

Abstract
We have previously shown that adenoviral expression of peroxisome proliferator-activated receptors (PPARs) leads to rapid establishment of transcriptionally active complexes and activation of target gene expression within 5–8 h after transduction. Here we have used the adenoviral delivery system combined with expression array analysis to identify novel putative PPARγ target genes in murine fibroblasts and to determine the role of the A/B-domain in PPARγ-mediated transactivation of genomic target genes. Of the 257 genes found to be induced by PPARγ2 expression, only 25 displayed A/B-domain dependency, i.e. significantly reduced induction in the cells expressing the truncated PPARγ lacking the A/B-domain (PPARγCDE). Nine of the 25 A/B-domain-dependent genes were involved in lipid storage, and in line with this, triglyceride accumulation was considerably decreased in the cells expressing PPARγCDE compared with cells expressing full-length PPARγ2. Using chromatin immunoprecipitation, we demonstrate that PPARγ binding to genomic target sites and recruitment of the mediator component TRAP220/MED1/PBP/DRIP205 is not affected by the deletion of the A/B-domain. By contrast, the PPARγ-mediated cAMP response element-binding protein (CREB)-binding protein (CBP) and p300 recruitment to A/B-domain-dependent target genes is compromised by deletion of the A/B-domain. These results indicate that the A/B-domain of PPARγ2 is specifically involved in the recruitment or stabilization of CBP- and p300-containing cofactor complexes to a subset of target genes.
The N-terminal domain of PPARγ is specifically required for full activation of a subset of lipogenic target genes and for p300/CBP recruitment to associated target sites.
The peroxisome proliferator-activated receptor (PPAR) subgroup of the nuclear receptor (NR) family of transcription factors consists of three subtypes, PPARα, PPARβ/δ, and PPARγ. The PPARs bind as heterodimers with the retinoid X receptor (RXR) to conserved PPAR response elements (PPREs) but display distinct tissue-specific expression patterns. PPARα is expressed at high levels in metabolically active tissues such as liver, muscle, and heart, whereas PPARβ/δ expression is ubiquitous (1). PPARγ exists in two isoforms: PPARγ2, which contains 30 additional amino acids at the N terminus and is fat selective, and PPARγ1, which is expressed at high levels in adipose tissue but also at low levels in many other tissues, including colon, liver, muscle, and macrophages (2). In keeping with the high degree of sequential and structural homology (1, 3), the PPARs activate overlapping sets of target genes. However, in addition, the different subtypes activate distinct sets of target genes some of which are involved in opposing metabolic pathways. Thus, whereas PPARα (4) and -β/δ (5, 6) are potent activators of genes involved in lipid oxidation, PPARγ stands out by its ability to activate lipogenic genes (7). Notably, this subtype specificity is maintained when the PPARs are overexpressed in different cell types (8, 9, 10).
Like most NRs, the PPARs encompass two transactivating domains, i.e. the N-terminal ligand-independent activation function 1 (AF-1) in the A/B-domain and the C-terminal ligand-dependent activation function (AF-2) in the E-domain (1). The latter is critically dependent on the C-terminal α-helix (helix 12) (11), which governs ligand-dependent interactions with several transcriptional cofactor complexes that modify and remodel chromatin to prime promoters for the transcriptional machinery (12). The cofactor interaction surfaces of the AF-1 are much less defined and are likely to be subtype specific because the PPAR subtypes have highly divergent A/B-domains (1). Thus, the PPAR A/B-domains have been demonstrated to limit nonselective target gene activation (13); however, it is controversial if they contribute positively to the transcriptional activity of the PPARs (14, 15), are completely dispensable (16), or may even be inhibitory (17).
The PPARγ AF-1 is the most well studied of the PPAR N-terminal transactivation functions. It has been shown that not only the coactivators HIV-1 Tat-interacting protein 60 (Tip60) (18), cAMP response element-binding protein (CREB)-binding protein (CBP), p300 (19), and PPARγ coactivator 2 (PGC-2) (14) but also the corepressor tribbles homolog 3 (TRB3) (20) interact with PPARγ exclusively or partly through the A/B-domain. Interestingly, the A/B-domain of PPARγ is also subject to posttranslational modifications. Small ubiquitin-like modifier (SUMO)-ylation of lysine 77/107 inhibits the transcriptional activity of PPARγ (21), whereas phosphorylation of serine 82/112 by MAPKs or cyclin-dependent kinase 9 (CDK9) has been reported to both decrease (22) and increase (23, 24) transactivation by PPARγ.
We have previously described a cellular system, based on acute ectopic expression of the PPARs by adenoviral vectors, that is ideal for analyzing PPAR subtype specificity of genomic target genes (9). This system allows us to induce rapid expression of PPARs and subsequently evaluate the immediate effects on target gene activity at the mRNA level within 8 h after transduction, whereby secondary effects (e.g. induction of endogenous PPARs) on gene expression are minimized. Here we have used this cellular system in combination with expression array analysis to identify direct PPARγ target genes in fibroblasts. We show that PPARγ2 activates a large number of known as well as novel target genes involved in lipid storage, fatty acid degradation, glucose metabolism, electron transport, and several other pathways. Surprisingly, using an A/B-domain-deficient version of PPARγ (PPARγCDE), we demonstrate that only a small subset of target genes displays A/B-domain dependency in the presence of full PPARγ agonist. The A/B-domain-dependent genes include several genes known to be involved in lipid storage. The reduced transcriptional activity of PPARγCDE was not due to decreased binding to the genomic PPREs but correlated with decreased recruitment of RNA polymerase II (RNAPII). Interestingly, we found that the PPARγ-mediated recruitment of CBP and p300 to A/B-domain-dependent target genes is decreased or abolished by deletion of the A/B-domain. These results define a novel role of the PPARγ A/B-domain in the recruitment of CBP and p300 to a subset of target genes.
Results
Identification of direct PPARγ target genes by adenoviral transduction
We have previously shown that adenoviral transduction of NIH-CAR cells, i.e. a clonal cell line of NIH-3T3 fibroblasts stably expressing the Coxsackie-adenovirus receptor (CARΔ1), leads to expression of PPAR and subsequent induction of several PPAR target genes within the first hours after the infection (9). PPAR-mediated recruitment of RNAPII to target gene promoters is established within 4–5 h after transduction, and immediate effects on target gene expression can be determined at the mRNA level within 6–8 h (9). To take advantage of this system for the identification of novel putative PPARγ target genes, we performed expression array analysis on three biological replicates of NIH-CAR cells transduced with adenovirus encoding N-terminally hemagglutinin (HA)-tagged PPARγ2 (AdHA-PPARγ2) in the presence of the potent agonist rosiglitazone (25). The adenovirus was titrated to obtain ectopic HA-PPARγ protein expression similar to the level found in fully differentiated 3T3-L1 adipocytes (Fig. 1B). The expression of endogenous PPARγ mRNA was very low and unaffected by ectopic HA-PPARγ2 expression (Fig. 1D). Similarly, the expression of PPARα and PPARβ/δ was not affected (results not shown).
Fig. 1.

Characterization of AdHA-PPARγ2 and AdHA-PPARγCDE. A, Schematic diagram of the HA-tagged full-length and A/B-domain-deleted PPARγ encoded by the recombinant adenoviruses. Numbers indicate amino acid positions of domain borders (Swiss-Prot; www.expasy.org). B, Western blot showing HA-PPARγ expression in NIH-CAR cells transduced for 8 h with AdEmpty (AdE), AdHA-PPARγ2 (AdHA-γ2), or AdHA-PPARγCDE (AdHA-γCDE) and treated with vehicle (DMSO) or PPARγ agonist rosiglitazone (Rosi) as indicated. Whole-cell extract from 3T3-L1 adipocytes (ADI) is used for comparison. Cell extracts were subjected to SDS-PAGE and immunoblotted using antibodies against PPARγ and TFIIB. C, Validation of HA-tags on PPARγ proteins. Whole-cell lysates were prepared as in B and immunoblotted using antibodies against HA, TFIIB, and PPARγ as indicated. D, Expression levels of adenoviral E4 (AdE4), total PPARγ, and endogenous PPARγ mRNA 8 h after transduction with the indicated adenoviral vector and treatment with vehicle (DMSO), rosiglitazone, or antagonist GW9662 (GW) as indicated. mRNA expression was determined by RT-qPCR and normalized to the corresponding TFIIB levels. E, Deletion of the A/B-domain does not affect the subcellular localization of HA-PPARγCDE. Cells were transduced as in B and fixed 8 h after transduction. Cells were stained with DAPI (blue), primary αPPARγ antibody, and Alexa flour 546-conjugated secondary antibody (red). Fluorescence was detected by confocal microscopy and images were merged. F, Lack of the A/B-domain does not affect in vitro DNA-binding activity of HA-PPARγ. Nuclear extracts were prepared 8 h after transduction of NIH-CAR cells as in B, subjected to SDS-PAGE, and immunoblotted using an antibody against PPARγ (left). EMSAs were performed using these nuclear extracts (right). All results are representative of at least three independent experiments.
Quantitative RT-PCR (RT-qPCR) was employed to validate the expression array data on selected target genes and to determine levels of transcripts that could not be detected due to the occurrence of nonfunctional probes in the Affymetrix Mouse430 2.0 array [e.g. the perilipin (PLIN) probe] as well as the levels of transcripts that were listed either as not present or nonsignificantly induced although they had previously been reported to be PPAR responsive. In total, 257 genes were significantly induced by acute HA-PPARγ2 expression and rosiglitazone as compared with infection with adenovirus containing empty vector (AdEmpty) (Table 1 and supplemental Table 1, published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). Of the 257 genes, 34 were already registered in the Ingenuity database (http://www. ingenuity.com) as up-regulated in response to PPARγ expression or administration of glitazones (Table 1 and supplemental Table 1).
Table 1.
Transcriptome analysis after short-term exposure to HA-PPARγ adenoviral vectors
| Gene | Accession no. | HA-PPARγ2 + Rosi, fold induction | HA-PPARyCDE + Rosi, fold induction | HA-PPARγ2 vs. HA-PPARγCDE, fold change | P value | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lipid storage | ||||||||||
| ADFP/ADRP1 | NM_007408 | 8.29 | 7.37 | 1.13 | 0.056 | |||||
| AQ712 | AB056091 | 61.47 | 20.70 | 2.97 | qPCR | |||||
| CD3612 | BB534670 | 636.92 | 28.53 | 21.97 | qPCR | |||||
| CIDEC/FSP2712 | BB221402 | 7353.53 | 680.90 | 10.80 | qPCR | |||||
| FABP4/A-FABP12 | NM_024406 | 194.96 | 110.16 | 1.77 | 0.044 | |||||
| LPL12 | AK017272 | 9.29 | 3.67 | 2.53 | 0.011 | |||||
| PCX12 | NM_008797 | 3.90 | 1.83 | 2.13 | 0.046 | |||||
| PLIN12 | BG065200 | 101.76 | 64.36 | 1.58 | qPCR | |||||
| SOAT12 | BG064396 | 3.40 | 2.06 | 1.65 | 0.002 | |||||
| STOM2 | AF093620 | 2.64 | 1.39 | 1.90 | 0.019 | |||||
| Lipolysis | ||||||||||
| ADCY6 | NM_007405 | 2.11 | 1.43 | 1.47 | 0.007 | |||||
| PNPLA2/ATGL | AK003207 | 1.75 | 1.42 | 1.23 | 0.071 | |||||
| Electron transport | ||||||||||
| CYB51 | NM_025797 | 1.87 | 1.35 | 1.38 | 0.013 | |||||
| ETFDH1 | BC012522 | 1.99 | 1.87 | 1.07 | 0.396 | |||||
| FDX1 | D43690 | 2.28 | 1.59 | 1.44 | 0.147 | |||||
| FOXRED2 | BM227994 | 1.92 | 1.11 | 1.73 | 0.051 | |||||
| HCCS | BB129992 | 2.44 | 2.45 | −1.01 | 0.895 | |||||
| Mitochondrial β-oxidation | ||||||||||
| ACAA21 | AK002555 | 1.99 | 1.87 | 1.06 | 0.396 | |||||
| ACADL1 | BB728073 | 1.69 | 1.58 | 1.07 | 0.126 | |||||
| ACADVL1 | BC026559 | 2.05 | 1.61 | 1.28 | 0.032 | |||||
| ACOT2 | BB500039 | 3.02 | 2.95 | 1.02 | 0.748 | |||||
| ALDH9A11 | AV028069 | 2.23 | 2.01 | 1.11 | 0.120 | |||||
| CPT1A1 | AI987925 | 3.27 | 3.46 | −1.06 | 0.147 | |||||
| CPT21 | NM_009949 | 1.98 | 1.86 | 1.06 | 0.320 | |||||
| HADH1 | BB114220 | 1.69 | 1.66 | 1.02 | 0.537 | |||||
| HADHA1 | AW107842 | 1.65 | 1.58 | 1.04 | 0.752 | |||||
| HADHB1 | BG866501 | 1.71 | 1.72 | −1.01 | 0.921 | |||||
| PDK41 | NM_013743 | 3.98 | 5.19 | −1.30 | 0.035 | |||||
| POR | NM_008898 | 1.63 | 1.32 | 1.23 | 0.056 | |||||
| UCP21 | AW108044 | 2.37 | 2.37 | 1.00 | 0.980 | |||||
| Peroxisomes | ||||||||||
| ABCD3 | BC009119 | 1.51 | 1.38 | 1.09 | 0.102 | |||||
| ACOX11 | AB034914 | 2.86 | 2.52 | 1.13 | 0.319 | |||||
| CAT1 | NM_009804 | 2.13 | 1.75 | 1.22 | 0.067 | |||||
| ECH11 | NM_016772 | 2.43 | 2.41 | 1.01 | 0.837 | |||||
| MLSTD2 | BG094874 | 1.69 | 1.25 | 1.35 | 0.008 | |||||
| PEX11A1 | NM_011068 | 3.81 | 3.36 | 1.14 | qPCR | |||||
| PEX13 | BB045429 | 2.25 | 1.59 | 1.42 | 0.010 | |||||
| PEX 14 | NM_019781 | 1.77 | 1.45 | 1.21 | 0.080 | |||||
| PEX16 | BC010822 | 1.67 | 1.40 | 1.20 | 0.117 | |||||
| Insulin/IGF-I signaling | ||||||||||
| IRS21 | BE199054 | 2.99 | 2.43 | 1.23 | 0.059 | |||||
| RAPGEFI1 | BB339051 | 1.71 | 1.49 | 1.15 | 0.110 | |||||
| Glycogen/glucose metabolism | ||||||||||
| GYS11 | NM 008195 | 1.62 | 1.16 | 1.39 | 0.026 | |||||
| PFKL1 | BE914497 | 1.56 | 1.25 | 1.25 | 0.039 | |||||
| PGM2 | BC00SS27 | 1.63 | 1.28 | 1.27 | 0.026 | |||||
| PHKA2 | BG076063 | 1.56 | 1.32 | 1.18 | 0.045 | |||||
| Inflammation | ||||||||||
| CLCF1 | BB825816 | 2.09 | 1.54 | 1.36 | 0.074 | |||||
| MGST3 | NM_025569 | 2.20 | 1.89 | 1.16 | 0.037 | |||||
| PTGES | BB730139 | 2.01 | 1.68 | 1.20 | 0.036 | |||||
| Vesicle formation and transport | ||||||||||
| ARF6 | B1248938 | 1.72 | −1.08 | 1.86 | 0.083 | |||||
| ATP6VOA1 | U13836 | 1.77 | 1.76 | 1.00 | 0.925 | |||||
| IQSECI | BF164393 | 2.14 | 1.73 | 1.23 | 0.060 | |||||
| ITSN1 | AA172344 | 1.52 | 1.23 | 1.23 | 0.074 | |||||
| VAMPS | AK009266 | 2.62 | 2.34 | 1.12 | 0.487 | |||||
| (Continued) | ||||||||||
Table 1A.
Continued
| Gene | Accession no. | HA-PPARγ2 + Rosi, fold induction | HA-PPARyCDE + Rosi, fold induction | HA-PPARγ2 vs. HA-PPARγCDE, fold change | P value |
|---|---|---|---|---|---|
| Growth/proliferation | |||||
| BTG1 | L16846 | 1.54 | 1.40 | 1.10 | 0.060 |
| BTG2 | NM_007570 | 5.35 | 3.11 | 1.72 | 0.053 |
| CDC25A | C76119 | 1.52 | 1.37 | 1.11 | 0.249 |
| CDK2AP2 | NM_026373 | 1.98 | 1.83 | 1.08 | 0.446 |
| CUL1 | BG070097 | 1.59 | 1.39 | 1.15 | 0.287 |
| CYR61 | BB533736 | 1.86 | 1.77 | 1.05 | 0.761 |
| EEF2K | C86191 | 1.51 | 1.38 | 1.09 | 0.287 |
| EPS8 | BM213788 | 1.72 | 1.52 | 1.13 | 0.529 |
| ERBB2IP | BB336138 | 1.84 | 1.82 | 1.01 | 0.836 |
| FGFRLI | AF321301 | 2.93 | 1.98 | 1.49 | 0.014 |
| FOSL11 | U34245 | 1.65 | 1.49 | 1.10 | 0.322 |
| GSPT1 | BB162021 | 1.59 | 1.24 | 1.28 | 0.154 |
| KLF10 | NM_013692 | 1.69 | 1.68 | 1.01 | 0.893 |
| NEK6 | BB528391 | 1.97 | 1.86 | 1.06 | 0.166 |
| NET1 | NM_019671 | 1.65 | 1.56 | 1.06 | 0.311 |
| NRP1 | AK011144 | 1.65 | 1.49 | 1.10 | 0.073 |
| NRP2 | BB409477 | 1.65 | 1.46 | 1.13 | 0.144 |
| P8 | NM_019738 | 1.60 | 1.27 | 1.25 | 0.106 |
| PFTK1 | AI327038 | 1.61 | 1.44 | 1.12 | 0.164 |
| PLK32 | BM947855 | 2.28 | 1.45 | 1.57 | 0.048 |
| POLD4 | AK010477 | 1.55 | 1.45 | 1.06 | 0.338 |
| RALGDS | NM_009058 | 1.61 | 1.47 | 1.09 | 0.302 |
| RBPJ | NM_009035 | 1.53 | 1.52 | 1.01 | 0.816 |
| RRM2 | NM_009104 | 1.73 | 1.57 | 1.11 | 0.259 |
| SPRY42 | BB080456 | 2.63 | 1.55 | 1.70 | 0.018 |
| STK1O | NM_009288 | 2.01 | 1.81 | 1.11 | 0.039 |
| TNK2 | NM_016788 | 1.63 | 1.17 | 1.39 | 0.097 |
| TOB1 | BQ266486 | 2.68 | 1.91 | 1.41 | 0.016 |
| TRIM25 | D63902 | 2.99 | 2.67 | 1.12 | 0.114 |
| VEGFC C | NM_009506 | 2.78 | 2.15 | 1.30 | 0.135 |
The table presents clustered genes induced more than 1.5-fold in NIH-CAR cells transduced with either AdHA-PPARγ2 or AdHA-PPARγCDE in the presence of rosiglitazone (Rosi). Background is defined as cells transduced with AdEmpty in the presence of vehicle (DMSO). The mRNA levels were detected by Affymetrix microarray analysis (GEO accession no. GSE15433) or RT-qPCR (indicated) performed in triplicate on independent biological experiments. Fold change in expression level between cells transduced with AdHA-PPARγ2 relative to cells transduced with AdHA-PPARγCDE and the corresponding P value are indicated. For the genes detected by RT-qPCR, a representative fold change is indicated.
Genes registered as regulated by PPARγ or PPARγ agonists in the Ingenuity database.
A/B-domain-dependent genes, i.e. genes displaying significantly reduced induction in the cells expressing HA-PPARγCDE.
A functional clustering of the genes induced by HA-PPARγ2 in combination with rosiglitazone revealed four major groups of genes involved in lipid storage, mitochondrial β-oxidation, peroxisomal functions, or growth/proliferation. The up-regulation of genes involved in lipid storage was not surprising given the role of PPARγ as a lipogenic transcription factor. This group includes annotated PPARγ target genes involved in fatty acid uptake such as CD36 (26), and lipoprotein lipase (LPL) (27), and aquaporin 7 (AQ7) involved in glycerol transport (28, 29) in addition to the adipocyte fatty acid binding protein (A-FABP) (30). The pyruvate carboxylase (PCX) (31), which in adipose tissue catalyzes the initial reaction converting pyruvate into the acetyl groups and NADPH required for synthesis of fatty acids (32), was also found to be up-regulated. Notably, we found induction of several lipid droplet-associated proteins; the cell death-inducing DNA fragmentation factor A-like effector C (CIDEC)/fat-specific protein 27 (FSP27) (33), required for the formation of large lipid droplets (34); PLIN, which effectively shields the stored lipid from basal lipolytic activity but facilitates hormonally stimulated lipolysis (35); the adipose differentiation-related protein (ADRP) which functions to limit lipase accessibility (36, 37), and stomatin (STOM) (38). Interestingly, we also saw up-regulation of sterol O-acetyltransferase 1 (SOAT-1), which catalyzes the esterification of cholesterol and fatty acids into cholesterol esters, another major component of lipid droplets (39). To our knowledge this is the first demonstration that STOM and SOAT-1 are PPARγ target genes.
In addition to genes involved in lipid storage, ectopic PPARγ expression induced genes involved in peroxisomal [ATP-binding cassette, sub-family D member 3 (ABCD3), acyl-coenzyme A oxidase 1 (ACOX1), catalase (CAT), and enoyl coenzyme A hydratase 1 (ECH1)] and mitochondrial [e.g. acetyl-coenzyme A acyltransferase 2 (ACAA2), acyl-CoA dehydrogenase, long-chain (ACADL), carnitine palmitoyltransferase 2 (CPT2), and hydroxyacyl-CoA dehydrogenase (HADH)] β-oxidation of fatty acids. This induction of fatty acid oxidation genes by PPARγ is in keeping with previous observations (9, 40, 41, 42). PPARγ also induced expression of the known target gene adipose triglyceride lipase (ATGL) (43). Furthermore, we also detected induction of genes involved in insulin/IGF-I signaling [insulin receptor substrate 2 (IRS2) and Rap guanine nucleotide exchange factor 1 (RAPGEF1)] and glycogen/glucose metabolism [glycogen synthase 1 (GYS1), phosphofructokinase (PFKL), phosphoglucomutase 2 (PGM2), phosphorylase kinase α2 (PHKA2)]. Interestingly, we also found up-regulation of genes involved in vesicle formation and transport, which potentially could play an important role in the formation and trafficking of lipid droplets.
These data represent the first transcriptome analysis after short-term exposure to PPARγ adenoviral vectors and show that this is an efficient procedure for identification of direct PPAR target genes. Furthermore, this approach is ideal for studying the contribution of individual domains or modifications to PPARγ transcriptional activity, because it allows equal expression of ectopic PPARs while induction of endogenous PPAR is avoided. Thus, we next employed this adenoviral delivery system to determine the direct role of the PPARγ A/B-domain in transactivation of genomic target genes.
Adenoviral vectors for investigation of the role of the PPARγ A/B-domain
To determine how the AF-1 activity affects the transcriptional profile of PPARγ, we constructed adenoviral vectors expressing HA-tagged PPARγ1 (AdHA-PPARγ1) and A/B-domain truncated PPARγ (AdHA-PPARγCDE) (Fig. 1, A and C) for comparison with HA-PPARγ2. Notably, equal protein expression (Fig. 1B and supplemental Fig. 1A) corresponded to equal mRNA levels of the HA-PPARγ transgenes and of the adenoviral AdE4 gene, a marker of transduction efficiency (Fig. 1D and supplemental Fig. 1B). These observations suggest that the protein stabilities of HA-PPARγ1, HA-PPARγ2, and HA-PPARγCDE are similar and show that the cells were subjected to equal adenovirus exposure. Addition of the specific PPARγ agonist rosiglitazone decreased protein levels of both HA-PPARγ2 and truncated HA-PPARγCDE (Fig. 1B). This observation is in agreement with previous studies reporting that agonists enhance the turnover rate of PPARγ (44).
To first determine whether the two PPARγ isoforms displayed any functional difference in our system, we compared the ability of HA-PPARγ1 and -γ2 to activate a number of known target genes. Interestingly, there was no difference in the transcriptional activity between the isoforms, neither in the absence nor in the presence of agonist (supplemental Fig. 1C). We therefore concentrated our subsequent studies on the comparison of full-length HA-PPARγ2 and AB-domain truncated HA-PPARγCDE.
Deletion of the PPARγ A/B-domain affects neither DNA binding in vitro nor nuclear localization
Immunostaining combined with confocal microscopy showed that the majority of the NIH-CAR cells (∼90%) were targeted by the adenovirus and that transduction efficiency was similar between the different cells in the culture.
There have been several reports on proteins that bind and induce nuclear export of the PPARs (45, 46, 47), but as shown in Fig. 1E, both HA-PPARγ2 and HA-PPARγCDE were primarily localized to the nucleus. To investigate whether deletion of the A/B-domain affects the DNA-binding activity of the adenovirally expressed HA-PPARγ, we performed EMSAs using nuclear extracts from infected NIH-CAR cells (Fig. 1F). These data clearly showed that in vitro DNA binding and heterodimerization on several different PPREs are fully maintained in the HA-PPARγCDE protein.
Thus, using adenoviral vectors, we are able to express equal and moderate levels of full-length and truncated HA-PPARγ in NIH-CAR cells and thereby compare their ability to acutely activate target genes. Both proteins localize to the nucleus in NIH-CAR cells and display equal binding to naked DNA in vitro.
The role of the A/B-domain in PPARγ-mediated transactivation is gene specific
In the absence of exogenous agonist, deletion of the A/B-domain inhibited HA-PPARγ-mediated transactivation of most genes investigated to levels close to the background expression (Fig. 2). Furthermore, addition of the PPARγ-specific antagonist GW9662 (48) did not suppress transactivation either by full-length or by truncated PPARγ (Fig. 2). These results corroborate previous findings that the concentration of endogenous agonist is low in NIH-CAR fibroblasts (9) and indicate that, at least in these cells, ligand-independent transactivation by the HA-PPARγ2:RXR heterodimer is primarily mediated through the PPARγ2 A/B-domain, rather than the ligand-binding domain (LBD) or RXR. Interestingly, however, in the presence of the potent PPARγ-specific agonist rosiglitazone, deletion of the A/B-domain only led to a decrease in the transactivation of some but not all target genes.
Fig. 2.
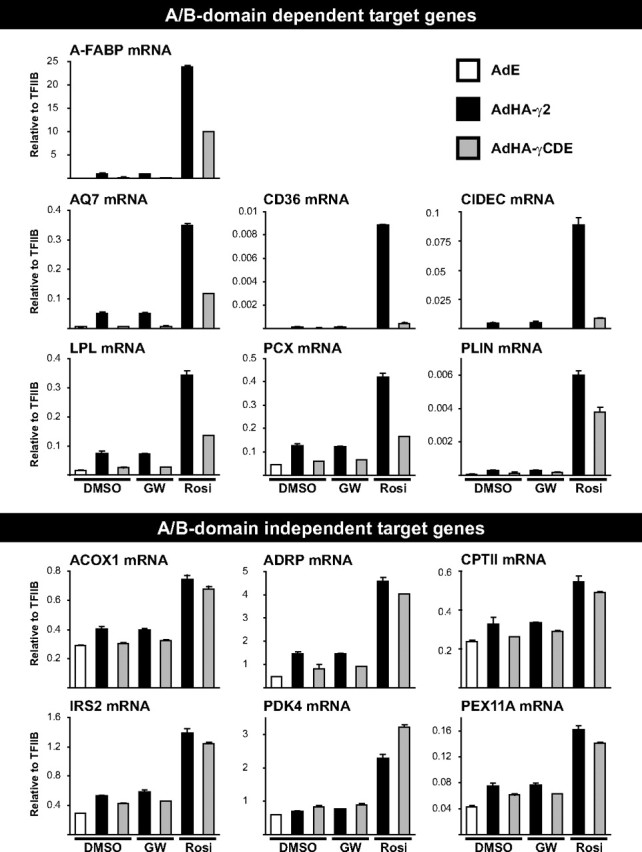
Comparison of the transcriptional activity of HA-PPARγ2 and HA-PPARγCDE on genomic target genes. NIH-CAR cells were transduced with AdEmpty (AdE), AdHA-PPARγ2 (AdHA-γ2), or AdHA-PPARγCDE (AdHA-γCDE) and treated with vehicle (DMSO), rosiglitazone (Rosi), or antagonist GW9662 (GW) as indicated. RNA was isolated 8 h after transduction, and mRNA expression was determined by RT-qPCR and normalized to the corresponding TFIIB levels. Results are representative of at least three independent experiments.
To further compare the ability of full-length and A/B-domain-deleted HA-PPARγ to acutely activate target genes in the presence of rosiglitazone, we performed transcriptome analyses of three independent biological replicas in parallel with the transcriptome analyses described above (Table 1, supplemental Table 3, and Fig. 2). For most PPARγ target genes, the A/B-domain appeared to have only very minor effects on gene expression in the presence of rosiglitazone. Notably, this group of A/B-domain-independent target genes included all the known and putative PPAR target genes involved in both mitochondrial and peroxisomal β-oxidation of fatty acids, which we found to be up-regulated in response to HA-PPARγ2 expression (Table 1 and Fig. 2). This was also the case for genes associated with lipolysis, insulin/IGF-I signaling, and glycogen/glucose metabolism (Table 1 and Fig. 2). In fact, only a small subset of 25 genes proved to be dependent on the A/B-domain to become fully activated. Interestingly, most of the PPARγ target genes involved in lipid storage are included in this group (Table 1 and Fig. 2). The only gene in this cluster that was induced equally well by full-length and truncated HA-PPARγCDE encodes ADRP. Like the products of the A/B-domain-dependent target genes CIDEC/FSP27 and PLIN, ADRP is localized to lipid droplets but is in contrast selectively associated with lipid droplets subjected to lipolysis (49). Whereas the expression of CIDEC/FSP27 and PLIN is limited to a few tissues that employ specific signaling pathways to regulate lipogenesis and lipolysis, ADRP is ubiquitously expressed (35, 50) and can be activated equally well by PPARα and PPARγ2 in contrast to other genes involved in lipid storage such as PLIN and A-FABP (9).
Activation of the PPARγ:RXR heterodimer with RXR agonist could not compensate for the lack of the A/B-domain activity in target gene regulation. However, in combination with rosiglitazone HA-PPARγCDE, transactivation of the highly A/B-domain-dependent genes CD36 and CIDEC/FSP27 was potentiated by the RXR agonist (supplemental Fig. 2).
It has previously been found that the A/B-domain confers subtype specificity to the PPARs in part by preventing nonselective target gene activation (13). In keeping with these results, RT-qPCR experiments demonstrated that in the presence of rosiglitazone, HA-PPARγCDE was approximately twice as potent as full-length HA-PPARγ2 in the transactivation of the gene encoding 3-hydroxy-3-methyl-glutaryl-coenzyme A synthase 2 (HMG-CoA S2) (Fig. 3C), which has previously been shown to be activated selectively by PPARα (9). This observation could not be explained by increased affinity of the A/B-domain-truncated receptor toward the HMG-CoA S2 PPRE in vitro (Fig. 1F, right). The induction of HMG-CoA S2 could be detected only by RT-qPCR analysis, and the array data did not reveal the existence of other genes where the A/B-domain significantly inhibited HA-PPARγ2 transactivation in this cell type.
Fig. 3.

Comparison of the transcriptional activity of HA-PPARγ2, HA-PPARγCDE, and HA-PPARαA/BγCDE on genomic target genes. A, Schematic diagram of the HA-tagged PPARα and -γ and the chimeric PPARαA/BγCDE encoded by the recombinant adenoviruses. Numbers indicate amino acid positions of domain borders (Swiss-Prot; www.expasy.org). B–D, NIH-CAR cells were transduced with AdEmpty (AdE), AdHA-PPARγ2 (AdHA-γ2), AdHA-PPARγCDE (AdHA-γCDE), or AdHA-PPARαA/BγCDE (AdHA-αA/BγCDE) and treated with vehicle (DMSO) or rosiglitazone (Rosi) as indicated. The cells were harvested 8 h after transduction and subjected to either SDS-PAGE and immunoblotted using antibodies against PPARγ and TFIIB or RNA purification. mRNA expression was determined by RT-qPCR and normalized to the corresponding TFIIB levels. Results are representative of at least three independent experiments.
Both PPARα and PPARγ have a strong N-terminal transactivation domain, whereas that of PPARδ is very weak (15). To investigate whether the A/B-domain of PPARα could substitute for the PPARγ2 A/B-domain, we constructed an adenoviral vector encoding an HA-tagged chimeric PPAR consisting of the PPARα A/B-domain fused to the CDE domains of PPARγ (HA-PPARαA/BγCDE) (Fig. 3, A and B). Notably, we found that the transcriptional activity and specificity of the chimeric PPAR was comparable to that of HA-PPARγCDE on all lipogenic target genes investigated (Fig. 3D), thereby clearly demonstrating that PPARα and -γ A/B-domains are functionally distinct and that PPARγ is unique in its ability to promote activation of the lipogenic subset of target genes. In contrast, the PPARα A/B-domain potentiated transactivation of the PPARα-selective target gene HMG-CoA S2 (Fig. 3C).
These results show that the PPARγ A/B-domain contributes considerably to the lipogenic subtype characteristics of PPARγ by enabling full transactivation of the subset of target genes involved in lipid storage. By contrast, although the PPARγ2 A/B-domain may account for some weak ligand-independent transactivation, it plays a minor role in the activation of the remaining PPARγ target genes. Finally, the A/B-domain renders PPARγ less efficient in activating the expression of the PPARα selective target gene HMG-CoA S2 and possibly other PPARα target genes yet to be identified.
PPARγ A/B-domain deletion results in decreased lipid accumulation 48 h after transduction
It has previously been demonstrated that NIH-3T3 fibroblasts constitutively expressing ectopic PPARγ2 can be induced to accumulate lipid and develop an adipocyte-like phenotype (17). Because the PPARγ2 A/B-domain proved important for complete induction of genes involved in lipid storage, we compared the ability of HA-PPARγ2 and HA-PPARγCDE to promote cellular lipid accumulation. By measuring the incorporation of [14C]acetate into triglyceride 48 h after adenoviral transduction, we established that even such brief exposure to AdHA-PPARγ was sufficient to induce lipid accumulation in the NIH-CAR cells. Notably, HA-PPARγ2 expression increased the incorporation of [14C]acetate by 62%, whereas the A/B-domain-truncated HA-PPARγCDE displayed a much lower lipogenic potential with an incorporation of only 28% above background (Fig. 4B).
Fig. 4.

The effect of A/B-domain deletion on the lipogenic potential of HA-PPARγ2. NIH-CAR cells were transduced with AdEmpty (AdE) and treated with vehicle (DMSO) or transduced with AdHA-PPARγ2 (AdHA-γ2) or AdHA-PPARγCDE (AdHA-γCDE) and treated with rosiglitazone (Rosi). A, Cells were harvested 48 h after transduction and subjected to SDS-PAGE and immunoblotted using antibodies against PPARγ and TFIIB. B, Lipid synthesis was assessed in triplicate by determining incorporation of [14C]acetate into total lipids 48 h after adenoviral transduction of which the last 24 h were in the presence of [14C]acetate. Extracted lipids were separated on silica thin-layer chromatography plates and radioactivity in the triglyceride fraction was quantified. Results are representative of two independent experiments.
Full-length and truncated PPARγ bind equally well to genomic target sites but differ in their ability to recruit RNAPII
To investigate the molecular mechanisms underlying the potentiation of transactivation by the PPARγ2 A/B-domain, we performed chromatin immunoprecipitation (ChIP) experiments. Similar to in vitro DNA binding (Fig. 1F), ChIP assays with antibodies against HA or RXR demonstrated that binding of the HA-PPARγ2:RXR and HA-PPARγCDE:RXR heterodimers to genomic PPREs is comparable (Fig. 5). The only exception is the A-FABP PPRE to which the truncated HA-PPARγCDE binds less well than full-length HA-PPARγ2. Thus, in general, the PPARγ2 A/B-domain does not appear to affect binding to genomic target sites, either for A/B- domain-dependent or for A/B-independent target genes.
Fig. 5.

HA-PPARγ2:RXR and HA-PPARγCDE:RXR heterodimers bind equally well to most genomic PPREs. Cross-linked chromatin was harvested from NIH-CAR cells 8 h after transduction with either AdEmpty (AdE) and treatment with vehicle (DMSO) or transduction with AdHA-PPARγ2 (AdHA-γ2) or AdHA-PPARγCDE (AdHA-γCDE) and treatment with rosiglitazone (Rosi). Relative occupancy at PPREs of interest was determined by ChIP-qPCR using antibodies against HA and RXR, respectively. Results are representative of at least three independent experiments. The figure shows means of two experiments with indication of range.
We next investigated the ability of full-length and truncated HA-PPARγ to recruit RNAPII to the corresponding target promoters. For all promoters analyzed, RNAPII recruitment (Fig. 6) was found to reflect the mRNA expression pattern (Fig. 2). Thus, for A/B-domain-dependent target promoters, deletion of the A/B-domain decreased RNAPII recruitment by HA-PPARγCDE, whereas RNAPII was recruited equally well or even slightly better by the truncated PPARγ on the A/B-domain-independent target genes.
Fig. 6.
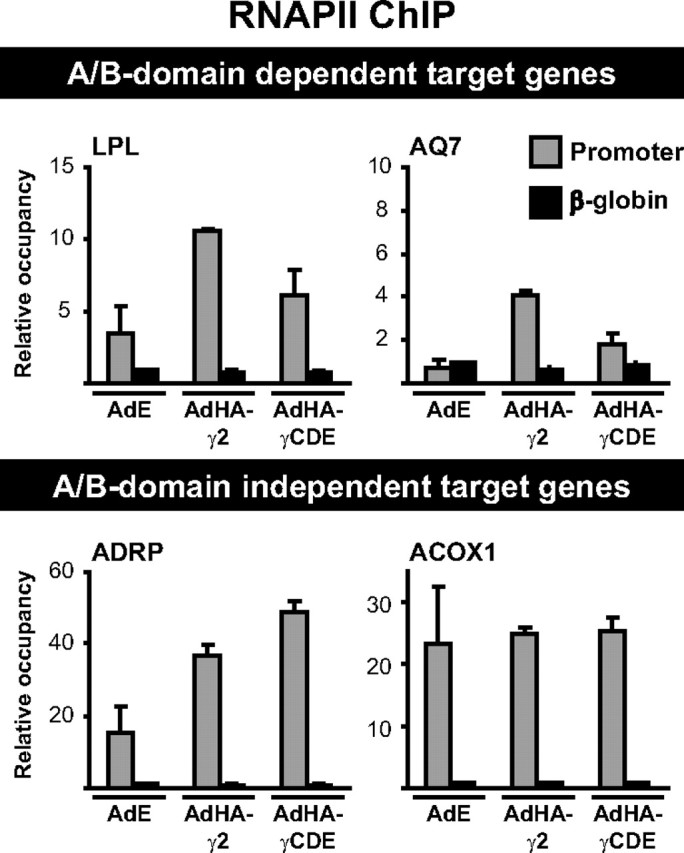
The HA-PPARγ2- and HA-PPARγCDE-induced RNAPII occupancy at target gene promoters reflects mRNA expression. Cross-linked chromatin was harvested from NIH-CAR cells 8 h after transduction with either AdEmpty (AdE) and treatment with vehicle (DMSO) or transduction with AdHA-PPARγ2 (AdHA-γ2) or AdHA-PPARγCDE (AdHA-γCDE) and treatment with rosiglitazone (Rosi). Relative occupancy at target gene promoters of interest was determined by ChIP-qPCR using an antibody against RNAPII. Results are representative of at least three independent experiments. The figure shows means of two experiments with indication of range.
The PPARγ A/B-domain is important for the recruitment of CBP and p300 to the A/B-domain-dependent target genes
Because receptor binding to target sites in chromatin was unaffected by the A/B-domain deletion, we sought to establish whether the PPARγ A/B-domain is important for recruitment and/or stabilization of specific coactivators. Recruitment of thyroid hormone receptor-associated protein 220 (TRAP220), a PPAR-interacting subunit of the mediator complex, was increased on all PPREs investigated upon HA-PPARγ expression although to different extents (Fig. 7). Interestingly, for all target genes except A-FABP, TRAP220 recruitment was not markedly dependent on the PPARγ2 A/B-domain. This indicates that the difference in TRAP220 recruitment at the A-FABP PPRE may simply reflect the difference in receptor binding, and we conclude that the deletion of the PPARγ A/B-domain does not interfere with TRAP220 recruitment, which is in keeping with the previously reported interaction through the PPARγ LBD (51).
Fig. 7.
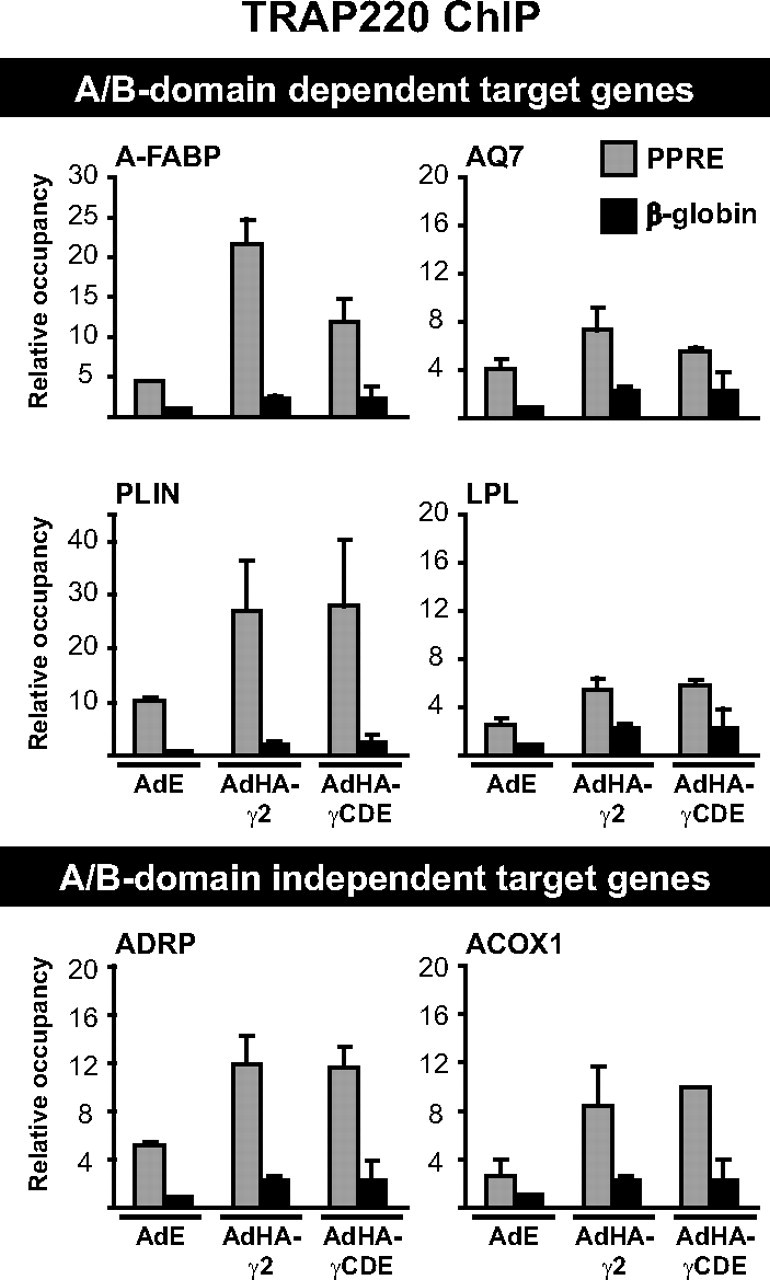
HA-PPARγ2 and HA-PPARγCDE recruit TRAP220 equally well to genomic PPREs. Cross-linked chromatin was harvested from NIH-CAR cells 8 h after transduction with either AdEmpty (AdE) and treatment with vehicle (DMSO) or transduction with AdHA-PPARγ2 (AdHA-γ2) or AdHA-PPARγCDE (AdHA-γCDE) and treatment with rosiglitazone (Rosi). Relative occupancy at PPREs of interest was determined by ChIP-qPCR using an antibody against TRAP220. Results are representative of at least three independent experiments. The figure shows means of two experiments with indication of range.
The coactivator Tip60 interacts with PPARγ through the A/B-domain and has been demonstrated to promote adipogenesis and lipid accumulation in 3T3-L1 cells (18). Nevertheless, we were unable to detect Tip60 recruitment to the PPREs of PPARγ target genes in NIH-CAR cells (results not shown). The acetyl transferase CBP and the highly homologous p300 have been shown to interact with both the A/B- and E-domains of PPARγ2 (19). Interestingly, we found that deletion of the A/B-domain severely decreased or abolished HA-PPARγ-mediated recruitment of CBP on A/B-domain-dependent but not independent target genes (Fig. 8). p300 was recruited to only a subset of targets, but recruitment followed the same pattern as for CBP, i.e. for A/B-domain-independent genes, recruitment of p300 was unaffected by deletion of the A/B-domain, whereas A/B-domain deletion completely abolished recruitment to the A/B-domain-dependent gene A-FABP (Fig. 8). These data indicate that a major function of the PPARγ A/B-domain is to potentiate recruitment or stabilization of coactivator complexes containing CBP and p300 to a subset of target genes.
Fig. 8.

HA-PPARγ2 and HA-PPARγCDE differ in their ability to recruit CBP and p300 occupancy to genomic PPREs. Cross-linked chromatin was harvested from NIH-CAR cells 8 h after transduction with either AdEmpty (AdE) and treatment with vehicle (DMSO) or transduction with AdHA-PPARγ2 (AdHA-γ2) or AdHA-PPARγCDE (AdHA-γCDE) and treatment with rosiglitazone (Rosi). Relative occupancy at PPREs of interest was determined by ChIP-qPCR using antibodies against CBP or p300, respectively. Results are representative of at least three independent experiments. The figure shows means of two experiments with indication of range.
Knockdown of CBP and p300 differentially affects expression of PPARγ target genes
The observation that HA-PPARγ2 induced recruitment of p300 only to the PPREs of A-FABP, ACOX1, and ADRP, whereas CBP was recruited to all target sites investigated (Fig. 8), indicated that CBP and p300 could play nonredundant roles in PPARγ transactivation. To further investigate this, we transfected NIH-CAR cells with small interfering RNA (siRNA) against either CBP, p300, or both 24 h before transduction with adenovirus (Fig. 9A). Of note, establishment of stable clones with short hairpin RNA-mediated knockdown of these coactivators was impossible due to effects on cellular growth. In keeping with the recruitment of CBP to all PPARγ target sites investigated, knockdown of CBP suppressed induction of most target genes (Fig. 9B and supplemental Fig. 3). The combination of p300 and CBP knockdown further suppressed the expression of several target genes indicating partial redundancy between CBP and p300 on these genes. By contrast, PPARγ induction of PDK4 and CPTII occurred independently of CBP and p300 (Fig. 9B and supplemental Fig. 3). In general, lack of CBP led to a more pronounced decrease in expression of PPARγ target genes than lack of p300, although both coactivators were efficiently knocked down. This indicates that CBP plays a more important role than p300 in PPARγ transactivation in NIH-CAR cells. It is notable, however, that p300 knockdown affected PPARγ-mediated induction of several target genes, although it was recruited in a PPARγ-dependent manner to only a subset of target genes (ADRP, ACOX1, and A-FABP) (Fig. 8). This suggests that p300 may be recruited to the target genes by other transcription factors and that this is necessary for full transactivation by PPARγ. Alternatively, p300 knockdown may indirectly affect these promoters.
Fig. 9.

CBP and p300 knockdown differentially affects the expression of PPARγ target genes. NIH-CAR cells were transfected with siRNA against CBP, p300, or both 24 h before transduction with either AdEmpty (AdE) and treatment with vehicle (DMSO) or transduction with AdHA-PPARγ2 (AdHA-γ2) and treatment with rosiglitazone (Rosi). The siRNA against luciferase (Luc) was used as a control. The cells were harvested 8 h after transduction. A, Protein expression was determined by SDS-PAGE and immunoblotting using antibodies against PPARγ, CBP, p300, or Sp1. B, mRNA expression was determined by RT-qPCR and normalized to the corresponding TFIIB levels. Results are representative of three independent experiments.
Discussion
PPARγ is an important regulator of lipogenesis as well as cellular differentiation and signaling, and PPARγ agonists are used clinically as insulin sensitizers. Here we have used the adenoviral delivery system to induce acute expression of HA-PPARγ in the NIH-CAR fibroblast cell line, which has very low levels of endogenous PPARs, and allow rapid induction of PPAR target genes upon AdHA-PPARγ transduction (9). Adenovirus has previously been employed to identify PPARγ target genes through ectopic overexpression of PPARγ1 in the liver of PPARα−/− mice for 2–6 d (42). Other approaches to identify novel target genes have likewise been based on prolonged activation of PPARγ, either by agonist or by exposure to ectopic PPARγ expression and include transcriptome analysis of either fully differentiated adipocytes treated with PPARγ agonist (40, 52) or NIH-3T3 fibroblasts transduced with retrovirus encoding PPARγ2 (13). The lists of putative target genes generated in these studies will inevitably include a number of genes regulated secondarily to PPARy activation. In contrast, our data represent the first identification of PPARγ target genes by acute adenoviral expression combined with expression array analysis. In this system, induction of a given gene will primarily be a direct effect of PPARγ2 expression. This approach allowed us to identify 257 putative target genes. (Table 1 and supplemental Table 3) of which 34 were registered in the Ingenuity database as induced by PPARγ expression or glitazone treatment. As expected, we found induction of a cluster of genes involved in lipid accumulation, but in keeping with observations by others, we also found increased expression of several genes involved in peroxisomal and mitochondrial β-oxidation (9, 40, 41, 42). These results corroborate previous findings from our lab that when expressed at adipocyte levels, PPARγ2 is a strong transactivator capable of activating most PPAR target genes, even if other subtypes expressed at the same level would be better activators (9). Notably, in the NIH-CAR setting, there is no difference between the transactivation ability of PPARγ1 and -γ2 (supplemental Fig. 1C). This indicates that the function of these subtypes are not fundamentally different, which is in keeping with the findings by Spiegelman and colleagues (7) showing that in terms of ability to induce adipocyte differentiation, these isoforms differ only slightly in their potency.
Although the different PPAR subtypes show significant structural homology and most PPREs bind all three PPAR subtypes (9), PPARγ is by far the most lipogenic. The A/B-domain is the least conserved domain, both among the PPAR subtypes (1) as well as for the NRs in general (53). The PPAR A/B-domains appear to be highly unstructured and have so far escaped crystallization. It has been speculated that cofactors, upon binding, could serve as templates thereby directing the A/B-domain into a more ordered structure (53). Whereas the function of the LBD in PPAR-mediated transactivation is well characterized, the role of the A/B-domains is controversial. The PPARα and -γ A/B-domains are potent transactivators when expressed as GAL4-fusion proteins (15); however, they are also the targets of both activating and inhibiting posttranslational modifications (21, 22, 23, 24, 54). The complexity is further increased by the observation that a handful of coactivators (Tip60, CBP, p300, and PGC-2) but also a corepressor (TRB3) interact with PPARγ2 partly or exclusively through the A/B-domain (14, 18, 19, 20). In addition, it has been reported that the A/B-domain of some NRs, including the PPARs, regulate the activity of the ligand-dependent AF-2 (22, 55). To determine the direct role of PPARγ AF-1 in transactivation of genomic target genes, we constructed adenoviral vectors expressing HA-tagged A/B-domain-truncated PPARγ (AdHA-PPARγCDE) (Fig. 1, A–D) for comparison with full-length HA-PPARγ2. HA-PPARγCDE remains localized to the nucleus (Fig. 1E) and maintained full heterodimerization with RXR and in vitro DNA binding on several different PPREs (Fig. 1F, right). Expression of HA-PPARγCDE in the absence of synthetic agonist or in the presence of an irreversible antagonist showed that the PPARγ2 A/B-domain generally is the primary contributor to the weak ligand-independent transcriptional activity of the PPARγ2:RXR heterodimer (Fig. 2). Conversely, expression array analysis demonstrated that in the presence of potent agonist, the majority of target genes were unaffected by the A/B-domain deletion with only a small subset displaying A/B-domain dependency (Table 1, supplemental Table 3, and Fig. 2). This target gene specificity could help explain why the A/B-domains inconsistently have been described as being very important or completely dispensable for the transcriptional activity of the PPARs (15, 16, 18).
Interestingly, nine of the 25 A/B-domain-dependent genes clustered as being involved in lipid storage, and triglyceride accumulation was considerably reduced in the cells expressing HA-PPARγCDE compared with cells expressing full-length HA-PPARγ2 (Fig. 4B). Notably, using an HA-tagged chimeric PPAR consisting of the PPARα A/B-domain fused to the PPARγ CDE domains, we show that the PPARα A/B-domain is incapable of substituting for the PPARγ A/B-domain in the transactivation of lipogenic target genes (Fig. 3D) but renders PPARγ more potent in the activation of a highly PPARα-selective target gene (Fig. 3C). Thus, although previous attempts to determine the role of the PPARγ A/B-domain has reported PPARγCDE to be more adipogenic/lipogenic than full-length PPARγ2 (17), our results clearly demonstrate that the PPARγ A/B-domain is specifically involved in and necessary for complete transactivation of lipogenic target genes and has rightfully deserved the designation as the lipogenic domain (14). In keeping with previous reports that the A/B-domain restricts the transcriptional activity of a given PPAR by preventing nonselective target gene activation (13), we found HA-PPARγCDE to be a better transactivator of the PPARα-selective gene HMG-CoA S2 (9) than HA-PPARγ2 (Fig. 3C). The induction of HMG-CoA S2 was not detected in the array experiment, possibly because this method is less sensitive than RT-qPCR. Thus, although the expression array data did not indicate the existence of other target genes with similar characteristics as HMG-CoA S2, we cannot exclude the existence of other genes, where the A/B-domain of PPARγ is inhibitory. Together, the above results demonstrate a dual role of the A/B-domains in PPARα- and -γ2-mediated transactivation and preservation of PPAR subtype characteristics by preventing nonselective target gene activation but enabling full transactivation of highly subtype-selective target genes.
Using ChIP assays combined with qPCR, we showed that the reduced transcriptional activity of HA-PPARγCDE on the A/B-domain-dependent target genes was generally not due to decreased binding to the genomic PPREs (Fig. 5). This demonstrated that the A/B-domain is not required for the establishment or stabilization of HA-PPARγ2 binding to chromatin. Instead, we found a correlation between mRNA data and RNAPII binding, because deletion of the A/B-domain results in decreased RNAPII binding to the promoters of A/B-domain-dependent target genes but not of A/B-domain-independent target genes (Fig. 6). These results suggested that the reduced transactivation potential of HA-PPARγCDE on the lipogenic target genes originated from reduced recruitment of specific coactivators.
TRAP220 recruitment was unaffected by deletion of the PPARγ A/B-domain (Fig. 7), although it has been demonstrated that the mediator complex binds the glucocorticoid receptor by bridging the AF-1 and AF-2 through interactions with the TRAP170 and TRAP220 subunits, respectively (56). Interestingly, the coactivator CBP and the highly homologous p300 have been shown to interact with both the A/B- and E-domains in glutathione S-transferase pull-down studies (19). The relative importance of the interaction with the A/B- and E-domains is not clear, but it has been suggested that CBP and p300 mainly are recruited to NRs indirectly through binding to steroid receptor coactivator-1 (SRC-1) interacting with the E-domain (57, 58). Studies on thyroid hormone receptor β2 (59) and the estrogen receptor (60) have nevertheless demonstrated that interaction with CBP/p300 through the A/B-domain is necessary to obtain full ligand-dependent transcriptional activity. Correspondingly, we found HA-PPARγ-mediated recruitment of CBP and p300 to the A/B-domain-dependent target sites to be abolished upon deletion of the A/B-domain. In contrast, the PPARγ A/B-domain is dispensable for the recruitment of CBP and p300 on the PPREs of the A/B-domain-independent target genes (Fig. 8). Thus, the A/B-domain of PPARγ is important for the recruitment of CBP and p300 containing cofactor complexes, specifically to the PPREs of the lipogenic subset of PPARγ target genes.
Notably, our ChIP data suggested that CBP and p300 play nonredundant roles in PPARγ induction of some target genes, and we addressed this using siRNA-mediated knockdown of p300 and CBP (Fig. 9 and supplemental Fig. 3). We failed to see a clear correlation between PPARγ-mediated recruitment and dependency of p300, suggesting that p300 may be recruited to the target genes by other transcription factors (or PPARγ at different noncharacterized PPREs) and that this is necessary for full transactivation by PPARγ. Alternatively, p300 knockdown may affect expression of other genes that indirectly affect PPARγ target promoters. In general, however, PPARγ transactivation was affected more by knockdown of CBP than of p300, suggesting that CBP is more important for PPARγ transactivation in NIH-CAR cells.
In conclusion, we demonstrate that adenoviral gene delivery efficiently allows identification of novel putative PPARγ target genes when combined with expression array analysis. We show that the A/B-domain is the primary mediator of the weak ligand-independent activity of HA-PPARγ, whereas only a small subset of genes is A/B-domain dependent in the presence of rosiglitazone. Several of these genes are involved in lipid storage, and triglyceride accumulation was impaired in cells expressing HA-PPARγCDE compared with HA-PPARγ2. These results could not be explained by decreased binding of the HA-PPARγCDE:RXR heterodimer or by decreased recruitment of the mediator component TRAP220 to the genomic PPREs but correlated with decreased recruitment of RNAPII and decreased or abolished recruitment of CBP and p300 selectively on the A/B-domain-dependent target genes.
Materials and Methods
Cell cultures
NIH-CAR cells obtained by retroviral transduction of NIH-3T3 fibroblasts with the Coxsackie-adenovirus receptor (CARΔ1) (9) were cultured in DMEM (GIBCO, Carlsbad, CA) supplemented with 10% calf serum (Fisher Scientific, Pittsburgh, PA). 293-HEK cells (ATCC CRL-1573) were cultured in DMEM supplemented with 10% fetal calf serum (Biochrom AG, Berlin, Germany), and 1% non essential amino acids (GIBCO). Furthermore, streptomycin (100 μg/ml) and penicillin (62.5 μg/ml) were added to the medium. Cells were grown in NUNC plates at 37 C, 95% humidity, and 10% CO2.
Adenovirus generation and purification
The generation of recombinant adenovirus encoding full-length HA-tagged mouse PPARγ2 and PPARα using the AdEasy cloning system (Stratagene, La Jolla CA) has previously been described (9). Similarly, PCR cloning was used to generate the construct encoding HA-tagged PPARγ1, the A/B-domain-deleted PPARγ (HA-PPARγCDE) and to create domain constructs, i.e. HA-PPARα-A/B and PPARγCDE, that were subsequently fused in a conserved region to avoid mutations in the chimeric HA-PPARαA/BγCDE. HA-PPARγ1, HA-PPARγCDE, and HA-PPARαA/BγCDE were cloned into pShuttle-CMV (Stratagene) (where CMV is cytomegalovirus) using the SalI/NotI, SalI/EcoRV, or SalI/NotI restriction sites, respectively. The adenoviruses were packed and amplified in 293-HEK cells (ATCC CRL-1573; American Type Culture Collection, Manassas, VA), and viral titers were estimated by the tissue culture infectious dose 50 (TCID50) plaque assay-based approach (as described in the AdEasy protocol; Stratagene).
Adenoviral transduction
Adenoviral transduction was performed as previously described (9). Briefly, adenovirus was suspended in medium and added to NIH-CAR cells at 80% confluency. After 2 h transduction, the virus-containing medium was removed and new medium containing the vehicle dimethylsulfoxide (DMSO), 1 μm of the PPARγ-specific agonist rosiglitazone/BRL49653 (Novo Nordisk Copenhagen, Denmark) (25), or 1 μm of the PPARγ-specific antagonist GW9662 (Sigma-Aldrich, St. Louis, MO) (48) was added for an additional 6 h.
Nuclear extraction
Nuclei were purified from the transduced NIH-CAR cells by washing them twice in PBS followed by resuspension in lysis buffer containing 20 mm Tris-Cl (pH 7.5), 10 mm NaCl, 3 mm MgCl2, 1 mm 1,4-dithioerythritol, 1 mm Na orthovanadate, 30 mm β-glycerol phosphate, and Complete. The cell membrane was disrupted by addition of 1% Nonidet P-40 followed by 12 strokes in a Dounce homogenizer. Nuclear extracts were prepared as previously described (61) and used for EMSA (62). The sequences of the annotated PPREs used as probes are listed in supplemental Table 2.
Western blotting
Adenovirally transduced NIH-CAR cells were harvested in hypotonic SDS sample buffer (63) and subjected to Western blotting as previously described (9). The membrane was probed with the following primary antibodies: anti-PPARγ (sc-7273; Santa Cruz Biotechnology, Santa Cruz, CA), anti-TFIIB (sc-225; Santa Cruz), anti-CBP (sc-369; Santa Cruz), anti-p300 (sc-584; Santa Cruz), anti-Sp1 (sc-59; Santa Cruz), and anti-HA (12CA5) (64). The secondary antibodies used for enhanced chemiluminescence detection (ECL; Amersham Phamacia Biotech, Piscataway, NJ) were horseradish peroxidase-conjugated goat antimouse IgG (P0447; Dako, Glostrup, Denmark) and swine antirabbit IgG (P0339; Dako).
Immunostaining and confocal microscopy
The NIH-CAR cells were seeded on glass coverslips and transduced with adenovirus. Subsequently the cells were washed in PBS supplemented with 2.5 mm Ca2+ and 4 mm Mg2+ and fixed with 4% formaldehyde. After incubation with 100 mm glycine, cells were permeabilized by incubation in 0.1% Triton X-100. Nonspecific binding sites were blocked by incubation in 3% BSA dissolved in PBS supplemented with Ca2+ and Mg2+. Cells were stained with αPPARγ (sc-7273; Santa Cruz) and subsequently with Alexa fluor 546-conjugated antimouse IgG (A11030; Molecular Probes, Eugene, OR) and 4′,6-diamidino-2-phenylindole (DAPI). Coverslips were mounted onto slides and observed through a Zeiss LSM 510 META confocal microscope. Two-photon excitation of the fluorophores was obtained using a MaiTai XF-W2S Ti:Sa laser (Broadband Mai Tai with 10-W Millennia pump laser, tunable excitation range 710–980 nm; Spectra Physics, Mountain View, CA). The excitation wavelength was 730 nm. Images were collected simultaneously in two different channels using band-pass filters of 460 ± 25 and 590 ± 25 nm for DAPI and Alexa fluor 546, respectively.
[14C]Acetate incorporation into cellular lipid
NIH-CAR cells were transduced with adenovirus and incubated with DMSO or rosiglitazone. Twenty-four hours after transduction, 35 nmol [14C]acetate (specific activity 57 mCi/mmol) was added to each well and incubation continued for another 24 h (this incubation period was chosen based on the results from a preliminary study, data not shown). Uptake was terminated by removal of the medium, followed by a wash with Krebs-Ringer buffer and ice-cold PBS. The cells were harvested in PBS and sonicated for four times for 30 sec each with 30-sec intervals at level 5 in the Bioruptor (Diagenode, Liege, Belgium). Protein concentration was determined on a Qbit (Invitrogen, Carlsbad, CA), and total lipid was extracted and purified using the Bligh and Dyer method (65). After drying in a stream of N2, samples were redissolved in 1:2 chloroform/methanol and lipids were separated on silica thin layer chromatography plates (HPTLC silica gel 60; Merck, Darmstadt, Germany) developed in hexane-diethylether-acetic acid (70:30:1, vol/vol). The incorporated radioactivity was detected by a Typhoon TRIO scanner (Amersham Biosciences) and quantified using the ImageQuant TL software.
RNA extraction and cDNA synthesis
The adenovirally transduced NIH-CAR cells were harvested in 500 μl TRIzol. Total RNA was extracted by addition of chloroform and precipitated with isopropanol and centrifugation. The pellet was washed with 75% ethanol and redissolved in diethyl pyrocarbonate-treated water. From each preparation, 1 μg RNA was subjected to deoxyribonuclease I (Invitrogen) treatment, and cDNA was synthesized using random deoxynucleic acid hexamers and reverse transcriptase (RT) (First-Strand Kit; Invitrogen) as previously described (63). All experiments were performed in duplicate.
Microarray analysis
RNA samples from three individual experiments in which NIH-CAR cells were transduced with AdEmpty followed by addition of vehicle (DMSO) or transduced with either AdHA-PPARγ2 or AdHA-PPARγCDE and treated with rosiglitazone were purified further on RNAeasy columns (QIAGEN, Valencia, CA). Two micrograms of total RNA was used to synthesize double-stranded cDNA with the one-cycle cDNA synthesis kit (Affymetix Inc., Santa Clara, CA) using an oligo(dT) primer containing a T7 RNA polymerase promoter (Affymetrix). The cDNA was purified using the GeneChip clean-up module (Affymetrix) and used as the template for an in vitro transcription reaction to synthesize biotin-labeled antisense cRNA (IVT labeling kit; Affymetrix). After fragmentation at 94 C for 35 min in fragmentation buffer (40 mm Tris, 30 mm magnesium acetate, and 10 mm potassium acetate), 15 μg labeled cRNA was hybridized for 16 h to the Mouse 430 2.0 array (Affymetrix) that contains 45,000 probe sets, which analyze the expression level of over 39,000 transcripts and variants from over 34,000 well characterized mouse genes. The raw intensity data was normalized using the RMA algorithm implemented in the Bioconductor package affy running under R version 2.8.1. The normalized expression data were exported to the DNA Chip Analyzer program (dChip, November 2007, www.dchip.org). Three group comparison analyses were performed, HA-PPARγ2-vs.-AdEmpty, HA-PPARγ2-vs.-HA-PPARγCDE, and HA-PPARγCDE-vs.-AdEmpty, respectively. A gene was defined as being differentially expressed if it had a P value below 0.05 in a paired Welch t test (variance not equal), an absolute fold change greater than 1.5, and a difference of group means greater than 50. Functional clustering of the up-regulated genes was performed using Ingenuity Pathway Analysis (Ingenuity Systems Inc., Redwood City, CA).
ChIP
The adenovirally transduced NIH-CAR cells were fixed with 1% formaldehyde for 10 min followed by incubation with 12.5 mm glycine. Cells were washed twice in PBS and harvested in lysis buffer containing 0.1% SDS, 1% Triton X-100, 0.15 m NaCl, 1 mm EDTA, and 20 mm Tris (pH 8). Cells were sonicated eight times for 30 sec each with 30-sec intervals at level 5 in the Bioruptor (Diagenode). The DNA concentrations were determined by measuring OD260 and subsequently adjusted to equal levels. Protein A beads (Amersham Pharmacia Biotech) were prepared by three washes in IP buffer containing 0.15% SDS, 1% Triton X-100, 0.15 m NaCl, 1 mm EDTA, and 20 mm HEPES (pH 7.6) followed by 2 h incubation in IP buffer supplemented with 1 μg/μl BSA. One hundred micrograms of chromatin were diluted three times in IP buffer and incubated overnight with 1–2 μg antibody and 100 μl prepared beads rotating at 4 C. Beads were washed at 4 C, twice with buffer 1 containing 0.1% SDS, 0.1% deoxycholic acid (DOC), 1% Triton X-100, 0.15 m NaCl, 1 mm EDTA, and 20 mm HEPES (pH 7.6); once in buffer 2 containing 0.1% SDS, 0.1% DOC, 1% Triton X-100, 0.5 m NaCl, 1 mm EDTA, and 20 mm HEPES (pH 7.6); once in buffer 3 containing 0.25 m LiCl, 0.5% DOC, 0.5% Nonidet P-40, 1 mm EDTA, and 20 mm HEPES (pH 7.6); and twice in buffer 4 containing 1 mm EDTA, and 20 mm HEPES (pH 7.6). DNA-protein complexes were eluted with elution buffer (1% SDS and 0.1 m NaHCO3) and de-cross-linked by adding 0.2 m NaCl followed by shaking overnight at 65 C. DNA was isolated by chloroform-phenol extraction. Immunoprecipitated DNA and 10% input DNA were analyzed by qPCR. Antibodies used for IP were anti-RXRα (sc-774; Santa Cruz), anti-CBP (sc-369; Santa Cruz), anti-p300 (sc-584; Santa Cruz), anti-TRAP220 (sc-8998; Santa Cruz), anti-polII (8WG16; Covance Research Products, Princeton, NJ), and anti-HA (12CA5) (64). For each ChIP experiment, the enrichment of target site and background control (β-globin promoter), respectively, was normalized to input DNA (i.e. recovery). To relate independent experiments, we subsequently determined the relative occupancy by normalizing the recovery of the target site and β-globin promoter in the individual samples to the recovery of the β-globin promoter in the control cells transduced with AdEmpty.
siRNA-mediated knockdown
Twenty-four hours before adenoviral transduction, NIH-CAR cells were switched to OptiMEM (Invitrogen) and transfected with combinations of predesigned siRNA against firefly luciferase [CGUACGCGGAAUACUUCGA(dT)(dT); Sigma-Aldrich, St. Louis, MO], CBP [GUAACUCUGGCCAUAGCUU(dT)(dT) Sigma-Aldrich], or p300 [GAAUGACUUUCUGAGGCGA(dT)(dT) Sigma-Aldrich] in a final concentration of 50 nm using RNAiMAX (Invitrogen). The transfection medium was replaced with normal culture medium 4 h before the adenoviral transduction.
qPCR
Quantitative three-step qPCR was performed on the Mx3000 real-time PCR instrument (Stratagene) using 2× SYBR Green Master Mix and Sigma passive reference (Sigma-Aldrich) according to the instructions from the manufacturer. All measurements were performed in duplicate. Primers for qPCR (supplemental Table 3) were designed using Primer Express 2.0 (Applied Biosystems, Foster City, CA) or Primer3 (http://frodo.wi.mit.edu). Specificity and efficacy were validated before use.
Acknowledgments
We thank P. Sauerberg (Novo Nordisk A/S) for the rosiglitazone/BRL49653 ligand, L. A. Bagatolli for expert assistance with the confocal microscope, and T. Obsen and N. J. Færgeman for instructions on lipid analyses.
NURSA Molecule Pages:
Coregulators: CBP | TRAP220 | p300;
Ligands: GW 9662 | Rosiglitazone;
Nuclear Receptors: PR.
Footnotes
This work was supported by grants from the Danish Natural Science Research Council and grants to the European Union FP6 Specific Targeted Project X-TRA-NET. M.M.A. was supported by the Novo scholarship program.
Disclosure Summary: None of the authors have any conflict of interest.
First Published Online March 19, 2009
Abbreviations: ADRP, Adipose differentiation-related protein; AF-1, activation function 1; CBP, cAMP response element-binding protein (CREB)-binding protein; CDK9, cyclin-dependent kinase 9; ChIP, chromatin immunoprecipitation; CIDEC, cell death-inducing DFFA-like effector C; DAPI, 4′,6-diamidino-2-phenylindole; DMSO, dimethylsulfoxide; DOC, deoxycholic acid; HA, hemagglutinin; HMG-CoA S2, 3-hydroxy-3-methyl-glutaryl-coenzyme A synthase 2; LBD, ligand-binding domain; NR, nuclear receptor; PGC-2, PPARγ coactivator 2; PLIN, perilipin; PPAR, peroxisome proliferator-activated receptor; PPRE, PPAR response element; RT-qPCR, quantitative RT-PCR; RXR, retinoid X receptor; siRNA, small interfering RNA; TRB3, tribbles homolog 3; Tip60, Tat-interacting protein 60; TRAP220, thyroid hormone receptor-associated protein 220.
References
- 1.Escher P, Wahli W2000. Peroxisome proliferator-activated receptors: insight into multiple cellular functions. Mutat Res 448:121–138 [DOI] [PubMed] [Google Scholar]
- 2.Bishop-Bailey D, Wray J2003. Peroxisome proliferator-activated receptors: a critical review on endogenous pathways for ligand generation. Prostaglandins Other Lipid Mediat 71:1–22 [DOI] [PubMed] [Google Scholar]
- 3.Xu HE, Lambert MH, Montana VG, Plunket KD, Moore LB, Collins JL, Oplinger JA, Kliewer SA, Gampe Jr RT, McKee DD, Moore JT, Willson TM2001. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA 98:13919–13924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kersten S, Desvergne B, Wahli W2000. Roles of PPARs in health and disease. Nature 405:421–424 [DOI] [PubMed] [Google Scholar]
- 5.Chevillotte E, Rieusset J, Roques M, Desage M, Vidal H2001. The regulation of uncoupling protein-2 gene expression by ω-6 polyunsaturated fatty acids in human skeletal muscle cells involves multiple pathways, including the nuclear receptor peroxisome proliferator-activated receptor β. J Biol Chem 276:10853–10860 [DOI] [PubMed] [Google Scholar]
- 6.Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM2003. Peroxisome-proliferator-activated receptor δ activates fat metabolism to prevent obesity. Cell 113:159–170 [DOI] [PubMed] [Google Scholar]
- 7.Mueller E, Drori S, Aiyer A, Yie J, Sarraf P, Chen H, Hauser S, Rosen ED, Ge K, Roeder RG, Spiegelman BM2002. Genetic analysis of adipogenesis through peroxisome proliferator-activated receptor γ isoforms. J Biol Chem 277:41925–41930 [DOI] [PubMed] [Google Scholar]
- 8.Brun RP, Tontonoz P, Forman BM, Ellis R, Chen J, Evans RM, Spiegelman BM1996. Differential activation of adipogenesis by multiple PPAR isoforms. Genes Dev 10:974–984 [DOI] [PubMed] [Google Scholar]
- 9.Nielsen R, Grøntved L, Stunnenberg HG, Mandrup S2006. Peroxisome proliferator-activated receptor subtype- and cell-type-specific activation of genomic target genes upon adenoviral transgene delivery. Mol Cell Biol 26:5698–5714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravnskjaer K, Boergesen M, Rubi B, Larsen JK, Nielsen T, Fridriksson J, Maechler P, Mandrup S2005. Peroxisome proliferator-activated receptor α (PPARα) potentiates, whereas PPARγ attenuates, glucose-stimulated insulin secretion in pancreatic β-cells. Endocrinology 146:3266–3276 [DOI] [PubMed] [Google Scholar]
- 11.Berger J, Patel HV, Woods J, Hayes NS, Parent SA, Clemas J, Leibowitz MD, Elbrecht A, Rachubinski RA, Capone JP, Moller DE2000. A PPARγ mutant serves as a dominant negative inhibitor of PPAR signaling and is localized in the nucleus. Mol Cell Endocrinol 162:57–67 [DOI] [PubMed] [Google Scholar]
- 12.Nagy L, Schwabe JW2004. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci 29:317–324 [DOI] [PubMed] [Google Scholar]
- 13.Hummasti S, Tontonoz P2006. The peroxisome proliferator-activated receptor N-terminal domain controls isotype-selective gene expression and adipogenesis. Mol Endocrinol 20:1261–1275 [DOI] [PubMed] [Google Scholar]
- 14.Castillo G, Brun RP, Rosenfield JK, Hauser S, Park CW, Troy AE, Wright ME, Spiegelman BM1999. An adipogenic cofactor bound by the differentiation domain of PPARγ. EMBO J 18:3676–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hi R, Osada S, Yumoto N, Osumi T1999. Characterization of the amino-terminal activation domain of peroxisome proliferator-activated receptor α. Importance of α-helical structure in the transactivating function. J Biol Chem 274:35152–35158 [DOI] [PubMed] [Google Scholar]
- 16.Hsu MH, Palmer CN, Song W, Griffin KJ, Johnson EF1998. A carboxyl-terminal extension of the zinc finger domain contributes to the specificity and polarity of peroxisome proliferator-activated receptor DNA binding. J Biol Chem 273:27988–27997 [DOI] [PubMed] [Google Scholar]
- 17.Tontonoz P, Hu E, Spiegelman BM1994. Stimulation of adipogenesis in fibroblasts by PPAR γ2, a lipid-activated transcription factor. Cell 79:1147–1156 [DOI] [PubMed] [Google Scholar]
- 18.van Beekum O, Brenkman AB, Grøntved L, Hamers N, van den Broek NJ, Berger R, Mandrup S, Kalkhoven E2008. The adipogenic acetyltransferase Tip60 targets activation function 1 of peroxisome proliferator-activated receptor γ. Endocrinology 149:1840–1849 [DOI] [PubMed] [Google Scholar]
- 19.Gelman L, Zhou G, Fajas L, Raspé E, Fruchart JC, Auwerx J1999. p300 interacts with the N- and C-terminal part of PPARγ2 in a ligand-independent and -dependent manner, respectively. J Biol Chem 274:7681–7688 [DOI] [PubMed] [Google Scholar]
- 20.Takahashi Y, Ohoka N, Hayashi H, Sato R2008. TRB3 suppresses adipocyte differentiation by negatively regulating PPARγ transcriptional activity. J Lipid Res 49:880–892 [DOI] [PubMed] [Google Scholar]
- 21.Shimizu M, Yamashita D, Yamaguchi T, Hirose F, Osumi T2006. Aspects of the regulatory mechanisms of PPAR functions: analysis of a bidirectional response element and regulation by sumoylation. Mol Cell Biochem 286:33–42 [DOI] [PubMed] [Google Scholar]
- 22.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK1997. Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem 272:5128–5132 [DOI] [PubMed] [Google Scholar]
- 23.Iankova I, Petersen RK, Annicotte JS, Chavey C, Hansen JB, Kratchmarova I, Sarruf D, Benkirane M, Kristiansen K, Fajas L2006. Peroxisome proliferator-activated receptor γ recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis. Mol Endocrinol 20:1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B, Berger J, Zhou G, Elbrecht A, Biswas S, White-Carrington S, Szalkowski D, Moller DE1996. Insulin- and mitogen-activated protein kinase-mediated phosphorylation and activation of peroxisome proliferator-activated receptor γ. J Biol Chem 271:31771–31774 [DOI] [PubMed] [Google Scholar]
- 25.Seimandi M, Lemaire G, Pillon A, Perrin A, Carlavan I, Voegel JJ, Vignon F, Nicolas JC, Balaguer P2005. Differential responses of PPARα, PPARδ, and PPARγ reporter cell lines to selective PPAR synthetic ligands. Anal Biochem 344:8–15 [DOI] [PubMed] [Google Scholar]
- 26.Sato O, Kuriki C, Fukui Y, Motojima K2002. Dual promoter structure of mouse and human fatty acid translocase/CD36 genes and unique transcriptional activation by peroxisome proliferator-activated receptor α and γ ligands. J Biol Chem 277:15703–15711 [DOI] [PubMed] [Google Scholar]
- 27.Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, Heyman RA, Briggs M, Deeb S, Staels B, Auwerx J1996. PPARα and PPARγ activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J 15:5336–5348 [PMC free article] [PubMed] [Google Scholar]
- 28.Kishida K, Shimomura I, Nishizawa H, Maeda N, Kuriyama H, Kondo H, Matsuda M, Nagaretani H, Ouchi N, Hotta K, Kihara S, Kadowaki T, Funahashi T, Matsuzawa Y2001. Enhancement of the aquaporin adipose gene expression by a peroxisome proliferator-activated receptor γ. J Biol Chem 276:48572–48579 [DOI] [PubMed] [Google Scholar]
- 29.Rojek A, Praetorius J, Frøkiaer J, Nielsen S, Fenton RA2008. A current view of the mammalian aquaglyceroporins. Annu Rev Physiol 70:301–327 [DOI] [PubMed] [Google Scholar]
- 30.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM1994. mPPAR γ2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 8:1224–1234 [DOI] [PubMed] [Google Scholar]
- 31.Jitrapakdee S, Slawik M, Medina-Gomez G, Campbell M, Wallace JC, Sethi JK, O'rahilly S, Vidal-Puig AJ2005. The peroxisome proliferator-activated receptor-γ regulates murine pyruvate carboxylase gene expression in vivo and in vitro. J Biol Chem 280:27466–27476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ballard FJ, Hanson RW1967. The citrate cleavage pathway and lipogenesis in rat adipose tissue: replenishment of oxaloacetate. J Lipid Res 8:73–79 [PubMed] [Google Scholar]
- 33.Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S, Gonzalez FJ2008. Hepatic steatosis in leptin-deficient mice is promoted by the PPARγ target gene Fsp27. Cell Metab 7:302–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keller P, Petrie JT, De Rose P, Gerin I, Wright WS, Chiang SH, Nielsen AR, Fischer CP, Pedersen BK, MacDougald OA2008. Fat-specific protein 27 regulates storage of triacylglycerol. J Biol Chem 283:14355–14365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brasaemle DL2007. Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res 48:2547–2559 [DOI] [PubMed] [Google Scholar]
- 36.Bildirici I, Roh CR, Schaiff WT, Lewkowski BM, Nelson DM, Sadovsky Y2003. The lipid droplet-associated protein adipophilin is expressed in human trophoblasts and is regulated by peroxisomal proliferator-activated receptor-γ/retinoid X receptor. J Clin Endocrinol Metab 88:6056–6062 [DOI] [PubMed] [Google Scholar]
- 37.Targett-Adams P, McElwee MJ, Ehrenborg E, Gustafsson MC, Palmer CN, McLauchlan J2005. A PPAR response element regulates transcription of the gene for human adipose differentiation-related protein. Biochim Biophys Acta 1728:95–104 [DOI] [PubMed] [Google Scholar]
- 38.Umlauf E, Csaszar E, Moertelmaier M, Schuetz GJ, Parton RG, Prohaska R2004. Association of stomatin with lipid bodies. J Biol Chem 279:23699–23709 [DOI] [PubMed] [Google Scholar]
- 39.Bartz R, Li WH, Venables B, Zehmer JK, Roth MR, Welti R, Anderson RG, Liu P, Chapman KD2007. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J Lipid Res 48:837–847 [DOI] [PubMed] [Google Scholar]
- 40.Wang P, Renes J, Bouwman F, Bunschoten A, Mariman E, Keijer J2007. Absence of an adipogenic effect of rosiglitazone on mature 3T3-L1 adipocytes: increase of lipid catabolism and reduction of adipokine expression. Diabetologia 50:654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Festuccia WT, Laplante M, Berthiaume M, Gélinas Y, Deshaies Y2006. PPARγ agonism increases rat adipose tissue lipolysis, expression of glyceride lipases, and the response of lipolysis to hormonal control. Diabetologia 49:2427–2436 [DOI] [PubMed] [Google Scholar]
- 42.Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ, Reddy JK2003. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor γ1 (PPARγ1) overexpression. J Biol Chem 278:498–505 [DOI] [PubMed] [Google Scholar]
- 43.Kershaw EE, Schupp M, Guan HP, Gardner NP, Lazar MA, Flier JS2007. PPARγ regulates adipose triglyceride lipase in adipocytes in vitro and in vivo. Am J Physiol Endocrinol Metab 293:E1736–E1745 [DOI] [PMC free article] [PubMed]
- 44.Hauser S, Adelmant G, Sarraf P, Wright HM, Mueller E, Spiegelman BM2000. Degradation of the peroxisome proliferator-activated receptor γ is linked to ligand-dependent activation. J Biol Chem 275:18527–18533 [DOI] [PubMed] [Google Scholar]
- 45.Sumanasekera WK, Tien ES, Turpey R, Vanden Heuvel JP, Perdew GH2003. Evidence that peroxisome proliferator-activated receptor α is complexed with the 90-kDa heat shock protein and the hepatitis virus B X-associated protein 2. J Biol Chem 278:4467–4473 [DOI] [PubMed] [Google Scholar]
- 46.Patel H, Truant R, Rachubinski RA, Capone JP2005. Activity and subcellular compartmentalization of peroxisome proliferator-activated receptor α are altered by the centrosome-associated protein CAP350. J Cell Sci 118:175–186 [DOI] [PubMed] [Google Scholar]
- 47.Burgermeister E, Chuderland D, Hanoch T, Meyer M, Liscovitch M, Seger R2007. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor γ. Mol Cell Biol 27:803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leesnitzer LM, Parks DJ, Bledsoe RK, Cobb JE, Collins JL, Consler TG, Davis RG, Hull-Ryde EA, Lenhard JM, Patel L, Plunket KD, Shenk JL, Stimmel JB, Therapontos C, Willson TM, Blanchard SG2002. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry 41:6640–6650 [DOI] [PubMed] [Google Scholar]
- 49.Brasaemle DL, Dolios G, Shapiro L, Wang R2004. Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J Biol Chem 279:46835–46842 [DOI] [PubMed] [Google Scholar]
- 50.Kim JY, Liu K, Zhou S, Tillison K, Wu Y, Smas CM2008. Assessment of fat-specific protein 27 in the adipocyte lineage suggests a dual role for FSP27 in adipocyte metabolism and cell death. Am J Physiol Endocrinol Metab 294:E654–E667 [DOI] [PubMed]
- 51.Yang W, Rachez C, Freedman LP2000. Discrete roles for peroxisome proliferator-activated receptor γ and retinoid X receptor in recruiting nuclear receptor coactivators. Mol Cell Biol 20:8008–8017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerhold DL, Liu F, Jiang G, Li Z, Xu J, Lu M, Sachs JR, Bagchi A, Fridman A, Holder DJ, Doebber TW, Berger J, Elbrecht A, Moller DE, Zhang BB2002. Gene expression profile of adipocyte differentiation and its regulation by peroxisome proliferator-activated receptor-γ agonists. Endocrinology 143:2106–2118 [DOI] [PubMed] [Google Scholar]
- 53.Wärnmark A, Treuter E, Wright AP, Gustafsson JA2003. Activation functions 1 and 2 of nuclear receptors: molecular strategies for transcriptional activation. Mol Endocrinol 17:1901–1909 [DOI] [PubMed] [Google Scholar]
- 54.Burns KA, Vanden Heuvel JP2007. Modulation of PPAR activity via phosphorylation. Biochim Biophys Acta 1771:952–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kumar R, Thompson EB2003. Transactivation functions of the N-terminal domains of nuclear hormone receptors: protein folding and coactivator interactions. Mol Endocrinol 17:1–10 [DOI] [PubMed] [Google Scholar]
- 56.Hittelman AB, Burakov D, Iñiguez-Lluhí JA, Freedman LP, Garabedian MJ1999. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J 18:5380–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen D, Huang SM, Stallcup MR2000. Synergistic, p160 coactivator-dependent enhancement of estrogen receptor function by CARM1 and p300. J Biol Chem 275:40810–40816 [DOI] [PubMed] [Google Scholar]
- 58.Li J, O'Malley BW, Wong J2000. p300 requires its histone acetyltransferase activity and SRC-1 interaction domain to facilitate thyroid hormone receptor activation in chromatin. Mol Cell Biol 20:2031–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian H, Mahajan MA, Wong CT, Habeos I, Samuels HH2006. The N-terminal A/B domain of the thyroid hormone receptor-β2 isoform influences ligand-dependent recruitment of coactivators to the ligand-binding domain. Mol Endocrinol 20:2036–2051 [DOI] [PubMed] [Google Scholar]
- 60.Kobayashi Y, Kitamoto T, Masuhiro Y, Watanabe M, Kase T, Metzger D, Yanagisawa J, Kato S2000. p300 mediates functional synergism between AF-1 and AF-2 of estrogen receptor α and β by interacting directly with the N-terminal A/B domains. J Biol Chem 275:15645–15651 [DOI] [PubMed] [Google Scholar]
- 61.Lavery DJ, Schibler U1993. Circadian transcription of the cholesterol 7 α-hydroxylase gene may involve the liver-enriched bZIP protein DBP. Genes Dev 7:1871–1884 [DOI] [PubMed] [Google Scholar]
- 62.Helledie T, Grøntved L, Jensen SS, Kiilerich P, Rietveld L, Albrektsen T, Boysen MS, Nohr J, Larsen LK, Fleckner J, Stunnenberg HG, Kristiansen K, Mandrup S2002. The gene encoding the Acyl-CoA-binding protein is activated by peroxisome proliferator-activated receptor γ through an intronic response element functionally conserved between humans and rodents. J Biol Chem 277:26821–26830 [DOI] [PubMed] [Google Scholar]
- 63.Hansen JB, Petersen RK, Larsen BM, Bartkova J, Alsner J, Kristiansen K1999. Activation of peroxisome proliferator-activated receptor γ bypasses the function of the retinoblastoma protein in adipocyte differentiation. J Biol Chem 274:2386–2393 [DOI] [PubMed] [Google Scholar]
- 64.Mitsiou DJ, Stunnenberg HG2000. TAC, a TBP-sans-TAFs complex containing the unprocessed TFIIAαβ precursor and the TFIIAγ subunit. Mol Cell 6:527–537 [DOI] [PubMed] [Google Scholar]
- 65.Bligh EG, Dyer WJ1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917 [DOI] [PubMed] [Google Scholar]