

Abstract
Steroid hormone receptors act directly in the nucleus on the chromatin organization and transcriptional activity of several promoters. Furthermore, they have an indirect effect on cytoplasmic signal transduction pathways, including MAPK, impacting ultimately on gene expression. We are interested in distinguishing between the two modes of action of progesterone receptor (PR) on the control of gene expression and cell proliferation. For this, we have stably expressed, in PR-negative breast cancer cells, tagged forms of the PR isoform B mutated at regions involved either in DNA binding (DNA-binding domain) or in its ability to interact with the estrogen receptor and to activate the c-Src/MAPK/Erk/Msk cascade (estrogen receptor-interacting domain). Both mutants impair PR-mediated activation of a well-understood model promoter in response to progestin, as well as hormone-induced cell proliferation. Additional mutants affecting transactivation activity of PR (activation function 2) or a zinc-finger implicated in dimerization (D-box) have also been tested. Microarrays and gene expression experiments on these cell lines define the subsets of hormone-responsive genes regulated by different modes of action of PR isoform B, as well as genes in which the nuclear and nongenomic pathways cooperate. Correlation between CCND1 expression in the different cell lines and their ability to support cell proliferation confirms CCND1 as a key controller gene.
Gene expression analysis in T47D cells stably expressing functionally deficient PR-B forms denotes the relevance of crosstalk between genomic and non-genomic receptor modes of action.
Ovarian steroid hormones (estrogens and progestins) control growth and differentiation of normal and transformed epithelial breast cells by virtue of their interaction with specific intracellular receptors. Steroid hormone receptors (SHRs) are classically seen as nuclear transcription factors that, upon activation by binding with their corresponding ligands, regulate the expression of different target genes. Ligand-activated SHRs can act by binding as dimers to their hormone-responsive elements (HREs) at promoters or by interaction with other DNA-bound factors. In both cases, the process results in the recruitment of coregulators, chromatin remodeling complexes, and the general transcriptional machinery (1).
However, SHRs can also modulate gene expression by activation of cytoplasmic signaling pathways (nongenomic actions) (2). Estrogen receptor (ER) binds to c-Src and to the regulatory subunit of phosphoinositol 3-kinase (PI3K), activating the c-Src/Ras/Erk and PI3K/Akt pathways, respectively (3, 4). In both cases, these rapid hormone-triggered effects have been associated with their proliferative role. The ultimate targets of these signaling cascades are not well defined and likely include transcription factors and coactivators.
Direct interaction and activation of c-Src by progesterone receptor (PR) has also been reported (5). Nonetheless, in the breast cancer cell line T47D, ligand-activated PR activates the c-Src/Ras/Erk pathway indirectly via an interaction with ERα in the absence of estrogens (6). Two regions of PR important for this interaction with ER have been mapped and named ERID (ER-interacting domain) I (residues 165-345) and II (456-546) (7). Activation of the ER/c-Src/Ras/Erk pathway is essential for progestin induction of cell proliferation in breast cancer cells, as ER antagonists and inhibitors of Erk activation block progestin-induced DNA synthesis and progression through the cell cycle (8). Furthermore, progestin has also been shown to activate the PI3K/Akt and Janus family of tyrosine kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathways in a manner dependent on c-Src in mammary tumor cells and to be correlated with progestin stimulation of growth (9, 10). In breast cancer cells, progestin has a biphasic effect on cell growth, with an initial proliferation burst, followed by an arrest of the cells in late G1 phase of the second cycle. This might be due to initial expression of several cyclins and other proliferation-associated genes and late induction of cyclin-dependent kinase (cdk) inhibitors (11, 12).
The nuclear action of steroid receptors as transcription factors binding to target promoters has been extensively studied, mostly with a reduced number of model promoters, such as mouse mammary tumor virus (MMTV) for PR or pS2 (trefoil factor-1) for ER. A more recent challenge has been to reveal the mechanisms by which receptors modulate extranuclear signaling pathways and how this impacts on gene expression. Three mechanisms have been proposed to link the activation of kinase cascades and initiation of transcription: 1) kinases may phosphorylate and activate nuclear transcription factors binding to promoters devoid of HREs without involvement of the nuclear steroid receptor, 2) steroid receptors may interact with a transcription factor, which first needs to be activated by a protein kinase, targeting the receptor to a specific promoter containing binding sites for such transcription factor (and, alternatively, also HREs), and 3) the transcriptional activity of a steroid receptor on a HRE-containing direct target gene may require direct phosphorylation of either the receptor itself or a receptor-interacting coactivator. The last two mechanisms involve a cross talk between the nuclear and extranuclear functions of steroid receptors.
Recently, we have reported examples of such cross talk between PR functions in breast cancer cells (13, 14). After progesterone treatment, Erk and Msk1 kinases are activated and recruited with phosphorylated PR to the MMTV promoter, where histone H3 is phosphorylated and acetylated locally (13). These H3 modifications seem to be a key switch for the exchange of a repressive complex containing HP1γ by coactivators, chromatin remodeling complexes, and RNA polymerase II. Thus, rapid kinase activation by progestin may participate in induction of PR direct target genes by preparing the chromatin for transcription, indicating that both PR actions cross talk with each other. Furthermore, we have recently described how activation of hydroxysteroid dehydrogenase (HSD)11B2 gene expression in response to progestin depends on JAK/STAT activation and STAT5A-mediated recruitment of PR to a distal promoter region, concomitant with histone modification, and transcription factor and RNA polymerase recruitment (14). The number of examples showing cooperation between SHRs genomic and nongenomic functions is rapidly increasing.
Steroid receptors share a characteristic modular structure with an N-terminal transactivation function (AF1), a highly conserved DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) that also contains a ligand-regulated transcriptional activation function (AF2) necessary for recruitment of coactivating proteins (Fig. 1A) (1). DBD is the domain responsible for the binding to hormone response elements (HRE) in DNA of target promoters. The DBD is a globular domain made up of two different zinc-finger structures. The DBD undergoes DNA-induced dimerization upon binding to palindromic HREs. The residues that define the dimer interface are located into the C-terminal zinc finger and constitute the D-box. The sequence-specific DNA binding residues are defined as the P-box in the N-terminal zinc-finger (Fig. 1A). Furthermore, DBD is an allosteric transmitter of information to other regions of the receptor molecule. The DBD is connected to the LBD via a short amino acid sequence termed “the hinge.” LBD is also a mediator of receptor dimerization, necessary for binding to HREs.
Fig. 1.

Construction of breast cancer cell lines stably expressing WT or functionally deficient, tagged forms of PRB. A, Schematic representation of recombinant PRB forms cloned into the pRAV-Flag bicistronic retroviral vector used for expression (see Ref. 44 for details). Predicted molecular weight and altered function of each variant are indicated. EGFP, enhanced green fluorescent protein; H, hinge region; IF, inhibitory function; IRES, internal ribosome entry site; LTR, long terminal repeat; TAP, tandem affinity purification peptide, constituted by a protein A fragment (IgG binding domain), two tobacco etch virus cleavage sites and a Flag tag. Lower panel, Schematic structure of PR Zn-fingers indicating residues at P- and D-boxes mutated to generate the different DBD mutants. Dimerization mutants contained mutations A604T (D4) or R606W (M6). B, Stable integration of an MMTV-Luc reporter construct into PR-defective T47D-YV breast cancer cells. T47D-YV cells were stably transfected with an MMTV-Luc construct and neomycin-resistant clones obtained. MMTV-Luc response to progestin was tested in three individual clones upon transient transfection with empty (EMP) or PRB-expressing pRAV-Flag plasmids. After transfection, media were replaced with serum-free media and 48 h later 10 nm R5020 or vehicle (EtOH diluted 1:10,000) was added. Luciferase activity was measured on cell extracts 12 h later, after protein determination. Relative luciferase units (RLU) are shown. The values represent mean ± sd of a representative experiment performed in triplicate. Clone 25 was further used and named TYML. C, FACS analysis of TYML cells infected with bicistronic retroviruses expressing PRB variants and GFP at low multiplicity. One week after infection, GFP-positive cells (gated) were sorted and cell lines established. A control cell line was established infecting TYML with the empty pRAV-Flag vector. D, Western blot analysis of recombinant PRB forms in GFP-positive sorted cells. Total protein extract (50 μg) from each of the cell lines generated was resolved in SDS-PAGE and hybridized against an anti-PR antibody (Ab11). T47D cells were used as a control of endogenous PRA/B levels. Tubulin antibody was used as a loading control. E, Immunofluorescence detection of PRB variants expression. Each established cell line was cultured over coverslips in 10% FBS-rich medium, cells were fixed, permeabilized, and incubated with anti-PR H190 antibody, followed by secondary antibody and DAPI staining. T47D and TYML/empty plasmid cells were used as controls. F, RT-PCR of PRB gene expression in the different cell lines, using real-time PCR and specific primers corresponding to the coding region (upper panel) or to the 3′-UTR present in the endogenous gene, but not in the recombinant PR form (lower panel). GAPDH expression was measured for normalization. The values, as relative units (RU), represent mean ± sd of an experiment performed in triplicate.
Two isoforms of the PR, A and B, are encoded from the same gene using two different promoters. Both isoforms differ only in their amino termini, PRB extending 164 amino acids further than PRA. Transcriptionally, the two isoforms differ, PRB being a much stronger transactivator in response to progestin (15, 16). The region of the protein that is unique to PRB contains a transcription activation function (AF)3, that has a distinct conformation and is likely to mask an inhibitory function domain (IF) that is active in the N terminus of PRA (17). The two PR isoforms regulate some distinct, nonoverlapping genes and functions, but also many of the same genes. The interplay between the two isoforms is determinant of the outcome of tumor progression in breast cancer (18, 19).
To further explore the role of PR acting either as a nuclear transcription factor or participating in the activation of kinase cascades through the interaction with ER in endogenous gene expression, we have constructed cell lines expressing PRB mutants that would presumably have affected one of the two abilities. Thus, we have stably introduced in the PR-defective breast cancer cell line T47D-YV, wild-type (WT) PRB and PRB mutants affected either in the ERID domain, one of the two zinc-fingers of DBD (P- and D-boxes), or AF2. By using microarrays and kinetic gene expression experiments, we have defined sets of hormone-induced genes that uniquely require signaling cascade activation through PR-ER interaction or the transcriptional capacity of PRB, or the synergistic collaboration of both mechanisms. We also define PR modes of action required to support progestin-induced cell proliferation, which correlate with those required for cyclin D1 gene induction.
Results
Characterization of breast cancer cell lines stably expressing functionally deficient forms of the human PR isoform B
With the aim to stably express in breast cancer cells PRB mutants defective in several functions, we have used a retroviral vector (pRAV-Flag) (see Materials and Methods). This vector has two interesting peculiarities: it creates an N-terminal fusion of the expressed protein with a peptide tag including the FLAG peptide, and coexpresses green fluorescent protein (GFP) for sorting of positively infected cells. Stable expression is accomplished without the need for antibiotic selection. In addition to WT PRB, we have cloned in the pRAV-Flag vector variant forms of the receptor affected in its ability to induce the ERα/c-Src/Ras/Erk signaling pathway, or in its transcriptional function (Fig. 1A). A deletion mutant of residues 166-372 of PRB (estrogen receptor interacting domain; ERID) has been previously described as being unable to interact with ERα and, consequently, to mediate MAPK activation in response to progestin in T47D cells (7). The point mutation E911A in the activation function 2 (AF2) domain of PRB was reported to have lost ligand-dependent activation of PRE-containing reporter constructs (20). The two functional zinc fingers that are formed at the DBD have been targeted with three different mutations (Fig. 1A). The P-box at the first zinc finger, involved in direct contacts with DNA at specific PRE sequences, has been altered with the triple mutation G585E-S586G-V589A (DBD) (21). The D-box present in the second zinc finger, involved in PR dimerization, was altered with mutations A604T (D4) and R606W (M6), based on previous reports of the effect of these changes in the context of other nuclear receptors on dimerization and DNA binding (22, 23).
All these different PRB mutants, as well as WT PRB and empty pRAV-Flag vector as control were introduced through retroviral infection as detailed in Materials and Methods into TYML cells. This is a derivative clone of the breast cancer cell line T47D-YV devoid of expression of endogenous PR isoforms A and B (24, 25), containing a single integrated copy of a MMTV-luciferase reporter construct (construction and testing of this cell clone is detailed in Materials and Methods) (Fig. 1B). Low multiplicity of infection (∼5–8%) was used to obtain a limited number of integrated copies of the PRB expression vector in the host genome (Fig. 1C). Successfully infected cells expressing GFP were cell sorted for maximal purity and long-term expression. Expression of each of the PRB variants was tested by Western blot, immunofluorescence, and RT-PCR (Fig. 1, D–F). Expression levels and cellular localization of all PRB variants was comparable to endogenous PRB in parental T47D cells (see details in Materials and Methods).
The functionality of the PRB variants stably transduced into TYML breast cancer cells was analyzed to confirm their known or expected behavior in terms of transcriptional activation of the MMTV reporter, binding to PRE-containing promoters, recruitment of coactivators, interaction with ERα, activation of MAPK, and cell proliferation induction, as well as to reveal new involvements of the altered domains in these processes.
To confirm that the DBD mutant we constructed (G585E-S586G-V589A) was unable to bind to DNA at a promoter containing well-characterized PREs, we investigated the recruitment of PRB variants to the integrated MMTV promoter shortly after R5020 addition in the TYML-derived cell lines constructed by chromatin immunoprecipitation (ChIP) (Fig. 2, A and B). WT, tagged PRB was recruited as early as 15 min after hormone addition to the MMTV nucleosome B (NucB), as it has been previously reported in T47D-MTVL cells expressing endogenous PRB (26). Furthermore, PRB-DBD was drastically affected with regard to its ability to interact with this promoter upon hormone treatment, confirming that the triple mutation at the P-box abolishes binding to PREs. The two D-box mutants showed reduced recruitment but were not equivalent; whereas PRB-mD4 was unable to bind to the promoter, PRB-mM6 retained partial recruitment (Fig. 2B). This indicates that the two point mutations might affect dimerization differently, with D4 generating a more drastic change.
Fig. 2.

Functional characterization of PRB variants stably expressed in breast cancer cells. A, Chromatin immunoprecipitation analysis of PRB variants recruitment to the integrated MMTV promoter in response to progestin. Indicated cell lines growing in serum-free media were untreated (0) or treated with R5020 (10 nm) for 15 or 30 min before chromatin preparation for ChIP. PR-containing chromatin fragments were immunoprecipitated with an anti-FLAG antibody. PCR primers were used to amplify the MMTV nucleosome B region by regular PCR followed by EtBr-stained 1.2% agarose gel loading. PCR of input DNA (representing 1% of immunoprecipitated material) and amplification of ß-globin gene were used for normalization. B, Similar to panel A, but immunoprecipitated material was amplified by real-time PCR with MMTV NucB-specific primers. Serum-starved cells were treated with R5020 or vehicle for 15 min. PCR amplification of actin gene was used for normalization. Relative units of NucB PCR amplification corrected by actin amplification are shown. The values represent the mean ± sd of a representative experiment performed in triplicate. C, ChIP analysis of SRC and RNA polymerase recruitment to the MMTV promoter in WT- and AF2-expressing cell lines. ChIP was performed as in panel A, after 15-min hormone treatment, with antibodies against FLAG-PR, SRC-1 and SRC-3 coactivators, total RNA polymerase II, and RNA polymerase phosphorylated at Ser5 of C-terminal domain. Material was amplified by real-time PCR with MMTV NucB or luciferase (position 49-255)-specific primers. One or two asterisks denote significant differences (P < 0.1 or P < 0.05, respectively) between hormone-treated and untreated data sets, as analyzed by Student’s t test. D, In vivo interaction between PRB and ERα. WT and ΔERID-I PRB-expressing cells growing in serum-free media were incubated with R5020 (10 nm) for 15 min before cell extraction. Cell lysates were immunoprecipitated with anti-FLAG antibody and material eluted from the beads was analyzed by Western blotting with anti-ERα and anti-FLAG antibodies (right panel). Input material is shown. Left panel, Low abundance of ERα in T47D-YV cells, in comparison with T47D and MCF7 cell lines, analyzed by Western blotting. Antitubulin was used as control. E, Transactivation of the endogenous MMTV-Luc reporter gene by progestin in PRB variant-expressing cells. Cells were cultured in serum-free media, and 48 h later R5020 (10 nm) or vehicle (EtOH diluted 1:10,000) was added. Luciferase activity was measured in cell extracts 16 h later. Fold induction of luciferase activity of R5020-treated cells with respect to vehicle is represented. The values represent the mean ± sd of a representative experiment performed in triplicate. RLU, Relative luciferase units. F, Transcriptional activity of PRB variants on the MMTV promoter in response to increasing time of hormone treatment. Indicated cell lines cultured in serum-free media for 48 h were treated with R5020 (10 nm) for the time indicated. Cells were harvested, RNA was extracted, and gene expression was measured by RT-qPCR with luciferase-specific primers. GAPDH expression was measured for normalization. The values represent the mean ± sd of a representative experiment performed in triplicate. G, Transcriptional activity of the PRB DBD mutant on the CDKN1A (p21) promoter measured by RT-PCR. Cells were treated as in panel E, except that R5020 was left for 9 or 12 h. CDKN1A expression was measured by RT-PCR with gene-specific primers. MMTV-luciferase and GAPDH expression was measured as controls. PCR products were run on a 1.2% agarose gel and visualized with ethidium bromide. H, Activation of Msk1 by progestin in PRB variant-expressing cell lines. Cells expressing the different PRB variants were cultured in serum-free media for 48 h on coverslips and treated for 10 min with R5020, permeabilized, and incubated with an antibody against phosphorylated mitogen- and stress-activated protein kinase. After incubation with an appropriate labeled secondary antibody and DAPI staining, fluorescence images were registered with a confocal laser microscopy system. IP, Immunoprecipitation; RNAP II, RNA polymerase II; WB, Western blot.
PRB-AF2 (E911A) was recruited to the MMTV promoter to the same extent as WT in response to a short hormone treatment (Fig. 2, A and B). The AF2 domain of steroid receptors is involved in recruiting coactivators. To test the defect of our AF2 point mutant, we performed a ChIP assay to test whether this mutation affected the previously described recruitment by PR of the steroid receptor coactivator (SRC)-1 to the MMTV NucB (26). SRC-1 was recruited to this promoter after hormone addition not only in WT PRB-expressing cells but also in cells expressing PRB-AF2 (Fig. 2C). Additionally, SRC-3, recently described to mediate PR activation of MMTV in breast cancer cells (27), was also recruited similarly in WT- and AF2-expressing cells. We also tested recruitment of total and activated (phospho-Ser5) RNA polymerase II in response to progestin. The polymerase was equally recruited to the MMTV promoter in WT and AF2 cells. Nonetheless, when an amplicon inside the luciferase gene was used, it showed that the polymerase was not progressing accordingly in the AF2 cells (Fig. 2C). Consequently, the AF2 point mutation does not impair SRC-1 or SRC-3 recruitment to MMTV but may affect recruitment of some other coactivator required for setting up a fully processive RNA polymerase complex.
The PRB-ERID mutant showed reduced recruitment to the MMTV promoter, indicating that MAPK activation mediated by PR-ER interaction in response to progestin may be required to gain full access to the integrated MMTV promoter by PR (Fig. 2, A and B). We have previously described that MAPK activation by progestin via PR-ER cross talk is required for the induction of MMTV (and other hormone target genes), through the involvement of activated Erk and Msk kinases and histone H3 phosphorylation at NucB (13).
Formal demonstration that stably expressed PRB-ERID could not interact with ERα is shown in Fig. 2D. PR was immunoprecipitated with a FLAG antibody from extracts of the stably expressing cell lines, and the interacting material was analyzed on Western blot with an ERα-specific antibody. ERα coimmunoprecipitated with the tagged WT PRB, but not with the ERID-I deletion mutant. We noticed that our TYML-derived cell lines, as well as parental T47D-YV, expressed a reduced amount of ERα protein in comparison with the PRA/PRB-expressing T47D cell line (Fig. 2D, left panel). This reduced ER expression was also observed at the transcriptional level by RT-PCR (data not shown). Despite the low amount of ERα present, PR-ER interaction could be detected when ERID was intact.
Transcriptional activity of PRB variants in response to progestin
After the initial characterization of the PRB variants stably transduced into TYML breast cancer cells, we investigated their ability to mediate activation of the model MMTV promoter by progestin, both measuring luciferase activity of the integrated MMTV-Luc construct, and its transcript accumulation by RT-PCR. For the luciferase assay, cells were serum starved for 48 h, after which 10 nm R5020 was added, and luciferase activity was measured 16 h later (Fig. 2E). The MMTV promoter was hormone unresponsive in the empty vector-containing line. Transcriptional activation was maximal in the presence of WT PRB. MMTV response to hormone was reduced in cells expressing mutants ERID and AF2 and completely abolished in DBD-expressing cells. Response of MMTV in the two D-box mutants was also reduced, more importantly in the D4 mutant. This confirmed that MMTV activation by PRB requires binding of the receptor to the promoter region and, to some extent, the recruitment of coactivators presumably blocked by the E911A mutation. Deletion of ERID also interfered with MMTV induction, indicating that PR-ER cross talk and kinase activation are also involved. In fact, MMTV response to hormone was also reduced when WT-expressing cells were preincubated with ER (ICI182780) or MAPK (PD98059) inhibitors (data not shown), as happens in T47D cells (13). The reduced activation observed with the ERID mutant was not further affected by PD98059 (supplemental Fig. S1 published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend. endojournals.org). It should be noted that the ERID mutant fully activates MMTV in transient transfection experiments in COS-7, 293T and T47D-Y cell lines (7) (supplemental Fig. S2). This indicates that PR-ER-mediated activation of MAPK is not required to stimulate the nonchromatinized MMTV promoter and, importantly, that the ERID deletion impairs neither DNA binding nor transactivation potential per se.
Similar results were obtained when analyzing transcript accumulation after hormone treatment by RT-PCR. We have analyzed MMTV expression in the different PRB variant-expressing cells over time from 1–10 h after hormone addition (Fig. 2F). MMTV-driven luciferase transcript accumulated progressively up to 10 h. This kinetic experiment confirmed previous conclusions on the MMTV regulation by the different variants, i.e. hormone induction was abolished in the presence of DBD and affected to different degrees in the remaining mutants.
To test the functionality of the DBD mutant in transcription of a gene devoid of PREs in its promoter, we analyzed induction of CDKN1A encoding for the cell cycle regulator p21. It has been proposed that PR is recruited to this promoter through interaction with the Sp1 transcription factor (28, 29). Because CDKN1A is a late progestin-responsive gene, we tested its expression at 9–12 h after hormone addition in the WT- and DBD-expressing cells. DBD supported its activation similar to the WT receptor (Fig. 2G, and additional data on Fig. 4A).
Fig. 4.

Kinetic analysis of gene response to R5020 in the different PRB-containing cell lines. A, Response to hormone over time of representative genes of groups I (STAT5A), II (BIRC3), III (CDKN1A), and IV (JUN), defined according to their dependence on DBDs and ERIDs. Cells cultured in serum-free media for 48 h were left untreated (0) or treated with R5020 (10 nm) for the indicated time points. Cells were harvested, RNA was extracted, and gene expression was measured by RT-qPCR with gene-specific primers. GAPDH expression was measured for normalization. The values represent mean ± sd relative units of two experiments performed in duplicate. Results for a total of 17 genes are shown in supplemental Fig. S4. RU, Relative units. B, Response to hormone of genes showing a better response in cells expressing the DBD PRB mutant than in WT. Experiment was performed as in panel A.
Kinase cascade activation by progestin in cells stably expressing PRB variants
Progestin treatment of mammalian cells rapidly activates different signaling pathways, including MAPK (5, 6, 10). Domains of PR interacting with ERα are required for progesterone activation of the c-Src/Ras/Erk pathway in T47D cells. We expected the ERID mutant to be the only PR variant not to activate this pathway. MAPK activation by progestin in T47D cells can be experimentally detected by examining phosphorylation of Erk1/2 or a downstream target, such as the Msk1 kinase, shortly after hormone treatment (6, 13). We have explored the phosphorylation of Msk1 (pMsk) in response to progestin by confocal immunofluorescence, as a measure of the functionality of the pathway ERα/c-Src/Ras/Erk (as reported in Ref. 13). In all mutant-expressing cells, pMsk was detected within the nuclei shortly after hormone addition similar to the WT-expressing cells, except for the ERID mutant, unequivocally indicating that with this deletion the ERα/c-Src/Ras/Erk/Msk pathway could not be activated (Fig. 2H). Involvement of this pathway in Msk activation was further tested by incubating WT-expressing TYML cells with ER, MAPK, and Msk1 inhibitors before hormone treatment. All inhibitors blocked progestin-induced phosphorylation of Msk1, as well as phosphorylation of PR at Ser294 (except for H89), a target of MAPK activation (see supplemental Fig. S1).
Phosphorylation of PRB mutant variants was tested by Western blot also after short (i.e. 10 min) R5020 treatment. All PRB variants were normally phosphorylated at Ser294, to a similar extent as the endogenous PR in T47D cells, except for the ERID mutant (supplemental Fig. S1). All these data support the normal functionality of PRB introduced into TYML cells, comparable to endogenous PRB in T47D cells.
Progestin-induced cell proliferation in T47D-YV cells expressing the different PRB mutants
Our data indicate that all PRB variants tested, except ERID, had a normal capacity to activate the ERα/MAPK pathway as previously described in parental T47D cells. Arrested breast cancer cells cultured in serum-deprived media conditions respond to progestin with a single cycle of proliferation and stop in the G1 phase of the next cycle (11, 12). Signaling pathways activation has been found to be required, as kinase inhibitors block this proliferative effect (8, 30). To further characterize our cell lines, we have investigated the proliferative effect of R5020 by measuring distribution of cells in S phase by fluorescence-activated cell sorting (FACS) analysis upon propidium iodide staining. Initially, we observed that WT and AF2 variants supported hormone-induced progression to S phase (Fig. 3A). As expected, PD98059 (PD), a specific inhibitor of MAPK kinase 1 (MKK1), blocked the proliferative effect of hormone in WT PRB-expressing cells (data not shown). Moreover, hormone-induced proliferation was not observed in cells expressing the ERID mutant, indicating that interaction with ER is required for cell signaling leading to cell proliferation, and confirming once more the expected functional defect of this mutant. Additionally, R5020 was also unable to induce cell proliferation in the DBD-containing cells. In a later experiment, we included the D-box mutants in the study, and the variation in the proportion of cells in S-phase was followed along several time points after hormone addition (Fig. 3B). Cells in S-phase were increased at 14 h after hormone, but diminished to the initial numbers at 24 h, in accordance with the reported induction of a single cell cycle. This experiment confirmed our previous results, i.e. ERID and DBD did not support cell proliferation. With regard to the D-box mutants, M6 supported the proliferative effect of R5020 normally, but D4 response was similar to the DBD mutant (Fig. 3B). Thus, intriguingly, the proliferative effect of progestin depends not only on signaling pathway activation, but also on the ability of PR to directly interact with some target promoter.
Fig. 3.
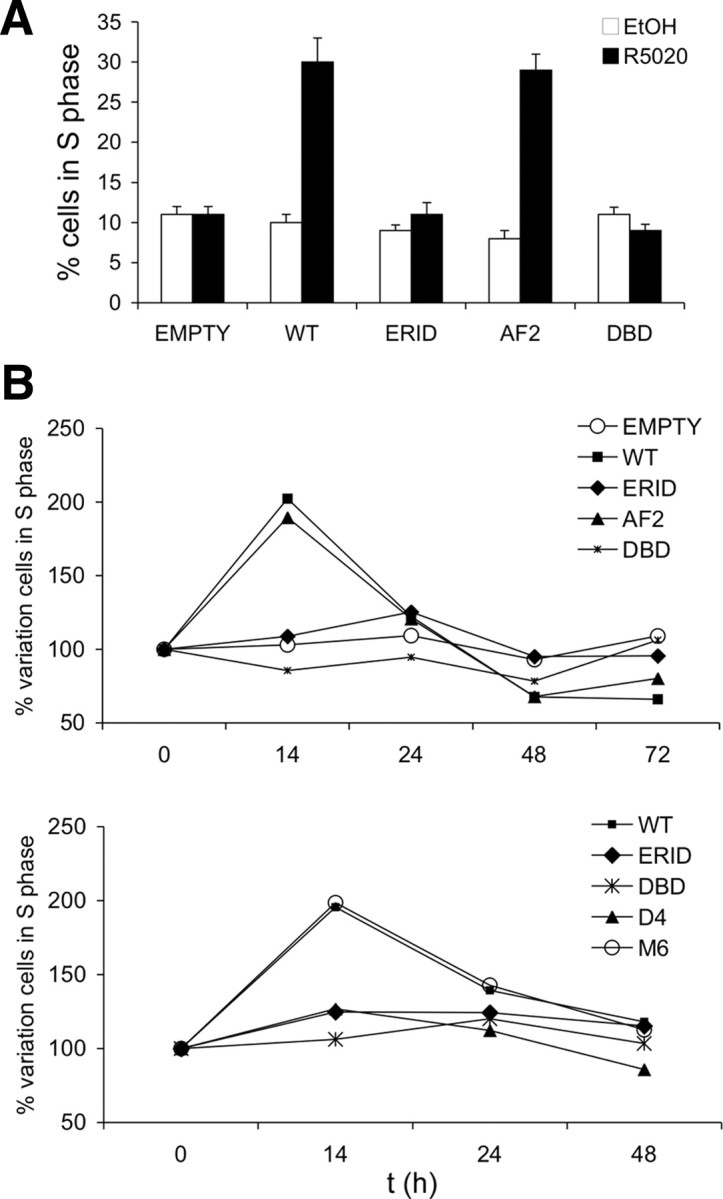
Progestin-induced cell proliferation in T47D-YV breast cancer cells expressing the different PRB mutants. A, Cells of the indicated cell lines were grown in serum-free media for 48 h, followed by a 12-h incubation with vehicle or R5020 (10 nm). Distribution of cells in the various phases of the cell cycle was evaluated by FACS analysis of cells stained with propidium iodide. The percentage of cells in S-phase is shown. The values represent mean ± sd of two experiments performed in duplicate. B, Cells of the indicated cell lines were grown in serum-free media for 48 h and, after R5020 addition, cells were analyzed at different time points (0–14–24–48–72 h). The distribution of cells in the various phases of the cell cycle was evaluated by FACS analysis of cells stained with propidium iodide. The percentage variation in the number of cells found in S-phase at each time point with respect to time zero is represented. Two different experiments performed in duplicate are shown.
All these results confirmed that tagged PRB mutants stably expressed in PR-deficient cells behaved as expected and identified new features that can help to understand PRB functions. For instance, the ERID mutant could not fully support the activation of a promoter (i.e. MMTV) in which PRB is directly recruited to DNA, and the DNA-binding capacity of PRB is required for hormone-induced cell proliferation. This can lead to the prediction that a number of genes regulated by PRB may depend on DNA binding and cell signaling, as recently shown for the MMTV promoter, but others may only depend on interaction with ER and signaling pathway activation, or exclusively on promoter binding and transcription factors recruitment. To investigate these possibilities, we performed a transcriptome analysis of the hormone response in the cell lines constructed.
Analysis of progestin-responsive gene expression in PRB mutant-expressing cells using a customized microarray
A customized human cDNA microarray containing 826 genes of interest in breast cancer or steroid hormone regulation was used to identify subsets of genes that retain response to progestin in cells expressing defective PRB variants. Previous kinetic experiments performed with T47D cells on this array platform have shown that an extensive number of genes change their expression at 6 h of R5020 treatment (Ballare, C., and M. Beato, unpublished results). This time point is a compromise between rapid and long-term effects of this hormone on gene expression. TYML cells containing the empty vector, WT PRB, AF2, DBD and ERID, and T47D parental cells were serum starved for 48 h and hormone- (10 nm R5020) or vehicle-treated for 6 h. Cells were collected, and RNA was extracted for microarray hybridization. Upon analysis of data, we obtained a group of 27 genes reproducibly activated by hormone in the WT cell line (supplemental Fig. S3).
Activation of these genes in the AF2-, DBD-, and ERID-containing cell lines was analyzed to establish whether these mutations impaired gene activation. The majority of genes showed a decreased activation in AF2 in comparison with WT, and only five genes were completely uninduced by hormone in the AF2 cells. Few genes were unaffected by the AF2 mutation.
The majority of genes were not induced in the DBD-expressing cells. Only a minority of genes were induced by hormone in the DBD cells, similar to WT, below the WT levels or, surprisingly, with a better response than in WT.
ERID deletion also completely affected the vast majority of genes. Only one gene retained a similar hormone response to WT, and few genes presented some reduced activation in comparison with WT. In summary, a majority of genes require intact DBD and ERID domains and only partially depend on the AF2 domain integrity. See Table 2 and supplemental Table S1 for a summary of genes being affected by each different mutation.
Table 2.
Summary of the effect of PRB mutations in the ERID, DBD and AF2 functional domains on progestin-induced gene expression in comparison to the response in WT PRB expressing cells
| Compromised by DBD | Noncompromised by DBD | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Kinetics | RT-PCR | Array | Kinetics | RT-PCR | Array | ||||||
| GRB14 | COL4A21 | CDC14B | GAS6b, c | AYTL2c | |||||||
| MMTVb, c | CXCL12b, c | CDKL1c | PCAFb, c | QSCN6L1 | |||||||
| SAP30b, c | CHES1a, c | HSD17B2c | TGFAc2 | ||||||||
| AF2 | STAT5Ac | DNAH1c | ING1a, c | ||||||||
| compromised | THBS11 | MAP3K3c | KPNA3c | ||||||||
| PLAUR1 | NEO1c | ||||||||||
| SOS1 | RASL10B | ||||||||||
| ERID | VEGFc | ||||||||||
| compromised | AF2 | AKAP13a, b | MUC2L | CDKN1A | |||||||
| noncompromised | CCND1 | PIK3CB | HSD11B2b | ||||||||
| MYCb | |||||||||||
| AF2 | BIRC3a | FOS2 | |||||||||
| ERID | compromised | ||||||||||
| noncompromised | AF2 noncompromised. | CXCR4 | DUSP12 JUN | ||||||||
Genes are classified in accordance with their response to progestin in the three different mutants, whether activation is reduced (<WT, no induction or becomes repressed) or not (≈WT or >WT) compared with expression in WT, according to criteria set forth in Tables 1 and S1. Also indicated is whether conclusions are extracted from microarray data solely, RT-qPCR at a single time point (6 h R5020), or time course experiments.
Indicate hormone response is retained partially in the DBDa, ERIDb, or AF2c mutants (<WT).
Indicates that the gene is overinduced in the DBD mutant (>WT).
The number of hormone-repressed genes was more limited, probably due to the eminently activating function of PRB in comparison with PRA (15, 16, 17), and have not been analyzed in detail in this report (supplemental Fig. S3).
Analysis of transcript accumulation in response to progestin by RT-PCR
Extensive validation of these results was performed using RT-PCR with gene-specific oligonucleotides. A total of 24 genes activated by hormone in the WT cells were analyzed: 18 were present in the previous array data, and six additional genes were added to the study (HSD11B2, STAT5A, FOS, MUC2L, SOS1, QSCN6L1) (Table 1). The majority of genes coincident in the microarray dataset were validated accordingly. Additionally, it was confirmed that AF2 mutation only partially affects the transcriptional activity of PR, whereas the majority of progestin-responsive genes require intact DBD and ERID domains. Here, as well as in the microarray analysis (supplemental Fig. S3), we observed significant differences in the response of some genes in the WT-expressing cell line in comparison with parental T47D. This might be due to the absence of PRA in our cell line, although other genetic differences between cell lines, e.g. ERα levels, may also exist.
Table 1.
Influence of PRB functional mutations on the progestin-induced gene expression profile
| Fold induction 6 h R5020/vehicle | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EMPTY | T47D | WT | AF2 | DBD1 | DBD2 | ERID-I | |||||||
| COL4A2 | −1.69 | 3.31 | 47.70 | 4.51 | 1.54 | 1.77 | 2.42 | ||||||
| PLAUR | 1.61 | 1.24 | 36.97 | 5.62 | 2.42 | 3.28 | 3.79 | ||||||
| STAT5A | 1.29 | 6.28 | 11.92 | 2.63 | 1.04 | 1.49 | 2.12 | ||||||
| THBS1 | 2.12 | 2.59 | 10.77 | 2.91 | 8.31 | 5.95 | 2.92 | ||||||
| DNAH1 | −1.19 | 1.04 | 8.54 | 1.41 | −1.35 | −1.41 | 1.20 | ||||||
| HSD11B2 | −1.39 | 8.50 | 8.22 | 6.15 | 2.49 | 4.87 | 2.57 | ||||||
| MMTV-LUC | 1.36 | 3.35 | 7.13 | 3.13 | −1.04 | −1.61 | 3.50 | ||||||
| CXCL12 | −1.06 | −1.52 | 6.76 | 1.83 | −1.54 | 1.54 | 2.39 | ||||||
| MAP3K3 | −1.43 | 1.53 | 5.17 | 1.44 | −1.72 | −1.28 | 1.31 | ||||||
| CHES1 | 1.19 | −1.08 | 4.21 | 2.34 | 1.63 | 1.58 | 1.24 | ||||||
| PCAF | −1.41 | 2.26 | 4.05 | 2.22 | 1.38 | −1.05 | 1.26 | ||||||
| CCND1 | −1.23 | 5.35 | 3.97 | 2.42 | −2.22 | −1.82 | 1.32 | ||||||
| TGFA | −1.03 | 5.63 | 3.96 | 1.90 | 3.82 | 6.73 | 1.38 | ||||||
| BIRC3 | 1.12 | −1.14 | 3.75 | −1.19 | 1.42 | −1.61 | 1.79 | ||||||
| AKAP13 | −1.39 | 1.52 | 3.37 | 1.09 | −1.64 | −1.18 | 1.43 | ||||||
| AYTL2 | −1.69 | 1.77 | 3.05 | 2.03 | 2.38 | 2.69 | 1.21 | ||||||
| SAP30 | −1.49 | 4.07 | 2.69 | 1.58 | −1.64 | −2.00 | 1.18 | ||||||
| GAS6 | −1.06 | 1.82 | 2.67 | 1.44 | 2.46 | 3.31 | 2.05 | ||||||
| GRB14 | −1.20 | −2.27 | 2.57 | 1.25 | −1.96 | −2.13 | 2.13 | ||||||
| SOS1 | −1.02 | 3.56 | 2.29 | 1.05 | −1.75 | −1.27 | 1.32 | ||||||
| PIK3CB | 1.08 | 1.81 | 2.17 | 1.78 | −1.16 | −1.06 | 1.01 | ||||||
| FOS | −1.15 | 2.34 | 1.99 | −1.41 | 5.35 | 6.74 | 1.80 | ||||||
| MUC2L | −1.23 | 3.12 | 1.78 | 1.60 | −1.54 | −1.19 | 1.15 | ||||||
| QSCN6L1 | 1.10 | 1.59 | 1.71 | 1.29 | 1.39 | 1.68 | 1.34 | ||||||
| CTSL | −1.15 | 1.03 | 1.21 | 1.00 | −1.92 | 1.10 | 1.40 | ||||||
| NOTCH3 | 1.04 | −1.85 | −1.22 | −1.45 | −2.13 | −1.89 | −1.10 | ||||||
| ZNF350 | −1.33 | 1.23 | −1.52 | −1.64 | −1.85 | −1.69 | 1.23 | ||||||
| NCOA3 | 1.12 | −1.37 | −1.54 | −1.56 | −1.69 | −1.79 | 1.05 | ||||||
| PGRB | −1.35 | −1.85 | −1.54 | −1.56 | −2.78 | −3.45 | −1.61 | ||||||
| CCNG2 | 1.13 | −2.76 | −3.55 | −1.88 | −2.86 | −3.58 | 1.12 | ||||||
T47D and PRB-expressing TYML-derived cells were serum-starved for 48 h and treated with ethanol or R5020 (10 nm) for 6 h. Cells were harvested, RNA extracted, and gene expression measured by RTqPCR with specific primers for selected genes. GAPDH expression was measured for normalization. Average fold induction in response to hormone compared to ethanol is shown. Fold changes of at least 1.4 in the WT cell line are in bold. In the PRB mutants columns, italic numbersmean that fold change is less than 70% of fold in WT (<WT), in boldwhen ≈WT (>70% of WT) or >WT, considering only fold-changes of at least 1.4. The values represent the average of two experiments performed in duplicate. Two PRB-DBDm containing cell lines were used:
DBD contained a PRB concentration similar to the PRB-WT cell line, whereas
DBD contained 50% more.
Because analysis of gene expression at a single time point (6 h) after hormone addition is not fully informative and might mask a more complex effect of a certain PRB mutant, such as a delay of the transcript accumulation, we next focused on a number of representative genes (17 genes) to further study their hormone response at different time points after hormone addition (Fig. 4 and supplemental Fig. S4). Here, we added three early-responsive genes not previously analyzed (MYC, JUN, and DUSP1) and the late-responsive CDKN1A, to a list of 13 of the genes previously studied. Table 2 (and supplemental Table S1) summarize the effect of the different functional mutants on the hormone response of analyzed genes, whether they have been analyzed only on the microarray experiment, by single time-point RT-PCR, or in time course experiments.
Taken all these data together, genes have been split into four groups (I–IV) according to their dependence on DBD and ERID domains. Figure 4A shows a representative example from each group. As previously indicated, the majority of genes have their hormone response compromised in cells expressing the DBD or ERID mutants (we shall refer to these as “group I” genes). Most of them are also partially affected in the presence of AF2. This includes genes such as SAP30, STAT5A, THBS1, and the previously mentioned MMTV promoter. Only BIRC3 and CXCR4 genes are affected by DBD, but not by the ERID deletion (group II). The number of genes affected by ERID deletion and unaffected by the DBD mutation is larger (group III), including PCAF, TGFA, HSD11B2, CDKN1A (p21), and MYC. Finally, a group of three genes (FOS, DUSP1, and JUN) was not compromised by DBD or ERID (group IV).
At the stage of time course experiments, we added the constructed D-box mutants to the analysis of the influence of the different functional domains. STAT5A hormone responsiveness was lost in ERID and DBD mutants (group I), as well as in the M6 D-box mutant, and diminished in AF2. On the other hand, hormone activation was normal in the D4 mutant (Fig. 4A). BIRC3 (group II) response was similar to WT in ERID and M6, but was affected in DBD, AF2, and D4. Activation of CDKN1A (group III) was normal in DBD and AF2, reduced in the two D-box mutants, and completely blocked in ERID. JUN (goup IV) activation occurred in all mutants and was more sustained in the D-box mutants than in WT (Fig. 4A). Early responsive genes FOS, JUN, and DUSP1 from group IV were not affected by any functional mutation of PRB. Dependence on PR was ascertained by confirming its lack of induction in TYML/empty cells after hormone addition (supplemental Fig. S5). A table in Fig. 5A summarizes the effect of each mutation on the activation by hormone of 17 analyzed genes (in groups I–IV), compared with expression in the WT cell line.
Fig. 5.

Summary of the effect of all PRB mutants constructed on expression of selected genes investigated in time course experiments. A, Four groups (I–IV) of progestin-activated genes are defined according to their dependence on the ERID-I and DBDs. Response to progestin in comparison with WT is shown for each gene in the different PRB mutant-expressing cell lines. Response to progestin may be enhanced compared with WT (>WT), similar to WT (≈WT), lower but still present (<WT), induction may be lost (NO IND.), or response to progestin become repressive (REPRESS.). Shaded results group those that involve no reduction of progestin activation of gene expression (≈WT or >WT). B, Schematic gene expression profiles over time in response to progestin in PRB-WT-expressing cells. Four groups (A–D) of progestin-activated genes are defined according to their profile in time course experiments: A, activation increases up to 6 h hormone treatment, decreasing afterward; B, constant increase along time up to 10 h; C, two phases of hormone response are observed; D, early response to hormone.
It is noteworthy that some genes show higher response to hormone in cells expressing the DBD mutant of PRB than in WT-expressing cells, i.e. TGFA, FOS and DUSP1 (Fig. 4B). This is especially visible in the activation of the FOS gene. Despite being an immediate-early gene, FOS transcript levels remained elevated several hours after hormone addition. Also in the D-box mutants D4 and M6, a few genes are slightly better induced by hormone than in WT cells, but not in ERID- or AF2-expressing cells (Fig. 5A and supplemental S4). When comparing the three PRB mutants in the DBD (P-box DBD, D-box D4, and M6), we observed that 10 of 12 genes affected by some of these mutations are also affected by a second one. Only two genes (i.e. MMTV and SAP30) are affected by all three mutations in the DBD.
Finally, according to the kinetics of transcript accumulation after hormone treatment, genes can be classified in four categories (Fig. 5B): A, genes with a late induction, with a maximum at 6 h, that was decreased at 10 h; B, genes with a sustained accumulation of mRNA up to 10 h; C, genes showing two phases of maximal accumulation, at 1 and 6 h (i.e. BIRC3); and D, early responsive genes, with maximum accumulation at 1 h.
Progestin induction of Cyclin D1 gene expression requires intact DBD and ERID domains
As mentioned above, progestin induces one cycle of cell proliferation in serum-starved T47D cells. Cyclin D1 has been previously described as a key player in the transition of G1-arrested cells into the DNA synthesis phase (S).
CCND1 gene expression is induced by progestin at the transcriptional level. In the WT PRB-expressing TYML cell line generated here, CCND1 mRNA was induced 4- to 5-fold upon 6 h of hormone treatment, comparable to induction achieved in parental T47D cells (Fig. 6A). In a time-course experiment, CCND1 transcript accumulation was maximal at 6 h and decreased at 10 h after hormone addition (Fig. 6B). CCND1 induction was normal in cells expressing AF2 and M6 mutants but drastically reduced in the ERID and D4 cells. In DBD-expressing cells, hormone produced a slight decrease of CCND1 expression compared with serum-starved, untreated cells (Fig. 6, A and B). Cyclin D1 protein accumulation in response to hormone was also observed by Western blotting in WT PRB-expressing cells, but not in the ERID cells (Fig. 6C). The behavior of CCND1 response to hormone in the presence of the different PRB mutants tested mirrored the ability of these cell lines to enter into proliferation in response to progestin, in accordance with the data presented in Fig. 3 (summarized in Table 3). This suggests that cyclin D1 induction may be key in supporting the transition of arrested cells into S phase in response to progestin.
Fig. 6.

PRB-mediated induction of CCND1 expression requires an intact DBD and progestin-induced cell signaling. A, CCND1 gene induction in response to progestin in TYML cells expressing the different PRB mutants. Cells cultured in serum-free media for 48 h were left untreated (0) or treated with R5020 (10 nm) for 6 h. Cells were harvested, RNA was extracted, and gene expression was measured by RT-qPCR with specific CCND1 primers. GAPDH expression was measured for normalization. Fold induction of CCND1/GAPDH expression of R5020-treated compared with vehicle is represented. The values represent mean ± sd of two experiments performed in duplicate. B, Time course analysis of CCND1 response to R5020 in the presence of the different PRB mutants. Cells cultured in serum-free media for 48 h were left untreated (0) or treated with R5020 (10 nm) for the indicated time points. Cells were harvested, RNA was extracted, and gene expression was measured by RT-qPCR with CCND1-specific primers. GAPDH expression was measured for normalization. The values represent mean ± sd relative units of two experiments performed in duplicate. C, Cyclin D1 accumulation after progestin addition. TYML cells expressing WT PRB or the ERID mutant were cultured in serum-free media and, 48 h later, cells were untreated (0) or treated with R5020 (10 nm) for the time indicated. Cells were harvested, and cell lysates were analyzed by Western blot with antibodies against cyclin D1. Tubulin antibody was used as a loading control. D, ChIP analysis of PRB recruitment to the CCND1 promoter in response to R5020. PRB-WT and DBD cell lines growing in serum-free media were treated with R5020 (10 nm) or vehicle (EtOH) for 15 min before chromatin preparation for ChIP. PR-containing chromatin fragments were immunoprecipitated with an anti-FLAG antibody. Seven CCND1 regions (promoter or coding) were analyzed by qPCR. Amplification of actin gene was used for normalization. Enrichment of recruitment in response to hormone compared with nontreated cell cultures is shown. Asterisks denote significant (P < 0.05) differences (fold-enrichment >2) between hormone-treated and untreated data sets, as analyzed by Student’s t test. E, Later, all PRB variant cell lines were used to investigate recruitment at −2000 and −50 (with respect to the transcription start site) CCND1 promoter regions in response to R5020. Cell culture and ChIP were performed as in panel D. Relative units of CCND1 amplification corrected by actin from cells treated with R5020 or vehicle (EtOH) are shown. One or two asterisks denote significant differences (P < 0.1 or P < 0.05, respectively) between hormone-treated and untreated data sets, as analyzed by Student’s t test. RU, Relative units.
Table 3.
Correlation between the proliferative response to progestin, CCND1 expression, and PRB recruitment to the CCND1 promoter in the different PRB variant-expressing cell lines
| WT | ERID | AF2 | DBD | D4 | M6 | |
|---|---|---|---|---|---|---|
| Proliferation | Yes | No | Yes | No | No | Yes |
| CCND1 expression | Yes | No | Yes | No | No | Yes |
| Recruitment to promoter | Yes | Yes | Yes | No | No | Yes |
PRB interacts with two distant regions on the CCND1 promoter and requires an intact DBD
Our results suggest that CCND1 induction by progestin-activated PRB requires the ability of the receptor to activate cytoplasmic signaling pathways (MAPK/Msk) through its interaction with ERα, as well as residues involved in contacting with hormone-responsive elements (HREs) in DNA. CCND1 gene activation in response to progestin is also impaired in T47D cells treated with ER, MAPK, and Msk inhibitors (Ballare, C., unpublished results). Interaction of PRB with the CCND1 promoter has not been reported before [except for some limited data published by Cicatiello et al. (31)], nor the presence of functional HREs. Therefore, we investigated recruitment of PRB to the CCND1 promoter by ChIP with the FLAG antibody in our cell lines. For this, several quantitative PCR (qPCR) amplicons reported elsewhere (32) covering relevant regions for CCND1 expression or response to estrogens were used. We observed a significant increase (>2-fold) in WT PRB recruitment in response to hormone (15 min R5020 10 nm) to two regions of the promoter, named −50 (amplicon −76/−23) and −2000 (amplicon −2033/−1967) relative to the transcription start site. This recruitment was not observed in cells expressing the DBD mutant of PR, indicating that recruitment may be due to direct contact of the receptor with DNA (Fig. 6D). Recruitment to these two regions was further investigated in cells expressing each of the five PRB mutants generated. Recruitment in response to hormone treatment was observed for the WT, ERID, AF2, and M6 mutants, but not for the DBD and D4 mutants (Fig. 6E). These results correlate with the ability of the different mutants to mediate hormone induction of CCND1 gene expression, except for the ERID mutant that, despite being recruited, is unable to mediate gene expression, probably due to the need for parallel signaling activation (Table 3).
In conclusion, our data suggest that CCND1 expression and cell proliferation in response to hormone depend on cytoplasmic signaling activation by the receptor, as well as its ability to act as a transcription factor through binding to promoter DNA, and proves the utility of cell lines expressing functional mutants of PR to define different combinatorial mechanisms for gene expression control.
Discussion
Steroid hormone receptors such as PR have dual functions acting in the nucleus as direct ligand-dependent transcription factors and outside the nucleus to interact with and modulate the activity of signal transduction pathways in response to hormone. Ultimately, ligand-dependent activation of a receptor causes its biological effects by influencing gene expression in the cell. To further explore the importance of each mechanism of PRB’s action in gene expression and how extensive the cross talk between the two is, we have chosen the strategy of introducing mutations in the PRB gene that effectively inactivate one or the other function of the receptor. Three of the PRB mutants used here (ERID, AF2, and DBD) have previously been reported (7, 20, 21), although neither in the context of stably expressing cell lines, nor in a study of the expression of an extensive number of progestin-responsive genes. The two D-box mutants (A604T and R606W) are inspired in equivalent mutations in other nuclear receptors (22, 23) but are new in the context of PR.
Properties of functional PRB mutants
The ERID deletion mutant of PRB is unable to interact with ERα and, consequently, cannot mediate the activation of the c-Src/MAPK signaling pathway in response to progestin in breast cancer cells expressing both receptors (7). Downstream of MAPK activation, the Msk1 kinase becomes activated, bridging the extranuclear effects of PR and the nuclear effects that alter gene expression, e.g. of the MMTV promoter (13). We confirm here that the ERID mutant does not support Msk1 phosphorylation upon progestin treatment. Moreover, ERID deletion greatly impairs MMTV activation in the context of chromatin, and this mutant receptor is recruited to a lesser extent to this promoter. In agreement with this, whereas MMTV activation by hormone in the presence of WT PRB is significantly reduced in the presence of the MAPK inhibitor PD98059 or ER antagonist ICI182780, the partial activation observed in cells expressing the ERID mutant was almost unaffected by PD (supplemental Fig. S1). This suggests that PD targets a step already abolished with this mutant, i.e. MAPK activation mediated by PR-ER interaction. We discard the possibility that, in the absence of ERID domain, direct interaction between PRB and c-Src leads to MAPK activation (5), because no activation of the downstream Msk1 kinase has been observed.
MAPK activation by progestin leads to phosphorylation of PR at particular residues, including Ser294 and Ser345. Ser294 phosphorylation is required for PRB to shuttle in and out of the nuclei, its transcriptional activity, and subsequent degradation of the receptor (33, 34). PRB phosphorylated at Ser345 appears to interact strongly with Sp1 and can mediate activation of Sp1 target genes such as p21 (28). The ERID deletion affects serine residues 294 and 345. Nonetheless, the observed deficiencies of this mutant are due to its inability to mediate MAPK activation, not to the lack of these important residues, which in any case would be phosphorylated. Single-point mutants at Ser294 and 345 do not by themselves impair MAPK activation, and the S294A mutant supports progestin-induced cell proliferation (28, 35).
Finally, we cannot discard the possibility that the effects observed with the ERID mutant are due not only to the absence of MAPK activation, but also to conformational changes imposed by the deletion affecting other receptor functions. What we can be sure of is that this mutant is not completely inactive, because it supports partial or total activation of some progestin-responsive genes. This includes activation of transiently transfected, unintegrated MMTV and HSD11B2 promoters (7, 14).
The single-point mutation E911A in the AF2 domain was predicted as affecting the transactivation function of PRB without affecting ligand and DNA binding (20). In the cell line created here, we observed normal recruitment of this mutant to the MMTV promoter upon hormone addition, but transcriptional activation was greatly impaired. The mutated glutamic residue, conserved in virtually all the nuclear receptors, may participate in the interaction with transcriptional coactivators. An equivalent mutant in glucocorticoid receptor (E773A) disrupts in vivo binding of glucocorticoid receptor to SRC-1 and affects transactivation of some target genes, although the mutant retained activity in a palindromic glucocorticoid response element (36). In the context of PR, SRC-1 appears to interact with both AF1- and AF2-containing domains (37, 38).
We tested whether SRC-1 and SRC-3 recruitment to the MMTV promoter was impaired in cells expressing the AF2 mutant and, to our surprise, we observed close to normal recruitment. Moreover, the RNA polymerase was also recruited to the MMTV promoter, but not to the luciferase-coding region. We conclude that E911A affects recruitment of additional factors involved in full activation of a processive polymerase complex, other than SRC-1 or SRC-3. We predict that AF2 may have dissimilar participation depending on promoter context, i.e. number of PREs, coactivators being recruited, chromatin organization, etc. Accordingly, we have detected reduced recruitment of SRC-3 to the SOS1 promoter in AF2 cells (supplemental Fig. S6).
The triple mutant affecting the P-box of DBD (G585E-S586G-V589A) is unable to bind to PREs in DNA, so it may be impaired in activating all those genes that depend on the nuclear action of the receptor through direct contact with the target promoter (21). We have shown that the DBD mutant is unable to bind to the HRE-containing MMTV promoter and activate its transcription. Nonetheless, DBD was able to normally activate expression of the p21-encoding gene (CDKN1A) in response to progestin. The p21 promoter has been described as lacking canonical PRE sequences. Progestin activation appears to be mediated by interaction of activated PR with Sp1 transcription factor and recruitment to Sp1-binding sites at the promoter (29).
Because interfering with dimerization has been suggested as precluding receptor binding to palindromic HREs, we expected that our D-box mutants would mimic the P-box DBD mutant in terms of recruitment to the MMTV promoter. The two D-box mutants (A604T and R606W) are inspired in equivalent mutations in other nuclear receptors but are new in the context of PR. The A604T mutation is equivalent to A596T of the androgen receptor (AR) (23). This mutation is found in patients with partial androgen-insensitive disorders (Reifenstein syndrome). Promoters with single isolated HREs are not transactivated by the mutant receptor. Promoters with closely positioned multiple regulatory elements for AR and other transcription factors are normally regulated, including a transiently transfected MMTV reporter (23). Nonetheless, in the context of stably expressed PRB and integrated MMTV, the A604T (D4) mutation greatly impaired recruitment to this promoter and its activation in response to progestin. Consequently, such behavior, observed in the context of AR and with transfected reporters, may not apply to endogenous gene expression regulated by PRB.
The R606W mutation is equivalent to R91W in the orphan receptor HNF4 (22). This D-box arginine residue, previously implicated in nuclear receptor dimerization, is methylated by PRMT1 after binding of this methyltransferase to the DBD, thereby enhancing the affinity of HNF4 for its binding site in promoters. Consequently, R91W HNF4, resistant to methylation, displayed reduced DNA binding and transcriptional activity (22). In the context of PRB, the equivalent mutation R606W (M6) presented reduced MMTV activation, but still we observed considerable recruitment to DNA measured by ChIP. Considering only the results with the MMTV promoter, D4 and M6 behave slightly differently, with D4 being a better mimicker of the P-box DBD mutant. When other genes were analyzed, stronger discrepancies appeared between the three mutants, suggesting that receptor methylation, dimerization, DNA binding, and transcriptional activity might not always be absolutely linked and may depend greatly on promoter context.
Cross talk between the nuclear and extranuclear actions of PRB in the control of progestin-induced gene expression
Gene expression in response to progestin treatment has been investigated by customized microarray hybridization and gene-specific RT-PCR in the different cell lines created. The ERID and P-box DBD mutants have been used to distinguish between the involvement of the receptor’s extranuclear and nuclear modes of action. Consequently, we have classified genes studied in four categories depending on the effect of deleting each of these two domains. Group I includes genes that depend on the ERID and DBDs for full activation by progestin. In some cases, activation is completely lost (STAT5A or CCND1); in other cases, there is some remaining induction (AKAP13 or THBS1). These are genes that depend on both PR action mechanisms, i.e. that require activation of a signaling pathway initiated by the interaction between PR and ER and direct binding of PR to DNA, as occurs for the MMTV model promoter. The majority of progestin-induced genes fall into this category, indicating that cross talk between PR’s two modes of action is not an exception but the predominant mechanism for progestin to control gene expression. The role of kinase cascade activation by extranuclear liganded PR may be to phosphorylate the receptor itself, an associated coactivator, or an accompanying transcription factor, resulting in positive regulation of receptor action via feedback regulation (see Fig. 7 for a schematic model of PR actions). Moreover, the majority of genes in this group are partially affected by the point mutation in AF2 (Table 2), supporting the involvement of this domain in the transcriptional activity of the receptor. Few genes are completely unaffected by this mutation (CCND1 or AKAP13), or totally impaired (GRB14). Furthermore, our data agree with the model that different AF domains of the receptor synergize to render full transcriptional activity and that the importance of this domain depends on the promoter context. Finally, when analyzing the effect of the two D-box mutants, we observed that the seven genes tested present in group I are affected by at least one of the two mutations, but only two genes are impaired in both mutants (Fig. 5A and supplemental Fig. S7). This suggests that dimerization might be involved in DNA binding as reported elsewhere, but also denotes that the two D-box mutants are functionally different to some extent. The importance of receptor dimerization in DNA binding might depend on the organization of HREs and on whether they are half-palindromic or palindromic sites.
Fig. 7.

Models for PR actions on gene expression control. In accordance with their dependence on the ERID and DBDs, four categories of progestin-induced genes have been defined. In group I genes, ligand-bound PR enters the nuclei and binds directly to PRE regions in the promoter of target genes. In the cytoplasm, PR-ER interaction initiates a signaling cascade. Activated kinases may phosphorylate the receptor in a positive feedback regulation that favors its nuclear localization or transcriptional activity. Alternatively, phosphorylation may activate receptor coactivators (CA), including chromatin-modifying enzymes (e.g. Msk1 kinase), or additional transcription factors (TF) with which PR synergizes. In group II genes, PR also acts in the genome binding to direct gene targets through its DBD, either purely or with the participation of alternative signaling pathways independent on PR-ER interaction and the ERID. Group III genes are regulated through the extranuclear action of PR upon binding to ER, activating signaling pathways that may promote activation of transcription factors, providing for regulation of gene targets lacking PREs in the promoter. PR may still have a transcriptional role by being recruited to promoters indirectly, as has been shown for several promoters. Group IV contains early-responsive genes regulated by an unknown mechanism, involving neither direct binding of PR to target promoters nor ERID-mediated signaling activation. Alternative signaling pathways activating transcription factors or the nucleosome response may be involved. Significant genes that can be used as examples for each category are shown. Our investigation into cell lines containing different functional PRB mutations suggests that cell proliferation induction is controlled by some gene falling into group I. P, phosphorylation; TFBS, transcription factor binding site.
Group II includes genes depending on the ability of PR to directly bind to DNA and that do not require signaling activation mediated by PR-ER interaction. Only two genes have been found to fall into this category, and only BIRC3 has been studied in some detail. Genes in this category would use the genomic mode of action of the receptor. Alternatively, activation of some other signaling pathway occurs in the ERID mutant-containing cell line by an alternative mechanism not depending on this domain.
Genes in group III are activated by the extranuclear action of PR on MAPK activation with no role played by the receptor directly binding to the regulated promoter. These promoters may rely on the activity of MAPK-targeted transcription factors such as the Ets family members, Elk-1, c-myc, fos, and jun (components of AP-1). These factors may act entirely independently of steroid receptor transcriptional activities or may associate and bring the receptor to the promoter (Fig. 7). In this group, the proportion of genes normally induced in the presence of the AF2 mutant is larger than in group I, denoting that the transcriptional activity of the receptor is rarely required. Despite the fact that they do not depend on DNA binding, dimerization mutants partially affect several of the genes in this category. Dimerization may also be important for PR functions not associated with binding to palindromic HREs. Worthy of note is the fact that two genes (TGFA and MYC) are independent of D-box mutations; this was not the case in groups I and II.
This group includes previously studied genes such as CDKN1A and HSD11B2. As mentioned before, the CDKN1A gene is regulated in response to progestin by the association between PR and the Sp1 transcription factor, depending on MAPK activation, and recruitment to Sp1-binding sites present in this promoter (28, 29), fitting with our data and the proposed model. We have recently described how the HSD11B2 gene is regulated by PR-mediated activation of JAK/STAT pathway, with STAT5A being the transcription factor that brings PR to a distal enhancer in the promoter, independently of the receptor’s ability to bind to DNA (14). Our results now suggest that the ERID deletion affects the ability of PR to activate the JAK/STAT pathway, or that MAPK activation is also involved.
Group IV is probably the most intriguing. It contains those genes the progestin response of which is not affected by any PR functional mutation, i.e. FOS, DUSP1, and JUN. Interestingly, genes in this category are early-responsive genes, showing maximal transcript accumulation 1 h after hormone addition. These immediate-early genes are characterized by their rapid and transient expression in response to extracellular stimuli and are expressed when a cell is stimulated to leave the G0 phase of the cell cycle and enter G1. We can only speculate that additional signaling activation occurs, independent of PR-ER interaction and not affected by ERID deletion, leading to activation of some transcription factor impacting on the promoter of these early-responsive genes or activating the so-called nucleosome response (39) (Fig. 7).
Progestin-induced cell proliferation depends on both the nuclear and extranuclear functions of PRB
Arrested breast cancer cells undergo one cycle of cell proliferation in response to progestin treatment, mediated by the extranuclear action of PR on the activation of cytoplasmic kinases, including MAPK and PI3K/Akt pathways. Addition of inhibitors of these pathways blocked S-phase entry (8, 9, 28, 30). The involvement of direct regulation of transcription by PR in cell proliferation was rejected because the transcriptionally impaired PRB harboring a point mutation at Ser294 is capable of promoting proliferation (35). Nonetheless, progestin-induced S-phase entry was attenuated in T47D cells stably expressing the DBD mutant C587A (21, 28). This was correlated with the inability of this mutant to tether Sp1 transcription factors and activate Sp1 target genes that may be involved in cell proliferation (p21, EGFR).
Here, we describe how, in TYML cells, progestin induction of S-phase entry depends not only on the integrity of PRB ERID domain and, consequently, on the activation of ERα/c-Src/MAPK pathway, but also on the ability of the receptor to bind DNA. Cells expressing the DBD and D4 mutants remain arrested upon hormone treatment, indicating that a key gene required for S-phase entry is regulated by the nuclear action of the receptor. Additionally, mutants AF2 and M6 supported normal progression through the cell cycle, indicating that this activation function is not required for regulation of that key gene.
Expression of functional mutants of PRB reveals CCND1 as a major player in the progestin induction of cell proliferation
It has been previously suggested that progestin-induced S-phase progression correlates with up-regulation of cyclin D1 expression via nongenomic mechanisms. Measurement of CCND1 expression in the different mutant-expressing cell lines showed that there is an absolute correlation between the ability of each PRB mutant to support CCND1 induction and its ability to promote S-phase entry (Table 3). This is highly suggestive of the key role of this cyclin in controlling cell cycle progression. CCND1-silencing experiments in breast cancer cells have further confirmed the correlation between CCND1 expression levels and cellular proliferation, emphasizing CCND1 as a potential therapeutic target for breast cancer (40, 41).
CCND1 has been previously reported as not requiring nuclear action of PR but as being induced by progestin activation of c-Src signaling pathways resulting, presumably, in activation of another transcription factor that induces CCND1 gene expression (9, 35, 42). The CCND1 enhancer has many potential transcription factor-binding sites that could fulfill this role. An analogous situation has been described for estrogen induction of CCND1, because it also lacks ERE sequences, and induction requires activation of Src/MAPK and PI3K/Akt signaling. However, estrogen-dependent ER recruitment by enhancer 1 and 2 of CCND1 has been reported (32).
In this work, we suggest that the DNA-binding ability of PRB is required for CCND1 induction, in addition to its action on cytoplasmic signaling pathway activation. Moreover, by ChIP we have found sensitive recruitment of PRB to at least two regions of the CCND1 promoter region shortly after hormone treatment, and this was dependent on the integrity of the P-box of the DBD. Further correlation between the ability of the different mutants to be recruited to the CCND1 promoter and to support its expression has been found (Table 3), except for the ERID mutant which, despite being recruited, cannot activate CCND1 expression. This indicates that MAPK activation and PR phosphorylation are not required for tethering the receptor to this promoter but may be involved in phosphorylation of additionally required factors or chromatin modifiers, as reported for MMTV.
We have found PR being recruited to two different regions of the CCND1 promoter. One of them (∼−2000) is coincident with the region enhancer 1 previously described by Eeckhoute et al. (32). as recruiting ERα, which contains binding sites for transcription factors Sp1, Oct1, nuclear factor-κB, and CCAAT enhancer binding protein-ß. The other region (∼−50) was also tested in that report, showing little ER recruitment and contains cAMP response element- and Sp1-binding sites. On the other hand, a region reported by Cicatiello et al. (31) as binding ER through an interaction with AP1 (−954) and, possibly, PR, did not show PR recruitment in our hands (data not shown).
Our results suggest a direct, nuclear involvement of PRB in progestin-induced CCND1 expression and cell proliferation, in addition to the participation of its nongenomic, extranuclear function extensively reported previously, extending the list of target genes in which the two modes of action of a steroid hormone receptor cross talk to one another.
In conclusion, expression of functionally mutated forms of a steroid hormone receptor in cells devoid of its endogenous counterpart is a useful tool to define subsets of hormone-regulated genes depending on each of the different modes of action of the receptor. Furthermore, expression of these mutated recombinant forms of the receptor in a stable manner allows for the dynamic study of endogenous genes and their promoters in the context of chromatin.
Materials and Methods
Reagents
R5020 was purchased from PerkinElmer Life Sciences (Wellesley, MA); PD98059 (PD) inhibitor was from Calbiochem (La Jolla, CA), ICI182780 was from Tocris Cookson (Ellisville, MO) and H89 from Alexis Biochemicals (Carlsbad, CA). The anti-FLAG-tag (M2) and antitubulin antibodies were from Sigma (St. Louis, MO); anti-PR (H190), ERα (HC20), and SRC-3 (NCoA-3 M-397) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); anti-phospho PR S294 (Ab12) and PR (Ab11) were from NeoMarkers; anti-SRC-1 (128E7) and phospho-Msk1 were from Cell Signaling Technology, Inc. (Danvers, MA); cyclin D1 was from Abcam, Inc. (Cambridge, MA); anti-RNA polymerase II and phospho RNA polymerase II Ser5 were from Covance Laboratories, Inc. (Madison, WI).
Cell lines, culturing conditions, and hormone treatment
T47D breast cancer cells and T47D-MTVL cells [carrying one stably integrated copy of luciferase reporter gene driven by the MMTV promoter (43)] were routinely grown in DMEM or RPMI 1640, respectively, supplemented with 10% fetal bovine serum (FBS), 2 mm l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
T47D-YV cells [PR-negative clonal derivative cell line of T47D (24, 25)], were used to generate TYML cells (T47D-YV-derived cell lines with one integrated copy of plasmid MMTV-Luc, see below). All T47D-YV-derived cell lines were routinely grown in MEM supplemented with 7% FBS, 2 mm l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
For the experiments, cells were plated in medium without phenol red supplemented with 10% dextran-coated charcoal-treated FBS and, 24 h later, medium was replaced by fresh medium without serum. After 48 h in serum-free conditions, cells were treated with R5020 (10 nm) or ethanol for different times at 37 C. When indicated, PD (50 μm) or H89 (13.5 μm) was added 1 h before hormone treatment. ICI (10 μm) was added at the same time with hormone.
Construction of TYML cells
To investigate the functionality of different PRB mutants on hormone-induced gene expression in breast cancer cells, we used an existing clone of the cell line T47D devoid of endogenous PR isoforms A and B expression, named T47D-YV (24, 25). Before transducing PRB mutants into this line, a MMTV-luciferase reporter plasmid [pAGE5MMTVLu (43)] was introduced by stable transfection for monitoring transcriptional response to progestin. Stable transfection of pAGE5MMTVLu was performed by electroporation of linear plasmid DNA as reported (43). After antibiotic selection and clone isolation, three clones showing single MMTV-Luc integrations as determined by semiquantitative real-time PCR (data not shown) were selected. MMTV-Luc response to progestin was tested upon transient transfection with empty or WT PRB-expressing pRAV-Flag plasmids, obtaining inductions of between 20- and 35-fold upon hormone addition, only when PRB was added (Fig. 1B). No response was obtained in the absence of PRB, confirming the lack of endogenous PR expression. Clone no. 25, hereafter named TYML (standing for T47D-YV MMTV-Luc), was chosen for further transduction with pRAV-Flag constructs containing the different PRB variants or the empty plasmid as a control.
Cloning tandem affinity purification (TAP)-tagged PRB mutants into a retroviral vector
To stably express in breast cancer cells PRB mutants defective in several functions, we have used a retroviral vector that, at low multiplicity of infection, allows for the controlled integration of a limited number of copies of the gene of interest in the host genome. In addition, stable expression is accomplished without the need for antibiotic selection. We have used the vector pRAV-Flag, kindly provided by Xuedong Liu (University of Colorado), which has two interesting peculiarities (44). It creates an N-terminal fusion of the cloned gene-expressed protein with a peptide tag, consisting of an IgG-binding domain, tobacco etch virus cleavage sites, and a FLAG tag, and it also allows the coexpression of GFP for sorting of positively infected cells (see schematic representation in Fig. 1A). Tagging of PRB forms allows easy immunodetection of expressed protein using a FLAG antibody and, in the future, will be useful for the TAP method of PRB-containing complexes.
WT PRB was obtained from plasmid pBAC-2cp/HA2-hPRB digested with EcoRI/NotI and cloned into pRAV-Flag digested with MfeI/NotI, to generate pRAV-Flag-PRB. Similarly, PRB-ΔERID-I (deletion of residues 166-372) was extracted from pSG5-PRB-ΔERID-I (7) and PRB-E911A (point mutation in AF2 domain) from pBK-PRB-E911A (20), and cloned into pRAV-Flag. The three DBD mutants were generated by PCR-mediated site-directed mutagenesis on the pRAV-Flag-PRB using the QuikChange Site-Directed Mutagenesis XL kit (Stratagene, La Jolla, CA). PRB-DBD carried the triple mutation G584E/S585G/V589A in the P-box. PRB-D4 carried the A604T mutation, and PRB-M6 an R606W change (D-box mutants). Oligonucleotides for mutagenesis were designed with the QuikChange Primer Design Program and are available upon request. All constructs were verified by sequencing. Transient transfection of pRAV-Flag-derived vectors was performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions.
Generation of stable cell lines by retroviral infection
For the production of viral particles containing the pRAV-Flag-derived vectors, 2.5 × 106 cells of the packaging cell line GP2-293 (CLONTECH Laboratories, Inc., Palo Alto, CA) were transfected with plasmids pRAV-Flag-PRB WT or mutants (10 μg) and pVSV-G (CLONTECH) encoding the vesicular stomatitis virus G envelope protein (3.5 μg) in 10-cm dishes using calcium phosphate. Medium was collected every 24 h for 2 d and centrifuged for 1.5 h at 26,000 rpm at 4 C in a 20% sucrose cushion to concentrate viruses. Pellet containing viral particles was dissolved in medium and used for cell infection. Cells (3.5 × 105) were infected at different multiplicities in six-well plates using the spinoculation method, i.e. plates were centrifuged at 1200 × g for 2 h at 25 C. Viral suspensions were titrated on TYML cells by infecting a fixed number of cells with different volumes of the viral suspension, and the proportion of infected cells was followed by FACS analysis of GFP expression. Low multiplicities of infection (∼5–8%) were selected to achieve close to one integrant per host genome to avoid overexpression of PRB variants (Fig. 1C). GFP-positive cells were isolated by two rounds of sorting in a FACSvantageSE (Becton Dickinson), in a 2- to 3-wk lapse, establishing the cell lines used hereafter. Two months after sorting, more than 95% of cells retained GFP expression (data not shown). Afterward, PRB and GFP expression showed long-term stability.
Expression levels of PRB variants in TYML cells
Expression of PRB variants in the established cell lines was tested by Western blot with PR antibodies and compared with the endogenous levels of T47D cells (Fig. 1D). Tagged PRB variants of expected molecular mass (130 kDa, except 115 kDa for ERID deletion) were detected at similar levels among cell lines and comparable to endogenous PRB expression in T47D cell extracts. No specific signal was found in cells containing the empty pRAV-Flag vector, confirming that TYML cells are devoid of endogenous PRA and PRB expression. Because of the lack of antibiotic pressure, the long-term stability of PRB expression was followed with the WT PRB-expressing cell line, and found to not diminish during 12 wk of cell culture (data not shown). Recombinant PRB expression and subcellular localization were also analyzed by immunofluorescence, showing no differences between endogenous PR (in T47D cells), recombinant WT PRB, and the different mutants (Fig. 1E). As reported previously, a vast majority of PR is in the cell nuclei at a given time.
PR expression in the different cell lines was also measured at the transcription level by RT-PCR with specific oligonucleotides designed to amplify either the PR coding region, or 3′-UTR (untranslated region) present only in the endogenous gene (Fig. 1F). The coding region was expressed similarly in all PR-expressing cell lines, and in a comparable amount to T47D cells. As expected, 3′-UTR was only detected in T47D cells, and not in TYML-derived cells.
Immunoblotting
Cells treated as previously indicated, were lysed in 25 mm Tris-HCl (pH 7.4), 1 mm EDTA, 1 mm EGTA, and 1% sodium dodecyl sulfate plus protease and phosphatase inhibitors. Cell lysates were obtained by centrifugation, and the protein concentration was determined by Micro BCA protein assays (Pierce Chemical Co., Rockford, IL). Lysates were adjusted to equivalent protein concentrations with lysis buffer, subjected to SDS-PAGE, and analyzed by Western blot using specific primary antibodies and secondary antibodies conjugated to peroxidase. Bands were visualized by chemiluminescence using ECL system (Amersham Pharmacia Biotech, Piscataway, NJ).
Immunofluorescence
Immunofluorescence assays were performed essentially as previously described (14). The fixation step was performed by incubation for 15 min at 4 C in PBS-paraformaldehyde 4%. PR expression was detected with PR H190 antibody in cells cultured in 10% FBS-containing media. Detection of PR and Msk1 activation in response to hormone treatment was performed with phospho-specific antibodies in cells cultured in serum-free conditions as indicated above. After incubation with the appropriate secondary antibody, cells were counterstained with 4,6-diamidino-2-phenylindole (DAPI) and mounted onto slides using SlowFade (Molecular Probes, Inc., Eugene, OR).
Immunoprecipitation of tagged PR
Cultured cells were washed twice with PBS, scraped in PBS-EDTA (1 mm), flash frozen in N2 liquid, and lysed in lysis buffer (50 mm Tris, pH 7.4; 150 mm NaCl; 1 mm EDTA; 1% Nonidet P-40; and 15% glycerol) supplemented with protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 0.7 μg/ml pepstatin A, 2 μg/ml aprotinin, 2 mm NaF, and 1 mm NaVO4). PR-interacting proteins were batch purified by adding anti-Flag M2 affinity gel resin (Sigma) to cell lysate and incubating for 4 h at 4 C, washing three times with 300 μl of wash buffer, and eluting with 1 mg/ml Flag peptide (Sigma) in wash buffer. Immunoprecipitated proteins were analyzed by immunoblotting.
Cell cycle analysis
Cells were washed with cold 1× PBS, fixed in 70% ethanol, and stained with analysis solution: 3% ribonuclease A (Sigma), 10 mg/ml; 3% solution A (38 mm sodium citrate, 500 μg/ml propidium iodide) in 1× PBS. Samples were analyzed using a FACS Calibur machine (Becton Dickinson and Co., Franklin Lakes, NJ), CellQuest analysis software, and ModFit program.
RNA extraction and RT-PCR
Total RNA was extracted using the RNeasy Kit (QIAGEN). The quality of the RNA was analyzed using the Agilent Bioanalyzer 2100 and the RNA 6000 LabChip Kit (Agilent Technologies, Palo Alto, CA) with the Eukaryote Total RNA Nano Assay. cDNA was generated from 100 ng of RNA using Superscript First Strand Synthesis System (Invitrogen). cDNA (1 μl) was used as template for RT-PCR. Indicated gene products were analyzed by PCR with specific oligonucleotides, followed by visualization in agarose gel. When indicated, quantification of gene products was performed by real-time PCR using LightCycler 480 SYBR Green I Master (Roche, Indianapolis, IN). Each value was corrected by human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and expressed as relative units. Gene-specific oligonucleotide sequences are available on request. Each experiment was performed in triplicate. One representative experiment of at least two experiments is shown.
Luciferase assays
Cells were seeded in triplicate at 3.5 × 105 cells per well in a six-well plate. After 1 d in serum-free conditions, cells were incubated with R5020 for 16 h. After incubation, cells were harvested in lysis buffer (Promega), and the protein amount was determined by Micro BCA protein assay (Pierce). Lysates were adjusted to equivalent protein concentrations with lysis buffer, and luciferase activity was determined with a luciferase assay kit (Promega) according to the manufacturer’s instructions.
Chromatin immunoprecipitation assays
ChIP assays were performed as described (45) by using chromatin from TYML cells expressing Flag-tagged WT or mutant PRB, cultured and treated as described previously. Chromatin was sonicated to an average fragment size of 400–500 bp, routinely, in a Branson Sonifier. Rabbit or mouse IgG (Sigma) was used as a control for nonspecific interaction of DNA. To determine the linear range of the amplification, different numbers of cycles and dilution series of input DNA were used for PCR analysis of each amplicon. Input was prepared with 10% of the chromatin material used for an immunoprecipitation. Later, a 1:10 dilution of input material was performed before PCR amplification. The human ß-globin gene amplicon was used as a loading control, detecting unspecific binding of genomic DNA to beads or IgGs. The number of PCR amplification cycles was: 30 for MMTV-NucB amplicon and 32 for ß-globin. PCR products were resolved in agarose gel. Alternatively, quantification of gene products was performed by real-time PCR using LightCycler 480 SYBR Green I Master (Roche). Then, values were corrected by the human actin gene and expressed as relative units. Primer sequences and specific PCR conditions are available on request.
Microarray hybridization and data analysis
Procedures for microarray hybridization and data analysis are described elsewhere (14) and detailed in the Supplemental Methods.
Microarray data accession number
Microarray data are available at GEO under accession no. GSE11888 (http://www.ncbi.nlm.nih.gov/geo/index.cgi).
Acknowledgments
We thank K. Horwitz and B. Jacobsen (University of Colorado, US), for providing T47D-YV cells; X. Liu (University of Colorado), for providing pRAV-Flag plasmid; M.J. Melià, B. Miñana and L. Sumoy (Centre de Regulació Genòmica), for the microarray platform setup; G. Vicent (Centre de Regulació Genòmica) for helpful discussion and sharing unpublished data; and O. Fornàs (Universitat Pompeu Fabra) for help with FACS.
NURSA Molecule Pages:
Coregulators: AIB1 | SRC-1;
Ligands: R5020;
Nuclear Receptors: PR.
Footnotes
This work was supported by grants from the Catalan Department for Universities, Research and the Information Society, and the Spanish Ministry of Science and Technology and Fondo Europeo de desarrollo regional (SAF2002-03320, BFU2008-00359). A.J. was recipient of a ‘Ramón y Cajal’ appointment from the Spanish Ministry of Science and Technology. I.Q. was recipient of a Formación personal universitario predoctoral fellowship from the Spanish Ministry of Education. L.M-A. was recipient of a predoctoral fellowship funded by Fundación para la investigación y prevención del SIDA en España.
Disclosure Summary: The authors have nothing to disclose.
First Published Online March 19, 2009
Abbreviations: AF, Activation function; AR, androgen receptor; ChIP, chromatin immunoprecipitation; DAPI, 4,6-diamidino-2-phenylindole; DBD, DNA binding domain; ER, estrogen receptor; ERID, ER-interacting domain; FACS, fluorescence-activated cell sorting; FBS, fetal bovine serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescent protein; HRE, hormone response element; HSD, hydroxysteroid dehydrogenase; JAK, Janus family of tyrosine kinase; LBD, ligand binding domain; Luc, luciferase; MMTV, mouse mammary tumor virus; NucB, nucleosome B; PD, PD98059; PI3K, phosphoinositol 3-kinase; PR, progesterone receptor; PRB, PR-isoform B; PRE, progesterone response element; qPCR, quantitative PCR; SHR, steroid hormone receptor; SRC, steroid receptor coactivator; STAT, signal transducer and activator of transcription; TAP, tandem affinity purification; UTR, untranslated region; WT, wild type.
References
- 1.Beato M, Herrlich P, Schütz G1995. Steroid hormone receptors: many actors in search of a plot. Cell 83:851–857 [DOI] [PubMed] [Google Scholar]
- 2.Edwards DP2005. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol 67:335–376 [DOI] [PubMed] [Google Scholar]
- 3.Migliaccio A, Di Domenico M, Castoria G, de Falco A, Bontempo P, Nola E, Auricchio F1996. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J 15:1292–1300 [PMC free article] [PubMed] [Google Scholar]
- 4.Castoria G, Migliaccio A, Bilancio A, Di Domenico M, de Falco A, Lombardi M, Fiorentino R, Varricchio L, Barone MV, Auricchio F2001. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J 20:6050–6059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boonyaratanakornkit V, Scott MP, Ribon V, Sherman L, Anderson SM, Maller JL, Miller WT, Edwards DP2001. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol Cell 8:269–280 [DOI] [PubMed] [Google Scholar]
- 6.Migliaccio A, Piccolo D, Castoria G, Di Domenico M, Bilancio A, Lombardi M, Gong W, Beato M, Auricchio F1998. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J 17:2008–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ballaré C, Uhrig M, Bechtold T, Sancho E, Di Domenico M, Migliaccio A, Auricchio F, Beato M2003. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Mol Cell Biol 23:1994–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ballaré C, Vallejo G, Vicent GP, Saragüeta P, Beato M2006. Progesterone signaling in breast and endometrium. J Steroid Biochem Mol Biol 102:2–10 [DOI] [PubMed] [Google Scholar]
- 9.Saitoh M, Ohmichi M, Takahashi K, Kawagoe J, Ohta T, Doshida M, Takahashi T, Igarashi H, Mori-Abe A, Du B, Tsutsumi S, Kurachi H2005. Medroxyprogesterone acetate induces cell proliferation through up-regulation of cyclin D1 expression via phosphatidylinositol 3-kinase/Akt/nuclear factor-κB cascade in human breast cancer cells. Endocrinology 146:4917–4925 [DOI] [PubMed] [Google Scholar]
- 10.Proietti C, Salatino M, Rosemblit C, Carnevale R, Pecci A, Kornblihtt AR, Molinolo AA, Frahm I, Charreau EH, Schillaci R, Elizalde PV2005. Progestins induce transcriptional activation of signal transducer and activator of transcription 3 (Stat3) via a Jak- and Src-dependent mechanism in breast cancer cells. Mol Cell Biol 25:4826–4840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lange CA, Richer JK, Horwitz KB1999. Hypothesis: Progesterone primes breast cancer cells for cross-talk with proliferative or antiproliferative signals. Mol Endocrinol 13:829–836 [DOI] [PubMed] [Google Scholar]
- 12.Groshong SD, Owen GI, Grimison B, Schauer IE, Todd MC, Langan TA, Sclafani RA, Lange CA, Horwitz KB1997. Biphasic regulation of breast cancer cell growth by progesterone: role of the cyclin-dependent kinase inhibitors, p21 and p27(Kip1). Mol Endocrinol 11:1593–1607 [DOI] [PubMed] [Google Scholar]
- 13.Vicent GP, Ballaré C, Nacht AS, Clausell J, Subtil-Rodríguez A, Quiles I, Jordan A, Beato M2006. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell 24:367–381 [DOI] [PubMed] [Google Scholar]
- 14.Subtil-Rodríguez A, Millán-Ariño L, Quiles I, Ballaré C, Beato M, Jordan A2008. Progesterone induction of the 11β-hydroxysteroid dehydrogenase type 2 promoter in breast cancer cells involves coordinated recruitment of STAT5A and progesterone receptor to a distal enhancer and polymerase tracking. Mol Cell Biol 28:3830–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobsen BM, Schittone SA, Richer JK, Horwitz KB2005. Progesterone-independent effects of human progesterone receptors (PRs) in estrogen receptor-positive breast cancer: PR isoform-specific gene regulation and tumor biology. Mol Endocrinol 19:574–587 [DOI] [PubMed] [Google Scholar]
- 16.Richer JK, Jacobsen BM, Manning NG, Abel MG, Wolf DM, Horwitz KB2002. Differential gene regulation by the two progesterone receptor isoforms in human breast cancer cells. J Biol Chem 277:5209–5218 [DOI] [PubMed] [Google Scholar]
- 17.Huse B, Verca SB, Matthey P, Rusconi S1998. Definition of a negative modulation domain in the human progesterone receptor. Mol Endocrinol 12:1334–1342 [DOI] [PubMed] [Google Scholar]
- 18.Sartorius CA, Shen T, Horwitz KB2003. Progesterone receptors A and B differentially affect the growth of estrogen-dependent human breast tumor xenografts. Breast Cancer Res Treat 79:287–299 [DOI] [PubMed] [Google Scholar]
- 19.Graham JD, Yeates C, Balleine RL, Harvey SS, Milliken JS, Bilous AM, Clarke CL1995. Characterization of progesterone receptor A and B expression in human breast cancer. Cancer Res 55:5063–5068 [PubMed] [Google Scholar]
- 20.Gong W, Chávez S, Beato M1997. Point mutation in the ligand-binding domain of the progesterone receptor generates a transdominant negative phenotype. Mol Endocrinol 11:1476–1485 [DOI] [PubMed] [Google Scholar]
- 21.Takimoto GS, Tasset DM, Eppert AC, Horwitz KB1992. Hormone-induced progesterone receptor phosphorylation consists of sequential DNA-independent and DNA-dependent stages: analysis with zinc finger mutants and the progesterone antagonist ZK98299. Proc Natl Acad Sci USA 89:3050–3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barrero MJ, Malik S2006. Two functional modes of a nuclear receptor-recruited arginine methyltransferase in transcriptional activation. Mol Cell 24:233–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaspar F, Klocker H, Denninger A, Cato AC1993. A mutant androgen receptor from patients with Reifenstein syndrome: identification of the function of a conserved alanine residue in the D box of steroid receptors. Mol Cell Biol 13:7850–7858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sartorius CA, Groshong SD, Miller LA, Powell RL, Tung L, Takimoto GS, Horwitz KB1994. New T47D breast cancer cell lines for the independent study of progesterone B- and A-receptors: only antiprogestin-occupied B-receptors are switched to transcriptional agonists by cAMP. Cancer Res 54:3868–3877 [PubMed] [Google Scholar]
- 25.Jacobsen BM, Richer JK, Schittone SA, Horwitz KB2002. New human breast cancer cells to study progesterone receptor isoform ratio effects and ligand-independent gene regulation. J Biol Chem 277:27793–27800 [DOI] [PubMed] [Google Scholar]
- 26.Vicent GP, Nacht AS, Smith CL, Peterson CL, Dimitrov S, Beato M2004. DNA instructed displacement of histones H2A and H2B at an inducible promoter. Mol Cell 16:439–452 [DOI] [PubMed] [Google Scholar]
- 27.Aoyagi S, Archer TK2008. Nicotinamide uncouples hormone-dependent chromatin remodeling from transcription complex assembly. Mol Cell Biol 28:30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faivre EJ, Daniel AR, Hillard CJ, Lange CA2008. Progesterone receptor rapid signaling mediates Ser345 phosphorylation and tethering to Sp1 transcription factors. Mol Endocrinol 22:823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB1998. Progesterone regulates transcription of the p21(WAF1) cyclin- dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem 273:10696–10701 [DOI] [PubMed] [Google Scholar]
- 30.Castoria G, Barone MV, Di Domenico M, Bilancio A, Ametrano D, Migliaccio A, Auricchio F1999. Non-transcriptional action of oestradiol and progestin triggers DNA synthesis. EMBO J 18:2500–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cicatiello L, Addeo R, Sasso A, Altucci L, Petrizzi VB, Borgo R, Cancemi M, Caporali S, Caristi S, Scafoglio C, Teti D, Bresciani F, Perillo B, Weisz A2004. Estrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol Cell Biol 24:7260–7274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eeckhoute J, Carroll JS, Geistlinger TR, Torres-Arzayus MI, Brown M2006. A cell-type-specific transcriptional network required for estrogen regulation of cyclin D1 and cell cycle progression in breast cancer. Genes Dev 20:2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu M, Olsen A, Faivre E, Horwitz KB, Lange CA2003. Mitogen-activated protein kinase regulates nuclear association of human progesterone receptors. Mol Endocrinol 17:628–642 [DOI] [PubMed] [Google Scholar]
- 34.Lange CA, Shen T, Horwitz KB2000. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA 97:1032–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skildum A, Faivre E, Lange CA2005. Progesterone receptors induce cell cycle progression via activation of mitogen-activated protein kinases. Mol Endocrinol 19:327–339 [DOI] [PubMed] [Google Scholar]
- 36.Kucera T, Waltner-Law M, Scott DK, Prasad R, Granner DK2002. A point mutation of the AF2 transactivation domain of the glucocorticoid receptor disrupts its interaction with steroid receptor coactivator 1. J Biol Chem 277:26098–26102 [DOI] [PubMed] [Google Scholar]
- 37.Onate SA, Boonyaratanakornkit V, Spencer TE, Tsai SY, Tsai MJ, Edwards DP, O'Malley BW1998. The steroid receptor coactivator-1 contains multiple receptor interacting and activation domains that cooperatively enhance the activation function 1 (AF1) and AF2 domains of steroid receptors. J Biol Chem 273:12101–12108 [DOI] [PubMed] [Google Scholar]
- 38.Tung L, Abdel-Hafiz H, Shen T, Harvell DM, Nitao LK, Richer JK, Sartorius CA, Takimoto GS, Horwitz KB2006. Progesterone receptors (PR)-B and -A regulate transcription by different mechanisms: AF-3 exerts regulatory control over coactivator binding to PR-B. Mol Endocrinol 20:2656–2670 [DOI] [PubMed] [Google Scholar]
- 39.Thomson S, Mahadevan LC, Clayton AL1999. MAP kinase-mediated signalling to nucleosomes and immediate-early gene induction. Semin Cell Dev Biol 10:205–214 [DOI] [PubMed] [Google Scholar]
- 40.Grillo M, Bott MJ, Khandke N, McGinnis JP, Miranda M, Meyyappan M, Rosfjord EC, Rabindran SK2006. Validation of cyclin D1/CDK4 as an anticancer drug target in MCF-7 breast cancer cells: Effect of regulated overexpression of cyclin D1 and siRNA-mediated inhibition of endogenous cyclin D1 and CDK4 expression. Breast Cancer Res Treat 95:185–194 [DOI] [PubMed] [Google Scholar]
- 41.Arnold A, Papanikolaou A2005. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol 23:4215–4224 [DOI] [PubMed] [Google Scholar]
- 42.Boonyaratanakornkit V, McGowan E, Sherman L, Mancini MA, Cheskis BJ, Edwards DP2007. The role of extranuclear signaling actions of progesterone receptor in mediating progesterone regulation of gene expression and the cell cycle. Mol Endocrinol 21:359–375 [DOI] [PubMed] [Google Scholar]
- 43.Truss M, Bartsch J, Schelbert A, Haché RJ, Beato M1995. Hormone induces binding of receptors and transcription factors to a rearranged nucleosome on the MMTV promoter in vivo. EMBO J 14:1737–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knuesel M, Wan Y, Xiao Z, Holinger E, Lowe N, Wang W, Liu X2003. Identification of novel protein-protein interactions using a versatile mammalian tandem affinity purification expression system. Mol Cell Proteomics 2:1225–1233 [DOI] [PubMed] [Google Scholar]
- 45.Strutt H, Paro R1999. Mapping DNA target sites of chromatin proteins in vivo by formaldehyde crosslinking. Methods Mol Biol 119:455–467 [DOI] [PubMed] [Google Scholar]