

Abstract
Nicotinamide adenine dinucleotide phosphate (NADPH) enhances Ca2+-induced exocytosis in pancreatic β-cells, an effect suggested to involve the cytosolic redox protein glutaredoxin-1 (GRX-1). We here detail the role of GRX-1 in NADPH-stimulated β-cell exocytosis and glucose-stimulated insulin secretion. Silencing of GRX-1 by RNA interference reduced glucose-stimulated insulin secretion in both clonal INS-1 832/13 cells and primary rat islets. GRX-1 silencing did not affect cell viability or the intracellular redox environment, suggesting that GRX-1 regulates the exocytotic machinery by a local action. By contrast, knockdown of the related protein thioredoxin-1 (TRX-1) was ineffective. Confocal immunocytochemistry revealed that GRX-1 locates to the cell periphery, whereas TRX-1 expression is uniform. These data suggest that the distinct subcellular localizations of TRX-1 and GRX-1 result in differences in substrate specificities and actions on insulin secretion. Single-cell exocytosis was likewise suppressed by GRX-1 knockdown in both rat β-cells and clonal 832/13 cells, whereas after overexpression exocytosis increased by approximately 40%. Intracellular addition of NADPH (0.1 mm) stimulated Ca2+-evoked exocytosis in both cell types. Interestingly, the stimulatory action of NADPH on the exocytotic machinery coincided with an approximately 30% inhibition in whole-cell Ca2+ currents. After GRX-1 silencing, NADPH failed to amplify insulin release but still inhibited Ca2+ currents in 832/13 cells. In conclusion, NADPH stimulates the exocytotic machinery in pancreatic β-cells. This effect is mediated by the NADPH acceptor protein GRX-1 by a local redox reaction that accelerates β-cell exocytosis and, in turn, insulin secretion.
The redox regulatory protein glutaredoxin-1 locates to the cellular membrane of β-cells and enhances insulin exocytosis through a local redox reaction with NADPH.
Aberrant regulation of the redox environment is relevant for the pathogenesis of many human diseases (1, 2, 3), including diabetic complications (4, 5). The intracellular redox environment is normally maintained in a highly reduced state, which is the net result of activities in redox pairs. The most important extramitochondric redox pairs are the oxidative/reduced forms of glutathione (GSSG/GSH), glutaredoxin-1 and thioredoxin-1 (6, 7). They use the reducing equivalent nicotinamide adenine dinucleotide phosphate (NADPH) to generate reductive power that is funnelled onto target proteins. However, because NADPH is also a substrate for oxidases that generate reactive oxygen species (ROS), the availability of NADPH might simultaneously also result in generation of oxidants (8).
In agreement with the fact that the NADPH/NADP+ ratio increases during glucose-stimulated insulin release (9), we have previously shown that intracellular addition of NADPH (EC50 45 μm) enhances Ca2+-induced exocytosis of insulin granules in pancreatic β-cells and suggested that glutaredoxin-1 is involved in this process (10). However, in that previous study it was not possible to determine whether the addition of NADPH resulted in a net oxidization or reduction of the cytosolic redox environment. Another possibility that could not be discarded was that a loss of cell integrity when using the standard whole-cell configuration of the patch-clamp technique could have shifted the redox balance toward an oxidized state, meaning that addition of NADPH simply restored the normal reduced intracellular redox environment.
Therefore, in this work we set out to determine the exact role of glutaredoxin-1 for NADPH-stimulated insulin granule exocytosis and insulin secretion. To this end we have manipulated glutaredoxin-1 expression in clonal insulin-releasing cells and primary rat islets, followed by assaying associated changes in intracellular redox environment as differences in thiol concentration, as well as studied effects on cell viability, insulin secretion, and single-cell exocytosis. These data lead us to the conclusion that the NADPH-dependent stimulation of insulin release is specifically mediated by glutaredoxin-1, which acts by a local redox reaction, rather than a general change in the β-cell redox environment. This redox reaction targets the β-cell exocytotic machinery and amplifies glucose-dependent insulin secretion.
Results
Silencing of glutaredoxin-1 suppresses glucose-induced insulin secretion
Expression of glutaredoxin-1 (GRX-1), the extramitochondrial isoform of glutaredoxin, was demonstrated in INS-1 832/13 cells by real-time PCR and Western blotting (Fig. 1, A and B). We then silenced GRX-1 expression using small interfering RNA (siRNA) oligos against GRX-1 [small interfering GRX-1 (siGRX-1)], which transfected the cells with 90–100% efficacy (data not shown). GRX-1 silencing was verified by real-time PCR and accounted to 78 ± 8%. Reduction of GRX-1 protein levels was shown by immunoblotting (Fig. 1B). Unspecific effects exerted by the siGRX-1 oligo were tested for by using a negative control siRNA oligo with an unspecific target sequence (control), as well as verifying unaltered expression of the related thioredoxin-1 protein (TRX-1) by immunoblotting (Fig. 1B). Using a TRX-1 siRNA oligo we could achieve 79 ± 6% down-regulation of mRNA expression (Fig. 1C).
Fig. 1.

Lipofection of INS-1 832/13 cells with specific siRNAs against GRX-1 (siGRX-1) or TRX-1 (siTRX-1) reduced mRNA levels relative to HPRT as revealed by real-time PCR analysis, when compared with an inactive control oligo (control; A and C). GRX-1 protein decreased after siGRX-1 treatment, but TRX-1 and actin protein levels remained unchanged as shown by Western blot analysis (B). D, Insulin secretion assays revealed a failure of glucose-stimulated insulin secretion (16.7 mm, gray bars) after GRX-1 knockdown (siGRX-1) in INS-1 832/13, whereas secretion was unchanged upon TRX-1 knockdown (siTRX-1) when compared with the ineffective control siRNA (control). Cells treated with the lipofection reagent only (mock) secreted comparable amounts of insulin in relation to the control group (D). Both thiol concentration (E) and cell viability, assayed by TB (F), did not differ between the knockdown (black bars) and the control groups of INS-1 832/13 cells (gray bars). The suppression of GRX-1 mRNA expression by siGRX-1 (G) resulted in significantly reduced insulin release also in rat pancreatic islets (H). Data shown represent averages ± sem. *, P < 0.05; **, P < 0.005.
We then measured insulin secretion after GRX-1 silencing. In cells treated with a nontargeting siRNA oligo (control), insulin release at basal glucose concentrations (2.8 mm) measured 5.2 ± 0.8 ng/mg · h, which increased 3-fold to 14.8 ± 0.9 ng/mg · h in high (16.7 mm) glucose (P < 0.001; n = 15, Fig. 1D). In cells treated with the lipofection reagent only (mock), insulin secretion increased from 5.4 ± 0.7 to 20.7 ± 3.4 ng/mg · h upon glucose stimulation (P < 0.001, n = 15). After silencing of GRX-1, high glucose failed to stimulate insulin release, which essentially remained unchanged in high compared with basal glucose concentrations (5.7 ± 0.9 and 6.2 ± 1 ng/mg · h, respectively; n = 10). Comparing GRX-1-silenced with control cells, insulin secretion in high glucose was reduced by 62% (siGRX-1 5.7 ± 0.9 vs. 14.8 ± 0.9 ng/mg · h in controls, P < 0.001; n = 10/15) whereas basal secretion was unchanged (siGRX-1 6.2 ± 1 vs. 5.2 ± 0.8 ng/mg · h in controls; n = 10/15).
By contrast, silencing of TRX-1, which is structurally and functionally related to GRX-1, did not affect insulin secretion. Insulin release stimulated by high glucose was comparable to that of the controls (siTRX-1 14.5 ± 1.4 vs. 14.8 ± 0.9 ng/mg/h in controls; n = 10/15).
We then investigated the effects of GRX-1 silencing on insulin release in primary rat islets. This was achieved by culture with siRNA oligos for 72 h, which permeated well into the islets (see Fig. 5M). The reduction in GRX-1 mRNA averaged 83 ± 10% (Fig. 1G). In controls, high glucose induced a 3.5-fold increase in insulin secretion over basal (26 ± 5.1 in high vs. 7.3 ± 1.9 ng/ml · h in low glucose, Fig. 1H), which was suppressed by 46% (14.2 ± 3.2 vs. 7.7 ± 1.4 ng/ml · h in high and low glucose, respectively; P = 0.02, n = 4) after GRX-1 knockdown. Insulin release at basal glucose concentrations was unaltered.
Fig. 5.

Role of GRX-1 knockdown for NADPH-mediated effects on single-cell exocytosis and whole-cell Ca2+ currents in primary rat β-cells. A, Depolarization-evoked (V) increases in cell capacitance (ΔC) in control cells with (+; in gray) or without (−; in black) NADPH (0.1 mm) added intracellularly. NADPH had a strong stimulatory effect on exocytosis (A). The effect on the response to the first pulse (ΔCdepol 1; panel B) was milder than on the response to later pulses (ΔCdepol2-10; panel C). Total exocytosis (ΔCTOT; panel D) was increased. E, Typical examples of whole-cell Ca2+ current recordings (E) in control cells with (in gray) or without (in black) NADPH added. F, Average peak Ca2+ currents (IPEAK) were significantly reduced. The knockdown of GRX-1 with siRNA (siGRX-1) reduced the exocytotic response significantly (G and J). The early response was unchanged (H), and especially the late response was affected (I). Whole-cell Ca2+ currents were similar (K and L). M, Confocal microscopy of pancreatic islets transfected with siGRX-1 and simultaneously a fluorescein-labeled dsRNA oligomer reveal the efficient uptake and distribution of siRNA throughout the islets (−20 to +20 μm in 10-μm steps from the islet center at 0 μm). The knockdown of GRX-1 now rendered NADPH an ineffective stimulator of exocytosis (N–Q). A mild inhibition on Ca2+ currents remained (R and S). Scale bar, 50 μm. Bar graphs represent average values ± sem. *, P < 0.05. ms, Milliseconds.
GRX-1 silencing does not influence cell viability or intracellular redox environment
Subsequently, we investigated how GRX-1 silencing affected the cellular redox state. This was accomplished by measuring the concentration of thiol (-SH) groups by colorimetry (Fig. 1E). GRX-1 silencing did not affect the thiol concentration in INS-1 832/13 cells. It measured 44.2 ± 17.4 nm in those treated with the control oligo and 44.9 ± 15.9 nm in GRX-1 silenced cells (P > 0.05; n = 3 in both conditions). We also determined cell viability by Trypan blue (TB) stainings. The fraction of viable, TB-negative cells in cultures treated with the control siRNA amounted to 92 ± 1.5% at d 1, 89 ± 1.9% at d 2, and 88 ± 2.1% at d 3 (n = 3). This was not significantly different from that observed in cell cultures treated with siGRX-1 and the corresponding fractions of viable, TB-negative cells were 92 ± 0.6%, 90 ± 1.2%, and 90 ± 0.9% (n = 3), at d 1, 2, and 3, respectively (Fig. 1F).
The subcellular localization of GRX-1 and TRX-1 differs
To compare the cellular localization of GRX-1 with the one of TRX-1, confocal immunocytochemical stainings in INS-1 832/13 cells and dispersed primary rat islet cells were made. Subsequent imaging revealed a distinct subcellular expression pattern. GRX-1 was found to be present in the cytosol but was mainly accumulated in close proximity to the cell membrane (Fig. 2), whereas TRX-1 was evenly distributed throughout the cytosol. Intensity profiles of the respective fluorescent dye distribution support these results (Fig. 2, D, E, I, and J).
Fig. 2.
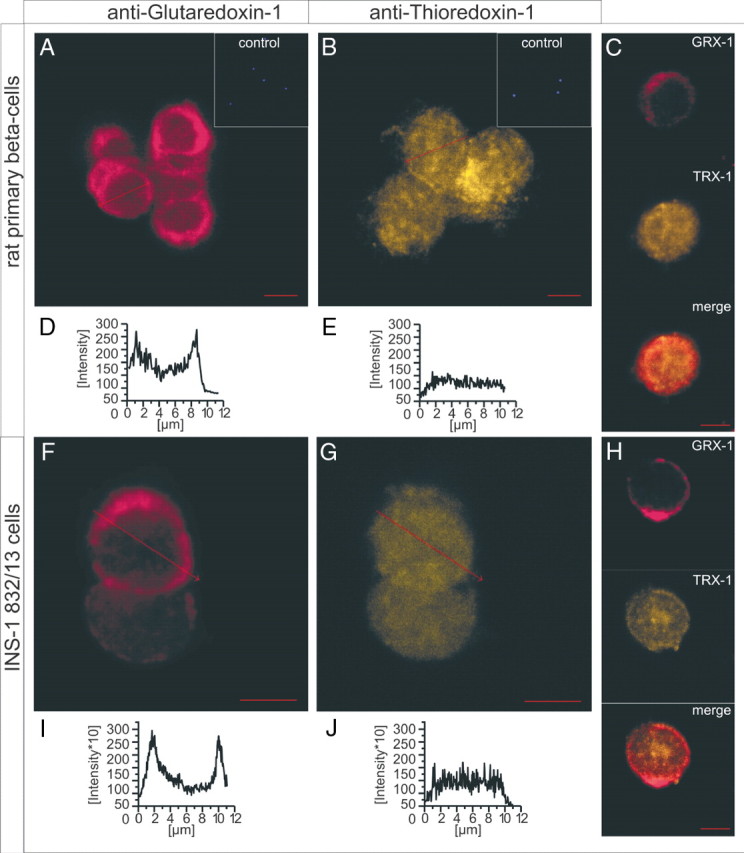
Confocal immunohistochemistry of GRX-1 and TRX-1 protein in rat primary (A–E) and clonal INS-1 832/13 (F–J) cells. GRX-1 is primarily located in structures close to the plasma membrane in rat primary (A), as well as in clonal INS-1 832/13 cells (F), as revealed by stainings and their respective intensity profiles (D and I). TRX-1 appears mainly as even distribution throughout the cytosol of primary rat (B) and clonal (G) cells, underlined by the respective intensity profiles (E and J). The same expression pattern is detected in single cells of rat islets (C) and clonal cells (H) double stained for GRX-1 (C and H, upper panels) and TRX-1 (middle panels). C and H (lower panels) show the merged picture. Insets (A and B) depict controls with omitted primary antibody and final DAPI staining to exclude unspecificity of the secondary antibody. Scale bars, 5 μm.
GRX-1 controls both NADPH-dependent and -independent exocytosis in INS-1 832/13 cells
To directly evaluate the role of GRX-1 for exocytosis in INS-1 823/13 cells, we next silenced GRX-1 expression by vector-based RNA interference. A short hairpin GRX-1 (shGRX-1) sequence incorporated in a cGFP [coral green fluorescent protein (GFP)]- expressing vector was transfected into the cells via lipofection. After 48 h, coral GFP-positive cells were selected for patch clamp experiments.
Single-cell exocytosis was measured by capacitance recordings that report net changes in membrane surface area and was elicited by a train of ten 500-msec voltage clamp depolarizations from −70 mV to zero.
In cells transfected with a scrambled nontargeting short hairpin RNA (shRNA), the train stimulus evoked an average increase in cell capacitance reporting exocytosis that amounted 182 ± 28 fF (femtofarads), to be compared with the 77 ± 9 fF in cells transfected with active shGRX-1 (P < 0.05; n = 7 and 4; Fig. 3, D and G), corresponding to a decrease in exocytosis by approximately 60%. The main effect was detected during late-phase exocytosis (Fig. 3F) whereas exocytosis after the first depolarization was not changed by GRX-1 knockdown (Fig. 3E).
Fig. 3.
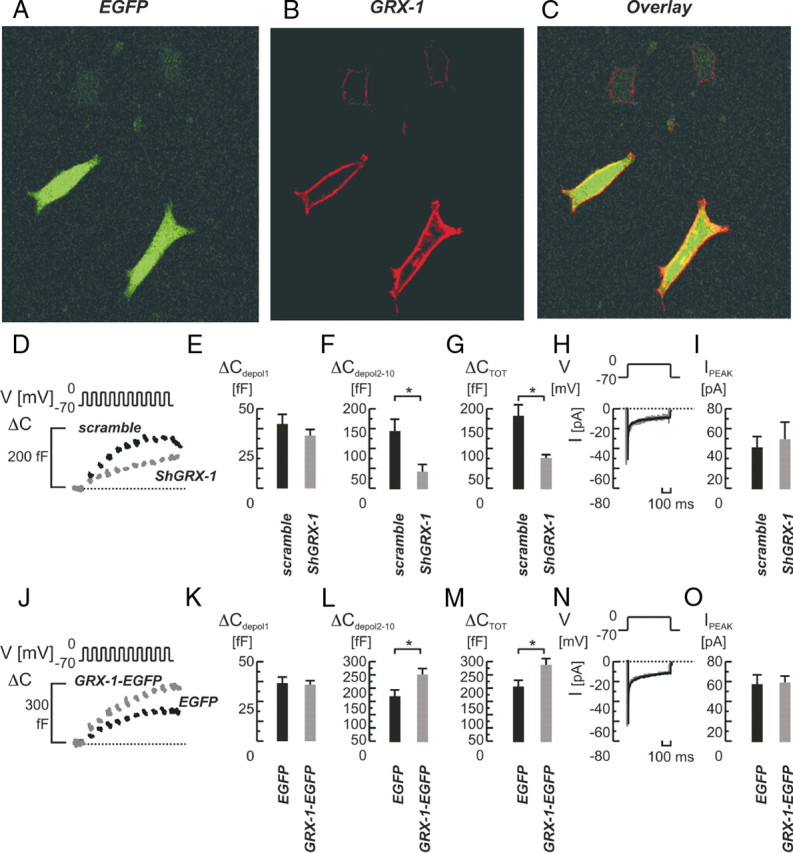
A–C, Confocal immunohistochemistry of GRX-1 protein in cells lipofected with a vector containing both EGFP and GRX-1. A, EGFP; B, GRX-1; C, overlay. Note the membranous localization of GRX-1 and the increased GRX-1 fluorescence in GFP-expressing (transfected) cells compared with the lower endogenous expression of GRX-1 in nontransfected cells (B, upper cells). D–O, Effects of GRX-1 knockdown (D–I) and overexpression (J–O) on single-cell exocytosis and whole-cell Ca2+ currents. D, Depolarization-evoked (V) increases in cell membrane capacitance (ΔC) in INS-1 832/13 cells lipofected with a vector expressing a shGRX-1 sequence (shGRX-1, in gray) or scrambled control sequence (scramble; in black). The early component of exocytosis in response to the train stimulus remained unaffected (ΔCdepol 1; panel E), whereas the late component was suppressed (ΔCdepol2-10; panel F), as well as the total exocytotic response (ΔCTOT; panel G). H, Whole-cell Ca2+-currents (I[Pa]) recorded under the respective condition. I, Average values of peak currents (IPEAK) were not significantly changed. J–O, as in panels D–I, but GRX-1 was overexpressed using a GRX-1-EGFP-expressing vector (GRX-1-EGFP; in gray) and the empty vector (EGFP) as control (in black). Note the increases in late-phase (ΔCdepol2-10; panel L) and total exocytosis (ΔCTOT; panel M). Bar graphs represent average values ± sem. *, P < 0.05. ms, Milliseconds.
Subsequently, we performed overexpression studies using an enhanced GFP (EGFP)-expressing vector with inserted GRX-1 sequence (GRX-1-EGFP) and the empty vector as control (EGFP). The GRX-1-EGFP-transfected cells expressed EGFP, and fluorescence intensities for GRX-1 coimmunostainings were typically more than 5-fold enhanced in those cells, demonstrating up-regulated GRX-1 expression compared with surrounding nontransfected cells (Fig. 3, A–C). In clonal control cells transfected with the empty EGFP-containing vector (EGFP), the train depolarization resulted in an average increase in cell capacitance of 207 ± 23 fF (n = 5; Fig. 3, J and M), not significantly different from that seen in cells using the scrambled inactive shRNA (182 ± 27 fF; Fig. 3, D and G). In contrast, GRX-1-EGFP transfected cells exhibited an increased exocytotic response, and increases in cell capacitance averaged 288 ± 24 fF (P < 0.05, EGFP vs. GRX-1-EGFP; n = 5 and 8, respectively; Fig. 3, J and M). Again, the effect was seen only during late-phase exocytosis (Fig. 3L), and exocytosis in response to the first depolarization remained unchanged (Fig. 3K). The stimulation of exocytosis in cells with up-regulated GRX-1 was not associated with any changes in whole-cell Ca2+ currents that remained 59 ± 7 pA, to be compared with the 58 ± 10 pA observed in cells expressing only EGFP (n = 8 and 5; Fig. 3, N and O).
We then investigated the role of GRX-1 in NADPH-stimulated exocytosis in INS-1 832/13 cells (Fig. 4). When adding NADPH (0.1 mm) intracellularly to cells treated with an inactive control siRNA (control), depolarization-evoked exocytosis was increased. The stimulation amounted to approximately 30% from an average 256 ± 29 to 329 ± 17 fF in the absence and presence of NADPH, respectively (P < 0.05; n = 9 for both; Fig. 4, A and D). The stimulatory effect of NADPH on exocytosis was consistently detected in response to the latter part (depolarizations 2–10) of the train stimulus (222 ± 28 fF in control vs. 292 ± 13 fF in NADPH-treated cells; Fig. 4C), whereas the first pulse delivered the same response in both groups (34 ± 3 fF in control vs. 37 ± 4 fF in NADPH-treated cells; Fig. 4B).
Fig. 4.

Role of GRX-1 for NADPH-mediated effects on single-cell exocytosis and whole-cell Ca2+ currents in INS-1 832/13 cells. A, Depolarization-evoked (V) increases in cell capacitance (ΔC) in cells treated with nontargeting siRNA (control), with (+; in gray) or without (−; in black) NADPH (0.1 mm) added intracellularly. On average, NADPH significantly stimulated total exocytosis (ΔCTOT; panel D), by increasing the exocytotic response from the second depolarizing pulse onwards (ΔCdepol2-10; panel C) whereas the response to the first pulse was unchanged (ΔCdepol 1; panel B). E, Whole cell Ca2+-currents (I) in control cells recorded with (in gray) or without (in black) NADPH added. F, Average peak Ca2+ currents (IPEAK) were significantly inhibited by NADPH. G–L, as in panels A–F, but after knockdown of GRX-1 (siGRX-1). Note the failure of NADPH to affect exocytosis, whereas the inhibitory action on Ca2+ currents remains intact. Bar graphs represent average values ± sem. *, P < 0.05. ms, Milliseconds.
The NADPH-dependent stimulation of exocytosis coincided with a significant 29% reduction in average whole-cell Ca2+ currents (59 ± 5 in control vs. 42 ± 6 pA in NADPH-treated cells; P < 0.05; n = 9 for both). Subsequently, we repeated the experiments with NADPH in GRX-1-silenced cells. Interestingly, the addition of NADPH was now ineffective, and capacitance increases remained comparable to those detected in control cells (133 ± 17 fF vs. 123 ± 14 fF in controls; Fig. 4, G and J). By contrast, silencing of GRX-1 did not prevent the inhibitory effect of NADPH on whole-cell Ca2+ currents, which were decreased by 24% (control 59 ± 5 pA vs. NADPH 45 ± 5 pA; P < 0.05; Fig. 4, K and L).
GRX-1 controls exocytosis and mediates effects of NADPH in rat β-cells
In a final approach we evaluated the effects of NADPH in primary rat β-cells and assessed the role of GRX-1 in this process. Addition of NADPH overall stimulated train-evoked exocytosis (Fig. 5, A–D), but, in contrast to the situation in INS-1 832/13 cells, this affected both the early and late components of exocytosis.
We then explored how GRX-1 silencing affected the exocytotic capacity. Islets were therefore cultured with the silencing siGRX-1 oligo and a fluorescein-labeled nontargeting double-stranded RNA (dsRNA) oligo for 48 h, followed by assessment of transfection efficacy by fluorescein intensity imaging (Fig. 5M). After the dispersal of islets into single cells, they were used for experiments within the next 24 h. GRX-1 silencing was also verified by real-time PCR (Fig. 1G) and reduced total exocytosis, evoked by a train stimulus, by 38% from 231 ± 42 fF observed in control cells to 143 ± 28 fF in siGRX-1-treated cells (Fig. 5, G and J). In contrast to the situation in INS-1 832/13 cells, mainly the early component of exocytosis was influenced (Fig. 5H).
The effects of NADPH on whole-cell Ca2+ currents were evident also in primary rat β-cells. A modest, but significant, 29% reduction in averaged whole-cell Ca2+ currents (P < 0.05; n = 6 and 8 in the absence or presence of NADPH, respectively; Fig. 5F) was observed. As in INS-1 832/13 cells, GRX-1 silencing did not affect Ca2+ currents (Fig. 5L).
Finally, we repeated the experiments adding NADPH to the pipette while patching GRX-1-silenced primary rat β-cells. As in clonal cells, the supplementation of NADPH was now ineffective, and capacitance increases remained comparable to those detected in the absence of the nucleotide (nt) (171 ± 37 fF vs. 143 ± 28 fF in the presence vs. absence of NADPH; Fig. 5, N–Q). The inhibitory effect on whole-cell Ca2+ currents in primary GRX-1-silenced cells was milder than in clonal cells but was also detectable here (168 ± 15 pA in control vs. 137 ± 15 pA in NADPH-treated cells; Fig. 5S).
Discussion
In this study, we have investigated redox regulation of insulin release considering the hypothesis that glucose- and Ca2+-dependent insulin secretion is controlled by the cytosolic redox environment, as suggested by previous studies performed in our laboratory (10). We here investigated whether the actions of NADPH are mediated by either the extramitochondrial redox protein GRX-1, the related TRX-1, or both. In the following, we will discuss our results and their implications for human disease and type 2 diabetes mellitus (T2DM) in which inadequate insulin release plays a major role.
GRX-1 controls the efficacy of insulin secretion
The present data clearly demonstrate that GRX-1 activity stimulates β-cell exocytosis and insulin secretion, both in clonal and primary β-cells (Figs. 1, 3, and 5). Moreover, GRX-1 mediates the stimulatory effect of NADPH on insulin exocytosis and secretion. The later action is exerted on the exocytotic machinery at a functional step distal to the elevation of [Ca2+]i, as previously demonstrated in our laboratory (10). This occurs in a manner similar to e.g. cAMP (11) and GTP derivatives (12, 13) that amplify Ca2+-dependent insulin exocytosis.
Interestingly, it appears that GRX-1 activity stimulates insulin secretion, whereas TRX-1 has no such effect. Why do the actions of these redox proteins differ? First of all, the proteins may have intrinsically different substrate recognition features, because structural studies of TRX show that although it shares the mechanism with GRX for correctly positioning substrate cysteine residues at the catalytic groups, it has its own unique structural element that allows recognition of protein disulfides (14).
Furthermore, we show that GRX-1 and TRX-1 proteins exhibit different subcellular localization patterns (Fig. 2). GRX-1 is accumulated in the cell periphery in the cellular region where exocytotic activities take place. This suggests that GRX-1 and TRX-1 target different proteins in insulin-secreting cells, and that the cellular localization of GRX-1 may underlie its particular importance for controlling the insulin-release machinery. A similar distribution pattern for GRX-1 has been reported in the neuropituitary in which GRX-1 localizes to the nerve terminals and controls secretory processes (15).
GRX-1 acts locally rather than globally
Silencing of GRX-1 expression did not result in a change in bulk intracellular thiol concentrations (Fig. 1E). The most likely explanation for this is the down-regulation of GRX-1 being indistinguishable from the background activity in other redox chains, which may even be up-regulated in a compensatory manner. This suggests, in turn, that the whole-cell thiol concentration does not determine the redox-dependent activity of the exocytotic machinery. Instead, this observation favors the idea that local GRX-1-catalyzed reactions and formation of thiol groups in specific proteins/compartments are relevant for the redox-dependent control of insulin exocytosis. This idea is even more likely in the light of the observation that TRX-1 and GRX-1 are differentially distributed in the β-cell and act differently on the insulin release machinery (Figs. 1 and 2). Local control of cellular processes by redox reactions has been reported previously. It has been demonstrated, for example, that TRX-1 expression is differentially changed in different cell compartments during oxidative stress responses (16).
GRX-1-independent effects of NADPH
In addition to the GRX-1-dependent action of NADPH that stimulates Ca2+-dependent exocytosis, we also observed that NADPH has a modest approximately 30% inhibitory effect on voltage-gated Ca2+ influx (Figs. 4 and 5) evoked by voltage-clamp depolarizations in both INS-1 832/13 cells and rat β-cells. The latter action appears not to rely on GRX-1, because NADPH remained an effective inhibitor of whole-cell Ca2+ currents in GRX-1-silenced INS-1 832/13 cells (Fig. 4L), and a similar tendency (albeit nonsignificant) was seen in primary rat cells. These findings do not mean that elevation of NADPH reduces intracellular Ca2+ concentrations upon glucose stimulation. Instead, this finding should be interpreted against the background that the ATP/ADP ratio remains the overruling determinant of β-cell electrical activity and Ca2+ influx (17). When extracellular glucose concentrations and intracellular ATP/ADP ratio increase, this activates electrical activity and Ca2+ influx through voltage-gated Ca2+ channels. The concomitant increase in NADPH/NADP+ ratio exerts a subordinate and modulatory action on both Ca2+ influx and Ca2+-evoked exocytosis of insulin granules [cf. with the triggering and amplifying actions of glucose (17)]. This subordinate action of the NADPH/NADP+ ratio leads to a modest suppression of Ca2+-currents through the voltage-gated Ca2+ channels, whereas Ca2+-evoked exocytosis of insulin granules is enhanced (10). Taken together, the GRX-1-dependent stimulation of the exocytotic machinery, and the GRX-independent inhibition of Ca2+ currents will enhance the efficacy of insulin release. This dual action of NADPH is advantageous for the β-cell because it will reduce the Ca2+ burden on the β-cell and thereby decrease the risk for excitotoxicity and apoptosis (18), while maintaining adequate amounts of insulin to be released. This is interesting in perspective to human T2DM where inadequate insulin secretion is a key finding (19, 20). Worthy of note, the oxidoreductase regulatory protein TXNIP/VDUP-1 is implicated in β-cell damage (21, 22), and genetic studies have identified the TXNIP gene to associate with T2DM (23). This underscores that detailed knowledge of redox processes in the β-cell are key for understanding the disease.
Materials and Methods
Reagents and siRNA
All chemicals were from Sigma (St. Louis, MO) if not stated otherwise. Small interfering RNAs (siRNAs) were 21-nt long duplexes designed, synthesized, and supplied by Ambion (Austin, TX). Three candidate sequences against GRX-1 were tested in preliminary experiments. The optimal sequence, used in all following experiments was: sense strand 5′-GGAUGUCAGUAUAUACCGGTT-3′ and antisense strand 5′-CCGGUAUAUACUGAC AUCCTT-3′. For TRX-1 the optimal sequence was: sense strand 5′-GGUUAAAAC CUGUACCUUUTT-3′ and antisense strand 5′-AAAGGUACAGGUUUUAACCTG-3′. As negative control (control) Silencer Negative Control no. 2 from Ambion was used. Cells were incubated with siRNA for 48 h before gene expression, thiol concentration, and cell viability were assessed.
Cell culture and islet preparation
The INS-1 832/13 cell line was used as a β-cell model because it exhibits a strong insulin-secretory response to increases in glucose concentration (24). They were seeded at a density of 5 × 104 cells/cm2 and cultured in 24-well plates (Nunc, Roskilde, Denmark) in RPMI 1640 containing 11.1 mm d-glucose supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 25 μm 2-mercaptoethanol, at 37 C in a humidified atmosphere containing 95% air and 5% CO2.
Primary rat pancreatic islets were isolated as described earlier (25) and hand picked under a stereo microscope. For indicated experiments they were dispersed into single cells with Ca2+-free buffer and allowed to adhere in petri dishes containing RPMI 1640 as above but substituted with 5 mm d-glucose and lacking 2-mercaptoethanol. Whole islets were kept similarly. All animal experimentation was conducted in accord with accepted standards of humane animal care and approved by the local ethics committee.
Vector design
The shRNA vectors were constructed by insertion of 72-nt long DNA sequences (sense and antisense) into the pRNAT H1.1 shuttle vector (GenScript Corp., Piscataway, NJ). The 72-nt DNA sequences were designed according to instructions from GenScript containing two 21-nt long sequences (antisense and sense) specific for GRX-1. For shGRX-1, the sense sequence was 5′-GCUACUAACAACACCA AUGTT-3′ and antisense was 5′-CAUUGGUGU UGUUAGUAGCTG-3′. For control, a scramble vector was used [5′-TCAACTCGTA TATGGCCTGTT-3′ (sense) and 5′-GCAGGCCA TATACGAGTTGATT-3′ (antisense)].
For overexpression of GRX-1 (GRX-1-EGFP), we cloned GRX-1 cDNA from an INS-1 832/13 cDNA library and inserted it into the pIRES2-EGFP vector (CLONTECH Laboratories, Inc., Palo Alto, CA). Empty pIRES2-EGFP was used for control (EGFP). All vectors were verified by sequencing.
Transfection
INS-1 832/13 ells and primary rat islets were transfected with siRNA (30 nm) using the Dharmafect 1 transfection reagent (Dharmacon, Lafayette, CO) according to the manufacturer’s recommendation. For islets, transfection efficacy was assessed by the fluorescein-labeled dsRNA-oligo BLOCK-iT (Invitrogen, Carlsbad, CA).
For transfection with pRNAT H1.1 vectors (sh-GRX-1 and scramble) and pIRES2EGFP vectors (GRX-1-EGFP and EGFP), we used Effectene Transfection Reagent (QIAGEN, Hilden, Germany) also according to the manufacturer’s protocol. Experiments were performed 48 h after transfection unless specified differently.
Gene expression analysis
RNA was extracted using the RNAeasy Kit (QIAGEN, Hilden, Germany). RNA (1 μg) was used for cDNA synthesis with SuperScript III (Invitrogen). Reaction mixture (5 μl) with 15 ng cDNA, 2.5 μl TaqMan mastermix (Applied Biosystems, Foster City, CA), and 900 nm TaqMan gene expression assay were run on a 7900HT Fast Real-Time System (Applied Biosystems). The real-time PCR was carried out as follows: 50 C for 2 min, 95 C for 10 min, 40 cycles of 95 C for 15 sec, and 60 C for 1 min. The amount of mRNA was calculated relative to the amount of hypoxanthine-guanine phosphoribosyltrans-ferase (HPRT) mRNA in the same sample by the formula X0/R0 = 2CtR−CtX, where X0 is the original amount of mRNA for the gene of interest, R0 is the original amount of HPRT mRNA, CtR the Ct value for HPRT, and CtX the Ct value for the gene of interest.
Western blotting
INS-1 832/13 cells were homogenized in 50 mm TES buffer, pH 7.4, containing 250 mm sucrose, 1 mm EDTA, 0.1 μm EGTA, and complete protease inhibitor cocktail (Roche, Stockholm, Sweden) on ice using a ULTRA-TURRAX (Janke and Kunkel, IKA-WERK, Staufen, Germany) or by repeated aspiration through a needle (Ø0.4 μm). After homogenization tissue debris was removed by centrifugation (20,000 × g, 15 min, 4 C) and the supernatant was collected. Protein content of extract was measured using a kit (DC protein assay, Bio-Rad Laboratories, Inc., Hercules, CA). Of total protein content 40 μg was electrophoresed on 12% SDS-PAGE, and the separated proteins were transferred onto a polyvinylidine difluoride membrane (Amersham Pharmacia Biotech, Uppsala, Sweden). The membrane was blocked with 5.0% nonfat dry milk in TBST (Tris-buffered saline with Tween 20) (pH 7.4; 0.15m NaCl, 10 mm Tris-HCl, and 0.1% Tween 20) for 1 h at room temperature or overnight at 4 C. After blocking, the membrane was incubated with polyclonal rabbit anti-GRX (1:250, lot. no. WXD01, R&D Systems Abington, UK) and monoclonal mouse anti-β actin (1:15,000; Sigma) in TBST for 1 h at room temperature. After washing with TBST, the membrane was incubated with peroxidase-linked antirabbit IgG (Amersham Bioscience; dilution 1:5000) or peroxidase-linked antimouse IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA; dilution 1:20.000) for 1 h at room temperature. Detection was done by an enzymatic chemiluminescence (ECL) kit (Amersham).
Insulin secretion assays
INS-1 832/13 cells were cultured in 24-well dishes and transfected with siRNA at 72 h before assay. Islets, after picking, were placed in 96-well plates in a batch of five and transfected likewise. When assayed, the cells/islets were washed in Krebs-Ringer bicarbonate buffer, pH 7.4, supplemented with 10 mm HEPES, 0.1% BSA and preincubated in 2.8 mmol/liter glucose in buffer for 120 min at 37 C. Insulin secretion was then induced by static incubation of the cells/islets for 1 h in 1 ml buffer containing 2.8 and 16.7 mm glucose, respectively. Insulin from INS-1 832/13 cells was measured by the Coat-a-Count RIA (Siemens Healthcare Diagnostics, Deerfield, IL) and normalized according to protein content per well. Protein content was determined using a BCA-assay. For islets, a rat insulin RIA was used (Linco Research, Inc., St. Charles, MO).
Redox status and cell viability assays
Cellular thiol concentration was measured using a Thiol and Sulfide quantification Kit (Invitrogen). Cells were trypsinized, counted, and washed and assimilated with 1 ml of the provided buffer followed by sonication on ice. A 3-μl sample was used for analysis. Cell viability was assessed using TB and a hemocytometer.
Immunocytochemistry and microscopy
INS-1 832/13 or dispersed primary rat islet cells cultured in glass-bottom petri dishes (Mattek Corp., Ashland, MA) were fixed in 4% formaldehyde in PBS and permeabilized with 0.1% Triton X-100 in PBS. After blocking nonspecific binding with 5% normal donkey serum, cells were first incubated for 2 h with the polyclonal rabbit anti-GRX-1 (5 μg/ml; lot no. 0001; AbFrontier, Seoul, Korea) or goat anti-TRX-1 (5 μg/ml; lot no. KOX0107041; R&D Systems, Abington, UK) or both. After further washing they were then incubated for 1 h with an antirabbit-Cy5 and/or antigoat-Alexa Fluor 546-conjugated secondary antibody (Jackson ImmunoResearch Laboratories) at a 1:500 dilution. Cells that were transfected with the GRX-1-containing vector were treated likewise but grown on coverslips and incubated with polyclonal rabbit anti-GRX (lot no. WXD01, R&D Systems) at a 1:50 dilution and antirabbit Cy3 (Pierce Chemical Co., Rockford, IL) at a 1:150 dilution. Immunoreactivity was visualized using the multitrack mode of a Zeiss 510 LSM confocal microscope (Carl Zeiss, Thornwood, NY) and a C-Apochromat 40×/1.2 WDICIII objective. Cy5 fluorescence was excited using the 633-nm line of the HeNe-laser, and emitted light was collected using a 650- to 710-nm bandpass filter. For Alexa Fluor 546 and Cy3 the 543-nm laser line and a 560- to 615-nm bandpass filter was used. EGFP and fluorescein fluorescence was excited using the 488-nm line of the Argon-laser, and emitted light was collected using a 505- to 530-nm bandpass filter.
Electrophysiology
INS-1 832/13 cells or primary cells were prepared, and whole-cell capacitance and whole cell Ca2+ current measurements were performed using EPC10 patch-clamp amplifiers with the software suite Pulse+X-chart extension (version 8.64; HEKA; Lambrecht-Pfalz, Germany), as previously described (10). Cells were seeded in plastic petri dishes (Nunc, Roskilde, Denmark) and were used for experiments 48 h after transfection. All chemicals were of analytical grade (Sigma). The extracellular solution contained 118 mm NaCl, 20 mm tetraethylammonium chloride, 5.6 mm KCl, 2.6 mm CaCl2, 1.2 mm MgCl2, 5 mm HEPES and 5 mm glucose (pH 7.4 with NaOH). Tetraethylammonium chloride was used to block outward K+ currents that otherwise obscured the (smaller) inward Ca2+ currents. The pipette (intracellular) solution for studying depolarization-evoked exocytosis in the standard whole-cell experiments contained 125 mm Cs-glutamate, 10 mm CsCl, 10 mm NaCl, 1 mm MgCl2, 5 mm HEPES, 3 mm Mg-ATP, 0.1 mm cAMP and 0.05 mm EGTA, with 0.1 mm NADPH added as indicated (pH 7.2 with CsOH). Cs+-salts were used to block outward K+-currents. Primary β-cells were identified by the absence of Na+-currents at physiological membrane potentials.
Acknowledgments
We thank Britt-Marie Nilsson for expert technical assistance. INS-1 832/13 cells were kindly provided by Dr. Hans Hohmeier, Division of Endocrinology, Metabolism, and Nutrition, Duke University (Durham, NC).
Footnotes
This work was supported by the Swedish Research Council (SRC), the Knut and Alice Wallenberg Foundation, the Novo Nordisk Foundation, and Albert Påhlsson foundation (to E.R); Tage Blücher’s memorial foundation (to R.I.); Lars Hierta’s memorial foundation (to R.I.); and Magnus Bergvalls stiftelse (to U.B. and R.I). T.R. and E.Z. are fellows at the European Union network CavNet. E.R. is a senior researcher at the SRC. U.B.’s fellowship is supported by a Linneaus grant from the SRC.
Disclosure Summary: The authors have no conflict of interest to disclose.
First Published Online March 19, 2009
Abbreviations: cGFP, Coral GFP; dsRNA, double-strand RNA; EGFP, enhanced GFP; fF, femtofarad; GFP, green fluorescent protein; GRX-1, glutaredoxin-1; HPRT, hypoxanthine-guanine phosphoribosyltrans-ferase; NADPH, nicotinamide adenine dinucleotide phosphate; nt, nucleotide; shGRX-1, short hairpin GRX-1; siGRX-1, small interfering GRX-1; siRNA, small interfering RNA; TB, Trypan blue; TBST, Tris-buffered saline with Tween 20; TRX-1, thioredoxin-1.
References
- 1.Rotilio J, Stella AM2006. Molecular basis of neurodegenerative diseases. Ital J Biochem 55:189–193 [PubMed] [Google Scholar]
- 2.Markesbery WR1997. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med 23:134–147 [DOI] [PubMed] [Google Scholar]
- 3.Mizuno Y, Ikebe S, Hattori N, Nakagawa-Hattori Y, Mochizuki H, Tanaka M, Ozawa T1995. Role of mitochondria in the etiology and pathogenesis of Parkinson’s disease. Biochim Biophys Acta 1271:265–274 [DOI] [PubMed] [Google Scholar]
- 4.Hayden MR, Tyagi SC2002. Islet redox stress: the manifold toxicities of insulin resistance, metabolic syndrome and amylin derived islet amyloid in type 2 diabetes mellitus. JOP 3:86–108 [PubMed] [Google Scholar]
- 5.Hayden MR, Tyagi SC2002. Intimal redox stress: accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovasc Diabetol 1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernandes AP, Holmgren A2004. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid Redox Signal 6:63–74 [DOI] [PubMed] [Google Scholar]
- 7.Holmgren A2000. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid Redox Signal 2:811–820 [DOI] [PubMed] [Google Scholar]
- 8.Babior BM1999. NADPH oxidase: an update. Blood 93:1464–1476 [PubMed] [Google Scholar]
- 9.Ashcroft SJ, Christie MR1979. Effects of glucose on the cytosolic ration of reduced/oxidized nicotinamide-adenine dinucleotide phosphate in rat islets of Langerhans. Biochem J 184:697–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivarsson R, Quintens R, Dejonghe S, Tsukamoto K, in 't Veld P, Renström E, Schuit FC2005. Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 54:2132–2142 [DOI] [PubMed] [Google Scholar]
- 11.Yajima H, Komatsu M, Schermerhorn T, Aizawa T, Kaneko T, Nagai M, Sharp GW, Hashizume K1999. cAMP enhances insulin secretion by an action on the ATP-sensitive K+ channel-independent pathway of glucose signaling in rat pancreatic islets. Diabetes 48:1006–1012 [DOI] [PubMed] [Google Scholar]
- 12.Vallar L, Biden TJ, Wollheim CB1987. Guanine nucleotides induce Ca2+-independent insulin secretion from permeabilized RINm5F cells. J Biol Chem 262:5049–5056 [PubMed] [Google Scholar]
- 13.Jonas JC, Li G, Palmer M, Weller U, Wollheim CB1994. Dynamics of Ca2+ and guanosine 5′-[γ-thio]triphosphate action on insulin secretion from α-toxin-permeabilized HIT-T15 cells. Biochem J 301:523–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maeda K, Hägglund P, Finnie C, Svensson B, Henriksen A2006. Structural basis for target protein recognition by the protein disulfide reductase thioredoxin. Structure 14:1701–1710 [DOI] [PubMed] [Google Scholar]
- 15.Padilla CA, Martínez-Galisteo E, López-Barea J, Holmgren A, Bárcena JA1992. Immunolocalization of thioredoxin and glutaredoxin in mammalian hypophysis. Mol Cell Endocrinol 85:1–12 [DOI] [PubMed] [Google Scholar]
- 16.Byrne BM, Welsh J2005. Altered thioredoxin subcellular localization and redox status in MCF-7 cells following 1,25-dihydroxyvitamin D3 treatment. J Steroid Biochem Mol Biol 97:57–64 [DOI] [PubMed] [Google Scholar]
- 17.Henquin JC2000. Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49:1751–1760 [DOI] [PubMed] [Google Scholar]
- 18.Del Río P, Massieu L2008. Mild mitochondrial inhibition in vivo enhances glutamate-induced neuronal damage through calpain but not caspase activation: role of ionotropic glutamate receptors. Exp Neurol 212:179–188 [DOI] [PubMed] [Google Scholar]
- 19.Groop L, Lyssenko V2008. Genes and type 2 diabetes mellitus. Curr Diabetes Rep 8:192–197 [DOI] [PubMed] [Google Scholar]
- 20.Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, Ardlie K, Bostrom KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P, Ding CJ, Doney AS, Duren WL, Elliott KS, Erdos MR, Frayling TM,et al 2008. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 40:638–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A2008. Thioredoxin-interacting protein: a critical link between glucose toxicity and β-cell apoptosis. Diabetes 57:938–944 [DOI] [PMC free article] [PubMed]
- 22.Corbett JA2008. Thioredoxin-interacting protein is killing my β-cells! Diabetes 57:797–798 [DOI] [PubMed]
- 23.Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJ, Jensen CB, Krook A, Björnholm M, Tornqvist H, Zierath JR, Ridderstrale M, Altshuler D, Lee RT, Vaag A, Groop LC, Mootha VK2007. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med 4:e158 [DOI] [PMC free article] [PubMed]
- 24.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB2000. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49:424–430 [DOI] [PubMed] [Google Scholar]
- 25.Fransson U, Rosengren AH, Schuit FC, Renstrom E, Mulder H2006. Anaplerosis via pyruvate carboxylase is required for the fuel-induced rise in the ATP:ADP ratio in rat pancreatic islets. Diabetologia 49:1578–1586 [DOI] [PubMed] [Google Scholar]