

Abstract
Liver X receptor (LXR) is a ligand-activated transcription factor that plays important roles in cholesterol and lipid homeostasis. However, ligand-induced posttranslational modification of LXR is largely unknown. Here, we show that ligand-free LXRα is rapidly degraded by ubiquitination. Without ligand, LXRα interacts with an ubiquitin E3-ligase protein complex containing breast and ovarian cancer susceptibility 1 (BRCA1)-associated RING domain 1 (BARD1). Interestingly, LXR ligand represses ubiquitination and degradation of LXRα, and the interaction between LXRα and BARD1 is inhibited by LXR ligand. Consistently, T0901317, a synthetic LXR ligand, increased the level of LXRα protein in liver. Moreover, overexpression of BARD1/BRCA1 promoted the ubiquitination of LXRα and reduced the recruitment of LXRα to the target gene promoters, whereas BARD1 knockdown reversed such effects. Taken together, these data suggest that LXR ligand prevents LXRα from ubiquitination and degradation by detaching BARD1/BRCA1, which might be critical for the early step of transcriptional activation of ligand-stimulated LXRα through a stable binding of LXRα to the promoters of target genes.
Liver X receptor ligands stabilize LXRα protein by preventing LXRα ubiquitination through dissociation of BARD1/BRCA1 complex, a novel ubiquitin E3-ligase of LXRα.
Liver X receptor (LXR) α and LXRβ play crucial roles in metabolic homeostasis including cholesterol efflux, lipogenesis, and inflammatory responses (1, 2, 3, 4, 5). LXRα is highly expressed in several tissues that are important for cholesterol and lipid metabolism such as the liver, adipose tissue, and macrophage, whereas LXRβ is ubiquitously expressed (6). Both LXRα and LXRβ are stimulated by several natural and synthetic ligands including 20(S)-hydroxycholesterol [20(S)-HC], 22(R)-HC, 24-HC, T0314407, T0901317, and GW3965 (7, 8). In the liver, macrophage, and intestine, LXR activation by ligands regulates cholesterol homeostasis through the expression of certain target genes such as ATP-binding cassette A1 (ABCA1), ABCG1, cholesterol 7α-hydroxylase, and apolipoprotein E, which are responsible for cholesterol efflux, transport, and bile acid excretion (9). In addition, activated LXR stimulates lipogenesis by enhancing fatty acid synthesis via increase of carbohydrate-response element-binding protein, sterol element-binding protein 1c (SREBP1c), and fatty acid synthase gene expressions (10, 11, 12). Due to the potent lipogenic activity of LXR, unwanted LXR stimulation by its ligands is associated with several disorders such as hyperlipidemia and β-cell dysfunction (13, 14).
In the presence of ligands, most nuclear receptors undergo a conformational change of C-terminal helix 12 [also known as activation function 2 (AF-2)] in the ligand-binding domain (LBD). This change results in the displacement of histone deacetylase-containing corepressor complex from nuclear receptors in exchange for histone acetylase-containing coactivators and in the subsequent transcriptional activation of the target gene expression (15, 16). Similarly, it has been reported that ligand-free LXR also interacts with corepressors such as nuclear receptor corepressor and silencing mediator of retinoid and thyroid hormone receptor to silence its target genes (17). In the presence of ligands, activated LXR rapidly induces conformational change in the LBD, which leads to interact with coactivators such as p300, steroid receptor coactivator 1, and peroxisome proliferator-activated receptor (PPAR)γ coactivator 1 (18). As well as the ligand-induced exchange of corepressors and coactivators, ligand treatment affects the transcriptional activities of many nuclear receptors through posttranslational modifications including acetylation, phosphorylation, sumoylation, and ubiquitination (19, 20, 21, 22). For example, DNA-binding domain (DBD) of estrogen receptor (ER)α is acetylated by p300 in the presence of ligand, which enhances the DNA-binding activity of ERα (23). Additionally, it has been reported that ERα is subjected to other posttranslational modifications including phosphorylation and ubiquitination in a ligand-dependent manner (21, 24). These posttranslational modifications of nuclear receptors often influence corepressor/coactivator recruitments, transcriptional activities, and protein stabilities of nuclear receptors in response to specific ligands (20, 24, 25).
LXRα has been reported to undergo posttranslational modifications such as phosphorylation and sumoylation. For instance, two groups have demonstrated that LXRα is phosphorylated. Tontonoz and associates (26) showed that LXRα is phosphorylated at serine 198 residue of human LXRα (equivalent to residue 196 of mouse LXRα). However, mutation of serine 198 residue affects neither its DNA binding ability nor the transcriptional activity of LXRα. On the contrary, Yamamoto et al. (27) have revealed that protein kinase A directly phosphorylates LXRα at multiple target sites (195/196 and 209/291 serine residues of mouse LXRα), which causes impaired DNA-binding ability and decreases transcriptional activity of LXRα. In addition, it has been reported that both LXRα and -β could be sumoylated by SUMO2 and SUMO3 in the presence of ligand, which appears to be important for the antiinflammatory response of LXRα/β (28, 29). Sumoylated LXRα/β forms protein complexes with nuclear receptor corepressor on the promoters of proinflammatory genes, which eventually suppresses toll-like receptor signaling-dependent proinflammatory gene expressions (28).
Although the phosphorylation and sumoylation events occur in LXRα, it has not been thoroughly understood whether other posttranslational modification(s) may control LXR activity in a ligand-dependent manner. In this study, we demonstrate that the ubiquitination and subsequent degradation of LXRα are tightly regulated in a ligand-dependent manner. Furthermore, BARD1 (BRCA1-associated RING domain 1)/BRCA1 (breast and ovarian cancer susceptibility 1) is involved in ubiquitination of ligand-free LXRα, whereas ligand-activated LXRα binds to the promoters of its target genes after dissociation with BARD1/BRCA1.
Results
LXR ligands inhibit ubiquitination of LXRα
To determine whether proteasomal degradation is involved in the LXRα protein stability, we first examined the degradation rate of LXRα in the presence of cycloheximide, which inhibits de novo protein synthesis. Compared with vehicle-treated cells, LXRα protein degradation was markedly inhibited in cells treated with MG132, an inhibitor of the 26S proteasome (Fig. 1A). To elucidate whether LXRα is ubiquitinated to facilitate degradation of LXRα protein, we conducted in vivo ubiquitination assays. As shown in Fig. 1B, coexpression of LXRα with ubiquitin produced high molecular weight protein species of LXRα, implying that LXRα appears to be modified by polyubiquitination for rapid degradation and clearance.
Fig. 1.

Ubiquitination and degradation of ligand-free LXRα. A, LXRα-expressing HEK293 cells were pretreated with MG132 (20 μm) for 30 min and harvested at 1, 3, and 6 h after cycloheximide treatment (10 μg/ml). After isolating total lysates, Western blottings were preformed. B, The ubiquitination of LXRα was examined by in vivo ubiquitination assays, as described in Materials and Methods. Briefly, LXRα and/or HA-tagged ubiquitin expression vector was cotransfected into HEK293 cells, and the cells were incubated with MG132 (20 μm) for 3 h before harvesting. After isolating total cell lysates, coimmunoprecipitation and Western blotting analysis were performed with indicated antibodies. All the experiments were independently repeated at least three times, and representative results were shown. CHX, Cycloheximide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IB, immunoblotting; IP, immunoprecipitation.
Next, we investigated whether LXR ligands might affect the stability of LXRα protein by altering ubiquitination status. Interestingly, treatment of several LXR ligands, including 22(R)-HC, T0901317, and GW3965, enhanced the levels of LXRα protein (Fig. 2A). Moreover, LXR ligands attenuated the degradation rate of LXRα protein after cycloheximide treatment (T1/2vehicle = 0.62 h vs. T1/2T0901317 = 0.85 h) (Fig. 2B). In accordance with these results, synthetic ligands of LXR, T0901317 and GW3965, significantly diminished the level of LXRα polyubiquitination (Fig. 2, C and D). These findings indicate that LXR ligands evidently suppress the ubiquitination and proteasomal degradation of LXRα and increase the stability of LXRα protein.
Fig. 2.

LXRα ubiquitination is decreased by LXR ligands. A, LXRα-expressing HEK293 cells were treated with LXR ligands such as 22(R)-HC, T0901317, and GW3965 for 3 h. Total lysates were subjected to Western blottings. Band intensities were calculated by the LabWorks software (UVP Bioimaging Systems), and fold induction of LXRα protein was normalized by glyceraldehyde-3-phosphate dehydrogenase protein level. B, T0901317 (5 μm) was pretreated to the LXRα-expressing HEK293 cells for 30 min and harvested at the indicated time periods after cycloheximide treatment. Protein level of LXRα was analyzed by Western blotting. C, Transfected HEK293 cells with LXRα and HA-tagged ubiquitin expression vectors were incubated with MG132 (20 μm) and/or T0901317 (5 μm) for 3 h. Total cell lysates were isolated and subjected to in vivo ubiquitination assays. D, Similarly, ubiquitination of LXRα was analyzed after GW3965 treatment (5 μm). All the experiments were independently repeated at least three times, and representative results were shown. CHX, Cycloheximide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IB, immunoblotting; IP, immunoprecipitation.
The LBD of LXRα is important for ubiquitination of LXRα
To examine the domain(s) responsible for the ubiquitination of LXRα, we generated two deletion mutants of LXRα, a C-terminal deletion mutant lacking the LBD (ΔLBD) and an N-terminal deletion mutant lacking the DBD (ΔDBD) (Fig. 3A). When in vivo ubiquitination assays were performed, wild-type (WT) and ΔDBD LXRα were heavily ubiquitinated without ligand whereas such polyubiquitination of WT and ΔDBD LXRα was suppressed by LXR ligand (Fig. 3B, lanes 3 vs. 4 and 9 vs. 10). Surprisingly, polyubiquitination was almost abolished in ΔLBD LXRα even without ligand (Fig. 3B, lane 6), implying that C-terminal region of LXRα seems to be indispensable for the ubiquitination of LXRα in a ligand-dependent manner. These observations could be explained by the following possibilities: 1) lysine residues in the C-terminal region with LBD of LXRα may be direct ubiquitination sites(s), and 2) this region in LXRα may serve as a binding domain for the interaction with ubiquitin E3-ligase or ubiquitination-related protein complexes.
Fig. 3.
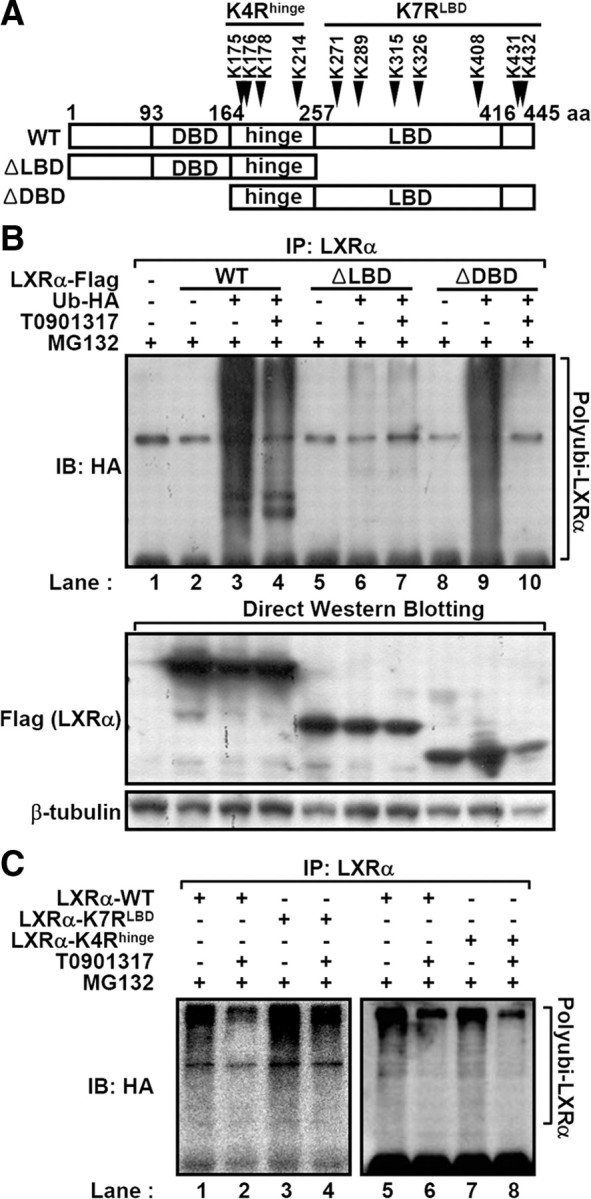
The LBD of LXRα is responsible for the interaction with the ubiquitin-proteasomal degradation complex. A, Schematic representation of ΔLBD and ΔDBD LXRα. Arrowhead indicates the seven lysine residues within C-terminal domain including LBD. B, The WT, ΔLBD, and ΔDBD LXRα expression vectors were individually transfected into HEK293 cells. The ubiquitination of each construct was examined by in vivo ubiquitination assays after treatment with T0901317 (5 μm) and MG132 (20 μm) for 3 h. C, Ubiquitination of the K7RLBD LXRα mutant and K4Rhinge. Equal amounts of WT, K7RLBD, and K4Rhinge LXRα expression vectors were transfected into HEK293 cells. After treatment with T0901317 and MG132 for 3 h, in vivo ubiquitination assays were conducted. IB, Immunoblotting; IP, immunoprecipitation.
To test the first possibility, each lysine residue in the LBD and AF-2 domain of LXRα was mutated into an arginine residue (K271R, K289R, K315R, K326R, K408R, or K431/432R) (Fig. 3A), and the degree of ubiquitination of each LXRα point mutant was examined by conducting in vivo ubiquitination assays. None of these LXRα point mutants significantly differed from WT LXRα in the aspect of polyubiquitination without ligand (data not shown). Additionally, we mutated all the above seven lysine residues in the C-terminal region of LXRα into arginine residue (K7RLBD) (Fig. 3A) and assessed the polyubiquitination of K7RLBD LXRα. Similar to other LXRα point mutants, K7RLBD LXRα was efficiently polyubiquitinated in the absence of ligand, and ubiquitination of K7RLBD LXRα was successfully inhibited by ligand treatment (Fig. 3C, lanes 3 and 4), indicating that the above seven lysine residues in the C-terminal region of LXRα are not crucial for LXRα ubiquitination in the absence of ligand. Instead, these data strongly suggest that the C-terminal region with the LBD of LXRα would be important for the association with ubiquitin-related protein complex in the absence of ligand.
To identify the ubiquitination target site(s) in LXRα, we performed in vivo ubiquitination assays with other LXRα mutants. Considering that ΔDBD LXRα containing the hinge, LBD, and AF-2 domain of LXRα was efficiently ubiquitinated (Fig. 3B, lane 9), it is possible to speculate that several lysine residues in the hinge region (between 164 and 257 amino acids in Fig. 3A) might be LXRα ubiquitination target site(s). When we examined the ubiquitination degree of every single K to R mutations in the hinge region (K175/176R, K178R, or K214R), all the mutants had successfully undergone the ubiquitination process. Furthermore, K4Rhinge mutant LXRα of which four lysine residues in the hinge region were mutated into arginine was also polyubiquitinated (Fig. 3C, lanes 7). Thus, it is likely that multiple lysine residues in LXRα protein might be involved in the process of LXRα polyubiquitination.
BARD1 interacts with LXRα in a ligand-dependent manner
Next, we attempted to identify LXRα-interacting protein(s), particularly, an LXRα-targeting ubiquitin E3-ligase. By performing a glutathione S-transferase-pulldown assay and mass spectrometry analysis, we identified an ubiquitin E3-ligase, breast and ovarian cancer susceptibility 1 (BRCA1)-associated RING-domain 1 (BARD1), which bound to unliganded LXRα recombinant protein (data not shown). To confirm the interaction between BARD1 and LXRα, we expressed both proteins in human embryonic kidney (HEK)293 cells with or without LXR ligand and examined their interactions by coimmunoprecipitation assays. Ligand-free LXRα preferentially associated with BARD1 (Fig. 4A), and the interaction between LXRα and BARD1 was notably decreased upon incubation periods with LXR ligand (Fig. 4B).
Fig. 4.

BARD1/BRCA1 interacts with LXRα in a ligand-dependent manner. A, HEK293 cells transfected with LXRα and BARD1/BRCA1 expression vectors were incubated with T0901317 (5 μm) and MG132 (20 μm) for 3 h, and total lysates were subjected to coimmunoprecipitation and Western blotting. B, Cotransfected HEK293 cells with LXRα and BARD1/BRCA1 were incubated with T0901317 (5 μm) and MG132 (20 μm) for the indicated periods. After harvesting, total cell lysates were coimmunoprecipitated with LXRα antibodies, and Western blotting was performed. C, The WT, ΔLBD, and ΔDBD LXRα were cotransfected with BARD1 and BRCA1. After treatment with T0901317 (5 μm) and MG132 (20 μm) for 3 h, coimmunoprecipitation assays were performed. Relative fold changes of immunoprecipitated BARD1 protein by T0901317 were indicated. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; IB, immunoblotting; IP, immunoprecipitation.
To manifest the finding that the C-terminal region of LXRα may serve as a docking region for BARD1, we examined the interaction between BARD1 and ΔLBD or ΔDBD LXRα in the absence or presence of LXR ligand. As shown in Fig. 4C, the deletion of both LBD and DBD in LXRα protein decreased the association with BARD1 compared with WT LXRα (lane 3 vs. lanes 6 and 9), implying that DBD and LBD of LXRα might contribute to stable interaction between LXRα and BARD1. Moreover, the interaction between BARD1 and ΔDBD LXRα was reduced by LXR ligand (lane 10) as observed in WT LXRα (lane 4). However, the binding of BARD1 to ΔLBD LXRα was not regulated by LXR ligand (lane 7), suggesting that C-terminal domain including LBD and AF-2 appears to be crucial for ligand-dependent interaction between BARD1 and LXRα.
BARD1/BRCA1 serves as a novel ubiquitin E3-ligase for LXRα
BARD1 forms a heterodimeric complex with BRCA1 that functions as an ubiquitin E3-ligase in DNA repair, transcriptional regulation, cell-cycle checkpoint, and centrosome dynamics (30, 31). Very recently, it has been reported that BARD1/BRCA1 complex ubiquitinates several nuclear receptors including ERα and progesterone receptor (22, 32). To examine the effects of BARD1/BRCA1 on the stability and ubiquitination of LXRα, BARD1/BRCA1 was ectopically expressed. As illustrated in Fig. 5A, degradation of LXRα protein was stimulated by overexpression of the BARD1/BRCA1. Concomitantly, the degree of LXRα polyubiquitination was augmented by BARD1/BRCA1 (Fig. 5B, lanes 1 vs. 5). Consistent with these results, suppression of BARD1 expression by BARD1 small interfering RNA (siRNA) increased LXRα protein stability [Fig. 5C and supplemental Fig. 1 (published as supplemental data on the Endocrine Society’s Journals Online web site at http://mend. endojournals.org)] and dramatically decreased its ubiquitination degree in the absence of ligand (Fig. 5D, lane 4). Similarly, we observed that BRCA1 knockdown attenuated ubiquitination of LXRα protein (supplemental Fig. 2). These data clearly indicate that BARD1/BRCA1 would be involved in the ubiquitination and degradation of LXRα in the absence of its ligand.
Fig. 5.

BARD1/BRCA1 induces ubiquitination of LXRα. A, LXRα-expressing HEK293 cells were successively transfected with BARD1/BRCA1 and then treated with cycloheximide for the indicated periods. B, Transfected HEK293 cells with LXRα- and HA-tagged ubiquitin expression vectors were sequentially transfected BRCA1 and/or BARD1. After treatment of T0901317 (5 μm) and MG132 (20 μm) for 3 h, total cell lysates were subjected to in vivo ubiquitination assays. C, LXRα-expressing HEK293 cells were transfected with siRNA duplex for negative control (siControl) and BARD1 (siBARD1), and the stability of LXRα protein was monitored by Western blotting. D, The siControl and siBARD1 siRNA duplexes were transfected into HEK293 cells expressing LXRα and ubiquitin, after which in vivo ubiquitination assay was performed. CHX, Cycloheximide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IB, immunoblotting; IP, immunoprecipitation.
LXR ligand stabilizes LXRα protein in vivo
To confirm whether protein stability of endogenous LXRα is indeed affected by LXR ligand treatment, we examined the changes of LXRα protein levels in several cell lines including RWPE1, THP1, Hepa1c1c7, and 3T3-L1 adipocytes with or without LXR ligands. However, we failed to detect enough amounts of endogenous LXRα protein even in the presence of MG132 and/or LXR ligands. Next, to investigate the level of LXRα protein in vivo, we treated mice with T0901317 and monitored the changes of endogenous LXRα protein levels in liver where LXRα mRNA is more abundantly expressed than in other tissues (33). With T0901317 administration, the level of LXRα protein was elevated in liver (Fig. 6). However, mRNA levels of both LXRα and LXRβ were not significantly altered (supplemental Fig. 3), as described previously (34). Together, these data clearly suggest that LXR ligand prevents LXRα from proteasomal degradation, leading to the stabilization of LXRα protein.
Fig. 6.

LXR ligand suppresses protein degradation of LXRα in vivo. T0901317 (50 mg/kg) was ip injected into C57BL/6N (B6) mice for 6 or 24 h. Total protein was extracted from liver tissue of vehicle- or T0901317-treated mice (n = 4 per each group) and subjected to Western blotting analysis. Antibody for SREBP1 was used for positive control of LXR ligand. GAPDH, Glyceraldehyde-3-phosphate dehydrogenase.
BARD1/BRCA1 modulates LXRα recruitments to its target gene promoters
It has been reported that LXR ligands increase the recruitments of LXR protein to the target gene promoters (35, 36). To delineate whether BARD1/BRCA1 is able to affect LXRα binding to the target gene promoters, we performed chromatin immunoprecipitation (ChIP) analysis with the RWPE1 cell line, which originates from normal human prostate epithelia (37). In several aspects, RWPE1 cells appear to be suitable for the analysis of functional relationship between BARD1/BRCA1 and LXRα. For example, because RWPE1 cells have normal cell characteristics [the RWPE1 cell has intact p53 and Rb proteins and does not form tumor (37)], endogenous BARD1/BRCA1 would be normally regulated during cell cycle progression. Moreover, the expression of LXR target genes involved in cholesterol and lipid metabolism is appropriately modulated by LXR ligands in RWPE1 cells (our unpublished data).
Consistent with previous reports (35, 36), we observed that LXR ligand increased the binding of LXRα protein to the promoters of its target genes such as ABCA1 and SREBP1c (Fig. 7A). When the level of BARD1/BRCA1 was increased by ectopic expression, the binding of LXRα protein to its target promoters was decreased in the absence of ligand (Fig. 7B, lanes 1 vs. 3 and 5 vs. 7). Interestingly, PCR amplicon was barely detected in the immunoprecipitated complex with Flag-tagged BARD1, implying that BARD1/BRCA1 is not recruited to LXR target gene promoters (Fig. 7B). In contrast to the overexpression of BARD1/BRCA1, knockdown of BARD1 substantially elevated the recruitment of LXRα protein to the target promoters under ligand-free basal state, probably due to increased LXRα protein stability (Fig. 7C, lanes 1 vs. 3 and 5 vs. 7). Taken together, these data suggest that stabilized LXRα with ligand is efficiently recruited to the promoter of target genes for the early step of target gene expression, which might be linked with displacement of BARD1/BRCA1 from LXRα in a ligand-dependent manner.
Fig. 7.
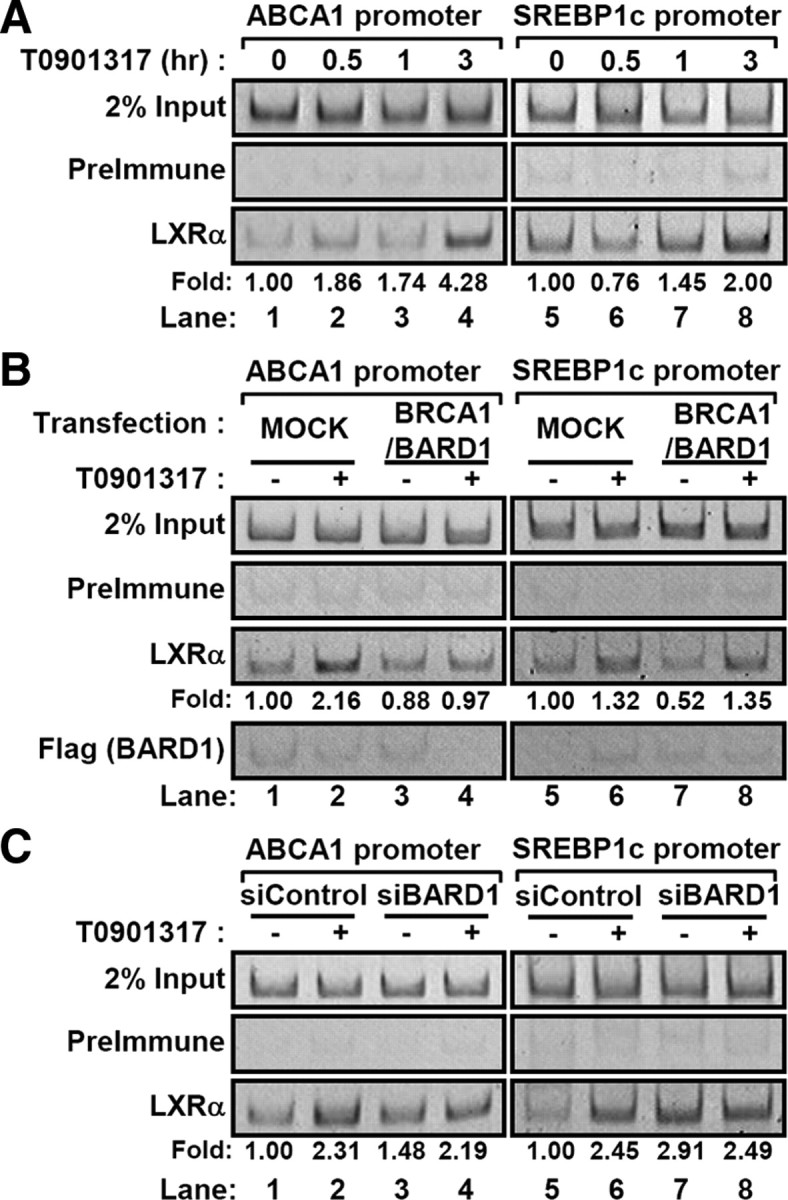
Regulation of LXRα recruitments to the target gene promoters. A, RWPE1 cells were treated with T0901317 (5 μm) for indicated periods (0.5, 1, and 3 h) and subjected to ChIP assays as described in Materials and Methods. B, RWPE1 cells were transfected with BRCA1/BARD1 expression vectors and incubated with T0901317 (5 μm) for 3 h. Then ChIP assays were performed. C, RWPE1 cells were transfected with siControl and siBARD1 RNA duplex. After treatment of T0901317 (5 μm) for 3 h, ChIP assays were conducted. Band intensities were calculated by the LabWorks software (UVP Bioimaging Systems), and net changes of amplified DNA from LXRα immunoprecipitates were normalized by input control of each sample.
Discussion
Because the cellular amounts of nuclear receptor proteins determine the level of ligand-induced target gene expression, the basal levels of nuclear receptors are tightly regulated through a balance between the synthesis and degradation of nuclear receptors. Further, the ligand-induced conformational changes of nuclear receptors dynamically alter the association with multiple protein complexes, which eventually results in regulating the protein stability and target gene expression. In the present study, we demonstrated that the ubiquitination of LXRα was sensitively controlled by various LXR ligands, which elevated the level of LXRα protein in vitro and in vivo (Figs. 2 and 6). In addition, we revealed that this ubiquitination process was mediated, at least in part, by the interaction between BARD1/BRCA1 and LXRα in a ligand-dependent manner (Figs. 4 and 5). Similar to LXRα, unliganded LXRβ was also ubiquitinated, and protein stability of LXRβ was increased by LXR ligand (supplemental Fig. 4, A and B). The observation that LXR ligand protected the polyubiquitination of LXRβ (supplemental Fig. 4C) implies that both LXRα and LXRβ may share a common regulatory mechanism(s) to coordinate LXR protein stability and target gene expression in a ligand-dependent manner.
Although LXR ligand and BARD1 knockdown successfully inhibited the ubiquitination of LXRα (Fig. 2, C and D, and Fig. 5D), degradation of LXRα protein in the presence of cycloheximide was not fully relieved (Figs. 2B and 5C). It raises the possibility that other ubiquitin E3-ligase might be involved in LXRα protein ubiquitination even in the presence of LXR ligand, which is somewhat consistent with the observations that residual polyubiquitinated LXRα protein was detected in the presence of LXR ligand and BARD1 knockdown (Fig. 2C, lane 5; Fig. 2D, lane 7; Fig. 3B, lane 4; Fig. 5D, lanes 4 and 5). Furthermore, we cannot exclude the possibility that other protein-degrading complexes such as calpain and the 20S proteasome might contribute to the degradation of LXRα. Indeed, we observed that the level of LXRα protein was increased by ALLN, a calpain protease inhibitor, as well as MG132 (supplemental Fig. 5), proposing that calpain protease might also affect LXRα protein stability.
Here, we clearly demonstrated that the ligand-mediated activation of LXRα was protected from the ubiquitination and subsequent degradation. Similar to our results, the vitamin D receptor, PPARα, and PPARδ are stabilized by their ligands (38, 39, 40), whereas other nuclear receptors such as ERα, the glucocorticoid receptor, and PPARγ are degraded upon their own ligands (41, 42, 43). These contradictory effects may be explained by the following reasons: 1) in certain nuclear receptors, the ligand-induced stabilization of nuclear receptor is required for the initiation of transcriptional activation, 2) whereas, in other nuclear receptors, ligand-induced degradation is required for eliminating used protein complexes to reinitiate transcription by recruiting new ones or for preventing hyperactivation to maintain proper levels of particular gene products (40, 44).
BARD1/BRCA1 was first isolated from breast, ovarian, and prostate cancer cells the mutation of which is closely linked to the hereditary early onset of certain cancers (45), indicating that its inactivation is involved in cancer development. The observation that LXRα ubiquitination was facilitated by BARD1/BRCA1 (Fig. 5) led us to propose the hypothesis that inactivation of this complex might induce the dysregulation of LXRα target genes such as SREBP1c and fatty acid synthase, resulting in elevated lipogenesis. In this regard, it is notable that hormone-sensitive breast and prostate cancer cells exhibit increased de novo fatty acid synthesis (46), although it needs to be investigated whether the BARD1/BRCA1 complex is mutated or dysregulated in these cancer cells.
To examine the direct effects of BARD1/BRCA1 on the transcriptional activity of LXRα, we tried to overexpress or knock down BARD1/BRCA1 in RWPE1 cells. However, overexpression or knockdown of BARD1 did not reveal meaningful changes in LXRα target gene expression (data not shown). This is probably due to the multiple roles of BARD1/BRCA1 in RNA polymerase II-dependent transcription. Because BARD1/BRCA1 is able to ubiquitinate general transcription machinery such as RNA polymerase II, SWI-SNF complex, histone deacetylases, and nucleosome core histones (47, 48, 49, 50), it is possible that overexpression or knockdown of BARD1/BRCA1 would cause pleiotropic effects, which might be difficult to clarify the specific change in LXRα target gene expression. Instead, we monitored the changes of endogenous LXRα protein upon BARD1/BRCA1 by use of ChIP assay (Fig. 7, B and C), which would reflect the relative level of LXRα protein on its target gene promoters.
While we have been preparing this manuscript, Li et al. (51) recently reported that LXRα interacts with SIRT1 in a ligand-dependent manner, which promotes the deacetylation and subsequent ubiquitination of LXRα. Although they suggested that LXRα/retinoic X receptor ligand [22(R)-HC and 9-cis-retinoic acid] may promote LXRα degradation via the deacetylation of a K432 residue, we clearly observed that various LXR ligands enhanced the level of LXRα protein in vitro and in vivo (Figs. 2 and 6 and supplemental Fig. 6). Additionally, we noticed that both LXRα K431/432R (data not shown) and K7RLBD mutants were efficiently ubiquitinated in the absence of its ligand (Fig. 3C). Although it is likely that they might overlook the effect of cycloheximide, which can solely decrease the level of LXRα protein (supplemental Fig. 6C), further studies are required to clarify these contradictory results.
Collectively, we propose a novel model that a basal state of ligand-free LXRα protein would associate with BARD1/BRCA1 to keep LXRα protein at a low level. However, when LXRα ligand is available, a conformational change in the LBD of LXRα is induced, which dissociates BARD1/BRCA1 from activated LXRα to enhance LXRα protein stability and to facilitate the expressions of its target genes. Therefore, this study provides a novel insight into the molecular mechanism explaining the early process of transcriptional activation of LXRα with its ligands.
Materials and Methods
Reagents
T0901317 and MG132 were purchased from Calbiochem (Darmstadt, Germany). Cycloheximide and 22(R)-HC were provided by Sigma Aldrich (St. Louis, MO). GW3965 was a generous gift from Dr. Peter Tontonoz (University of California, Los Angeles, CA). A small-interference RNA duplex was obtained from Bioneer (Daejeon, Korea; siControl, SN-1003; siBARD1, catalog no. 1011382).
Cell culture and mouse experiment
HEK293 cells were maintained in DMEM supplemented with 10% fetal bovine serum at 37 C with 5% CO2. RWPE1, a prostate epithelial cell line, was cultured in keratinocyte-serum-free medium supplemented with 5 ng/ml human recombinant epidermal growth factor and 0.05 mg/ml bovine pituitary extract (Invitrogen, Carlsbad, CA; catalog no. 17005). T0901317 was dissolved at dimethylsulfoxide (5 mg/ml) and diluted 1:4 in distilled water. C57BL/6N (B6) male mice (7 wk) were ip injected with T0901317 (50 mg/kg) for 6 or 24 h.
Cloning and mutagenesis
Full-length mouse LXRα construct was subcloned into the myc/his-tagged pCDNA3.1 (LXRα-MH; Invitrogen) or the p3xFlag (LXRα-Flag; Sigma Aldrich) vector. Truncated forms of LXRα (ΔLBD and ΔDBD) were engineered by PCR amplification using internal primers. The LXRα K7RLBD and K4Rhinge mutant was generated by site-directed mutagenesis of pCDNA3.1-LXRα performed using the QuikChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). The Ub-HA expression vector was kindly provided by Dr. Kunsoo Rhee (Seoul National University, Seoul, Korea), and the expression vectors for BARD1-Flag and BRCA1-HA were a generous gift from Dr. Hyunsook Lee (Seoul National University). The expression vector for LXRβ was kindly gifted by Dr. Christopher K. Glass (University of California, San Diego, CA).
Transient transfection
For transient transfection, HEK293 cells were grown to 70% confluence and subsequently transfected with several DNA constructs according to the previously described calcium-phosphate method (52). To minimize the possibility of different transfection efficiency, LXRα-transfected HEK293 cells were split and subjected to secondary transfection with BARD1/BRCA1 and/or drug treatment. Transfection of the BARD1/BRCA1 or siRNA duplex for BARD1 into a RWPE1 cell was carried out by using the Lipofectamine2000 reagent according to the manufacturer’s instructions (Invitrogen).
Immunoprecipitation and Western blotting
The cells were lysed with radioimmunoprecipitation assay (RIPA) buffer [1% Triton X-100, 0.1% sodium deoxycholate, 140 mm NaCl, and Complete protease inhibitor cocktail (Roche, Rotkreuz, Switzerland)] and subjected to immunoprecipitation or Western blotting. For immunoprecipitation, equal amounts of the total cell extracts were incubated with the LXRα antibody, and the immunoprecipitated complexes were collected by using protein A-sepharose beads (GE Healthcare, Piscataway, NJ). Further, the pellets were washed three times with 1 ml RIPA buffer and subjected to Western blotting according to previously described protocols (14). The following antibodies were used in the Western blotting: glyceraldehyde-3-phosphate dehydrogenase (LabFrontier Co., Ltd., Seoul, Korea), β-tubulin (Sigma Aldrich), Flag tag (Sigma Aldrich), hemagglutinin (HA) tag (Covance Laboratories, Inc., Madison, WI), and Myc tag (Cell Signaling Technology, Beverly, MA). The LXRα and SREBP1 antibodies were produced by LabFrontier, using full-length LXRα and nuclear form of SREBP1c as an immunogen. Relative amounts of each protein were calculated by the LabWorks software (UVP Bioimaging Systems, Upland, CA).
In vivo ubiquitination assay
HEK293 cells were cotransfected with LXRα, HA-tagged ubiquitin expression vectors. The transfected cells were treated with MG132 (20 μm) for 3 h, and the total cell lysate was prepared with the RIPA buffer. The lysates cleared by centrifugation were immunoprecipitated with the LXRα antibody for 18 h at 4 C and protein A-sepharose beads for additional 3 h. After washing with the RIPA buffer, Western blotting was performed using the HA antibody.
ChIP
Transfected or T0901317-treated RWPE1 cells were cross-linked in 1% formaldehyde at room temperature for 10 min, and cross-linking was terminated by adding glycine (125 mm) for 2 min. After a rinse with PBS, cells were collected with buffer 1 [100 mm Tris-HCl (pH 9.4) and 10 mm dithiothreitol (DTT)]. For the isolation of crude nuclei, cell pellets were resuspended with buffer 2 [10 mm Tris-HCl (pH 8.0), 0.25% Triton X-100, 0.5% Nonidet P-40, 10 mm EDTA, 0.5 mm EGTA, and 1 mm DTT] for 10 min and precipitated with centrifugation. Collected nuclei was washed with buffer 3 [10 mm Tris (pH 8.0), 0.2 m NaCl, 1 mm EDTA, 0.5 mm EGTA, and 1 mm DTT] twice and resuspended in buffer 3 without NaCl for sonication step. After sonicated, equal amounts of chromatin solution were incubated in 1× RIPA buffer with LXRα antibody at 4 C for overnight. The immunoprecipitates were collected by adding protein A-sepharose beads and sequentially washed with modified TSE 1 [0.1% sodium dodecyl sulfate (SDS), 0.5% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl (pH 8.0), and 100 mm NaCl], mTSE 2 [0.1% SDS, 0.5% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl (pH 8.0), and 200 mm NaCl], mTSE3 [0.25 M LiCl, 0.5% Nonidet P-40, 1 mm EDTA, 10 mm Tris-HCl (pH 8.0)] and TE [10 mm Tris-HCl (pH 8.0) and 1 mm EDTA]. Then, immune complexes were eluted with elution buffer (1% SDS and 0.1 m NaHCO3), and the protein-DNA cross-linking was reversed by incubating at 65 C for 12 h with 200 mm of NaCl. DNA was extracted with phenol/chloroform and precipitated with ethanol and 20 μg of glycogen. Precipitated DNA was amplified by PCR and resolved in 6% polyacrylamide/1× Tris-borate-EDTA buffer. Primers used in this study were as follows: human ABCA1 sense, 5′-CCCAACTCCCTAGATGTGTC-3, and antisense, 5′-CCACTCACTCTCGCTCGCA-3′; and human SREBP1c sense, 5′-GCCAGCGAACCAGTGATTTC-3′, and antisense, 5′-GGGGGCTCGAGTTTCACC-3′.
Acknowledgments
We thank Drs. Kunsoo Rhee (Seoul National University), Hyunsook Lee (Seoul National University), Peter Tontonoz (University of California, Los Angeles), and Christopher Glass (University of California, San Diego) for helpful discussions and sharing material/plasmids. We thank Drs. Joo Won Lee, Yun Sok Lee, and Hyun Sun Jo for the critical reading of the manuscript.
NURSA Molecule Pages:
Ligands: 22α-Hydroxycholesterol | GW 3965 | T0901317;
Nuclear Receptors: LXRα.
Footnotes
This work was supported by the Korea Science and Engineering Foundation (KOSEF) through the BioDiscovery Research Program (M10748000258-08N4800-25810), the Stem Cell Research Center of the 21st Century Frontier Research Program (M108KL010006-08K1201-00630), and the National Research Laboratory Program (R0A-2004-000-10359-0). K.H.K., J.M.Y., and A.H.C. were supported by the BK21 Research Fellowship from the Ministry of Education, Science and Technology. J.B.K. was a Yonam Foundation Scholar.
Disclosure Summary: The authors have nothing to disclose.
First Published Online January 22, 2009
Abbreviations: ABC, ATP-binding cassette; AF-2, activation function 2; BARD1, BRCA1-associated RING domain 1; BRCA1, breast and ovarian cancer susceptibility 1; ChIP, chromatin immunoprecipitation; DBD, DNA-binding domain; DTT, dithiothreitol; ER, estrogen receptor; HA, hemagglutinin; HC, hydroxycholesterol; HEK, human embryonic kidney; LBD, ligand-binding domain; LXR, liver X receptor; PPAR, peroxisome proliferator-activated receptor; SDS, sodium dodecyl sulfate; RIPA, radioimmunoprecipitation assay; siRNA, small interfering RNA; SREBP1c, sterol regulatory element-binding protein 1c; WT, wild type.
References
- 1.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O'Connell RM, Cheng G, Saez E, Miller JF, Tontonoz P2004. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell 119:299–309 [DOI] [PubMed] [Google Scholar]
- 2.Kalaany NY, Gauthier KC, Zavacki AM, Mammen PP, Kitazume T, Peterson JA, Horton JD, Garry DJ, Bianco AC, Mangelsdorf DJ2005. LXRs regulate the balance between fat storage and oxidation. Cell Metab 1:231–244 [DOI] [PubMed] [Google Scholar]
- 3.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ1998. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR α. Cell 93:693–704 [DOI] [PubMed] [Google Scholar]
- 4.Laffitte BA, Repa JJ, Joseph SB, Wilpitz DC, Kast HR, Mangelsdorf DJ, Tontonoz P2001. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc Natl Acad Sci USA 98:507–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naik SU, Wang X, Da Silva JS, Jaye M, Macphee CH, Reilly MP, Billheimer JT, Rothblat GH, Rader DJ2006. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation 113:90–97 [DOI] [PubMed] [Google Scholar]
- 6.Annicotte JS, Schoonjans K, Auwerx J2004. Expression of the liver X receptor α and β in embryonic and adult mice. Anat Rec A Discov Mol Cell Evol Biol 277:312–316 [DOI] [PubMed] [Google Scholar]
- 7.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ1996. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature 383:728–731 [DOI] [PubMed] [Google Scholar]
- 8.Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB, Parks DJ, Wilson JG, Tippin TK, Binz JG, Plunket KD, Morgan DG, Beaudet EJ, Whitney KD, Kliewer SA, Willson TM2002. Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J Med Chem 45:1963–1966 [DOI] [PubMed] [Google Scholar]
- 9.Tontonoz P, Mangelsdorf DJ2003. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol 17:985–993 [DOI] [PubMed] [Google Scholar]
- 10.Yoshikawa T, Shimano H, Amemiya-Kudo M, Yahagi N, Hasty AH, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Kimura S, Ishibashi S, Yamada N2001. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol Cell Biol 21:2991–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, Collins JL, Osborne TF, Tontonoz P2002. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J Biol Chem 277:11019–11025 [DOI] [PubMed] [Google Scholar]
- 12.Cha JY, Repa JJ2007. The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J Biol Chem 282:743–751 [DOI] [PubMed] [Google Scholar]
- 13.Chisholm JW, Hong J, Mills SA, Lawn RM2003. The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res 44:2039–2048 [DOI] [PubMed] [Google Scholar]
- 14.Choe SS, Choi AH, Lee JW, Kim KH, Chung JJ, Park J, Lee KM, Park KG, Lee IK, Kim JB2007. Chronic activation of liver X receptor induces β-cell apoptosis through hyperactivation of lipogenesis: liver X receptor-mediated lipotoxicity in pancreatic β-cells. Diabetes 56:1534–1543 [DOI] [PubMed] [Google Scholar]
- 15.Xu L, Glass CK, Rosenfeld MG1999. Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev 9:140–147 [DOI] [PubMed] [Google Scholar]
- 16.Treuter E, Johansson L, Thomsen JS, Warnmark A, Leers J, Pelto-Huikko M, Sjoberg M, Wright AP, Spyrou G, Gustafsson JA1999. Competition between thyroid hormone receptor-associated protein (TRAP) 220 and transcriptional intermediary factor (TIF) 2 for binding to nuclear receptors. Implications for the recruitment of TRAP and p160 coactivator complexes. J Biol Chem 274:6667–6677 [DOI] [PubMed] [Google Scholar]
- 17.Hu X, Li S, Wu J, Xia C, Lala DS2003. Liver X receptors interact with corepressors to regulate gene expression. Mol Endocrinol 17:1019–1026 [DOI] [PubMed] [Google Scholar]
- 18.Svensson S, Ostberg T, Jacobsson M, Norstrom C, Stefansson K, Hallen D, Johansson IC, Zachrisson K, Ogg D, Jendeberg L2003. Crystal structure of the heterodimeric complex of LXRα and RXRβ ligand-binding domains in a fully agonistic conformation. EMBO J 22:4625–4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK2005. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 437:759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C, Lisanti MP, Katzenellenbogen BS, Kato S, Hopp T, Fuqua SA, Lopez GN, Kushner PJ, Pestell RG2001. Direct acetylation of the estrogen receptor α hinge region by p300 regulates transactivation and hormone sensitivity. J Biol Chem 276:18375–18383 [DOI] [PubMed] [Google Scholar]
- 21.Cui Y, Zhang M, Pestell R, Curran EM, Welshons WV, Fuqua SA2004. Phosphorylation of estrogen receptor α blocks its acetylation and regulates estrogen sensitivity. Cancer Res 64:9199–9208 [DOI] [PubMed] [Google Scholar]
- 22.Eakin CM, Maccoss MJ, Finney GL, Klevit RE2007. Estrogen receptor α is a putative substrate for the BRCA1 ubiquitin ligase. Proc Natl Acad Sci USA 104:5794–5799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL2006. Acetylation of estrogen receptor α by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol 20:1479–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tateishi Y, Kawabe Y, Chiba T, Murata S, Ichikawa K, Murayama A, Tanaka K, Baba T, Kato S, Yanagisawa J2004. Ligand-dependent switching of ubiquitin-proteasome pathways for estrogen receptor. EMBO J 23:4813–4823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan M, Nakshatri H, Nephew KP2004. Inhibiting proteasomal proteolysis sustains estrogen receptor-α activation. Mol Endocrinol 18:2603–2615 [DOI] [PubMed] [Google Scholar]
- 26.Chen M, Bradley MN, Beaven SW, Tontonoz P2006. Phosphorylation of the liver X receptors. FEBS Lett 580:4835–4841 [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto T, Shimano H, Inoue N, Nakagawa Y, Matsuzaka T, Takahashi A, Yahagi N, Sone H, Suzuki H, Toyoshima H, Yamada N2007. Protein kinase A suppresses sterol regulatory element-binding protein-1C expression via phosphorylation of liver X receptor in the liver. J Biol Chem 282:11687–11695 [DOI] [PubMed] [Google Scholar]
- 28.Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK2007. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARγ. Mol Cell 25:57–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P2003. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med 9:213–219 [DOI] [PubMed] [Google Scholar]
- 30.Baer R, Ludwig T2002. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr Opin Genet Dev 12:86–91 [DOI] [PubMed] [Google Scholar]
- 31.Starita LM, Parvin JD2003. The multiple nuclear functions of BRCA1: transcription, ubiquitination and DNA repair. Curr Opin Cell Biol 15:345–350 [DOI] [PubMed] [Google Scholar]
- 32.Poole AJ, Li Y, Kim Y, Lin SC, Lee WH, Lee EY2006. Prevention of Brca1-mediated mammary tumorigenesis in mice by a progesterone antagonist. Science 314:1467–1470 [DOI] [PubMed] [Google Scholar]
- 33.Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ1995. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev 9:1033–1045 [DOI] [PubMed] [Google Scholar]
- 34.Ulven SM, Dalen KT, Gustafsson JA, Nebb HI2004. Tissue-specific autoregulation of the LXRα gene facilitates induction of apoE in mouse adipose tissue. J Lipid Res 45:2052–2062 [DOI] [PubMed] [Google Scholar]
- 35.Wagner BL, Valledor AF, Shao G, Daige CL, Bischoff ED, Petrowski M, Jepsen K, Baek SH, Heyman RA, Rosenfeld MG, Schulman IG, Glass CK2003. Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol Cell Biol 23:5780–5789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seo JB, Moon HM, Kim WS, Lee YS, Jeong HW, Yoo EJ, Ham J, Kang H, Park MG, Steffensen KR, Stulnig TM, Gustafsson JA, Park SD, Kim JB2004. Activated liver X receptors stimulate adipocyte differentiation through induction of peroxisome proliferator-activated receptor γ expression. Mol Cell Biol 24:3430–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bello D, Webber MM, Kleinman HK, Wartinger DD, Rhim JS1997. Androgen responsive adult human prostatic epithelial cell lines immortalized by human papillomavirus 18. Carcinogenesis 18:1215–1223 [DOI] [PubMed] [Google Scholar]
- 38.Genini D, Catapano CV2007. Block of nuclear receptor ubiquitination. A mechanism of ligand-dependent control of peroxisome proliferator-activated receptor δ activity. J Biol Chem 282:11776–11785 [DOI] [PubMed] [Google Scholar]
- 39.Li XY, Boudjelal M, Xiao JH, Peng ZH, Asuru A, Kang S, Fisher GJ, Voorhees JJ1999. 1,25-Dihydroxyvitamin D3 increases nuclear vitamin D3 receptors by blocking ubiquitin/proteasome-mediated degradation in human skin. Mol Endocrinol 13:1686–1694 [DOI] [PubMed] [Google Scholar]
- 40.Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C2002. Peroxisome proliferator-activated receptor α (PPARα) turnover by the ubiquitin-proteasome system controls the ligand-induced expression level of its target genes. J Biol Chem 277:37254–37259 [DOI] [PubMed] [Google Scholar]
- 41.Nawaz Z, Lonard DM, Dennis AP, Smith CL, O'Malley BW1999. Proteasome-dependent degradation of the human estrogen receptor. Proc Natl Acad Sci USA 96:1858–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wallace AD, Cidlowski JA2001. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem 276:42714–42721 [DOI] [PubMed] [Google Scholar]
- 43.Hauser S, Adelmant G, Sarraf P, Wright HM, Mueller E, Spiegelman BM2000. Degradation of the peroxisome proliferator-activated receptor γ is linked to ligand-dependent activation. J Biol Chem 275:18527–18533 [DOI] [PubMed] [Google Scholar]
- 44.Blanquart C, Mansouri R, Fruchart JC, Staels B, Glineur C2004. Different ways to regulate the PPARα stability. Biochem Biophys Res Commun 319:663–670 [DOI] [PubMed] [Google Scholar]
- 45.Rosen EM, Fan S, Pestell RG, Goldberg ID2003. BRCA1 in hormone-responsive cancers. Trends Endocrinol Metab 14:378–385 [DOI] [PubMed] [Google Scholar]
- 46.Kuhajda FP2000. Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition 16:202–208 [DOI] [PubMed] [Google Scholar]
- 47.Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, Wang W, Kashanchi F, Shiekhattar R2000. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell 102:257–265 [DOI] [PubMed] [Google Scholar]
- 48.Kleiman FE, Wu-Baer F, Fonseca D, Kaneko S, Baer R, Manley JL2005. BRCA1/BARD1 inhibition of mRNA 3′ processing involves targeted degradation of RNA polymerase II. Genes Dev 19:1227–1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mallery DL, Vandenberg CJ, Hiom K2002. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J 21:6755–6762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yarden RI, Brody LC1999. BRCA1 interacts with components of the histone deacetylase complex. Proc Natl Acad Sci USA 96:4983–4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L2007. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 28:91–106 [DOI] [PubMed] [Google Scholar]
- 52.Kim KH, Choi SH, Lee TS, Oh WK, Kim DS, Kim JB2006. Selective LXRα inhibitory effects observed in plant extracts of MEH184 (Parthenocissua tricuspidata) and MEH185 (Euscaphis japonica). Biochem Biophys Res Commun 349:513–518 [DOI] [PubMed] [Google Scholar]