

Abstract
It is well established that epidermal growth factor (EGF) induces the cytoskeleton reorganization and cell migration through two major signaling cascades: phospholipase C-γ1 (PLC-γ1) and Rho GTPases. However, little is known about the cross talk between PLC-γ1 and Rho GTPases. Here we showed that PLC-γ1 forms a complex with Rac1 in response to EGF. This interaction is direct and mediated by PLC-γ1 Src homology 3 (SH3) domain and Rac1 106PNTP109 motif. This interaction is critical for EGF-induced Rac1 activation in vivo, and PLC-γ1 SH3 domain is actually a potent and specific Rac1 guanine nucleotide exchange factor in vitro. We have also demonstrated that the interaction between PLC-γ1 SH3 domain and Rac1 play a significant role in EGF-induced F-actin formation and cell migration. We conclude that PLC-γ1 and Rac1 coregulate EGF-induced cell cytoskeleton remodeling and cell migration by a direct functional interaction.
PLC-γ1 binds to the Rac1 106PNTP109 motif and stimulates Rac1 activity acting as a GEF through its SH3 domain leading to regulation of EGF-induced cytoskeleton remodeling and cell migration.
Activation of the epidermal growth factor (EGF) receptor (EGFR) signals many biological responses including cell proliferation, cell differentiation, cell survival, and cell motility (1, 2). Binding of EGF to EGFR at the plasma membrane induces dimerization of EGFR, which results in the activation of EGFR tyrosine kinase and trans-autophosphorylation (3). Sites of tyrosine autophosphorylation in activated EGFR bind downstream signaling proteins to form receptor-signaling protein complexes that then initiate the activation of various signaling pathways (3). Cell motility elicited by EGF requires EGFR kinase activation and autophosphorylation (4). At least two signaling pathways downstream of EGFR can be linked to cell motility. Phospholipase C-γ1 (PLC-γ1) has been implicated in the pathway responsible for the reorganization of the cytoskeleton (5). On the other hand, EGFR activation also leads to membrane ruffling and reorganization of cytoskeleton and focal adhesions through activation of members of the Rho subfamily of GTP-binding proteins (6, 7).
PLC-γ1, a 145-kDa protein, contains two Src homology 2 (SH2) domains, one Src homology 3 (SH3) domain, and two pleckstrin homology (PH) domains and catalyzes the hydrolysis of phosphatidylinositol-4,5-bis-phosphate (PIP2), creating inositol 1,4,5-triphosphate and diacylglycerol. PLC-γ1 forms a complex in vivo with EGFR through its SH2 domain interaction (1, 8, 9, 10, 11). Complex formation leads to phosphorylation of PLC-γ1 on tyrosine residues and an increase in its enzymatic activity (12, 13, 14). PLC-γ1 has been implicated in many growth factor-induced cell signaling including cell proliferation, differentiation, receptor endocytosis, cell motility, membrane ruffle formation, and branching tubulogenesis (15, 16, 17, 18, 19, 20, 21).
All of the PLC-γ1 domains have been implicated in regulating the cellular localization of PLC-γ1 and in regulating GF-induced cell signaling. For example, both PLC-γ1 SH2 and pleckstrin homology (PH) domains have been implicated in regulating GF-induced translocation of PLC-γ1 (22, 23, 24, 25, 26). Recent studies have shown that PLC-γ1 is involved in much broader cell signaling than previously revealed (17, 27, 28). Interestingly, most recently identified interactions between PLC-γ1 and its binding proteins are mediated by its SH3 domain. EGF stimulates the interaction between PLC-γ1 and phospholipase D2 (PLD2) to potentiate EGF-induced inositol 1,4,5-triphosphate formation and Ca2+ increase. The interaction between PLC-γ1 and PLD2 is mediated by PLC-γ1 SH3 domain (27). PLC-γ1 is essential for the activation of calcium entry into cells after stimulation on cell-surface receptors, and PLC-γ1 SH3 domain is required for this effect (29). We recently showed that PLC-γ1 binds directly to Akt in response to EGF. The PLC-γ1-Akt interaction results in the serine phosphorylation of PLC-γ1 (30). One very interesting finding reported recently is that PLC-γ1 SH3 domain acts as a guanine nucleotide exchange factor (GEF) PIKE and dynamin-1. PLC-γ1 SH3 domain acts as a GEF for PIKE to regulate nerve growth factor-induced cell mitogenesis (28). PLC-γ1 SH3 domain acts as a GEF for dynamin-1 to regulate EGFR endocytosis, and the interaction between PLCγ-1 SH3 domain and dynamin-1 is EGF dependent (17).
It has been reported that PLC-γ1 is required for cell migration induced by many growth factors, including EGF (4), platelet-derived growth factor (31), and hepatocyte growth factor (2, 32). Although the mechanisms by which PLC-γ1 regulates cell migration are not yet clear, it was suggested that the activation of PLC-γ1 by EGF resulted in the reorganization of the cytoskeleton (5). PLC-γ1 hydrolyzes PIP2, which leads to the release of profilin, a cytoplasmic actin-binding protein (33). The role of PLC-γ1 in the regulation of cell motility has been shown in a variety of cell types, especially carcinoma cells (5).
Rho GTPases make up a large subfamily of the Ras superfamily and include Cdc42, Rac, and Rho proteins. Rho GTPases are guanine nucleotide-binding proteins that cycle between an active, GTP-bound and an inactive, GDP-bound state. The activity of Rho proteins is controlled by three distinct families of proteins: the activator or guanine nucleotide exchange factors (GEFs), and two families of suppressors, the GTPase-activating proteins (GAPs), and the guanine nucleotide dissociation inhibitors (34, 35). Rho proteins are able to induce the reorganization of the actin cytoskeleton (36). Because actin cytoskeletal changes are required for the migratory behavior of cells in response to growth factor stimulation or matrix interactions, Rho proteins play significant role in regulating cell migration. In fact, all aspects of cellular motility and invasion, including cellular polarity, cytoskeletal organization, and transduction of signals from the outside environment, are controlled through an interplay between the Rho-GTPases (37, 38). It has been shown that Cdc42 regulates the polarity of cell migration. RhoA is required for the generation of contractile force leading to rounding of the cell body (36, 39). Within the Rac subfamily, Rac1, Rac2, and Rac3 share significant sequence identities (∼88%). These three diverge primarily in the C-terminal 15 residues. All the Rac-related proteins stimulate the formation of lamellipodia and membrane ruffles, presumably through the interaction with the PIR121-Nap125-HSPC300-WAVE complex (40). A lack of Rac1 results in embryonic lethality (41). The in vivo and in vitro studies in the last decades have firmly established the role of Rac1 in cardiomyocyte signaling and hypertrophy (42, 43, 44).
Given that both PLC-γ1 and Rho GTPases control cell motility by regulating the reorganization of the cytoskeleton in response to EGF, it would be interesting to examine whether there is a direct functional link between PLC-γ1 and Rho GTPase in cell migration induced by EGF. Here we showed that EGF stimulates the association between PLC-γ1 and Rac1. The interaction between PLC-γ1 and Rac1 is direct and mediated by PLC-γ1 SH3 domain and Rac1 proline-rich motif 106PNTP109. We further showed that EGF-induced PLC-γ1 and Rac1 interaction resulted in the activation of Rac1, which suggest that PLC-γ1 is a GEF for Rac1 in vivo. Moreover, we demonstrated by in vitro GEF assay that PLC-γ1 SH3 domain is a strong and specific GEF for Rac1. Finally, we showed that the interaction between PLC-γ1 and Rac1 plays an important role in EGF-induced cytoskeleton remodeling and cell migration.
Results
Association between PLC-γ1 and Rac 1 in response to EGF
We first determined whether PLC-γ1 and Rac1 are physically associated in response to EGF by coimmunoprecipitation. Cos7 cells were treated with EGF for the indicated time, and the Rac1 was immunoprecipitated with mouse anti-Rac1 antibody. The coimmunoprecipitation of PLC-γ1 was examined by immunoblotting with antibodies to both Rac1 and PLC-γ1. As shown in Fig. 1, PLC-γ1 was coimmunoprecipitated with Rac1 after EGF stimulation, and the amount of coimmunoprecipitated PLC-γ1 reached maximum at 5–15 min after EGF stimulation. This indicates that EGF stimulates the association between PLC-γ1 and Rac1.
Fig. 1.

EGF-induced association of PLC-γ1 and Rac1 in Cos7 cells. Cos7 cells were treated with EGF (100 ng/ml) for the indicated time, and the Rac1 was immunoprecipitated with mouse anti-Rac1 antibody. The immunoprecipitates were then examined by immunoblotting with a monoclonal antibody to PLC-γ1 and a polyclonal antibody to Rac1. B, Quantification of the data from at least three independent experiments as described in panel A. The intensity of the bands in the PLC-γ1 blot was normalized against the intensity of the corresponding bands in the Rac1 blot. The data of a serum-free sample (−EGF) was set as 100%. The error bar is se. IP, Immunoprecipitation; SF, serum free.
PLC-γ1 SH3 domain mediates the association between PLC-γ1 and Rac1
We next determined which domain of PLC-γ1 is responsible for binding to Rac1. PLC-γ1 contains two SH2 domains, one SH3 domain, and one intact PH domain. We had previously fused each of these domains to glutathione S-transferase (GST) (23, 45). These GST-fusion proteins were used to pull down Rac1 from Cos7 cell lysates with or without EGF stimulation. As shown in Fig. 2A, with or without EGF stimulation only PLC-γ1 SH3 domain interacted with Rac1. This indicates that PLC-γ1 SH3 domain specifically interacts with Rac1.
Fig. 2.

The SH3 domain of PLC-γ1 is required for PLC-γ1 association with Rac1. A, The association of Rac1 with various GST-fusion PLC-γ1 domains by GST-fusion protein pulldown. COS-7 cells were not treated or treated with 100 ng/ml EGF for 15 min. The cell lysates were incubated with various GST-fusion PLC-γ1 proteins bound to glutathione-agarose beads. Bound proteins were analyzed by immunoblotting with anti-Rac1 antibody. Immunoblotting with anti-GST antibody was used as loading control. B, COS-7 cells were transfected with wt PLC-γ1, or PLC-γ1ΔSH3. The cells were either not treated or treated with 100 ng/ml EGF for 15 min. The cell lysates were subjected to immunoprecipitate with monoclonal anti-Rac1 antibody, and the resulting immunoprecipitates were subjected to immunoblotting with a polyclonal anti-GFP antibody and a polyclonal anti-Rac1 antibody. IP, Immunoprecipitation.
To determine whether the PLC-γ1 SH3 domain mediates the interaction between PLC-γ1 and Rac1 in vivo, we transfected Cos7 cells with either yellow fluorescent protein (YFP)-tagged wild-type (wt) PLC-γ1 or YFP-tagged mutant PLC-γ1 lacking of the SH3 domain (PLC-γ1ΔSH3). Both constructs were generated previously (23). After the transfection, EGF-stimulated interaction between Rac1 and PLC-γ1 was examined by coimmunoprecipitation. Rac1 was immunoprecipitated by mouse anti-Rac1 antibody, and the coimmunoprecipitation of YFP tagged PLC-γ1 was revealed by immunoblotting with anti-YFP and Rac1 antibodies. We showed that EGF stimulated strong interactions between wt PLC-γ1 and Rac1; however, EGF did not stimulate the association between PLC-γ1ΔSH3 and Rac1 (Fig. 2B). This indicates that PLC-γ1 SH3 domain is required for EGF-induced interaction between PLC-γ1 and Rac1.
Rac1 proline-rich motif 106PNTP109 mediates its interaction with PLC-γ1
We then determined which sequences of Rac1 interact with PLC-γ1. The Rac1 sequences that interact with PLC-γ1 SH3 domain likely contain PXXP motifs. Analysis of Rac1 sequence reveals the presence of one PXXP motif 106PNTP109. To determine whether this motif indeed mediates the interaction between PLC-γ1 and Rac1, we mutated the two prolines to alanines by site-directed mutagenesis. The green fluorescent protein (GFP)-tagged mutant (Rac1PP/AA) and wt Rac1 were expressed in Cos7 cells by transient transfection. EGF-stimulated interaction between PLC-γ1 and the mutant was examined by coimmunoprecipitation. We immunoprecipitated GFP-tagged Rac1 with anti-GFP antibody. Immunoblotting with antibodies to GFP and PLC-γ1 showed that PLC-γ1 only coimmunoprecipitated with wt Rac1 after EGF stimulation (Fig. 3, A and B). PLC-γ1 did not coimmunoprecipitate with Rac1PP/AA regardless of EGF stimulation (Fig. 3, A and B). This suggests that the interaction between PLC-γ1 and Rac1 is mediated by Rac1 106PNTP109.
Fig. 3.

Rac1 106PNTP109 motif is required for its association with PLC-γ1. A, Cos-7 cells were transfected with GFP-tagged wt Rac1 or Rac1PP/AA. The cells were either not treated or treated with EGF (15 min). The transfected Rac1 was immunoprecipitated with GFP antibody, and the resulting immunoprecipitates were immunoblotted with antibodies to GFP and PLC-γ1. B, Quantification of the data from at least three independent experiments as described in panel A. C, The effects of Rac1 106PNTP109 on its association with the PLC-γ1 SH3 domain. COS-7 cells were transfected with GFP-tagged wt Rac1 or Rac1PP/AA. With or without EGF treatment (15 min), the cells were lysed and incubated with GST or GST-fusion PLC-γ1 SH3 domain bound to glutathione-agarose beads. Bound proteins were analyzed by immunoblotting with anti-GFP antibody.
We further examined the interaction between PLC-γ1 SH3 domain and Rac1 106PNTP109 motif by GST pulldown. Cos7 cells were transfected with either wt Rac1 or mutant Rac1PP/AA, and the cell lysates were incubated with GST-fusion PLC-γ1 SH3 domain conjugated to glutathione beads. We showed that GST-fusion PLC-γ1 SH3 domain specifically pulled down wt Rac1, but not the Rac1PP/AA with or without EGF stimulation (Fig. 3C). Together, our data indicate that PLC-γ1 SH3 domain specifically interacts with Rac1 106PNTP109.
The interaction between PLC-γ1 and Rac1 is direct and specific
To further test whether the interaction between SH3 and Rac1 is direct and specific, we generated a GST fusion, loss of function mutant PLC-γ1 SH3 domain P842L (PLC-γ1 SH3P842L) with a single mutation of proline to leucine. We incubated PLC-γ1 SH3P842L and the other GST-fusion domains with purified Rac1 in a pure buffer system. As shown in Fig. 4A, only the intact SH3 domain can interact with purified Rac1 in vitro but not the nonfunctional PLC-γ1 SH3P842L. Consistent with our results in Fig. 2, other domains did not associate with Rac1 in vitro. Together, the results suggest that the SH3 domain of PLC-γ1 interacts with Rac1 directly in vitro.
Fig. 4.

The interaction between PLC-γ1 and Rac1 is direct and specific. A, Interaction between purified Rac1 and PLC-γ1 SH3 domain. Purified GST-fusion domains of various PLC-γ1 were immobilized on GST beads and the GST domain-immobilized beads were incubated with pure Rac1 purchased from Cytoskeleton Inc., as described in Materials and Methods. The beads with bound proteins were washed three times with binding buffer after which the bound proteins were resolved in 10% SDS-PAGE followed by immunoblotting. Top panel, Bound Rac1 revealed by anti-Rac1 antibody; bottom panel, various GST-fusion proteins revealed by Coommassie blue stain. B, Interaction between PLC-γ1 and Rac1, RhoA, and Cdc42. Cos7 cells were transfected with YFP-tagged wt PLC-γ1, PLC-γ1ΔSH3, or PLC-γ1P842L. After EGF treatment (15 min) transfected PLC-γ1 was immunoprecipitated (IPed) with GFP antibody, and the coimmunoprecipitated (Co-IPed) Rac1, Cdc42, and RhoA were revealed by antibodies to Rac1, Cdc42, and RhoA.
We next determined whether PLC-γ1 only specifically interacts with Rac1 or broadly interacts with other Rho proteins. We generated an YFP-tagged PLC-γ1 mutant with the mutation of P842 to L (PLC-γ1P842L) to disrupt the function of SH3 domain. Cos7 cells were transfected with YFP-tagged wt PLC-γ1, PLC-γ1ΔSH3, or PLC-γ1P842L. After EGF stimulation for 15 min, YFP tagged PLC-γ1, and the mutants were immunoprecipitated with anti-YFP antibodies, and bound Rac1, Cdc42, and RhoA were analyzed by immunoblotting with antibodies to Rac1, Cdc42, and RhoA. As shown in Fig. 4B, Rac1 was coimmunoprecipitated only with wt PLC-γ1 after EGF stimulation but not with PLC-γ1ΔSH3 and PLC-γ1P842L as expected, which further indicates that PLC-γ1 SH3 domain is required for EGF-induced interaction between PLC-γ1 and Rac1. Interestingly, no Cdc42 and RhoA were detected in the PLC-γ1 immunoprecipitates, which indicates that PLC-γ1 specifically interacts with Rac1, but not Cdc42 and RhoA.
Direct interaction between PLC-γ1 and Rac1 is critical for EGF-induced Rac1 activation in vivo
We examined whether the interaction between PLC-γ1 and Rac1 contributes to Rac1 activation in response to EGF. Activation of Rac1 was determined by its ability to bind to GST-fusion p21-activated protein kinase (PAK) Rho-binding domain (GST-PAK). We first determined the effects of EGF on Rac1 activation in both BT20 and Cos7 cells. As shown in Fig. 5A, Rac1 was activated by EGF, and the Rac1 activation reached maximum at 15 min of EGF stimulation for both BT20 and Cos7 cells.
Fig. 5.

Interaction between PLC-γ1 and Rac1 is critical for EGF-induced Rac1 activation. The activation of Rac1 was determined by pulldown with GST-fusion PAK Rac1-binding domain as described in Materials and Methods. Briefly, the cell lysates were incubated with GST-PAK bound to glutathione-agarose beads. Bound proteins were analyzed by immunoblotting with anti-Rac1 antibody. Immunoblotting with anti-GST antibody was used as loading control. A, EGF-induced activation of Rac1 in BT20 and Cos-7 cells. BT20 and Cos7 cells were stimulated with EGF for the indicated times, and the activation of Rac1 was determined as above. B, The effects of the PLC-γ1 SH3 domain on EGF-induced activation of Rac1. Cos7 cells were transfected with YFP-tagged wt PLC-γ1 or mutant PLC-γ1ΔSH3. 293T cells were transfected with wt EGFR and YFP-tagged wt PLC-γ1 or mutant PLC-γ1ΔSH3. The cells were either not treated or treated with EGF (100 ng/ml) for 30 min. The Rac1 activation was determined by GST-PAK pulldown. C, The effects of Rac1 106PNTP109 motif on EGF-induced activation of Rac1 in Cos7 cells. Cells were transfected with GFP-tagged wt Rac1 or mutant Rac1PP/AA. The cells were either not treated or treated with EGF (100 ng/ml) for 30 min. The Rac1 activation was determined by GST-PAK pulldown. D, The effects of PLC-γ1 SH3 domain on EGF-induced activation of Rac1 in 293T cells. Cells were transfected with wild-type EGFR and YFP-tagged wt PLC-γ1 or mutant PLC-γ1ΔSH3. The cells were either not treated or treated with EGF (100 ng/ml) for 30 min. The Rac1 activation was determined by GST-PAK pulldown. E, The effects of Rac1 106PNTP109 motif on EGF-induced activation of Rac1 in 293T cells. Cells were transfected with wild-type EGFR and GFP-tagged wt Rac1 or mutant Rac1PP/AA. The cells were either not treated or treated with EGF (100 ng/ml) for 30 min. The Rac1 activation was determined by GST-PAK pulldown.
We then determined whether EGF-induced Rac1 activation requires the direct interaction between PLC-γ1 and Rac1. We overexpressed either YFP-tagged wt PLC-γ1 or PLC-γ1ΔSH3 in Cos7 cells by transient transfection. Rac1 activation was determined by pulldown with GST-PAK. As shown in Fig. 5B, expression of wt PLC-γ1 but not the mutant PLC-γ1ΔSH3 significantly enhanced EGF-induced activation of Rac1. These data suggest that PLC-γ1 mediates EGF-induced Rac1 activation by its SH3 domain.
PLC-γ1 SH3 domain has been shown to participate in various cell signaling. To exclude the possibility that PLC-γ1 SH3 domain interacts with other proteins to indirectly regulate Rac1 activity, we determined whether Rac1 106PNTP109 that mediates Rac1 interaction with PLC-γ1 SH3 domain is also required for EGF-induced activation of Rac1. Both GFP-tagged wt Rac1 and Rac1PP/AA were expressed in Cos7 cells. Rac1 activation was determined by pulldown with GST-PAK. As shown in Fig. 5C, wt Rac1, but not Rac1PP/AA, was strongly activated by EGF stimulation. Together, our data indicate that EGF-induced Rac1 activation is mostly mediated by the interaction between PLC-γ1 SH3 domain and Rac1 106PNTP109.
To determine whether our observations also apply to other cells, we repeated the above experiment in 293T cells. Because 293T cells express only very low levels of EGFR, we cotransfected 293T cells with EGFR and YFP-tagged wt PLC-γ1 or PLC-γ1ΔSH3. The effects on Rac1 activation were very similar to that in Cos7 cells. Expression of wt PLC-γ1, but not the mutant PLC-γ1 P842L, significantly enhanced EGF-induced activation of Rac1 (Fig. 5D). We also cotransfected 293T cells with EGFR- and GFP-tagged wt Rac1 and Rac1PP/AA. We also showed by GST-PAK pulldown that wt Rac1, but not Rac1PP/AA, was strongly activated by EGF stimulation (Fig. 5E).
To exclude the possibility that PP/AA mutation of Rac1 altered its binding property to GTP and thus resulted in a nonfunctional Rac1, we determined the binding property of Rac1PP/AA to GTP. We transfected COS-7 cells with Rac1PP/AA and wt Rac1. The cell lysates were incubated with GTP-γS in the presence of EDTA for 15 min. The active form (GTP loaded form) of Rac1 was pulled down by GST-PAK. As shown in Fig. 6A, GST-PAK pulled down similar amounts of wt Rac1 and Rac1PP/AA, which indicates that both wt Rac1 and Rac1PP/AA have a similar ability to bind to GTP-γS. As controls, we also generated a GFP-tagged constitutive active Rac1 L61 with a single mutation of Q61 to L and a GFP-tagged dominant-negative Rac1 N17 (6). These two mutants were expressed in Cos7 cells by transient transfection, and their ability to bind to GTP-γS were determined as above. As expected, L61 showed strong binding affinity with GTP-γS, but N17 did not bind to GTP-γS. Therefore, the inability of Rac1PP/AA from getting activated under EGF stimulation is likely due to its inability to bind to PLC-γ1.
Fig. 6.

Direct interaction between PLC-γ1 and Rac1, not the other factors, is critical for EGF-induced Rac1 activation. A, Binding affinity of wt Rac1 and mutants Rac1 PP/AA, N17, and L61 to GTP-γS. COS-7 cells were transfected with GFP-tagged wt Rac1 or the mutants. The cell lysates were treated with GTP-γS for 15 min and were then pulled down by GST-PAK. Active GTP-Rac1 is visualized by immunoblotting with GFP antibody. B, Inhibition of PLC-γ1 phospholipase activity and the effects on EGF-induced Rac1 activation. Cos7 cells were transfected with PLC-γ1 and PLC-γ1ΔSH3. The cells were treated with U73122 and stimulated with EGF. The Rac1 activation was determined by GST-PAC pulldown. C, Quantification of the data from at least four independent experiments as described in panel B.
It is well documented that PLC-γ1 regulates various EGF-induced cell signaling through its phospholipase activity (16). To determine the contribution of PLC-γ1 phospholipase activity in EGF-induced Rac1 activation, we inhibited PLCγ1 phospholipase activity by U73122. We transfected Cos7 cells with wt PLC-γ1 and PLC-γ1ΔSH3. The cells were treated with U73122 and stimulated with EGF. As shown in Fig. 6B, wt PLC-γ1, but not PLC-γΔSH3, still strongly stimulates Rac1 activation. These data strongly suggest that PLC-γ1 SH3 domain, but not its phospholipase activity, is required for EGF-induced Rac1 activation. It is interesting to note that EGF-induced Rac1 activation seemed weaker when both PLC-γ1 SH3 domain was deleted and PLC-γ1 phospholipase activity was inhibited.
PLC-γ1 SH3 domain is a GEF for Rac1 in vitro
PLC-γ1 SH3 domain acts as a guanine nucleotide exchange factor (GEF) for PIKE and dynamin-1 (17, 28). To determine whether PLC-γ1 SH3 domain is a GEF for Rac1, an in vitro nucleotides-exchange assay was performed by using RhoGEF exchange assay kit (Cytoskeleton, Inc., Denver, CO). As shown in Fig. 7, A and C, the fluorescence intensity increased dramatically when purified PLC-γ1 SH3 domains were added to Rac1 but not when added to Cdc42 and RhoA. The GEF activity of Dbl on Cdc42 was used as a positive control, and water was used as a negative control. The exchange activity of PLC-γ1 SH3 domain on Rac1 is approximately 60% of the GEF activity of Dbl on Cdc42. However, the exchange activity of PLC-γ1 SH3 domain is almost 6-fold lower on RhoA than on Rac1 and is 50% lower on Cdc42 than on Rac1. These data indicate that PLC-γ1 SH3 domain is a specific GEF for Rac1.
Fig. 7.

The PLC-γ1 SH3 domain functions as a GEF for Rac1 in vitro. A, The GEF activity of the PLC-γ1 SH3 domain on Rac1, RhoA, and Cdc42 was determined by using RhoGEF Exchange Assay Biochem Kit as described in Materials and Methods. Experiments were performed in duplicate, and data from one experiment were presented. B, The GEF activity of other SH3 domains on Rac1. The same method was used as described in panel A. C, A bar chart showing the statistic analysis from three repeats shown in panels A and B.
We next examined whether only the SH3 domain from PLC-γ1 is a specific GEF for Rac1. We purified several SH3 domains from other proteins including p120 ras GAP, p85 subunit of phosphatidylinositol 3-kinase (PI3K), and Grb2. In vitro GEF activity assay showed that none of these SH3 domains had GEF activity on Rac1 (Fig. 7, B and C).
We next determined the binding affinity of PLC-γ1 SH3 domain with activated and inactivated Rac1. We generated two GFP-tagged Rac1 mutants: a dominant-negative mutant N17 and a constitutively activated mutant Q61. These two mutants and wt Rac1 were expressed in Cos7 cells by transient transfection. After serum starvation, the cells were either left nonstimulated or stimulated with EGF for 15 min. The PLC-γ1 GST-SH3 domain attached to glutathione beads was incubated with the cell lysates to pull down Rac1. As shown in Fig. 8, the PLC-γ1 SH3 domain bound most strongly to N17 regardless of EGF treatment. It also bound to GFP-Rac1 in both nonstimulated and stimulated conditions, but with a slightly higher affinity under nonstimulated conditions. The PLC-γ1 SH3 domain showed very weak binding to L61. These data indicated that like most GEFs, PLC-γ1 SH3 domain as a Rac1 GEF binds preferably to inactivated Rac1.
Fig. 8.

The binding affinity of PLC-γ1 SH3 domain to active and inactive Rac1. A, Cos7 cells were transfected by GFP-tagged wt Rac1, Rac1N17, or Rac1L61. With or without EGF stimulation for 15 min, the cell lysates were incubated with GST-fusion PLC-γ1 SH3 domain attached to glutathione-agarose beads. Bound proteins were analyzed by immunoblotting with antibodies to GFP. B, Quantification of the data from three independent experiments as described in panel D.
Regulation of EGF-induced F-actin formation and cell migration by PLC-γ1 SH3 domain and Rac1 interaction
We showed above that PLC-γ1 is a GEF for Rac1 in vitro and controls Rac1 activity in vivo by its SH3 domain interaction with Rac1 106PNTP109. We next examined whether this interaction regulates the cytoskeleton reorganization and cell migration. Cytoskeleton reorganization was assessed by F-actin formation. To examine F-actin formation, cells were incubated with 100 nm rhodamine-conjugated phalloidin. As shown in Fig. 9A, the addition of EGF for 15 and 30 min strongly stimulated the F-actin formation in Cos7 cells. We then disrupted the interaction between PLC-γ1 and Rac1 by transfecting Cos7 cells with YFP-tagged mutant PLC-γ1ΔSH3 or mutant PLC-γ1P842L. YFP-tagged wt PLC-γ1 was used as control. As shown in Fig. 9B, the transfected cells were green due to the expression of YFP-tagged PLC-γ1. We examined F-actin formation in these positively transfected green cells. After EGF stimulation for 15 min, F-actin formation (red fibers indicated by arrowhead) was prominent in cells transfected with wt PLC-γ1; however, F-actin formation was blocked in cells transfected with either mutant PLC-γ1ΔSH3 or mutant PLC-γ1P842L.
Fig. 9.
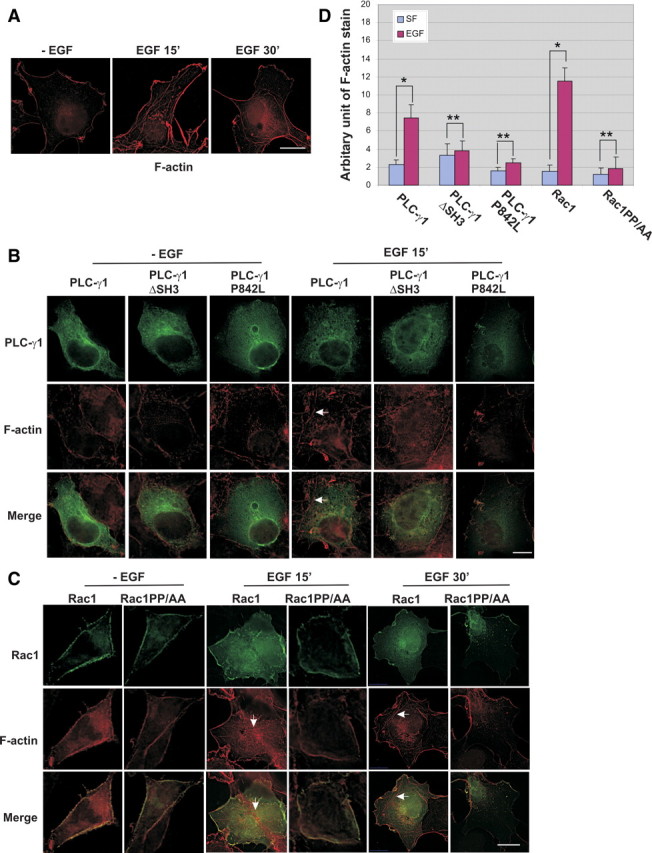
Effects of PLC-γ1 and Rac1 interaction on EGF-induced F-actin formation. F-actin was stained with 100 nm rhodamine-conjugated phalloidin as described in Materials and Methods. A, EGF-induced F-actin (red) formation in cos-7 cells. B, EGF-induced F-actin (red) formation in Cos7 cells transfected with GFP-tagged wt Rac1 or Rac1PP/AA. C, EGF-induced F-actin (red) formation in Cos7 cells transfected with YFP-tagged wt PLC-γ1, PLC-γ1ΔSH3, or PLC-γ1P842L. Arrows indicate F-actin. Scale bar, 20 μm. D, Quantification of the F-actin stain from images obtained from the experiments described in panels B and C. Quantification method is described in Materials and Methods. Each value is the mean of at least four experiments with more than three cells analyzed for each experiment. The error bar is the se. *, P < 0.01; **, P > 0.5. SF, Without EGF stimulation after incubation with serum-free medium; EGF, stimulation with EGF for 15 min.
We further disrupt the interaction between PLC-γ1 and Rac1 by transfecting Cos7 cells with GFP-tagged mutant Rac1PP/AA and examined the effects on EGF-induced F-actin formation. Cos7 cells transfected with GFP-tagged wt Rac1 were used as controls. As shown in Fig. 9C, both wt Rac1 and Rac1PP/AA (green) localized to the plasma membrane. After EGF stimulation, the F-actin formation (red fibers indicated by arrowhead) was prominent in cells transfected with wt Rac1. However, the expression of mutant Rac1PP/AA blocked EGF-induced F-actin formation. These data clearly indicate that the direct interaction between PLC-γ1 and Rac1 plays an important role in EGF-induced F-actin formation.
The images from the experiments described in Fig. 9, B and C, were quantitatively analyzed and the data were shown in Fig. 9D. Inhibition of the direct interaction between PLC-γ1 and Rac1 by mutation significantly reduced EGF-induced F-actin formation (Fig. 9D).
We then examined the effects of PLC-γ1 and Rac1 interaction in EGF-induced cell migration by wound-healing assay. We transfected Cos7 cells with either wt PLC-γ1 or PLC-γ1ΔSH3 tagged with YFP. Wound-healing assay showed that overexpression of wt PLC-γ1 significantly enhanced EGF-induced cell migration. However, deletion of PLC-γ1 SH3 domain not only abolished this enhancement but also significantly inhibited EGF-induced cell migration (Fig. 10A). These results suggest that the PLC-γ1 SH3 domain accounts for a significant part of cell migration induced by PLC-γ1. We then examined whether overexpression of wt Rac1 in Cos7 cells increases the cell’s migration and whether the effects are dependent on its interaction with PLC-γ1. Cos7 cells were transfected with either GFP-tagged wt Rac1 or mutant Rac1PP/AA. Wound-healing assay showed that overexpression of wt Rac1 increased cell migration by 30% (Fig. 10B). However, transfection of Cos7 cells with mutant Rac1PP/AA that does not bind to PLC-γ1 significantly inhibited EGF-induced cell migration (Fig. 10B).
Fig. 10.

Effects of PLC-γ1 and Rac1 interaction on EGF-induced cell motility. A and B, After transfection with wt or mutant PLC-γ1 or Rac1, confluent monolayers of serum-starved Cos7 cells were treated with EGF (100 ng/ml) as indicated. The cell migration was examined by wound-healing assay as described in Materials and Methods. C and D, After cotransfection with EGFR and wt or mutant PLC-γ1 or Rac1, confluent monolayer of serum-starved 293T cells were treated with EGF (100 ng/ml) as indicated. The cell migration was examined by wound-healing assay as described in Materials and Methods. Control: transfection of YFP vector (panels A and C) or GFP vector (panels B and D). Each value is the mean of at least three experiments with more than 50 cells counted for each experiment. The error bar is the se. *, Differences are statistically significant with P < 0.01.
We repeated the same experiments in 293T cells. As 293T cells express only very low level EGFR, we coexpressed EGFR with YFP-tagged PLC-γ1 or GFP-tagged Rac1. As shown in Fig. 10, C and D, we obtained very similar results as in Cos7 cells. These data again suggest that the enhancement of Rac1 on EGF-induced cell migration is dependent on its interaction with PLC-γ1. Together, our data indicate that interactions between PLC-γ1 and Rac1 are essential for EGF-induced cell migration.
Discussion
PLC-γ1 and Rac1 are two major players in EGF-induced cell migration (4, 5, 6, 7, 36, 37, 38). Although it is known that PLC-γ1 and Rac1 coordinate EGF-induced cell migration, no direct interactions between the two proteins have been reported. In this study, we, for the first time, demonstrated that PLC-γ1 and Rac1 coregulate EGF-induced cytoskeleton reorganization and cell migration by a direct functional interaction.
We first examined whether PLC-γ1 and Rac1 interact in response to EGF in vivo. Coimmunoprecipitation experiments showed that PLC-γ1 formed a complex with Rac1 in an EGF-dependent manner, and the complex formation reached maximum at 5–15 min of EGF stimulation (Fig. 1). It is well documented that Rac1 is localized to the plasma membrane (46) and PLC-γ1 translocated to the plasma membrane in response to EGF (23). This suggests that EGF-induced interaction between Rac1 and PLC-γ1 likely occurs at the plasma membrane. Further experiments were carried out to determine the structural requirement for the binding. PLC-γ1 contains two SH2 domains, one SH3 domain, and two PH domains. We conducted in vitro GST-pull-down experiments and showed that only GST-fusion PLC-γ1 SH3 domain pulled down Rac1, but not any other GST-fusion PLC-γ1 domain (Fig. 2A). We then showed that in vivo mutant PLC-γ1 lacking SH3 domain (PLC-γ1ΔSH3) failed to coimmunoprecipitate with Rac1 after EGF stimulation; however, the wt PLC-γ1 showed strong interactions with Rac1 (Fig. 2B). Together, these data suggest that PLC-γ1 SH3 domain is responsible for binding to Rac1.
We have also identified the 106PNTP109 of Rac1 as the motif responsible for binding to the PLC-γ1 SH3 domain. Mutation of the two proline residues to alanines completely abolished the interaction between PLC-γ1 and Rac1 (Fig. 3). When expressed in Cos7 cells, the mutant Rac1PP/AA failed to complex with PLC-γ1 in response to EGF (Fig. 3A). In the GST pull-down experiment, the PLC-γ1 SH3 domain failed to pull down the mutant Rac1PP/AA (Fig. 3B).
We further showed that purified PLC-γ1 SH3 domain is able to bind to purified Rac1; however, the nonfunctional mutant, PLCγ1 SH3 domain P842L, did not bind to purified Rac1 (Fig. 4). This demonstrates that the interaction between PLC-γ1 and Rac1 is direct. We also showed that the interaction between PLC-γ1 and Rac1 is very specific. No interaction between PLC-γ1 and RhoA or Cdc42 was observed (Fig. 4B).
It is interesting to note that the PLC-γ1 SH3 domain interaction with Rac1 is dependent on EGF stimulation in vivo but not in vitro. Although the in vivo and in vitro data seem at odds, similar observations have been reported previously. For example, the interaction between PLC-γ1 SH3 domain and PLD2 (27), PIKE (28), dynamin (17), and Emt (47) are all dependent on EGF stimulation in vivo. In fact, we recently observed similar interaction between PLC-γ1 and Akt (30). In that study our data suggest a model that in vivo, the full-length PLC-γ1 adopted a conformation that restricts the interaction of its SH3 domain with Akt. Phosphorylation of PLC-γ1 Y771 and/or Y783 by EGF stimulation releases the restriction on SH3 domain. However, in vitro, the GST-fusion PLC-γ1 SH3 domain does not contain any other parts of the protein, and the SH3 domain is always exposed for interaction with Akt with or without EGF stimulation. Therefore, the in vitro interaction between the PLC-γ1 SH3 domain and Akt is independent of EGF (30). It is likely that the interaction between the PLC-γ1 SH3 domain and Rac1 proline motif could also be explained by this model.
We have provided multiple evidence in this study to demonstrate that the interaction between PLC-γ1 and Rac1 results in the activation of Rac1. We showed that EGF strongly activated Rac1 activity, and the maximum activation of Rac1 is at 15 min, which is coincident with the maximum interaction between PLC-γ1 and Rac1 (Figs. 1 and 5A). We then showed that EGF-induced Rac1 activation is dependent on the interaction between PLC-γ1 SH3 domain and Rac1 106PNTP109 motif. When expressed in both Cos7 and 293T cells, mutant PLC-γ1ΔSH3 or PLC-γ1 P842L did not enhance EGF-induced Rac1 activation (Fig. 5B), which suggests that PLC-γ1 mediates EGF-induced Rac1 activation by its SH3 domain. Because the PLC-γ1 SH3 domain has been shown to participate in various cell signaling (27, 29, 30) and has been identified as a GEF for PIKE GTPase and dynamin GTPase (17, 28), it is possible that the role of PLC-γ1 SH3 domain in EGF-induced Rac1 activation may be due to its interaction with other proteins to indirectly regulate Rac1 activity. We excluded this possibility by showing that the expression of mutant Rac1PP/AA that failed to bind to PLC-γ1 blocked EGF-induced Rac1 activation (Fig. 5C). We also showed that the inability of Rac1PP/AA from getting activated under EGF stimulation is likely due to its inability to bind to PLC-γ1, rather than its inability to load GTP (Fig. 6A).
It is well documented that PLC-γ1 regulates various EGF-induced cell signaling through its phospholipase activity (16). It was reported that PLC-γ1 phospholipase activity is important in EGF-induced Rac1 activation (48). To exclude the possibility that the observed effects of PLC-γ1 on EGF-induced Rac1 activation is through its phospholipase activity, we inhibited PLCγ1 phospholipase activity by using a specific inhibitor U73122. Inhibition of PLC-γ1 phospholipase activity will block the production of PKC and DAG, and thus eliminate the indirect activation of Rac1 by PLC-γ1. In this case, the observed effects of PLC-γ1 on Rac1 activation will be likely due to the direct function of PLC-γ1 SH3 domain as a Rac1 GEF. Indeed, our data showed that inhibition of PLC-γ1 phospholipase activity did not inhibit PLC-γ1-mediated Rac1 activation after EGF stimulation (Fig. 6B). These data strongly suggest that PLC-γ1 SH3 domain, but not its phospholipase activity, plays important role in EGF-induced Rac1 activation. However, our data did not exclude the possibility that PLC-γ1 phospholipase activity may regulate Rac1 activation in a physiological condition.
Moreover, we demonstrated that PLC-γ1 SH3 domain is a specific and potent GEF for Rac1 (Fig. 7). This finding provides a logical explanation for our in vivo data that the direct interaction between PLC-γ1 and Rac1 results in Rac1 activation. Most likely, PLC-γ1 acts as a Rac1 GEF in vivo in response to EGF. Interestingly, we showed that the PLC-γ1 SH3 domain had a strong GEF activity on Rac1, but only slightly activated Cdc42 and RhoA. So far, most of the identified GEFs for Rho GTPases have been shown to activate Rac1, Cdc42, and RhoA. For example, the best-characterized Rho GEFs are Vav proteins. Both Vav1 and Vav2 have been shown to act on Rac1, Cdc42, and RhoA in vitro (49, 50). It is reported that Tiam1 is a GEF for RhoA, Rac1, and Cdc42 (51, 52). More importantly, we showed that PLC-γ1 only interacts with Rac1, but not RhoA and Cdc42 in response to EGF in vivo (Fig. 4B). Together these data indicate that by acting as a specific GEF for Rac1, PLC-γ1 provides a means for cells to selectively activate Rac1 without activating the other Rho proteins.
Consistent with previous reports (17, 28), the GEF activity of SH3 domain seems to be restricted only to PLC-γ1, as we showed that SH3 domains of Grb2, PI3K p85, and p120ras GAP did not show any GEF activity on Rac1, Cdc42, and Rho A (Fig. 7). Moreover, although it was shown to be a GEF for PIKE and dynamin, our finding is the first report that PLC-γ1 SH3 domain is a GEF for small GTPases.
We also showed that PLC-γ1 SH3 domain bound to GFP-Rac1 in both nonstimulated and stimulated conditions, but with a higher affinity under nonstimulated conditions (Fig. 8). PLC-γ1 SH3 domain bound most strongly to N17, the dominant-negative Rac1 regardless of EGF treatment, and it showed no binding to Q61, the constitutively activated Rac1, with or without EGF treatment (Fig. 8). These data indicated that like most GEFs, the PLC-γ1 SH3 domain as a Rac1 GEF binds preferably to inactivated Rac1. In light of this finding, it is interesting to note that we are able to show fairly strong coimmunoprecipitation of PLC-γ1 and Rac1 after EGF stimulation. One simplistic explanation may be that the activation process of PLC-γ1 on Rac1 is slow, which allows the coimmunoprecipitation of PLC-γ1 and Rac1 complex as we showed in the study. The reason that we did not observe the interaction between PLC-γ1 and Rac1 in the absence of EGF treatment is not that the PLC-γ1 SH3 domain does not interact with inactivated Rac1; instead, is that PLC-γ1 and Rac1 do not localize in the same subcellular compartment, and the PLC-γ1 SH3 domain is restricted from interaction as discussed above. Finally, we showed that the interaction between PLC-γ1 and Rac1 is critical in EGF-induced cytoskeleton reorganization and cell migration (Figs. 9 and 10). This indicates that the interaction between PLC-γ1 and Rac1 is physiologically relevant.
Before our reported interaction between PLC-γ1 and Rac1 in regulating EGF-induced F-actin remodeling and cell migration, both PLC-γ1 and Rac1 have been shown to regulate EGF-induced F-actin remodeling and cell migration. It was shown that EGF stimulates the PLC-γ1 phospholipase activity that, in turn, hydrolyzes PIP2, which leads to the release of profilin, a cytoplasmic actin-binding protein (33). It was also shown EGF activates Rac1 by stimulating Rac1 GEF Tiam-1 through the activation of phosphoinositide 3-kinases (53). Here, we not only provide a direct functional link between PLC-γ1 and Rac1 in EGF-induced cell migration but also revealed the mechanism of this functional link. PLC-γ1 acts as a GEF for Rac1 in response to EGF to activate Rac1. However, as discussed above, the interaction between PLC-γ1 and Rac1 is not the only mechanism for both of them to regulate EGF-induced cell migration. The multiple mechanisms may ensure the proper control of the cell on EGF-induced cell migration when one mechanism fails.
Materials and Methods
Cell culture, transfection, and treatment
COS-7 cells were grown at 37 C in DMEM containing 10% fetal bovine serum, and were maintained in a 5% CO2 atmosphere. Before transfection, COS-7 cells were seeded into 100-mm plates and incubated until cells were 40–60% confluent. The transfections were performed using Lipofectin Reagent (Invitrogen, Carlsbad, CA; 18292) according to the manufacturer’s instructions. For the EGF treatments, COS-7 cells were serum starved for 12 h, and EGF was added to a final concentration of 100 ng/ml.
Antibodies and chemicals
Mouse monoclonal anti-PLC-γ1 and mouse monoclonal anti Rac1 antibodies were from Upstate Biotechnology, Inc. (Lake Placid, NY). Rabbit anti-GFP antibody, pEGFP-C3, and pEYFP-C1 vectors were from CLONTECH (Mountain View, CA). Rho GEF Exchange assay biochem kit was purchased from Cytoskeleton. Mouse anti-GST antibodies and rabbit anti-Rac1 antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Glutathione cross-linked to 4% agarose, goat antimouse IgG conjugated with agarose, protein A conjugated with agarose, and factor Xa were purchased from Sigma-Aldrich (St. Louis, MO). Unless otherwise specified, all the chemicals were from Sigma.
Plasmids
The YFP-tagged full-length and SH3 domain deletion mutant of PLC-γ1 and pcDNA3-EGFR plasmid were generated previously (23). Various GST-fusion proteins (including PLC-γ1N-SH2, PLC-γ1C-SH2, PLC-γ1SH3, and PLC-γ1N-PH, PLC-γ1 N-SH2, C-SH2, SH3, Grb2 SH3, p120ras GAP SH3, and p85α subunit of PI3K SH3 domains) were generated previously in the laboratory (45). GFP-Rac1 was a gift from Dr. Mark R. Philips (New York University School of Medicine). GST-fusion PAK Rho binding domain (GST-PAK) construct was a gift from Dr. Gary Eitzen (University of Alberta). All the mutants with point mutation were created with the QuikChange Multiple Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) with GFP-tagged wt Rac 1 (a gift from Dr. Mark R. Philips, NYU School of Medicine) as a template. These mutants include a GFP-tagged mutant Rac 1 with mutation T17 to asparagine (termed N17), a GFP-tagged mutant with mutation of Q61 to leucine (termed L61), and a mutant of GFP-tagged Rac 1 with mutations of both P106 and P109 to alanine (termed Rac1PP/AA).
Expression and purification of GST-fusion proteins
To purify various GST-fusion proteins including PLC-γ1 N-SH2, C-SH2, SH3, Grb2 SH3, p120ras GAP SH3, p85α subunit of PI3K SH3, and GST-PAK, the pGEX plasmids containing these domains were transformed into Escherichia coli DH5α. Bacteria were grown to an optical density (OD)600 of 0.3–0.4 and induced with 1 mm isopropyl-1-thio-β-d-galactopyranoside for 2 h at 37 C. After pelleting, bacterial cells were lysed by sonication in 50 mn Tris (pH 8.0), 100 mm NaCl, 10% glycerol, 1 mm dithiothreitol, containing protease inhibitors [0.02% NaN3, 0.1 mm 4-(2-aminoethyl)-benzenesulfonyl fluoride, 10 μg/ml aprotinin, and 1 μm pepstatin A]. Triton X-100 was added to a final concentration of 1%, and particulates were removed by centrifugation for 10 min at 10,000 rpm in a JA-17 (Beckman Coulter, Fullerton, CA) rotor. The cleared lysate was incubated with glutathione-agarose beads (Sigma-Aldrich) for 1 h at 4 C, washed three times with ice-cold GST-wash buffer containing 1 mm dithiothreitol plus protease inhibitors, and protein bound to the glutathione-agarose beads was stored at 4 C. To purify the PH domain of PLC-γ1, E. coli BL-21, a strain that is defective in OmpT and Lon protease production and expresses the fusion protein in a more soluble and intact form, was used for transformation. Cells were grown to an OD600 of 0.3–0.4 and induced with 0.1 mm isopropyl-1-thio-β-d-galactopyranoside for 8–12 h at 22 C. The later steps were the same as for the other GST-fusion proteins.
In vitro GEF activity assay
We first prepared SH3 domains of PLC-γ1, p120ras GAP, p85a subunit of PI3K, and Grb2 by cleaving the GST with Factor Xa or thrombin. Briefly, the GST-fusion SH3 domains bound to glutathione-agarose beads were washed three times with Factor Xa or thrombin cleavage buffer containing 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 1 mm CaCl2. Factor Xa or thrombin was added to 50% slurry beads with final concentration of 50 μg/ml followed by agitation for 16 h at room temperature to cleave SH3 domains from GST-SH3 immobilized on beads. The supernatant of beads was recovered, and cleaved SH3 domains were visualized by Coomassie blue staining.
In vitro GEF activity assay was then carried out by using RhoGEF Exchange Assay Biochem Kit (Cytoskeleton, Inc.) according to the manufacturer’s instructions. Briefly, fluorescence spectroscopic analysis of N-methylanthraniloyl (mant)-GTP incorporation into purified His-Rac 1 was carried out using PTI QM-4 SE spectrometer at 20 C. Exchange reaction assay mixtures containing 20 mm Tris (pH 7.5), 50 mm NaCl, 10 mm MgCl2, 50 μg/ml BSA, 0.75 μm mant-GTP, and 2 μm of Rac 1, Cdc42, or RhoA GTPase were prepared and allowed to equilibrate with slow continuous stirring. After equilibration, the mixtures were placed into four sample holders, and fluorescence measurements were taken approximately every 30 sec (0–40 μsec integration) with excitation and emission wavelengths of 360 nm and 440 nm, respectively, and 10-nm bandwidth. After five readings (150 sec), purified SH3 domains/Dbls/Water was added to 0.8 μm, and the relative mant fluorescence (λex = 360 nm, λem = 440 nm) was monitored. Experiments were performed in duplicate.
In vitro GST pull-down assay
COS-7 cells were treated with or without EGF and then lysed into BOS buffer [50 mm Tris-HCl (pH 7.4), 200 mm NaCl, 1% Nonidet P-40, 10% glycerol, 10 mm MgCl2, and 1 mm EDTA] with protease inhibitors. The lysates were centrifuged at 21,000 × g at 4 C for 30 min. Supernatants were used in the binding assay. GST-fusion PLC-γ1 proteins bound to glutathione-agarose-beads in BOS buffer were added and incubated at 4 C for 1 h. Beads were collected by centrifugation and washed three times with BOS buffer after which loading buffer was added. The pull-down proteins were resolved on SDS-PAGE and analyzed by immunoblotting with anti-GST and anti-Rac1 antibodies.
In vitro binding of purified PLC-γ1 domains and Rac1
Purified GST domains of various PLC-γ1 were immobilized on GST beads, and the GST domain-immobilized beads were incubated with pure Rac1 purchased from Cytoskeleton Inc. in binding buffer containing 20 mm Tris (pH 7.5), 50 mm NaCl, 20 mm MgCl2, 50 μg/ml BSA, and 0.75 μm GTP at room temperature for 1 h. The beads with bound proteins were washed three times with binding buffer after which the bound proteins were resolved in 10% SDS-PAGE followed by immunoblotting.
Immunoprecipitation
Immunoprecipitation experiments were carried out as described previously (54). Briefly, cells were lysed with immunoprecipitation buffer [20 mm Tris (pH 7.5), 150 mm NaCl, 1% Nonidet P-40, 0.1% sodium deoxycholate, 100 mm NaF, 5 mm MgCl2, 0.5 mm Na3VO4, 0.02% NaN3, 0.1 mm 4-(2-aminoethyl)-benzenesulfonyl fluoride, 10 μg/ml aprotinin, and 1 μm pepstatin A] overnight at 4 C. Cell lysates were centrifuged at 22,000 × g for 30 min to remove debris. The supernatants, containing 1 mg of total protein, were precleared with the agarose beads and then were used to incubate with 1 μg of specific antibody for 2 h with gentle mixing by inverting. Then, goat antimouse IgG conjugated with agarose or protein A conjugated with agarose was added to each fraction and incubated for 2 h with agitation. Finally, both the agarose beads and the nonprecipitated supernatant were collected by centrifugation. For the controls, mouse or rabbit IgG was used to replace the primary antibodies. The agarose beads were washed three times with immunoprecipitation buffer, and 1× loading buffer was added. The sample was boiled for 5 min and prepared for SDS-PAGE followed by Western blot.
Immunofluorescence and F-actin formation assay
Indirect immunofluorescence was carried out as described previously (54). Cells were grown on glass cover slips to subconfluence and serum starved for 24 h. After treatment with 100 ng/ml EGF for the indicated time, the cells were fixed by immersion in −20 C methanol for 5 min. After removal of the methanol and washing with PBS, the cells were permeabilized with 0.5% Triton X-100 for 10 min at room temperature. The cover slips were incubated for 1 h at room temperature with the primary antibody, followed by 45-min incubation with the second antibody, respectively. For F-actin formation assay, cells were incubated with 100 nm rhodamine-conjugated phalloidin after permeablilization. The stained cells were analyzed by Delta Vision Deconvolution microscopic systems (Applied Precision, Issaquah, WA). Color photographs were taken with a digital camera by superimposing the monochrome graphs of two channels, and the data were analyzed using Delta Vision softWoRx software. To quantify the F-actin formation, the boundary of the cells were determined by using differential interference contrast images, after which the total intensity of F-actin in the cytosol was calculated by the software. Each value is the mean of at least three experiments.
In vivo GEF activity assay
In vivo PLC-γ1 GEF activity was measured by its ability to activate Rac1. Rac1 activity was determined by using an assay developed by Ren and Schwartz (55). The Rac1binding domain of PAK, a Rac1 effector, was used as GST fusion to pull down active Rac1. Briefly, COS-7 or 293T cells with or without transfections were serum starved for 12 h followed by EGF (100 ng/ml) stimulation for different time period. The cells were lysed into GST-PAK buffer (50 mm Tris-HCl, pH 7.6; 150 mm NaCl; 1% Triton X-100; and 10 mm MgCl2) with protease inhibitors. The lysates were centrifuged at 21,000 × g at 4 C for 15 min. Supernatants were used in the binding assay. GST-PAK-fusion proteins bound to glutathione-agarose-beads in GST-PAK buffer were added and incubated at 4 C for 1 h. Beads were collected by centrifugation, washed three times with GST-PAK buffer, after which SDS loading buffer was added. The pull-down active Rac 1 were resolved on SDS-PAGE and analyzed by immunoblotting with anti-GST and anti-Rac 1 antibodies. For the GTP-γS positive control, COS-7 cells were lysed in GST-PAK buffer, and lysates were centrifuged at 21,000 × g at 4 C for 15 min. Supernatants were collected. EDTA and GTP-γS were added to cell lysates with final concentration of 10 mm and 100 μm, respectively. The mixture then was incubated at 30 C for 15 min followed by adding MgCl2 to final 60 mm. The lysates were then used for GST-PAK binding assay.
Wound-healing assay
COS-7 cells were grown on 24-well plate at 40–60% confluence and transfected with different GFP-tagged PLC-γ1 constructs. After 24–48 h, the cells reached 100% confluence, and a wound was created with a glass pipette. A nearby reference point was created by a needle. The plate was washed once with serum free medium and replaced with the desired medium. The cells were observed under a fluorescence microscope to ensure that enough cells in the leading edge of the wound were positively transfected. Both phase-contrast and fluorescence images were acquired every 2 h by matching the reference point until the wound had completely closed. To calculate the rate of migration of the transfected cells, we measured the distance traveled toward the center of the wound after 8 h. We performed wound-healing assays only at 8 h, which can limit the effect of the DNA synthesis. At least eight to 10 randomly chosen areas including at least 50 cells were quantified. Experiments were repeated three to four times, and an individual photograph was chosen as the example. The relative distance to the reference line was calculated and normalized to the nontransfected cells, and the data are expressed as means ± ses of the percentage of the nontransfected cells from at least eight to 10 randomly chosen areas from three to four separate experiments.
Acknowledgments
We thank Dr. Mark R. Philips (NYU School of medicine) for providing GFP-Rac1 and Dr. Gary Eitzen (University of Alberta) for providing GST-PAK.
Footnotes
This work was supported in part by grants from the Canadian Institutes of Health Research, Natural Sciences and Engineering Research Council of Canada, and the Alberta Heritage Foundation for Medical Research (AHFMR). Z.W. is an AHFMR Senior Scholar.
Disclosure Summary: Z.W. receives research grants from Natural Sciences and Engineering Research Council and AHFMR to support the research reported here. S.L., Q.W., Y.W., and X.C. have nothing to declare.
First Published Online March 5, 2009
Abbreviations: EGF, Epidermal growth factor; EGFR, epidermal growth factor receptor; GAP, GTPase-activating protein; GEF, guanine nucleotide exchange factor; GF, growth factor; GFP, green fluorescent protein; GST, glutathione S-transferase; GST-PAK, GST-fusion PAK Rho-binding domain; PAK, p21-activated protein kinase; PH domain, pleckstrin homology domain; PI3K, phosphatidylinositol 3-kinase; PIKE, PI3kinase enhancer; PIP2, phosphatidylinositol-4,5-bis-phosphate; PLD2, phospholipase D2; PLC-γ1, phospholipase C-γ1; Rac1PP/AA, GFP-tagged mutant Rac; RhoA, rhodamine A; SH2 domain, Src homology 2 domain; SH3 domain, Src homology 3 domain; wt, wild type; YFP, yellow fluorescent protein.
References
- 1.Pawson T1995. Protein modules and signalling networks. Nature 373:573–580 [DOI] [PubMed] [Google Scholar]
- 2.Wells A, Gupta K, Chang P, Swindle S, Glading A, Shiraha H1998. Epidermal growth factor receptor-mediated motility in fibroblasts. Microsc Res Tech 43:395–411 [DOI] [PubMed] [Google Scholar]
- 3.Schlessinger J, Ullrich A1992. Growth factor signaling by receptor tyrosine kinases. Neuron 9:383–391 [DOI] [PubMed] [Google Scholar]
- 4.Chen P, Murphy-Ullrich JE, Wells A1996. A role for gelsolin in actuating epidermal growth factor receptor-mediated cell motility. J Cell Biol 134:689–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wells A, Kassis J, Solava J, Turner T, Lauffenburger DA2002. Growth factor-induced cell motility in tumor invasion. Acta Oncol 41:124–130 [DOI] [PubMed] [Google Scholar]
- 6.Ridley AJ, Hall A1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70:389–399 [DOI] [PubMed] [Google Scholar]
- 7.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A1992. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70:401–410 [DOI] [PubMed] [Google Scholar]
- 8.Margolis B, Zilberstein A, Franks C, Felder S, Kremer S, Ullrich A, Rhee SG, Skorecki K, Schlessinger J1990. Effect of phospholipase C-γ overexpression on PDGF-induced second messengers and mitogenesis. Science 248:607–610 [DOI] [PubMed] [Google Scholar]
- 9.Meisenhelder J, Suh PG, Rhee SG, Hunter T1989. Phospholipase C-γ is a substrate for the PDGF and EGF receptor protein-tyrosine kinases in vivo and in vitro. Cell 57:1109–1122 [DOI] [PubMed] [Google Scholar]
- 10.Wahl MI, Daniel TO, Carpenter G1988. Antiphosphotyrosine recovery of phospholipase C activity after EGF treatment of A-431 cells. Science 241:968–970 [DOI] [PubMed] [Google Scholar]
- 11.Anderson D, Koch CA, Grey L, Ellis C, Moran MF, Pawson T1990. Binding of SH2 domains of phospholipase C γ 1, GAP, and Src to activated growth factor receptors. Science 250:979–982 [DOI] [PubMed] [Google Scholar]
- 12.Kim HK, Kim JW, Zilberstein A, Margolis B, Kim JG, Schlessinger J, Rhee SG1991. PDGF stimulation of inositol phospholipid hydrolysis requires PLC-γ 1 phosphorylation on tyrosine residues 783 and 1254. Cell 65:435–441 [DOI] [PubMed] [Google Scholar]
- 13.Rönnstrand L, Mori S, Arridsson AK, Eriksson A, Wernstedt C, Hellman U, Claesson-Welsh L, Heldin CH1992. Identification of two C-terminal autophosphorylation sites in the PDGF β-receptor: involvement in the interaction with phospholipase C-γ. EMBO J 11:3911–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rotin D, Margolis B, Mohammadi M, Daly RJ, Daum G, Li N, Fischer EH, Burgess WH, Ullrich A, Schlessinger J1992. SH2 domains prevent tyrosine dephosphorylation of the EGF receptor: identification of Tyr992 as the high-affinity binding site for SH2 domains of phospholipase C γ. EMBO J 11:559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wahl M, Carpenter G1991. Selective phospholipase C activation. Bioessays 13:107–113 [DOI] [PubMed] [Google Scholar]
- 16.Kamat A, Carpenter G1997. Phospholipase C-γ1: regulation of enzyme function and role in growth factor-dependent signal transduction. Cytokine Growth Factor Rev 8:109–117 [DOI] [PubMed] [Google Scholar]
- 17.Choi JH, Park JB, Bae SS, Yun S, Kim HS, Hong WP, Kim IS, Kim JH, Han MY, Ryu SH, Patterson RL, Snyder SH, Suh PG2004. Phospholipase C-γ1 is a guanine nucleotide exchange factor for dynamin-1 and enhances dynamin-1-dependent epidermal growth factor receptor endocytosis. J Cell Sci 117:3785–3795 [DOI] [PubMed] [Google Scholar]
- 18.Gual P, Giordano S, Williams TA, Rocchi S, Van Obberghen E, Comoglio PM2000. Sustained recruitment of phospholipase C-γ to Gab1 is required for HGF-induced branching tubulogenesis. Oncogene 19:1509–1518 [DOI] [PubMed] [Google Scholar]
- 19.Bivona TG, Pérez De Castro, I, Ahearn IM, Grana TM, Chiu VK, Lockyer PJ, Cullen PJ, Pellicer A, Cox AD, Philips MR2003. Phospholipase Cγ activates Ras on the Golgi apparatus by means of RasGRP1. Nature 424:694–698 [DOI] [PubMed] [Google Scholar]
- 20.Chou J, Burke NA, Iwabu A, Watkins SC, Wells A2003. Directional motility induced by epidermal growth factor requires Cdc42. Exp Cell Res 287:47–56 [DOI] [PubMed] [Google Scholar]
- 21.Meyer RD, Latz C, Rahimi N2003. Recruitment and activation of phospholipase Cγ1 by vascular endothelial growth factor receptor-2 are required for tubulogenesis and differentiation of endothelial cells. J Biol Chem 278:16347–16355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsuda M, Paterson HF, Rodriguez R, Fensome AC, Ellis MV, Swann K, Katan M2001. Real time fluorescence imaging of PLC γ translocation and its interaction with the epidermal growth factor receptor. J Cell Biol 153:599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Wang Z2003. Regulation of EGF-induced phospholipase C-γ1 translocation and activation by its SH2 and PH domains. Traffic 4:618–630 [DOI] [PubMed] [Google Scholar]
- 24.Bar-Sagi D, Rotin D, Batzer A, Mandiyan V, Schlessinger J1993. SH3 domains direct cellular localization of signaling molecules. Cell 74:83–91 [DOI] [PubMed] [Google Scholar]
- 25.Gout I, Dhand R, Hiles ID, Fry MJ, Panayotou G, Das P, Truong O, Totty NF, Hsuan J, Booker GW1993. The GTPase dynamin binds to and is activated by a subset of SH3 domains. Cell 75:25–36 [PubMed] [Google Scholar]
- 26.Seedorf K, Kostka G, Lammers R, Bashkin P, Daly R, Burgess WH, van der Bliek AM, Schlessinger J, Ullrich A1994. Dynamin binds to SH3 domains of phospholipase C γ and GRB-2. J Biol Chem 269:16009–16014 [PubMed] [Google Scholar]
- 27.Jang IH, Lee S, Park JB, Kim JH, Lee CS, Hur EM, Kim IS, Kim KT, Yagisawa H, Suh PG, Ryu SH2003. The direct interaction of phospholipase C-γ 1 with phospholipase D2 is important for epidermal growth factor signaling. J Biol Chem 278:18184–18190 [DOI] [PubMed] [Google Scholar]
- 28.Ye K, Aghdasi B, Luo HR, Moriarity JL, Wu FY, Hong JJ, Hurt KJ, Bae SS, Suh PG, Snyder SH2002. Phospholipase C γ 1 is a physiological guanine nucleotide exchange factor for the nuclear GTPase PIKE. Nature 415:541–544 [DOI] [PubMed] [Google Scholar]
- 29.Patterson RL, van Rossum DB, Ford DL, Hurt KJ, Bae SS, Suh PG, Kurosaki T, Snyder SH, Gill DL2002. Phospholipase C-γ is required for agonist-induced Ca2+ entry. Cell 111:529–541 [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Wu J, Wang Z2006. Akt binds to and phosphorylates phospholipase C-γ1 in response to epidermal growth factor. Mol Biol Cell 17:2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kundra V, Escobedo JA, Kazlauskas A, Kim HK, Rhee SG, Williams LT, Zetter BR1994. Regulation of chemotaxis by the platelet-derived growth factor receptor-β. Nature 367:474–476 [DOI] [PubMed] [Google Scholar]
- 32.Derman MP, Chen JY, Spokes KC, Songyang Z, Cantley LG1996. An 11-amino acid sequence from c-met initiates epithelial chemotaxis via phosphatidylinositol 3-kinase and phospholipase C. J Biol Chem 271:4251–4255 [DOI] [PubMed] [Google Scholar]
- 33.Goldschmidt-Clermont PJ, Kim JW, Machesky LM, Rhee SG, Pollard TD1991. Regulation of phospholipase C-γ 1 by profilin and tyrosine phosphorylation. Science 251:1231–1233 [DOI] [PubMed] [Google Scholar]
- 34.Symons M, Settleman J2000. Rho family GTPases: more than simple switches. Trends Cell Biol 10:415–419 [DOI] [PubMed] [Google Scholar]
- 35.Moon SY, Zheng Y2003. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol 13:13–22 [DOI] [PubMed] [Google Scholar]
- 36.Malliri A, Collard JG2003. Role of Rho-family proteins in cell adhesion and cancer. Curr Opin Cell Biol 15:583–589 [DOI] [PubMed] [Google Scholar]
- 37.Sahai E, Marshall CJ2002. RHO-GTPases and cancer. Nat Rev Cancer 2:133–142 [DOI] [PubMed] [Google Scholar]
- 38.Lin M, van Golen KL2004. Rho-regulatory proteins in breast cancer cell motility and invasion. Breast Cancer Res Treat 84:49–60 [DOI] [PubMed] [Google Scholar]
- 39.Etienne-Manneville S, Hall A2002. Rho GTPases in cell biology. Nature 420:629–635 [DOI] [PubMed] [Google Scholar]
- 40.Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW2002. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 418:790–793 [DOI] [PubMed] [Google Scholar]
- 41.Sugihara K, Nakatsuji N, Nakamura K, Nakao K, Hashimoto R, Otani H, Sakagami H, Kondo H, Nozawa S, Aiba A, Katsuki M1998. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene 17:3427–3433 [DOI] [PubMed] [Google Scholar]
- 42.Loirand G, Guilluy C, Pacaud P2006. Regulation of Rho proteins by phosphorylation in the cardiovascular system. Trends Cardiovasc Med 16:199–204 [DOI] [PubMed] [Google Scholar]
- 43.Brown JH, Del Re DP, Sussman MA2006. The Rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circ Res 98:730–742 [DOI] [PubMed] [Google Scholar]
- 44.Lezoualc'h F, Métrich M, Hmitou I, Duquesnes N, Morel E2008. Small GTP-binding proteins and their regulators in cardiac hypertrophy. J Mol Cell Cardiol 44:623–632 [DOI] [PubMed] [Google Scholar]
- 45.Wang Z, Glück S, Zhang L, Moran MF1998. Requirement for phospholipase C-γ1 enzymatic activity in growth factor-induced mitogenesis. Mol Cell Biol 18:590–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt A, Hall A2002. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev 16:1587–1609 [DOI] [PubMed] [Google Scholar]
- 47.Perez-Villar JJ, Kanner SB1999. Regulated association between the tyrosine kinase Emt/Itk/Tsk and phospholipase-Cγ1 in human T lymphocytes. J Immunol 163:6435–6441 [PubMed] [Google Scholar]
- 48.Nogami M, Yamazaki M, Watanabe H, Okabayashi Y, Kido Y, Kasuga M, Sasaki T, Maehama T, Kanaho Y2003. Requirement of autophosphorylated tyrosine 992 of EGF receptor and its docking protein phospholipase C γ 1 for membrane ruffle formation. FEBS Lett 536:71–76 [DOI] [PubMed] [Google Scholar]
- 49.Bustelo XR1996. The VAV family of signal transduction molecules. Crit Rev Oncog 7:65–88 [DOI] [PubMed] [Google Scholar]
- 50.Bustelo XR2000. Regulatory and signaling properties of the Vav family. Mol Cell Biol 20:1461–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Habets GG, Scholtes EH, Zuydgeest D, van der Kammen RA, Stam JC, Berns A, Collard JG1994. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell 77:537–549 [DOI] [PubMed] [Google Scholar]
- 52.Mertens AE, Roovers RC, Collard JG2003. Regulation of Tiam1-Rac signalling. FEBS Lett 546:11–16 [DOI] [PubMed] [Google Scholar]
- 53.Schmitz AA, Govek EE, Böttner B, Van AL2000. Rho GTPases: signaling, migration, and invasion. Exp Cell Res 261:1–12 [DOI] [PubMed] [Google Scholar]
- 54.Wang Z, Zhang L, Yeung TK, Chen X1999. Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol Biol Cell 10:1621–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ren XD, Schwartz MA2000. Determination of GTP loading on Rho. Methods Enzymol 325:264–272 [DOI] [PubMed] [Google Scholar]