G-ECM 11.2 mm (absolute value, 3.63 ± 0.23). *, P < 0.01 vs. respective 2.8 mm glucose control; **, P = 0.01 vs. PLL at 11.2 mm glucose. B, Insulin secreted in the medium during 24 h culture was measured by RIA. *, P < 0.01 vs. respective 2.8 mm glucose control. C, Cells were cultured under standard conditions (11.2 mm glucose) as described for A but with addition of diazoxide (200 μm) or SR-7037 (1 μm), as indicated; n = 3–5. *, P < 0.05 vs. PLL control; **, P < 0.05 vs. 804G-ECM control (804G-ECM control absolute value, 2.74±0.63).
Glucose metabolism results in the production of ATP, with subsequent closure of KATP channels, membrane depolarization, and opening of voltage-operated Ca2+ channels. To investigate whether the KATP channel influences glucose-induced β-cell proliferation, we examined the effect of the sulfonamide diazoxide on β-cell proliferation. Diazoxide opens KATP channels, thereby preventing depolarization and Ca2+ entry. The KATP channel opener diazoxide decreased proliferation of β-cells attached to 804G matrix and abolished the difference between cells on PLL and 804G (Fig. 2C). To further address the role of calcium influx through the L-type voltage-gated Ca2+ channels in β-cell proliferation, these channels were blocked with the specific agent SR-7037. As shown in Fig. 2C, 1 μm SR-7037 decreased 804G-ECM-induced proliferation.
Activation of NF-κB by 804G-ECM is calcium dependent and is involved in rat β-cell proliferation
As previously shown (8), 804G-ECM increases significantly NF-κB nuclear translocation as well as IκBα gene expression, a target of NF-κB. Because Ca2+ signaling is known to be involved in activation of this pathway, we examined the impact of Ca2+ entry blockers on NF-κB activation. Treatment of β-cells with the KATP channel opener diazoxide or the L-type Ca2+ channel blocker SR-7037 prevented ECM-induced NF-κB nuclear translocation (Fig. 3A). Furthermore, ECM-induced IκBα gene expression was also significantly reduced in the presence of the two inhibitors of calcium influx (Fig. 3B). The inhibitor of NF-κB activation Bay 11-7082 (5 μm) blocked almost completely β-cell proliferation on 804G-ECM but also on PLL (Fig. 3C). Because IL-1β is a known target of NF-κB that is secreted from rat β-cells on ECM and appears to act on them in a positive feedback loop in this particular setting (9), we blocked the IL-1R with IL-1R antagonist (IL-1Ra) (2 μg/ml), and this indeed resulted in a significant decrease in β-cell proliferation on 804G-ECM (Fig. 3D).
Fig. 3.

Calcium-dependent NF-κB activity is involved in rat β-cell proliferation. A, Rat β-cells were fixed after 1 h exposure to PLL or 804G-ECM in presence or not of diazoxide (200 μm) or SR-7037 (1 μm), and NF-κB was detected by immunofluorescence. The number of cells with nuclear NF-κB localization was quantified as a percentage of total. *, P < 0.05 vs. PLL; **, P < 0.05 relative to control 804G-ECM (n = 3) with a minimum of 100 β-cells examined for each condition in each experiment. B, Cells were cultured on PLL or on 804G-ECM-coated dishes in the presence or not of inhibitors for 24 h; mRNA was extracted, and quantitative real-time RT-PCR was performed to quantify levels of IκBα mRNA vs. L3 mRNA (internal control). *, P < 0.05 vs. PLL; **, P < 0.05 vs. 804G-ECM (n = 6). C and D, Rat β-cells were attached either on PLL or on 804G-ECM in the presence or not of inhibitors of NF-κB (C, Bay11-7082 5 μm) or IL-1β (D, IL1-Ra 2 μg/ml). After an initial period of 24 h, cells were cultured with BrdU (10 μm) for another 24 h in continued presence of the inhibitors. β-Cell replication was determined as described in Fig. 2 (804G-ECM control absolute value, 2.87 ± 0.5 for C and 3.2 ± 0.8 for D); n = 3–4. *, P < 0.05 vs. PLL control; **, P < 0.05 vs. 804G-ECM control.
Analysis of the calcium signaling pathways involved in ECM-induced β-cell proliferation
NFAT signaling has been shown to regulate pancreatic β-cell growth and function (13). To determine the importance of the NFAT pathway on ECM-induced proliferation, we tested the effect of two agents known to act on this pathway (Fig. 4). Cyclosporin A (5 μm), an inhibitor of calcineurin, prevented ECM-induced β-cell proliferation without significantly affecting proliferation of cells plated on PLL (Fig. 4A). Furthermore, the direct inhibition of NFAT by VIVIT peptide partially inhibited 804G-ECM-induced β-cell proliferation (Fig. 4B), again with no impact on proliferation on PLL.
Fig. 4.
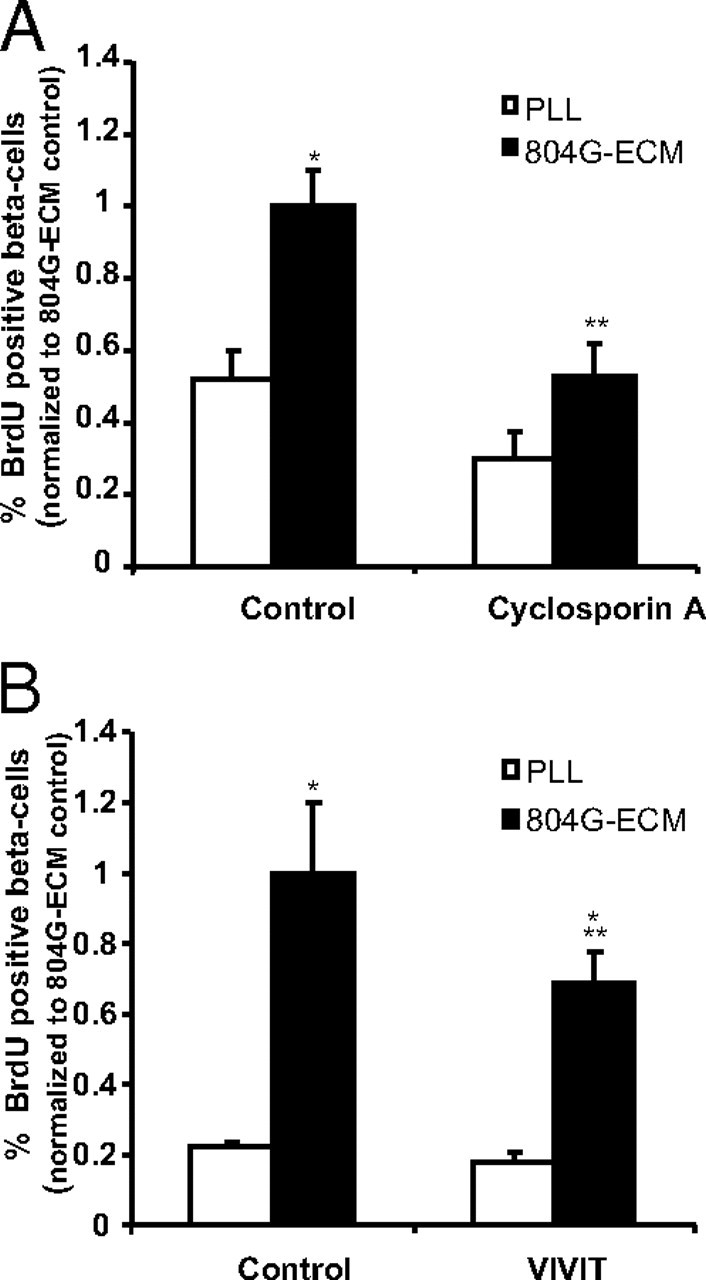
Analysis of the calcium signaling pathways involved in ECM-induced β-cell proliferation. Rat β-cells were attached on PLL or 804G-ECM in the presence or not of inhibitors of calcineurin (5 μm cyclosporin A) (A) or of NFAT (1 μm VIVIT) (B). After an initial period of 24 h, cells were cultured with BrdU (10 μm) for another 24 h in the continued presence of the inhibitors. β-Cell replication was determined as described in Fig. 2 (804G-ECM control absolute value, 3.21 ± 0.32 for A and 3.02 ± 0.88 for B); n =3–5. *, P < 0.05 vs. PLL control; **, P < 0.05 vs. 804G-ECM control.
Involvement of phosphatidylinositol 3-kinase (PI3K) and MAPK signaling pathways in 804G-ECM-induced β-cell proliferation
NF-κB and NFAT are also modulated by non-calcium-dependent pathways, including the MAPK and PI3K pathways. Furthermore, it was previously shown that 804G-ECM activates intracellular pathways in β-cells involving the signaling proteins Akt/PKB and ERK (8). Therefore, β-cells were cultured 48 h on PLL or on 804G-ECM in the presence of a PI3K inhibitor, LY294002, or a MAPK inhibitor, PD98059, during the last 24 h. Treatment of β-cells with LY294002 (50 μm) completed prevented increased proliferation on 804G-ECM (Fig. 5A) with no significant decrease on PLL. By contrast, addition of PD98059 for the last 24 h of culture had no effect on proliferation (Fig. 5B). To investigate the effects of inhibiting MAPK signaling for a longer period of time, various inhibitors were included in the culture medium throughout the 48-h culture period. Interestingly, with this longer period of inhibition of MAPK kinase (MEK)/ERK by PD98059, the percentage of bromodeoxyuridine (BrdU)-positive β-cells was significantly increased on 804G-ECM when compared with control (Fig. 5C). However, no effect of this MAPK inhibitor was observed on cells plated on PLL (Fig. 5C). Similar results were obtained when cells were incubated with SP600125 (10 μm), a c-Jun N-terminal kinase (JNK) inhibitor (Fig. 5D) and with SB20358 (10 μm), a p38 MAPK inhibitor (Fig. 5E). However, with this last inhibitor, the increase in proliferation was very high (4.3-fold vs. control on ECM) and was also observed on PLL (7-fold vs. control on PLL). These latter data suggest that when p38 MAPK is activated, it serves as a brake for β-cell proliferation and that this process is independent of the ECM effect. However, we observed no differences in phospho-p38 and phospho-JNK between cells plated on PLL or on 804G-ECM for 48 h (data not shown).
Fig. 5.
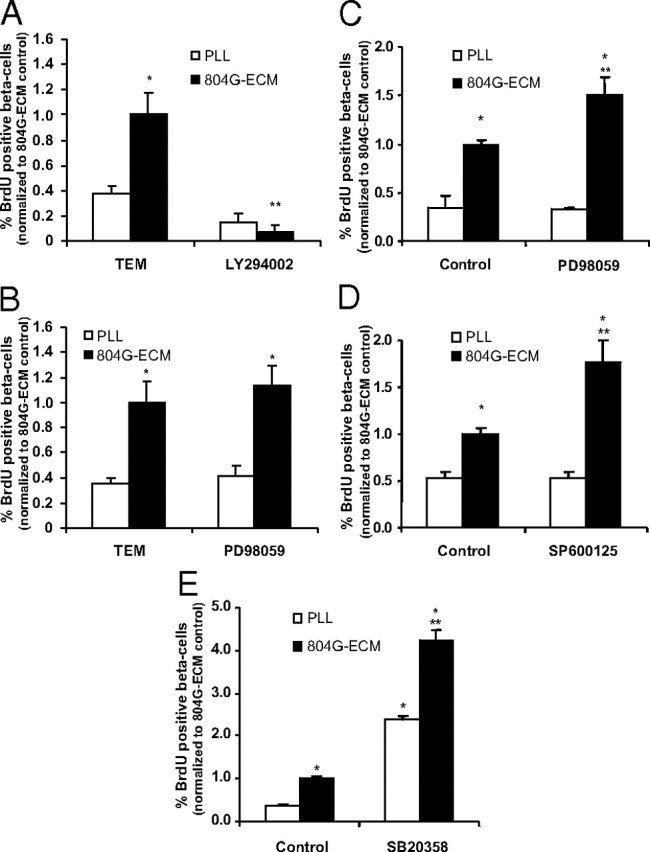
Involvement of Akt/PKB and MAPK pathways in 804G-ECM-induced β-cell proliferation. A and B, Rat β-cells were attached either on PLL or on 804G-ECM, and after an initial culture period of 24 h, cells were cultured with BrdU (10 μm) in presence of inhibitors of PI3K (50 μm LY294002) (A) or of MEK1 (50 μm PD98059) (B). C–E, Rat β-cells were attached either on PLL or on 804G-ECM in the presence or not of inhibitors MEK1 (50 μm PD98059) (C), JNK (10 μm SP600125) (D), or p38 (10 μm SB20358) (E). After 24 h, β-cell replication was determined as described in Fig. 2 (804G-ECM control absolute value, 2.35 ± 0.39 for A, 2.12 ± 0.35 for B, 3.57 ± 0.15 for C, 3.49 ± 0.23 for D, and 2.26 ± 0.38 for E); n =3–5. *, P < 0.05 vs. PLL control; **, P < 0.05 vs. 804G-ECM control.
Finally, we analyzed the effect of 804G-ECM on the PI3K/Akt signaling cascade. A 30-min attachment of cells on 804G-ECM-induced phosphorylation on Ser9 of GSK3β, a target of Akt (Fig. 6A), with no effect on the amount of total GSK3β. We also tested the effect of 6-bromoindirubin-3′-oxime (BIO), a GSK3β inhibitor on β-cell proliferation. Treatment of cells with BIO (1 μm) for 48 h significantly increased proliferation of cells plated on 804G-ECM but not on PLL (Fig. 6B). PKA is known to phosphorylate and thereby inhibit GSK3β. Inhibition of PKA should thus activate GSK3β and induce the opposite effects seen with its inhibition by BIO. Indeed, when PKA was inhibited by 48 h treatment with 10 μm H89, β-cell proliferation on 804G-ECM was significantly decreased (Fig. 6C).
Fig. 6.

Involvement of PI3K signaling in 804G-ECM-induced β-cell proliferation. A, Lysates of rat β-cells attached on PLL or 804G matrix for 30 min were immunoblotted with anti-phospho-GSK3β (Ser9) and anti-actin antibodies. Then, the immunoblot was stripped and reprobed for total GSK3β protein. Representative results are shown from one experiment. B, Rat β-cells were attached either on PLL or on 804G-ECM in the presence or not of inhibitors of GSK3 (1 μm BIO) (B) or of PKA (10 μm H89) (C). After an initial period of 24 h, cells were cultured with BrdU (10 μm) for another 24 h. β-Cell replication was determined as described in Fig. 2 (804G-ECM control absolute value, 3.32 ± 0.34 for B and 2.26 ± 0.38 for C); n =3–5. *, P < 0.05 vs. PLL control; **, P < 0.05 vs. 804G-ECM control.
Discussion
ECM has been reported to enhance cell attachment and proliferation as well as to induce differentiation in vitro (15). Recently, we demonstrated that primary rat β-cell proliferation is significantly increased by ECM (7). Here we show that ECM increases expression of two cell cycle proteins, PCNA and cyclin D1, in β-cells. PCNA is a nuclear nonhistone antigen that appears in the nucleus during late G1 phase, increases during S phase, and decreases during G2 and M phases (16). PCNA expression is thus an indicator for cell proliferation and has been used to determine β-cell mass expansion in rats (17). Three types of cyclin D, namely D1, D2, and D3, play an essential role in promoting cell cycle progression from G1 to S phase (18). Cyclin D1 and D2 are expressed in β-cells and are important regulators of β-cell proliferation, whereas cyclin D3 is expressed at low levels (19, 20, 21, 22). In our study, we show that ECM increases expression of cyclin D1 but not of cyclin D2. This could be explained by the fact that expression of the D-type cyclins is dependent on mitogenic stimulation. Indeed, cyclin D1 but not cyclin D2 expression is induced by glucagon-like peptide 1 (GLP-1) (23). The signaling pathway regulating cyclin D1 expression in β-cells is unknown even if overexpression of cyclin D1 in cultured islets increases β-cell proliferation (24).
Our results show that the ability of ECM to increase rat β-cell proliferation is glucose dependent as reported previously for cell spreading (11). It has long been known that glucose is a potent β-cell mitogen (25). In rats, glucose infusion results in an approximately 50% increase in both β-cell proliferation and mass (26, 27, 28). In our study, we confirm that glucose significantly increases the proliferation of rat β-cells in vitro and further show that ECM can stimulate proliferation only at 11.2 mm glucose and not at 2.8 mm. An inherent complication in studies of glucose effects on β-cells is the difficulty in separating the effect of glucose from those of insulin. A recent study has thus demonstrated that insulin secreted by β-cells in response to elevated glucose exerts autocrine effects to stimulate proliferation (29). However, the addition of exogenous insulin (at a concentration similar to that found in surrounding cells at high glucose) to β-cells cultured at 2.8 mm glucose increased cell proliferation but to a lower extent than that observed with 11.2 mm glucose (data not shown). There was, however, no impact of ECM on the total amount of insulin accumulating in the medium during the test culture period. These results suggest that glucose itself may be considered as a mitogen in our study, but we do not exclude a synergistic contribution from insulin. It was not the purpose of the present study to elaborate further on this.
Glucose metabolism induces an increase in intracellular Ca2+, and we have shown that increased intracellular calcium is required for spreading of rat islet β-cells on extracellular matrix (11). Here we show that 804G-ECM-induced NF-κB activation is Ca2+ dependent. Activation of NF-κB by depolarization and Ca2+ influx has been reported in MIN6 insulinoma cells (30). 804G-ECM-induced rat β-cell proliferation was prevented by inhibition of Ca2+ influx. Furthermore, calcineurin, a ubiquitous calcium-activated serine phosphatase, seems to be implicated because its inhibition by cyclosporin A decreased β-cell proliferation induced by 804G-ECM. Recently, calcineurin/NFAT signaling has been shown to regulate pancreatic β-cell growth and function (13, 31). Calcineurin is also known to activate the canonical NF-κB/NFAT pathway induced by inflammation in astrocytes and lymphocytes (32, 33). We further show that inhibition of NF-κB with Bay 11-7082 significantly decreased β-cell proliferation. The association between normal growth and NF-κB activation has been noted in many cells and tissues (34, 35). Further experiments in many cell types now indicate that NF-kB acts through increasing the abundance of cyclin D1 (34). Therefore, in our condition, the increase in cyclin D1 expression could similarly be due to 804G-ECM-induced NF-κB activation. IL-1β, which is secreted by β-cells plated on this particular ECM, can activate NF-κB (9). It is important to stress that in this particular context, the modest and relatively short-lived activation of NF-κB induced in rat β-cells by ECM is thought to be beneficial (8, 9). Now we show that this activation of NF-κB by IL-1β may contribute toward increased β-cell proliferation on ECM because treatment with IL-1Ra reduced significantly the ECM effect. This correlates with a finding that low concentrations of IL-1β increase human islet cell replication, whereas high levels induce apoptosis in the same cells (36).
The response of cells to ECM attachment is mediated primarily by the integrin family of adhesion receptors. It has been shown that 804G-ECM effects on pancreatic β-cells (spreading, glucose-stimulated insulin secretion, and survival) are mediated by the engagement of β1 integrins to laminin-5 (6, 37), which leads to focal adhesion kinase (FAK) phosphorylation and downstream activation of the PI3K/Akt pathway. Therefore, integrin signaling may induce the same signaling transduction cascades as growth factors and consequently may have comparable effects on cell cycle progression. Results of the present study provide evidence that ECM induces β-cell proliferation by activating the PI3K/Akt pathway, which could lead to NFAT nuclear translocation. We have previously demonstrated that ECM induces phosphorylation of Akt (6). Here we demonstrate that ECM induces also phosphorylation of its downstream target GSK3β (thereby inhibiting its activity and so decreasing phosphorylation and export of NFAT from the nucleus). Moreover, we show that inhibition of GSK3β increases β-cell proliferation, whereas PKA inhibition, which induces activation of GSK3β, inhibits β-cell replication. Our results are in accordance with recent reports showing that overexpression of GSK3β reduced mouse β-cell mass and proliferation (38, 39). Interestingly, activation of GSK3β (by inhibiting Akt or PKA) and therefore export of NFAT from the nucleus has a more pronounced impact on β-cell proliferation than direct inhibition of NFAT translocation to the nucleus (by inhibiting calcineurin or with VIVIT peptide). This may suggest that GSK3β regulates other signaling molecules than NFAT implicated in β-cell proliferation. Indeed, GSK3β regulates cell growth by inhibition and/or degradation of a large number of signaling molecules implicated in gene transcription, cell metabolism, and protein synthesis (40). One of them could be NF-κB, because it has been shown that GSK3β negatively regulates NF-κB (41).
The increase of ECM-induced β-cell proliferation by (48 h) inhibition of the MAPK pathway was not expected because it has been found to be involved in crucial functions implicated in proliferation. Furthermore, ERK has been demonstrated to be essential in triggering proliferation in response to growth factors (42, 43). However, it is known that depending on the cell type and the stimuli, these enzymes can be either inhibited or activated by the same secondary messenger. Therefore, MAPK signaling proteins are dynamic and may play different roles at different times (44). Indeed, in vascular smooth muscle cells, cell proliferation in three-dimensional matrices was inversely correlated to ERK activation (45). Inhibition of p38 MAPK increased β-cell proliferation regardless of the substrate used. It has been already reported that p38 appears to negatively influence cell cycle progression in many cells (46, 47). A conditionally activated form of MEK kinase 3 arrests fibroblast cell cycle, and this effect could be mediated via p38 MAPK, a downstream effector of MEK kinase 3 (48). Interestingly, p38 MAPK inhibition has been shown to enable proliferation of adult mammalian cardiomyocytes (49), cells that were considered terminally differentiated and incapable of proliferation, like β-cells a few years ago.
In summary, our work describes possible mechanisms for regulation of β-cell proliferation in response to ECM. Increased levels of intracellular Ca2+ are necessary for 804G-ECM increased β-cell proliferation, with involvement of downstream activation of NF-κB. IL-1β may amplify this process by feed-forward activation of NF-κB. Calcium entry can also activate the calcineurin/NFAT signaling to induce β-cell proliferation. Furthermore, activation of the PI3K cascade by integrin-ECM interaction may promote β-cell proliferation through phosphorylation and inhibition of GSK3ß. This study shows that β-cell proliferation can be enhanced by ECM via activation or inhibition of multiple signaling pathways. Due to the difficulty to increase human β-cell proliferation in vitro (7), it will be critical to learn in future studies if pharmacologic inhibition or activation of these specific pathways can be used to induce human β-cell regeneration.
Materials and Methods
Materials
Bay 11-7082 was from BioMol Research Laboratories (Hamburg, Germany) and IL-1Ra (Kineret) from Amgen (Europe B.V., Breda, The Netherlands). PD98059, LY294002, diazoxide, SR-7037, cyclosporin A, VIVIT peptide, SB20358, SP600125, and H89 were purchased from Calbiochem (Darmstadt, Germany). BIO was the kind gift from Rockefeller University (New York, NY). Primary antibodies for immunofluorescence were polyclonal anti-p65 subunit of NF-κB (C-20) from Santa Cruz Biotechnology (Santa Cruz, CA) and guinea pig antiinsulin serum (Domenico Bosco; University of Geneva, Geneva, Switzerland). Primary antibodies for Western blot were monoclonal anti-PCNA (Signet, Alexis Corp., Lausen, Switzerland); polyclonal anti-GSK3β, and anti-phospho-GSK3β (Ser9) (Cell Signaling Technology-Bioconcept, Allschwil, Switzerland); monoclonal anti-cyclin D1, monoclonal anti-cyclin D2, and polyclonal anti-CDK4 (Santa Cruz Biotechnology), monoclonal anti-actin (Sigma Chemical Co., St. Louis, MO). Secondary antibodies were Alexa fluor 488-conjugated anti-guinea pig from Molecular Probes (Eugene, OR) and fluorescein isothiocyanate-conjugated goat antirabbit (Sigma) for immunofluorescence and antimouse horseradish peroxidase and antirabbit horseradish peroxidase (Amersham Pharmacia Biotech, Dübendorf, Switzerland) for Western blot. Hoechst 33342 was from Sigma Fluka (Buchs, Switzerland).
Islet isolation and β-cell purification
All experiments were performed on primary pancreatic β-cells sorted from adult rat islet cells by autofluorescence-activated flow cytometry. Islets of Langerhans were isolated by collagenase digestion of pancreas from male Wistar rats (weighing 150–200 g), followed by Ficoll purification. Islets were trypsinized and β-cells purified using a fluorescence-activated cell sorter (FACStar-Plus; Becton Dickinson, Sunnyvale, CA) as described (37, 50), by autofluorescence to yield a population of more than 95% β-cells.
804G-ECM matrix preparation
The 804G cells were the kind gift of Desmos (San Diego, CA). ECM from medium conditioned by 804G cells was prepared and used as described previously (51). They were grown in DMEM (GIBCO, Invitrogen, Basel, Switzerland), containing 10% FCS and 5.6 mm glucose. At confluence, cells were rinsed and maintained for another 3 d in the same medium in the absence of FCS. Conditioned medium (referred to hereafter as 804G-ECM) was collected, centrifuged at 120 × g for 10 min to remove any detached cells and debris, filtered through a 0.22-μm Millipore filter, and frozen at −20 C for later use.
Coating of plastic dishes with poly-l-lysine and 804G-ECM
Aliquots (60 μl) of PLL (100 μg/ml) or of 804G-ECM were layered at the center of 35-mm culture petri dishes (adherent dishes for mammalian cell culture). Dishes were kept in a damp box at 37 C for 18–20 h before being rinsed three times with sterile H2O and air dried. Dishes coated with PLL were used as controls.
Cell culture
Sorted β-cells were washed twice in 10–15 ml sterile DMEM (GIBCO, Invitrogen) containing 10% FCS, 11.2 mm glucose, and 110 μg/ml sodium pyruvate and supplemented with 110 U/ml penicillin, 110 μg/ml streptomycin, and 50 μg/ml gentamycin, followed by centrifugation for 10 min at 130 × g. Aliquots of 3 × 105 cells were seeded in nonadherent 100-mm-diameter petri dishes containing 9 ml medium. Cells were then incubated for 20 h at 37 C to allow full recovery of any cell surface molecules that may have been lost or damaged during islet isolation or cell purification. After recovery, cells were resuspended at a density of 4 × 105 cells/ml in control DMEM supplemented or not with inhibitors at the indicated concentration. Aliquots (50 μl) of this suspension were plated as droplets at the center of petri dishes coated with 804G-ECM or PLL and were incubated at 37 C. One day later, cells were incubated with 10 μm BrdU for 24 h under control conditions or in the continued presence of the inhibitor. The inhibitors were dissolved in dimethylsulfoxide, which was also added to control cultured cells at the same concentration. In addition, medium (∼50 μl) was collected and centrifuged to remove any detached cells and debris and stored at −20 C for subsequent insulin measurement.
Detection of proliferation by immunofluorescence
Cell replication was determined by BrdU incorporation and the identity of β-cells confirmed by insulin immunofluorescence. β-Cells incubated with BrdU were fixed (1% paraformaldehyde, 1 h room temperature) and DNA denatured (1.5 m HCl, 1 h room temperature). After permeabilization (0.5% Triton X-100, 4 min), proliferation was estimated using an immunohistochemical assay kit as described by the manufacturer (BrdU labeling and detection kit; Roche, Bale, Switzerland). Cells were also costained for insulin (guinea pig anti-insulin serum 1:500). After extensive washing, cells were incubated with Alexa fluor 488-conjugated antiguinea pig Ig. Nuclei were stained with 10 μg/ml Hoechst 33342. The percentage of cells double positive for insulin and BrdU per total number of insulin single-positive cells was calculated.
Insulin measurement
Insulin release during the 24 h of culture was measured by RIA with rat insulin as the standard.
NF-κB activation
The cellular localization of NF-κB in various conditions was analyzed by immunofluorescence. Cells were fixed with 4% paraformaldehyde (20 min, room temperature) and permeabilized with 0.5% Triton X-100 (5 min, room temperature), and immunofluorescence for NF-κB was performed. The percentage of cells with dominant nuclear NF-κB staining was determined (five different fields were examined for each condition).
Quantitative real-time PCR
RNA was isolated using RNeasy mini kit (QIAGEN, Hombrechtikon, Switzerland). cDNA was synthesized with Superscript II (Invitrogen), using 1 μg total RNA in a 20-μl reaction volume. The double-stranded DNA-specific dye SYBR Green I (Eurogentech, Seraing, Belgium) and fluorescein (Bio-Rad, Bâle, Switzerland) were incorporated into the PCR buffer (qPCR core kit; Eurogentech) to allow for quantitative detection of the PCR product. The results were analyzed using the iCycler iQ System (Bio-Rad). The housekeeping gene L3 was used as an internal control. The primers used were as follows: IκBα forward 5′-TGC TGA GGC ACT TCT GAA AGC-3′ and IκBα reverse 5′-TCC TCG AAA GTC TCG GAG GTC-3′.
Western blot analysis
To analyze phospho-GSK3β, total GSK3β, PCNA, cyclin D1, and CDK4 protein expression, attached cells were washed with ice-cold PBS supplemented with 1 mm sodium vanadate and protease inhibitors and lysed in sample buffer 1× [62 mm Tris-HCl (pH 6.8), 2% SDS, 5% glycerol, 1% 2-mercaptoethanol]. Protein concentrations were determined with the amido black method (52), and equal amounts of total protein were loaded for SDS-PAGE. All samples, after separation on an SDS-polyacrylamide gel, were electroblotted onto nitrocellulose membranes (Schleicher & Schuell, Keene, NH) for immunoblotting with the appropriate antibody. An ECL protein detection kit (Amersham Biosciences) and a Kodak image station were used for visualization of the bands.
Presentation of data and statistical analysis
Unless stated otherwise, data are presented as mean ± se for n independent experiments, and levels of significance for differences between groups were assessed by Student’s t test for unpaired groups.
Acknowledgments
We thank Caroline Rouget and Nadja Perriraz for expert technical assistance.
Footnotes
This work was supported by the JDRF Program for Regeneration of β-Cell Function (Grant No. 1-2005-826) and by the JDRF Grant No. 7-2005-1158.
Disclosure Summary: The authors have nothing to disclose.
First Published Online May 14, 2009
G.P. and E.H. contributed equally to the study.
Abbreviations: BIO, 6-Bromoindirubin-3′-oxime; BrdU, bromodeoxyuridine; CDK4, cyclin-dependent kinase 4; ECM, extracellular matrix; FCS, fetal calf serum; GSK, glycogen synthase kinase; IL-1R, IL-1 receptor; IL-1Ra, IL-1R antagonist; JNK, c-Jun N-terminal kinase; MEK, MAPK kinase; NF, nuclear factor; NFAT, nuclear factor of activated T cells; PCNA, proliferating cell nuclear antigen; PI3K, phosphatidylinositol 3-kinase; PKB, protein kinase B; PLL, poly-l-lysine.
References
- 1.Roskelley CD, Srebrow A, Bissell MJ1995. A hierarchy of ECM-mediated signalling regulates tissue-specific gene expression. Curr Opin Cell Biol 7:736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lelièvre S, Weaver VM, Bissell MJ1996. Extracellular matrix signaling from the cellular membrane skeleton to the nuclear skeleton: a model of gene regulation. Recent Prog Horm Res 51:417–432 [PMC free article] [PubMed] [Google Scholar]
- 3.Li S, Edgar D, Fässler R, Wadsworth W, Yurchenco PD2003. The role of laminin in embryonic cell polarization and tissue organization. Dev Cell 4:613–624 [DOI] [PubMed] [Google Scholar]
- 4.Hisaoka M, Haratake J, Hashimoto H1993. Pancreatic morphogenesis and extracellular matrix organization during rat development. Differentiation 53:163–172 [DOI] [PubMed] [Google Scholar]
- 5.Boudreau NJ, Jones PL1999. Extracellular matrix and integrin signalling: the shape of things to come. Biochem J 339(Pt 3):481–488 [PMC free article] [PubMed]
- 6.Hammar E, Parnaud G, Bosco D, Perriraz N, Maedler K, Donath M, Rouiller DG, Halban PA2004. Extracellular matrix protects pancreatic β-cells against apoptosis: role of short- and long-term signaling pathways. Diabetes 53:2034–2041 [DOI] [PubMed] [Google Scholar]
- 7.Parnaud G, Bosco D, Berney T, Pattou F, Kerr-Conte J, Donath MY, Bruun C, Mandrup-Poulsen T, Billestrup N, Halban PA2008. Proliferation of sorted human and rat β-cells. Diabetologia 51:91–100 [DOI] [PubMed] [Google Scholar]
- 8.Hammar EB, Irminger JC, Rickenbach K, Parnaud G, Ribaux P, Bosco D, Rouiller DG, Halban PA2005. Activation of NF-κB by extracellular matrix is involved in spreading and glucose-stimulated insulin secretion of pancreatic β-cells. J Biol Chem 280:30630–30637 [DOI] [PubMed] [Google Scholar]
- 9.Ribaux P, Ehses JA, Lin-Marq N, Carrozzino F, Böni-Schnetzler M, Hammar E, Irminger JC, Donath MY, Halban PA2007. Induction of CXCL1 by extracellular matrix and autocrine enhancement by interleukin-1 in rat pancreatic β-cells. Endocrinology 148:5582–5590 [DOI] [PubMed] [Google Scholar]
- 10.Böni-Schnetzler M, Thorne J, Parnaud G, Marselli L, Ehses JA, Kerr-Conte J, Pattou F, Halban PA, Weir GC, Donath MY2008. Increased interleukin (IL)-1β messenger ribonucleic acid expression in β-cells of individuals with type 2 diabetes and regulation of IL-1β in human islets by glucose and autostimulation. J Clin Endocrinol Metab 93:4065–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bosco D, Gonelle-Gispert C, Wollheim CB, Halban PA, Rouiller DG2001. Increased intracellular calcium is required for spreading of rat islet β-cells on extracellular matrix. Diabetes 50:1039–1046 [DOI] [PubMed] [Google Scholar]
- 12.Whitaker M2006. Calcium microdomains and cell cycle control. Cell Calcium 40:585–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, Kim SK2006. Calcineurin/NFAT signalling regulates pancreatic β-cell growth and function. Nature 443:345–349 [DOI] [PubMed] [Google Scholar]
- 14.Rao A, Luo C, Hogan PG1997. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15:707–747 [DOI] [PubMed] [Google Scholar]
- 15.Adams JC, Watt FM1993. Regulation of development and differentiation by the extracellular matrix. Development 117:1183–1198 [DOI] [PubMed] [Google Scholar]
- 16.Moldovan GL, Pfander B, Jentsch S2007. PCNA, the maestro of the replication fork. Cell 129:665–679 [DOI] [PubMed] [Google Scholar]
- 17.Vasilijevic A, Buzadzic B, Korac A, Petrovic V, Jankovic A, Korac B2007. Beneficial effects of l-arginine nitric oxide-producing pathway in rats treated with alloxan. J Physiol 584:921–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boonstra J2003. Progression through the G1-phase of the on-going cell cycle. J Cell Biochem 90:244–252 [DOI] [PubMed] [Google Scholar]
- 19.Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocaña A, Vasavada R, Stewart AF2006. Molecular control of cell cycle progression in the pancreatic β-cell. Endocr Rev 27:356–370 [DOI] [PubMed] [Google Scholar]
- 20.Georgia S, Bhushan A2004. β-Cell replication is the primary mechanism for maintaining postnatal β-cell mass. J Clin Invest 114:963–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF2005. Cyclins D2 and D1 are essential for postnatal pancreatic β-cell growth. Mol Cell Biol 25:3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martín J, Hunt SL, Dubus P, Sotillo R, Néhmé-Pélluard F, Magnuson MA, Parlow AF, Malumbres M, Ortega S, Barbacid M2003. Genetic rescue of Cdk4 null mice restores pancreatic β-cell proliferation but not homeostatic cell number. Oncogene 22:5261–5269 [DOI] [PubMed] [Google Scholar]
- 23.Friedrichsen BN, Neubauer N, Lee YC, Gram VK, Blume N, Petersen JS, Nielsen JH, Møldrup A2006. Stimulation of pancreatic β-cell replication by incretins involves transcriptional induction of cyclin D1 via multiple signalling pathways. J Endocrinol 188:481–492 [DOI] [PubMed] [Google Scholar]
- 24.Cozar-Castellano I, Takane KK, Bottino R, Balamurugan AN, Stewart AF2004. Induction of β-cell proliferation and retinoblastoma protein phosphorylation in rat and human islets using adenovirus-mediated transfer of cyclin-dependent kinase-4 and cyclin D1. Diabetes 53:149–159 [DOI] [PubMed] [Google Scholar]
- 25.Chick WL, Lauris V, Flewelling JH, Andrews KA, Woodruff JM1973. Effects of glucose on β-cells in pancreatic monolayer cultures. Endocrinology 92:212–218 [DOI] [PubMed] [Google Scholar]
- 26.Bernard C, Thibault C, Berthault MF, Magnan C, Saulnier C, Portha B, Pralong WF, Pénicaud L, Ktorza A1998. Pancreatic β-cell regeneration after 48-h glucose infusion in mildly diabetic rats is not correlated with functional improvement. Diabetes 47:1058–1065 [DOI] [PubMed] [Google Scholar]
- 27.Bonner-Weir S, Deery D, Leahy JL, Weir GC1989. Compensatory growth of pancreatic β-cells in adult rats after short-term glucose infusion. Diabetes 38:49–53 [DOI] [PubMed] [Google Scholar]
- 28.Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A2003. Specific and combined effects of insulin and glucose on functional pancreatic β-cell mass in vivo in adult rats. Endocrinology 144:2717–2727 [DOI] [PubMed] [Google Scholar]
- 29.Muller D, Huang GC, Amiel S, Jones PM, Persaud SJ2006. Identification of insulin signaling elements in human β-cells: autocrine regulation of insulin gene expression. Diabetes 55:2835–2842 [DOI] [PubMed] [Google Scholar]
- 30.Bernal-Mizrachi E, Wen W, Shornick M, Permutt MA2002. Activation of nuclear factor-κB by depolarization and Ca2+ influx in MIN6 insulinoma cells. Diabetes 51(Suppl 3):S484–S488 [DOI] [PubMed]
- 31.Heit JJ2007. Calcineurin/NFAT signaling in the β-cell: from diabetes to new therapeutics. Bioessays 29:1011–1021 [DOI] [PubMed] [Google Scholar]
- 32.Fernandez AM, Fernandez S, Carrero P, Garcia-Garcia M, Torres-Aleman I2007. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J Neurosci 27:8745–8756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fisher WG, Yang PC, Medikonduri RK, Jafri MS2006. NFAT and NFκB activation in T lymphocytes: a model of differential activation of gene expression. Ann Biomed Eng 34:1712–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B, Pestell RG2001. NF-κB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev 12:73–90 [DOI] [PubMed] [Google Scholar]
- 35.Obata H, Biro S, Arima N, Kaieda H, Kihara T, Eto H, Miyata M, Tanaka H1996. NF-κB is induced in the nuclei of cultured rat aortic smooth muscle cells by stimulation of various growth factors. Biochem Biophys Res Commun 224:27–32 [DOI] [PubMed] [Google Scholar]
- 36.Maedler K, Schumann DM, Sauter N, Ellingsgaard H, Bosco D, Baertschiger R, Iwakura Y, Oberholzer J, Wollheim CB, Gauthier BR, Donath MY2006. Low concentration of interleukin-1β induces FLICE-inhibitory protein-mediated β-cell proliferation in human pancreatic islets. Diabetes 55:2713–2722 [DOI] [PubMed] [Google Scholar]
- 37.Parnaud G, Hammar E, Rouiller DG, Armanet M, Halban PA, Bosco D2006. Blockade of β1 integrin-laminin-5 interaction affects spreading and insulin secretion of rat β-cells attached on extracellular matrix. Diabetes 55:1413–1420 [DOI] [PubMed] [Google Scholar]
- 38.Liu Z, Tanabe K, Bernal-Mizrachi E, Permutt MA2008. Mice with β-cell overexpression of glycogen synthase kinase-3β have reduced β-cell mass and proliferation. Diabetologia 51:623–631 [DOI] [PubMed] [Google Scholar]
- 39.Mussmann R, Geese M, Harder F, Kegel S, Andag U, Lomow A, Burk U, Onichtchouk D, Dohrmann C, Austen M2007. Inhibition of GSK3 promotes replication and survival of pancreatic β-cells. J Biol Chem 282:12030–12037 [DOI] [PubMed] [Google Scholar]
- 40.Doble BW, Woodgett JR2003. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 116:1175–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bournat JC, Brown AM, Soler AP2000. Wnt-1 dependent activation of the survival factor NF-κB in PC12 cells. J Neurosci Res 61:21–32 [DOI] [PubMed] [Google Scholar]
- 42.Chambard JC, Lefloch R, Pouysségur J, Lenormand P2007. ERK implication in cell cycle regulation. Biochim Biophys Acta 1773:1299–1310 [DOI] [PubMed] [Google Scholar]
- 43.Katz M, Amit I, Yarden Y2007. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim Biophys Acta 1773:1161–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raman M, Chen W, Cobb MH2007. Differential regulation and properties of MAPKs. Oncogene 26:3100–3112 [DOI] [PubMed] [Google Scholar]
- 45.Hong H, McCullough CM, Stegemann JP2007. The role of ERK signaling in protein hydrogel remodeling by vascular smooth muscle cells. Biomaterials 28:3824–3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ambrosino C, Nebreda AR2001. Cell cycle regulation by p38 MAP kinases. Biol Cell 93:47–51 [DOI] [PubMed] [Google Scholar]
- 47.Lavoie JN, L'Allemain G, Brunet A, Müller R, Pouysségur J1996. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem 271:20608–20616 [DOI] [PubMed] [Google Scholar]
- 48.Ellinger-Ziegelbauer H, Kelly K, Siebenlist U1999. Cell cycle arrest and reversion of Ras-induced transformation by a conditionally activated form of mitogen-activated protein kinase kinase kinase 3. Mol Cell Biol 19:3857–3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT2005. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev 19:1175–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rouiller DG, Cirulli V, Halban PA1990. Differences in aggregation properties and levels of the neural cell adhesion molecule (NCAM) between islet cell types. Exp Cell Res 191:305–312 [DOI] [PubMed] [Google Scholar]
- 51.Bosco D, Meda P, Halban PA, Rouiller DG2000. Importance of cell-matrix interactions in rat islet β-cell secretion in vitro: role of α6β1 integrin. Diabetes 49:233–243 [DOI] [PubMed] [Google Scholar]
- 52.Schaffner W, Weissmann C1973. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal Biochem 56:502–514 [DOI] [PubMed] [Google Scholar]