

Abstract
Endometriosis is a prevalent gynecological disease characterized by growth of endometriotic tissue outside the uterine cavity. MicroRNAs (miRNAs) are naturally occurring posttranscriptional regulatory molecules that potentially play a role in endometriotic lesion development. We assessed miRNA expression by microarray analysis in paired ectopic and eutopic endometrial tissues and identified 14 up-regulated (miR-145, miR-143, miR-99a, miR-99b, miR-126, miR-100, miR-125b, miR-150, miR-125a, miR-223, miR-194, miR-365, miR-29c and miR-1) and eight down-regulated (miR-200a, miR-141, miR-200b, miR-142-3p, miR-424, miR-34c, miR-20a and miR-196b) miRNAs. The differential expression of six miRNAs was confirmed by quantitative RT-PCR. An in silico analysis identified 3851 mRNA transcripts as putative targets of the 22 miRNAs. Of these predicted targets, 673 were also differentially expressed in ectopic vs. eutopic endometrial tissue, as determined by microarray. Functional analysis suggested that the 673 miRNA targets constitute molecular pathways previously associated with endometriosis, including c-Jun, CREB-binding protein, protein kinase B (Akt), and cyclin D1 (CCND1) signaling. These pathways appeared to be regulated both transcriptionally as well as by miRNAs at posttranscriptional level. These data are a rich and novel resource for endometriosis and miRNA research and suggest that the 22 miRNAs and their cognate mRNA target sequences constitute pathways that promote endometriosis. Accordingly, miRNAs are potential therapeutic targets for treating this disease.
This study uses microarray technology, qRT-PCR and in silico analyses, to identify miRNA-regulated molecular pathways that are likely to contribute to the pathogenesis of endometriosis.
Endometriosis is a debilitating disease, causing chronic pelvic pain, painful periods, and infertility, that affects 10–15% of reproductive aged women. The disease is attributed to retrograde passage of endometrial fragments in menstrual fluid through the fallopian tubes, which then implant at ectopic sites, usually in the pelvis (1). There is evidence that the hormonal milieu, a genetic predisposition, an aberrant immune response, and environmental factors also contribute to endometriosis susceptibility and progression. Current medications ameliorate symptoms but do not cure endometriosis and cause significant side effects such as osteoporosis (reviewed in Ref. 2). Additionally, many women require extensive and repeated surgery for this condition. There is a need for novel therapeutic agents that suppress endometriotic lesion establishment and growth to reduce the high burden conferred by this disease (3).
Microarray analyses have identified a large number of mRNA transcripts differentially expressed in the eutopic endometrium of women with and without endometriosis (4, 5) and between ectopic and eutopic endometrial samples from the same women (6, 7, 8, 9). A species-specific microarray analysis of xenografts from the nude mouse model of endometriosis suggested the importance of the cellular and molecular dialogue between endometrial and peritoneal tissues in endometriotic lesion development. This cross talk appeared to be mediated by four key molecular networks involving the proteosome, nuclear factor κB (NFκB), TGFβ, and Ras. A comparison with differentially expressed transcripts in ectopic vs. eutopic endometrium in women with endometriosis indicated that these molecular networks may also underpin human disease (10).
However, an in silico study suggested that these data are incomplete, because many transcripts known to be associated with endometriosis are not identified in the published microarray studies of endometriosis (11). One explanation might be that alterations in mRNA transcript abundance reflect only a subset of the pathogenic mechanisms, and that posttranscriptional regulatory molecules such as microRNAs (miRNAs) also play an important role.
miRNAs are highly conserved, 20- to 24-nucleotide single-stranded RNAs that bind to single or multiple 7- to 8-mer motifs situated predominantly, but not exclusively, in the 3′-untranslated regions of target mRNAs. In mammalian cells, this generally leads to mRNA destabilization and/or translational repression, depending on the degree of sequence homology between the miRNA and target RNA transcript. Two recent proteomic studies showed that a single miRNA might curtail the synthesis of proteins encoded by hundreds of genes (12, 13). There may be up to 1000 miRNAs (14) targeting approximately 30% of the human genome and thereby regulating transcripts from as many as 8000 genes (15).
miRNAs are potent negative feedback regulators in a broad array of cellular processes that occur in endometriosis, such as cell differentiation and proliferation (16, 17, 18, 19), cell migration (20, 21, 22, 23), and myogenesis (16, 19, 24). Aberrant miRNA expression has been associated with complex disorders such as hematopoietic disease (18), heart failure (25), and several cancers including estrogen-regulated ovarian (26) and breast cancer (27). Therapeutic antagonism of miRNAs has been successful in nonhuman primates (28) and may therefore become a novel means of treating miRNA-associated human disease.
We hypothesized that miRNAs mediate posttranscriptional regulation in endometriotic lesions and regulate transcripts in previously identified endometriosis-associated molecular networks (10). Therefore, miRNA expression in paired eutopic and ectopic endometrial biopsies from patients with endometriosis was compared. Using microarray, quantitative RT-PCR (qRT-PCR), and bioinformatic techniques, we have identified both novel and previously characterized miRNA-regulated molecular pathways that are likely to contribute to the pathogenesis of endometriosis. These findings constitute a comprehensive foundation for future research into the role of miRNAs in this disease.
Results
miRNAs are differentially expressed in ectopic endometrial tissue
To determine the miRNA profile in endometrial tissue, miRNA microarray analysis was performed on sets of paired samples of eutopic and peritoneal ectopic endometrial tissue from seven patients with endometriosis. Three distinct computational analyses, Independent Component Analysis (29) (ICA, supplemental Fig. 1A published as supplemental data on The Endocrine Society’s Journals Online web site at http:// mend.endojournals.org), ANOVA, and Linear models for microarray data (Limma) (30) all identified 22 miRNAs that were differentially expressed with a fold change ±≥1.5 at P ≤ 0.05 in ectopic vs. eutopic endometrium (supplemental Fig. 1B). Fourteen miRNAs were up-regulated, and eight were down-regulated (Table 1) in ectopic tissue.
Table 1.
Differentially expressed miRNAs in paired ectopic vs. eutopic endometrial tissue from patients with endometriosis
| Human (hsa-) miRNA gene | Fold Change | Accession no. | Chr | Chromosomal location | miRNA family | miRNA cluster |
|---|---|---|---|---|---|---|
| miR-145 | 4.47 | MIMAT0000437 | 5 | Intergenic | mir-145 | 1 |
| miR-143 | 2.84 | MIMAT0000435 | 5 | Intergenic | mir-143 | 1 |
| miR-99a | 2.46 | MIMAT0000097 | 21 | Overlapping transcripts | mir-99 | |
| miR-99b | 2.43 | MIMAT0000689 | 19 | Intergenic | mir-99 | 2 |
| miR-126 | 2.33 | MIMAT0000445 | 9 | Overlapping transcripts | mir-126 | |
| miR-100 | 2.25 | MIMAT0000098 | 11 | Intergenic | mir-99 | |
| miR-125b | 1.96 | MIMAT0000423 | 11,21 | intergenic | mir-125 | |
| miR-150 | 1.95 | MIMAT0000451 | 19 | Overlapping transcripts | mir-150 | |
| miR-125a | 1.79 | MIMAT0000443 | 19 | Intergenic | mir-125 | 2 |
| miR-223 | 1.72 | MIMAT0000280 | X | Intergenic | mir-223 | |
| miR-194 | 1.67 | MIMAT0000460 | 1,11 | Overlapping transcripts | mir-194 | |
| miR-365 | 1.65 | MIMAT0000710 | 16,17 | Overlapping transcripts | mir-365 | |
| miR-29c | 1.63 | MIMAT0000681 | 1 | Intergenic | mir-29 | |
| miR-1 | 1.53 | MIMAT0000416 | 20,18 | Overlapping transcripts | mir-1 | |
| miR-196b | −1.60 | MIMAT0001080 | 7 | Overlapping transcripts | mir-196 | |
| miR-20a | −1.80 | MIMAT0000075 | 13 | Overlapping transcripts | mir-17 | |
| miR-34c | −1.91 | MIMAT0000686 | 11 | Intergenic | mir-34 | |
| miR-424 | −2.27 | MIMAT0001341 | X | Overlapping transcripts | mir-322 | |
| miR-142–3p | −2.27 | MIMAT0000434 | 17 | Intergenic | mir-142 | |
| miR-200b | −2.28 | MIMAT0000318 | 1 | Intergenic | mir-8 | 3 |
| miR-141 | −2.34 | MIMAT0000432 | 12 | intergenic | mir-8 | |
| miR-200a | −2.67 | MIMAT0000682 | 1 | Intergenic | mir-8 | 3 |
miRNA genes significantly (P ≤ 0.05) differentially regulated (fold change ± ≥1.5) determined by ANOVA from microarray analysis of ectopic compared with eutopic endometrial tissue from seven patients with endometriosis are listed in order of their fold change. The accession number is for the mature miRNA sequence; Chr, Chromosome.
An ANOVA analysis on the miRNA microarray data according to menstrual cycle phase was also performed using Limma. This failed to show any significant differences in miRNA profiles between tissues taken from women during proliferative (n = 4) and secretory (n = 3) phases (data not shown).
qRT-PCR analysis of endometrial miRNA expression
To validate the microarray methods, three up-regulated (miR-99a, miR-126, miR-145) and three down-regulated (miR-141, miR-200b, and miR-424) miRNAs were quantified using qRT-PCR analysis (Fig. 1). In concordance with the microarray, qRT-PCR showed significant differential expression of these miRNAs in ectopic vs. eutopic endometrium (P ≤ 0.05). miR-145 and miR-141 were the most differentially expressed of the six miRNAs (P ≤ 0.001), up- and down-regulated, respectively. The qRT-PCR ΔCt values were normalized to miR-let-7a and miR-let-7d, which were both highly abundant and invariably expressed (fold change = 1.09 and 1.05, respectively; P = 0.5) according to the microarrays.
Fig. 1.

qRT-PCR analysis of miRNA expression in endometriosis. miRNA expression in paired ectopic and eutopic endometrial samples from women in endometriosis, as determined by microarray (n = 7, black) and qRT-PCR (n = 8). In qRT-PCR analysis, miRNA expression was normalized to hsa-miR-let-7a (dark gray) or hsa-miR-let-7d (light gray). Significant fold changes in ectopic vs. eutopic endometrium are marked by *, P ≤ 0.05; **, P ≤ 0.01; or ***, P ≤ 0.001. Data are mean ± sem.
miRNA gene families and clusters
The mean microarray fold changes, chromosomal locations, family memberships and cluster affiliations for the 22 differentially expressed miRNAs are presented in Table 1. These include both inter- and intragenic miRNAs. Two up-regulated (miR-125 and miR-99) and one down-regulated (miR-8) miRNA gene families, and three clusters (defined as groups of miRNAs located within 10 kb of each other) were identified among the differentially regulated miRNAs.
Identification of miRNA targets in endometriosis
The predicted target mRNAs of the 22 differentially expressed miRNAs were identified in silico. A conservative approach was undertaken, limiting the targets to those predicted by two algorithms, TargetScanS (31) and four-way PicTar (32), hereafter referred to as the two-way intersection. Although no targets were listed for miR-365 and miR-424 in the four-way PicTar algorithm, 3851 target transcripts corresponding to 2340 unique official gene symbols were present in this intersection for the remaining 20 miRNAs (supplemental Table 1 published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). The number of mRNA targets predicted for each of the differentially expressed miRNAs varied substantially (Fig. 2A).
Fig. 2.

Functional analysis of all predicted mRNA targets of the 22 differentially expressed miRNAs in endometriosis. A, The number of mRNA transcripts predicted to be targeted by the 22 differentially expressed miRNAs in ectopic vs. eutopic endometrium from women with endometriosis. Seven miRNAs may each repress more than 200 mRNA transcripts, whereas five miRNAs had less than 30 anticipated targets. B, One of the molecular networks from IPA of all predicted targets of the 22 miRNAs incorporated molecules involved in cardiovascular system development and function, cellular movement, and cell morphology, including the validated miRNA target MEF2C. C, TGFβ2, SPARC, COL1A1, and COL1A2 are confirmed miRNA targets involved in extracellular matrix remodeling and cell invasion in an IPA network converging on interferon-γ. The gene names in these networks may be identified on the HUGO Gene Nomenclature Committee home page (http://www.genenames.org).
Functional analysis of miRNA targets in endometriosis
The functions of the 3851 predicted miRNA target transcripts and the molecular pathways they potentially constitute were assessed using Ingenuity Pathway Analysis (IPA) software. The predicted targets were significantly enriched for several biological functions known to be involved in endometriosis, including: cellular movement, assembly and organization; cell-to-cell signaling and interaction; nervous system development and function; embryogenesis; cell cycle; cell death; connective tissue development and function and endocrine system development and function (supplemental Table 2).
IPA identified 39 molecular networks constituted by the 3851 predicted targets, exemplified by two networks with well-characterized miRNA targets in Fig. 2.
The network in Fig. 2B includes myocyte enhancer factor 2C (MEF2C), a confirmed target (18) of one of the up-regulated miRNAs in ectopic endometrium in this study (miR-223). IPA suggested that 22 of the 3851 predicted targets of the differentially expressed miRNAs had molecular associations with MEF2C in the published literature (supplemental Fig. 2A). It is possible that groups of molecular associations such as these occur by chance. Therefore, as proof of principle, a permutation study was performed. This showed that only 4.4% of 1000 randomly selected lists of 22 miRNAs had more predicted targets associated with MEF2C in the IPA database than the 22 differentially expressed miRNAs (supplemental Fig. 2B). A corresponding permutation analysis focused on c-Jun, another target of interest in our study, showed similar results (supplemental Fig. 2, C and D).
Another molecular network constituted by the 3851 predicted targets converged on interferon-γ and included TGFβ2, secreted protein, acidic, cysteine-rich, osteonectin (SPARC), and collagens COL1A1 and COL1A2, which are all confirmed targets of the ectopically dysregulated miR-141 (33), and miR-29c (23), respectively. Mechanisms mediated by these molecules have been implicated in endometrial lesion development (10, 34), and the network in Fig. 2C suggests that these pathways may also be regulated by miRNAs.
Gene Ontology (GO) analysis suggested that the predicted targets of the 22 differentially expressed miRNAs were relatively enriched for GO paths associated with transcription. However, targets of five randomly chosen sets of 22 miRNAs were equally enriched for this category of GO paths (data not shown).
mRNAs differentially expressed in endometriosis are predicted targets of the 22 endometrial miRNAs
After identification of the 22 differentially expressed miRNAs and their predicted targets, we further restricted our analysis to a subset of putative miRNA targets that were also differentially expressed at the mRNA level (summarized in supplemental Fig. 3). In an mRNA microarray study of paired eutopic and peritoneal ectopic endometrium from women with endometriosis (10), hereafter referred to as the peritoneal comparison, 4034 transcripts were differentially expressed (fold change ±≥1.5, P ≤ 0.05). Of these transcripts, 673 (572 unique official gene symbols) were both differentially expressed and predicted to be targeted by at least one, but often several, of the 22 differentially expressed miRNAs (supplemental Table 3). IPA suggested that the 673 transcripts were significantly enriched for functional annotations associated with endometriosis, including: cell death; connective tissue, nervous and muscular system development and function; cellular movement; cell proliferation and the cell cycle; angiogenesis and reproductive/endocrine system disorders (supplemental Table 4).
These 673 mRNA transcripts were overrepresented in 12 IPA networks converging on 1) JUN; 2) platelet-derived growth factor β polypeptide)/chemokine (C-X-C motif) ligand 12 (PDGF BB/CXCL12); 3) CREBBP; 4) CCND1; 5) calmodulin; 6) NFkB; 7) MAPK; 8) caveloin 1/vinculin (CAV1/VCL); 9) c-AMP dependent protein kinase (PKA); 10) phosphoinositide-3-kinase (PI3K); 11) protein kinase C (PKC); and 12) AKT (Fig. 3, A–C; supplemental Fig. 4, A–I; and supplemental Table 4). Although each of these was originally identified as a distinct network, there was cross-interaction between the networks, exemplified by JUN in Fig. 3A, which is a transcriptional target of MEF2C in Fig. 3B. Overlap was also found between networks of all predicted targets of the 22 miRNAs and networks of the differentially expressed predicted targets of the 22 miRNAs, illustrated by TGFβ2, SPARC, COL1A1, and COL1A2 in Figs. 2C and 3A.
Fig. 3.
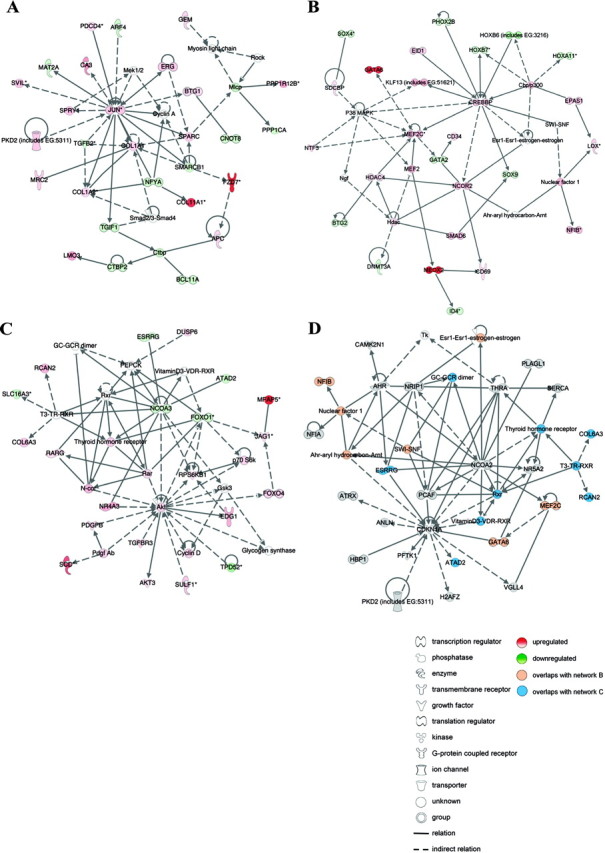
Functional analysis of differentially expressed miRNA targets in endometriosis. A–C, Networks identified by IPA of differentially expressed mRNAs that are also predicted targets of the 22 differentially expressed miRNAs in paired peritoneal ectopic vs. eutopic endometrial tissues from women with endometriosis. A, c-Jun is central in a network comprising extracellular matrix proteins that are confirmed targets of miRNAs differentially expressed in endometriosis. This network overlapped considerably to analysis of all predicted targets of the 22 miRNAs (Fig. 2C), suggesting a role for miRNAs in regulating cell migration during endometrial lesion development. B, MEF2C, histone deacetylase 4 (HDAC4), and DNA (cytosine-5-) methyltransferase 3α (DNMT3A) are validated miRNA targets represented in a network converging on CREBBP, proposing a role for miRNAs in the regulation of myogenesis, angiogenesis, and epigenetic modulation of gene expression in endometrial tissue remodeling. C, The IPA network converging on AKT indicated a role for miRNAs in regulating retinoic acid signaling, the cell cycle, and cellular growth and proliferation. D, IPA network from functional analysis of a second microarray data set of differentially expressed miRNA targets in paired ovarian ectopic vs. eutopic endometrial tissue from women with endometriosis. The network exemplifies the substantial overlap between the ovarian and the peritoneal comparisons, illustrated in orange and blue nodes. Red and green nodes depict up- and down-regulated miRNA targeted transcripts in ectopic vs. eutopic endometrial tissue, respectively. Lines represent the biological relationship between two nodes. The gene names in these networks may be identified on the HUGO Gene Nomenclature Committee home page (http://www.genenames.org).
To verify that the endometriosis-specific functions and networks identified here were not limited to the specific endometrial lesion types and patients examined, we also defined the intersection of miRNA data with a second mRNA microarray study of paired eutopic and ovarian ectopic endometriotic tissues (9), hereafter referred to as the ovarian comparison. This revealed 684 transcripts that were both differentially expressed (fold change ±≥1.5, P ≤ 0.05) and putatively targeted by one or more of the 22 differentially expressed miRNAs (supplemental Fig. 5 and supplemental Table 5).
The IPA networks identified for these 684 ovarian endometrial transcripts showed a remarkable overlap with those associated with the peritoneal comparison as exemplified in Fig. 3D, including those converging on AP1 (c-JUN/c-FOS), PI3K/AKT, CCND1, NFκB, and PDGF/BB (data not shown).
c-Jun mRNA expression
c-Jun is centrally located in one of the IPA networks (Fig. 3A) and is up-regulated at the mRNA level in the ectopic peritoneal endometrial tissue study (10). We used qRT-PCR to confirm the significant up-regulation of c-Jun mRNA (P = 0.015) in six of the same RNA samples used in the miRNA microarray and qRT-PCR analyses. Median normalized c-Jun expression in ectopic and eutopic tissue was 2.03 and 0.11, respectively (Fig. 4).
Fig. 4.

c-Jun mRNA expression was significantly increased in endometriosis. c-Jun expression was significantly increased (P = 0.015) in ectopic (Ec) vs. eutopic (Eu) endometrium from six women with endometriosis, as determined by qRT-PCR. Data are median and range of normalized c-Jun mRNA expression from three independent experiments for each patient.
Discussion
This study used microarray technology, qRT-PCR, and in silico analyses in a novel strategy to assess miRNA expression in peritoneal endometrial tissues from women with endometriosis. Twenty-two miRNAs that were differentially expressed in paired ectopic vs. eutopic endometrium were identified that putatively regulate the expression of 2340 genes. A subset of these target transcripts was previously found to be differentially expressed between endometrial ectopic and eutopic endometrium in two separate mRNA microarray studies (9, 10). The specific expression profiles of the 22 miRNAs and their predicted targets may cause alterations to molecular pathways that are pivotal in the endometriotic process. Therefore, the inhibition or reactivation of these pathways by synthetic antisense antagomiRs or over expression of these miRNAs are potential therapeutic interventions in endometriosis.
Because ectopic endometrial tissue is less susceptible to steroid hormone-driven fluctuations than eutopic endometrium from women with endometriosis (reviewed in Ref. 35), miRNA analysis in different phases of the cycle would be expected to mainly reflect alterations in eutopic tissue. Therefore, miRNA expression was studied in endometrial tissues during both secretory (n = 3) and proliferative (n = 4) phases. The 22 differentially expressed miRNAs are hence consistently associated with endometriosis across the menstrual cycle. No effect on menstrual cycle phase on endometrial miRNA expression could be demonstrated in the present study. Cycle-related effects may be revealed by analysis of a larger study group in the future.
The dataset of 22 differentially regulated miRNAs in endometriosis was defined conservatively by the concordance of three methods of analysis. The combination of two parametric statistical methods (linear models that use the pooled correlation of replicate spots on the arrays, as well as a nested linear model design) with an independent nonparametric method (ICA) as well as qRT-PCR confirmation of three up- and three down-regulated miRNAs lends weight to the validity of this dataset.
Although all the 22 miRNAs have overlapping target lists, seven had the potential to individually target up to 494 mRNAs. Each of the 22 miRNAs was predicted to suppress, on average, 175 target mRNAs, with some mRNAs predicted to be coregulated by multiple miRNAs. This indicates that miRNAs, like transcription factors, may be considered master regulators in cellular processes in endometriosis.
Proximally paired miRNA genes located up to 50 kb apart are frequently coexpressed (36). The finding of three miRNA clusters among the differentially expressed miRNAs provides further confidence in the results of this analysis. miRNAs in the same cluster have common predicted mRNA targets and might consequently have an additive repressive effect on target expression. The clustered and multitargeting miRNAs may be especially powerful regulators in ectopic endometrial lesions.
Approximately 80% of miRNA genes are located within introns (37), and these often have expression profiles that correlate with their host genes (36). We identified significant up-regulation in ectopic endometrial tissue of both miR-126 and epidermal growth factor (EGF)-like-domain, multiple 7 (EGFL7), a gene expressed in endothelial cells promoting vascular tubulogenesis. Because miR-126 is embedded in the EGFL7 gene, it is likely that these two genes are coexpressed in endometriosis, as previously described for miR-10a and homeobox B4 (38).
Correct identification of miRNA targets is central to determining the biological function of miRNAs. In this study, miRNA targets were identified at a conservative two-way intersection of TargetScanS and PicTar (four-way), as recommended in a recent consensus review (39). These are sensitive algorithms with substantial overlap in their predicted targets (40). miRNA targets with binding sites that were not conserved across species are excluded in this intersection, and we acknowledge that the impact of these miRNA targets in endometriosis was not explored here. However, in vitro studies suggested that this two-way intersection had a true positive rate of up to 90% (41) and that these two prediction models showed superior performance in a high-throughput examination of miRNA repression at protein level (12).
Frequent revision of miRNA target algorithms as well as target mRNA ENSEMBL annotations can dramatically alter predicted target transcript lists over time; therefore experimental confirmation of miRNA targets remains the gold standard. Despite the lack of large-scale screening methods for target verification, the number of validated miRNA targets is escalating, and there is growing evidence that miRNA targets can be confidently predicted in silico.
A large number of the mRNA targets of the 22 endometrial miRNAs discussed here are already experimentally confirmed in cellular systems similar to those involved in endometriosis. In addition, many of the molecular pathways these targets constitute are known to play important roles in the pathological processes involved in this condition. Therefore, we are confident that the molecular networks identified here represent conservative and well-founded hypotheses on which to base further experiments. Future studies are also necessary to determine the specific miRNA profiles of stromal and glandular epithelial cells as well as populations of infiltrating leukocytes, because the ratio of these cell types are altered in ectopic compared with eutopic endometrial tissue.
To appraise the general principle of identifying molecular networks using a systems biology database such as IPA, the associations with MEF2C and JUN of predicted targets of random miRNA datasets were assessed. These individual permutation studies suggested that, at least for these two genes, the results of the functional analysis were unlikely to occur by chance.
Functional analysis of miRNA targets was limited to the convergence of miRNA and mRNA microarray data. This method focuses the analysis not only on the potential actions of miRNAs in translational repression, but also on transcripts with a demonstrated regulation of abundance in endometriotic disease. We acknowledge that this approach may omit important molecules that are posttranscriptionally regulated by the 22 miRNAs, but that are not differentially expressed in endometriosis. Nevertheless we prefer to use this conservative approach to reduce false discovery. Predicted miRNA targets in two microarray studies (9, 10) identified pathways previously found to be critical in endometriosis (5, 6, 10). These two different mRNA data sets examined different sites of endometriosis and revealed distinct, but overlapping, transcript lists. The substantial correlation observed between these two independent comparisons lends confidence to the methodology used in evaluating the function of differentially expressed miRNAs in endometriosis.
Two distinct patient groups provided tissue for the mRNA and miRNA microarray analyses; therefore, it is possible that these individuals had distinct mRNA profiles. qRT-PCR of c-Jun confirmed that, at least for this transcript, the two independent tissue sets showed similar extents of dysregulation.
This is the first study demonstrating increased c-Jun mRNA expression by RT-PCR in ectopic vs. eutopic endometrial tissue, supporting our previous findings in a mRNA microarray study (10). Others identified reduced c-Jun mRNA levels in eutopic endometriotic compared with normal endometrial tissue (42). Endometrial c-Jun expression is induced by estrogen (43, 44, 45), a hormone required for endometriotic lesion development, whereas Danazol, an ethisterone derivative used to treat endometriosis, down-regulates c-Jun transcript levels in the lesions (46). Altogether this suggests a functional role for c-Jun at ectopic sites during endometriosis.
Functional analysis identified several molecular networks containing many transcripts linked to endometriosis. Whereas the cellular functions of these networks are consistent with the current view on endometriotic lesion pathogenesis, a comprehensive discussion of these pathways is beyond the scope of this paper. Instead, examples of the most prominent molecular networks are presented in Table 2 along with confirmed regulatory functions of the differentially expressed miRNAs, including regulation of cell migration and proliferation, wound contraction, extracellular matrix remodeling, and angiogenesis.
Table 2.
Experimentally confirmed regulatory functions of the 22 differentially expressed miRNAs in endometriosis
| IPA Network (figure) | miRNA | miRNA target | Regulatory feedback loops | Confirmed biological functions | Proposed role in endometriosis | Ref. nos. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | FC | mRNA | Reg | |||||||
| CREBBP (Fig. 2B) | miR-223 | ↑ | MEF2C | ↑ | MEF2C→ miR-1→| HDAC4→| MEF2C | miR-1 controls myogenesis and vascular development in association with MyoD family members | Cell migration, wound contraction, neovascularization in endometrial stromal myofibroblasts and pericytes | 10 16 18 19 24 25 | ||
| miR-1 | ↑ | HDAC4 | ↑ | 10 16 | ||||||
| miR-29c | ↑ | DNMT3A | ↓ | DNA methylation, tumorigenicity in lung cancer | DNA methylation, epigenetic regulation, gene expression | 10 54 | ||||
| JUN (Fig 2A) | miR-29c | ↑ | SPARC, COL1A1, COL1A2 | ↑ | Cell migration cancer metastasis | Extracellular matrix remodeling, cell invasion | 10 23 | |||
| miR-141 | ↓ | TGFβ2 | ↓ | TGFβ-induced EMT | TGFβ-regulated stromal proliferation | 10 33 55 56 57 | ||||
| CCND1 (S Fig. 3B) | miR-141, miR-200b | ↓ | ZEB1 | ZEB1→| miR-141 and miR-200b→| ZEB1 | TGFβ-induced EMT in cancer cell lines via targeting of TGFβ2, ZEB1 and ZEB2 | Stromal fibroblast migration and invasion | 20 21 22 33 58 59 | |||
| miR-141, miR-200b | ↓ | ZEB2 | ↑ | 10 20 21 22 59 | ||||||
| miR-20a | ↓ | CCND1 | ↑ | miR-20a→| CCND1→ miR-20a | Epithelial cell proliferation | 10 60 | ||||
| miR-20a | ↓ | E2F3 | ↑ | E2F3→ miR-20a→| E2F3 | Cell proliferation and apoptosis | 10 61 | ||||
| miR-20a | ↓ | RUNX1 | ↑ | miR-20a→| RUNX1→| miR-20a | Monocytopoiesis | Monocytic differentiation, proliferation, angiogenesis | 10 62 | |||
| PDGFBB/CXCL12 (S Fig. 3A) | miR-1 | ↑ | FN1 | ↑ | Cardiac hypertrophy, myocyte proliferation | Cell proliferation and migration, wound healing | 10 63 | |||
| NFκB (S Fig. 3D) | miR-20a | ↓ | CTGF | ↑ | Neovascularization in cancer | Neovascularization | 10 64 | |||
| miR-1 | ↑ | HSPD1 | ↓ | Cardiomyocyte apoptosis | Apoptosis | 10 65 | ||||
S, Supplemental; C, fold change; Reg, miRNA regulation; ZEB1 and ZEB2, zinc finger E-box binding homeobox 1 and 2; E2F3, E2F transcription factor 3; RUNX1, runt-related transcription factor 1; FN1, fibronectin 1; CTGF, connective tissue growth factor; HSPD1, heat shock 60-kDa protein 1; EMT, epithelial to mesenchymal transition; ↑, up-regulated; ↓, down-regulated; →, positive regulation; →|, inhibition.
To our knowledge only one other study has explored miRNAs in endometriosis. Pan et al. (47) identified 48 miRNAs that were differentially expressed in eutopic and ectopic endometrium from a small study group of women with and without endometriosis; however, a direct comparison between endometriotic ectopic and eutopic tissues was not undertaken. Nevertheless, despite differences in study design, methods of analysis, measures of stringency, and RT-PCR endogenous controls, eight miRNAs concurred with the 22 miRNAs described in our analysis. This concordance underlines the likely importance of these miRNAs in the pathogenesis of endometriosis.
In summary, this study suggests that both miRNAs and the molecular pathways they target are differentially expressed between ectopic endometrial lesions and eutopic endometrium in women with endometriosis. Many of the miRNA-regulated molecular networks and biological processes identified here corroborate known endometriosis associations in the literature. These findings propose an intricate control of gene expression in endometriosis both transcriptionally and by miRNAs at the posttranscriptional level. The microarray data and molecular pathways presented here constitute a comprehensive resource on which to base future investigations into the role of specific miRNAs in endometriosis. In particular, the endometriosis-associated miRNAs and the molecular pathways they target should be evaluated as new candidates for the diagnosis and treatment of endometriosis.
Materials and Methods
Tissue collection and ethics approval
All procedures for collection of tissue were approved by the Central and Northwestern Human Ethics Committee, South Australia, or the Children’s Youth and Women’s Human Research Ethics Committee, South Australia. Eutopic and ectopic endometrial tissue was obtained with written informed consent from eight women with AFS stage II–IV endometriosis during laparoscopic surgical procedures. Endometrial biopsies were collected using Pipelle suction curettes (Pipelle de Cornier, Laboratoire C.C.D, Paris, France). All women had regular menstrual cycles (28–30 d) and were not taking any medication. Four were in the follicular phase of the cycle (d 9-d 14) and four in the secretory phase (d 17-d 28). The cycle phase of all biopsies was histologically confirmed using Noyes criteria (48). Samples from seven women were used for the miRNA microarray analysis, and an additional sample was used in the miRNA qRT-PCR analysis. All biopsies were immediately placed in RNA Later (Invitrogen, Carlsbad, CA) and stored at −80 C.
RNA extraction
Total RNA was extracted from endometrial tissues homogenized in Trizol solution (Invitrogen) according to the manufacturer’s instructions. RNA concentration was assessed using a Nanodrop spectrophotometer, accepting a ratio of 2.0 for sample absorbance at 260/280.
Microarray hybridization
The microarrays consisted of 377 antisense miRNA oligonucleotide probes (mirVana miRNA probe set; catalog no. 1564V1, Applied Biosystems, Foster City, CA) printed in triplicate onto epoxide-coated microarray slides (Corning, Inc., Corning, NY) with a VersArray ChipWriter Pro system (Bio-Rad Laboratories, Inc., Hercules, CA) using tungsten pins (PointTech, Barreal de Heredia, Costa Rica). Included on the array were three negative control probes, which were small-interfering RNAs of bacterial origin. For detection on the array, 5 μg total RNA was labeled by the ligation of a fluorescently modified RNA dimmer as previously described by Thomson et al. (49). Competitive hybridizations were performed, in each case comparing RNA extracted from eutopic vs. ectopic tissues from the same woman. To reduce effects of dye bias, dye swaps were performed, with RNA from each tissue labeled in both combinations (50). RNA was hybridized to the arrays for 2 h at 42 C using LifterSlips (Erie Scientific, Northbrook, IL) in 25 μl of hybridization buffer (400 mm Na2HPO4, pH 7.0; 0.8% BSA; 5% sodium dodecyl sulfate; 12% formamide). Slides were placed in Corning hybridization chambers, incubated in the dark for 2 h, then washed twice in 2× standard sodium citrate (SSC)/0.025% sodium dodecyl sulfate and three times in 0.8× SSC at room temperature, and finally twice in 0.4× SSC at 4 C. Slides were scanned at 10 μm resolution with a Genepix 4000B Scanner (Molecular Devices, Sunnyvale, CA) using photomultiplier tube (PMT) settings that approximately balanced the channel intensity histograms.
Computational microarray analysis
Median spot pixel intensity values in scanned images were determined using the Spot version 3 plugin (Commonwealth Scientific and Industrial Research Organization, Campbell Australian Capital Territory, Australia) for the statistical environment R. After subtraction of median background intensities and Loess normalization, mean intensities were log2 transformed, and ratios (Cy5/Cy3) were obtained. To select differentially expressed miRNAs, these data were analyzed using three different statistical methods. 1) A pooled correlation of triplicate spots for each miRNA was calculated for each chip using linear models and empirical Bayesian moderation of standard errors (Limma R package, Walter and Eliza Hall Institute, Parkville Victoria, Australia). 2) Alternatively, the three measurements for each transcript on each chip were analyzed using a nested ANOVA approach without empirical Bayesian moderation. 3) Data for the three measurements for each transcript on each chip (generated in method 2 above) were reanalyzed using an Ensembl learning implementation of Independent Component Analysis (ICA), which summarizes the overall transcript abundance patterns in the data. The 1% of miRNAs that contributed most strongly to the component associated with eutopic vs. ectopic tissue differences were identified. miRNAs that differentially expressed between the paired eutopic and ectopic tissues (fold change ≥1.5, P ≤ 0.05) according to all three methods (1–3) above were identified by Venn diagram analysis. Heat map analysis and hierarchical clustering were performed using R.
qRT-PCR analysis of miRNA expression
Transcript levels of three up-regulated and three down-regulated miRNAs were measured by qRT-PCR in four proliferative and four secretory paired eutopic and ectopic endometrial total RNA samples (total n = 16).
Total RNA (5 μg) was reverse transcribed on a GeneAmp PCR System 9700 using the Taqman MicroRNA Reverse Transcription kit and primers from TaqMan MicroRNA Assays (Applied Biosystems). Water substituted for transcript and reverse transcriptase samples served as negative controls. Assay-specific cDNA and probes (TaqMan MicroRNA Assays) were combined with TaqMan 2× Universal PCR Master Mix, No AmpErase UNGb (Applied Biosystems), and real-time PCR amplification was performed in triplicates using an ABI Prism 7000 (Applied Biosystems).
ΔΔCt values were calculated, and the data were normalized to hsa-miR-let-7a, which has been used as a control previously (51, 52), and to hsa-miR-let-7d. Kolmogorov-Smirnov test followed by nonparametric Mann-Whitney U test (asymptomatic significance, correlated for ties) determined the significant up- or down-regulation of ΔCt values, respectively, in ectopic compared with eutopic endometrium. Mann-Whitney U test of Ct and ΔCt [Ct(let-7a)-Ct(let-7d) values] validated the use of hsa-miR-let7a and hsa-miR-let-7d as invariable and ubiquitous endogenous controls. The mean and sem of fold change from eutopic endometrium was determined and the data were presented by GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA).
miRNA target prediction
Target mRNAs of the 22 differentially expressed miRNAs were determined using the miRGen web tool (53) for the algorithms TargetScanS (release 3.1) (31) and PicTar (four-species conservation) (32). Targets predicted by the two-way intersection of these algorithms were further analyzed. The Ensembl gene identification nos. of putative targets retrieved from miRGen were referenced to Ensembl build 46. The convergence of these miRNA targets with mRNAs that were differentially expressed in a microarray study of ectopic and eutopic peritoneal endometrial tissue (10) was determined. According to quality control analysis in the DChip application (http://biosun1.harvard.edu/complab/dchip/), the mRNA microarray data from patient 7 contained relative outliers; therefore, this patient was excluded from further analysis. mRNA probe sets were considered to be differentially expressed at fold change ≥±1.5 at cyberT P ≤ 0.05. To control for potential confounding due to variation between the different patient groups contributing tissues for the miRNA and mRNA microarray analyses, a second mRNA data set comparing ovarian endometriosis with eutopic endometrium (9) was studied. mRNA targets of the 22 miRNAs that were differentially expressed in ectopic endometrium were identified in both data sets.
Permutation study of ingenuity pathway analysis of miRNA targets
Manual pathway analysis showed that 82 molecules are associated with transcription factor MEF2C in the IPA database (http://www.ingenuity.com; May 25, 2008). Of 82 molecules, 22 are also putative targets of the 22 differentially expressed miRNAs. A permutation analysis was performed to verify the high degree of association between MEF2C and other miRNA targets in IPA: 1000 random sets of 22 miRNAs were selected from the 281 miRNAs within the Ambion human microarray probe set that were human in origin, unique, and fully annotated, and their targets were identified using the two-way intersection method described above. The intersect of the targets of each of these 1000 randomly chosen lists of 22 miRNAs, and the 82 molecules related to MEF2C in the IPA database, were calculated and plotted as histograms using R. This analysis was repeated for JUN. IPA identified 632 molecules associated with JUN (November 11, 2008), of which 147 are also putative targets of the 22 differentially expressed miRNAs.
Functional analysis of predicted mRNA targets of the 22 miRNAs
IPA was performed to identify the molecular pathways and functional groupings based on published literature, to which predicted targets of the 22 differentially expressed miRNAs belong. The targets were uploaded into IPA and overlaid onto a global molecular network developed from information contained in the application. Networks of these genes were generated by IPA based on their connectivity, each ranked by a score. This score is based on the hypergeometric distribution, calculated with the right-tailed Fisher’s Exact Test, and corresponds to the negative log of this P value. Networks with a score more than or equal to 20 were selected for further analysis. Functional analysis in IPA identified the published biological functions that were most significantly associated with the genes in the network. Genes or gene products are represented as nodes, and the biological relationship between two nodes is represented as an edge (line). All lines are supported by at least one reference in literature, textbook, or from canonical information stored in the Ingenuity Pathways knowledge database.
The GOStat application (http://gostat.wehi.edu.au/cgibin/goStat.pl) was used to determine the biological processes annotated to the putative miRNAs targets in the GO database (http://www.geneontology.org). The GOA human database was used, only considering the biological processes GO hierarchy, and GO paths with length more than or equal to 3. As a cut-off for significance, Fisher’s Exact Test (P ≤ 0.01) with Benjamini and Hochberg False Discovery Rate assessment was used to estimate the proportion of GO terms that would be identified by chance alone.
qRT-PCR analysis of c-Jun expression
Standard SYBR Green qRT-PCR was carried out to detect c-Jun transcript levels in total cellular RNA from the eutopic and ectopic endometrial tissues. After treatment with ribonuclease-free deoxyribonuclease I (Ambion Turbo DNAse, Applied Biosystems; 60 min/37 C), 1 μg RNA of the individual samples was reverse transcribed using the Superscript III Reverse Transcriptase kit (Invitrogen). Real-time PCR was performed in a RotorGene 6000 (Corbett Life Science, Concorde, New South Wales, Australia; 95 C for 10 min, 95 C for 15 sec followed by 45 cycles at 60 C for 60 sec) on 2 μg cDNA with SYBR Green PCR Master Mix (Applied Biosystems) and 5 μg primers to c-Jun [sense (5′-GGAGAGGAAGCGCATGAGG-3′; and antisense 5′-CATGTTGGCCGTGGACG-3′]. These primers were specific for the published c-JUN cDNA sequence (GenBank accession no. NM_002228), designed using Primer Express version 2 software (Applied Biosystems) and optimized for use at a final concentration of 0.5 μm. The resulting 136-base amplicon spans nucleotides 1801–1943 and was analyzed by melt curve analysis to ensure lack of nonspecific products and primer dimmers; 18S rRNA was analyzed in the same run with primers as recommended by the manufacturer (Applied Biosystems).
Linear standard curve analysis for each primer pair at a maximum 1:4096 dilution of control pooled cDNA and primer amplification efficiency (E) greater than 0.96 was obtained for all PCR experiments. c-Jun data were normalized to 18S RNA and expressed in arbitrary mRNA units as median values of three independent PCR runs. The data were analyzed by two-tailed Mann-Whitney U test (95% confidence interval) in GraphPad software.
Acknowledgments
We thank Professor Julie Owens and Ms. Patricia Grant for technical advice and critical reading of this manuscript; the Adelaide Microarray Centre; Professor Yee Khong at the Department of Histopathology, Women’s and Children’s Hospital, Adelaide, Australia, for histologically dating endometrial tissue samples relative to the menstrual cycle; staff in the Discipline of Obstetrics and Gynaecology that assisted in this study; and finally all patients and nursing staff of Women’s and Children’s Hospital, Adelaide, Australia.
Footnotes
This work was supported by Women’s and Children’s Hospital Research Foundation Grant, Adelaide, Australia, The Arthur Wilson Fellowship, The Royal Australian and New Zealand College of Obstetricians and Gynaecologists Research Foundation, and, D.A. and J.S. Ballantyne Medical and Surgical Research Trust Grant (to M.L.H.).
Disclosure Statement: The authors have nothing to disclose.
First Published Online December 12, 2008
Abbreviations: Akt, Protein kinase B; CCND1, cyclin D1; COL1A1 and COL1A2, collagen type I, α-1 and -2; CREB, cAMP response element binding protein; CREBBP, CREB binding protein; EGF, epidermal growth factor; IPA, Ingenuity Pathway Analysis; MEF2C, myocyte enhancer factor 2C; miRNA, micro-RNA; NFκB, nuclear factor-κB; qRT-PCR, quantitative RT-PCR; SSC, standard sodium citrate; SPARC, secreted protein, acidic, cysteine-rich, osteonectin.
References
- 1.Sampson JA1927. Peritoneal endometriosis due to menstrual dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet Gynecol 14:442–469 [Google Scholar]
- 2.Giudice LC, Kao LC2004. Endometriosis. Lancet 364:1789–1799 [DOI] [PubMed] [Google Scholar]
- 3.Gao X, Outley J, Botteman M, Spalding J, Simon JA, Pashos CL2006. Economic burden of endometriosis. Fertil Steril 86:1561–1572 [DOI] [PubMed] [Google Scholar]
- 4.Kao LC, Germeyer A, Tulac S, Lobo S, Yang JP, Taylor RN, Osteen K, Lessey BA, Giudice LC2003. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology 144:2870–2881 [DOI] [PubMed] [Google Scholar]
- 5.Matsuzaki S, Canis M, Vaurs-Barriere C, Boespflug-Tanguy O, Dastugue B, Mage G2005. DNA microarray analysis of gene expression in eutopic endometrium from patients with deep endometriosis using laser capture microdissection. Fertil Steril 84(Suppl 2):1180–1190 [DOI] [PubMed] [Google Scholar]
- 6.Arimoto T, Katagiri T, Oda K, Tsunoda T, Yasugi T, Osuga Y, Yoshikawa H, Nishii O, Yano T, Taketani Y, Nakamura Y2003. Genome-wide cDNA microarray analysis of gene-expression profiles involved in ovarian endometriosis. Int J Oncol 22:551–560 [PubMed] [Google Scholar]
- 7.Eyster KM, Boles AL, Brannian JD, Hansen KA2002. DNA microarray analysis of gene expression markers of endometriosis. Fertil Steril 77:38–42 [DOI] [PubMed] [Google Scholar]
- 8.Wu Y, Kajdacsy-Balla A, Strawn E, Basir Z, Halverson G, Jailwala P, Wang Y, Wang X, Ghosh S, Guo SW2006. Transcriptional characterizations of differences between eutopic and ectopic endometrium. Endocrinology 147:232–246 [DOI] [PubMed] [Google Scholar]
- 9.Hever A, Roth RB, Hevezi P, Marin ME, Acosta JA, Acosta H, Rojas J, Herrera R, Grigoriadis D, White E, Conlon PJ, Maki RA, Zlotnik A2007. Human endometriosis is associated with plasma cells and overexpression of B lymphocyte stimulator. Proc Natl Acad Sci USA 104:12451–12456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hull ML, Rangel Escareno C, Godsland JM, Doig JR, Johnson CM, Phillips SC, Smith SK, Tavaré S, Print CG, Charnock-Jones DS2008. Endometrial-peritoneal interactions during endometriotic lesion establishment. Am J Pathol 173:700–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wren JD, Wu Y, Guo SW2007. A system-wide analysis of differentially expressed genes in ectopic and eutopic endometrium. Hum Reprod 22:2093–2102 [DOI] [PubMed] [Google Scholar]
- 12.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP2008. The impact of microRNAs on protein output. Nature 455:64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N2008. Widespread changes in protein synthesis induced by microRNAs. Nature 455:58–63 [DOI] [PubMed] [Google Scholar]
- 14.Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z2005. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 37:766–770 [DOI] [PubMed] [Google Scholar]
- 15.Bartel DP2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297 [DOI] [PubMed] [Google Scholar]
- 16.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ2006. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38:228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I2005. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPα regulates human granulopoiesis. Cell 123:819–831 [DOI] [PubMed] [Google Scholar]
- 18.Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, Brummelkamp TR, Fleming MD, Camargo FD2008. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 451:1125–1129 [DOI] [PubMed] [Google Scholar]
- 19.Zhao Y, Samal E, Srivastava D2005. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 436:214–220 [DOI] [PubMed] [Google Scholar]
- 20.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ2008. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601 [DOI] [PubMed] [Google Scholar]
- 21.Korpal M, Lee ES, Hu G, Kang Y2008. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283:14910–14914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park SM, Gaur AB, Lengyel E, Peter ME2008. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22:894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sengupta S, den Boon JA, Chen IH, Newton MA, Stanhope SA, Cheng YJ, Chen CJ, Hildesheim A, Sugden B, Ahlquist P2008. MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proc Natl Acad Sci USA 105:5874–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu N, Williams AH, Kim Y, McAnally J, Bezprozvannaya S, Sutherland LB, Richardson JA, Bassel-Duby R, Olson EN2007. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc Natl Acad Sci USA 104:20844–20849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D2007. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1–2. Cell 129:303–317 [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Volinia S, Bonome T, Calin GA, Greshock J, Yang N, Liu CG, Giannakakis A, Alexiou P, Hasegawa K, Johnstone CN, Megraw MS, Adams S, Lassus H, Huang J, Kaur S, Liang S, Sethupathy P, Leminen A, Simossis VA, Sandaltzopoulos R, Naomoto Y, Katsaros D, Gimotty PA, DeMichele A, et al 2008. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci USA 105:7004–7009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM2005. MicroRNA gene expression deregulation in human breast cancer. Cancer Res 65:7065–7070 [DOI] [PubMed] [Google Scholar]
- 28.Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjarn M, Hansen HF, Berger U, Gullans S, Kearney P, Sarnow P, Straarup EM, Kauppinen S2008. LNA-mediated microRNA silencing in non-human primates. Nature 452:896–899 [DOI] [PubMed] [Google Scholar]
- 29.Saidi SA, Holland CM, Kreil DP, MacKay DJ, Charnock-Jones DS, Print CG, Smith SK2004. Independent component analysis of microarray data in the study of endometrial cancer. Oncogene 23:6677–6683 [DOI] [PubMed] [Google Scholar]
- 30.Smyth GK2005. Limma: linear models for microarray data. In: Gentleman RVC, Dudoit S, Irizarry R, Huber W, eds. Bioinformatics and computational biology solutions using R and Bioconductor. New York: Springer; 397–420
- 31.Lewis BP, Burge CB, Bartel DP2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20 [DOI] [PubMed] [Google Scholar]
- 32.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N2005. Combinatorial microRNA target predictions. Nat Genet 37:495–500 [DOI] [PubMed] [Google Scholar]
- 33.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T2008. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 9:582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klemmt PA, Carver JG, Koninckx P, McVeigh EJ, Mardon HJ2007. Endometrial cells from women with endometriosis have increased adhesion and proliferative capacity in response to extracellular matrix components: towards a mechanistic model for endometriosis progression. Hum Reprod 22:3139–3147 [DOI] [PubMed] [Google Scholar]
- 35.Bulun SE, Cheng YH, Yin P, Imir G, Utsunomiya H, Attar E, Innes J, Julie Kim J2006. Progesterone resistance in endometriosis: link to failure to metabolize estradiol. Mol Cell Endocrinol 248:94–103 [DOI] [PubMed] [Google Scholar]
- 36.Baskerville S, Bartel DP2005. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11:241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A2004. Identification of mammalian microRNA host genes and transcription units. Genome Res 14:1902–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mansfield JH, Harfe BD, Nissen R, Obenauer J, Srineel J, Chaudhuri A, Farzan-Kashani R, Zuker M, Pasquinelli AE, Ruvkun G, Sharp PA, Tabin CJ, McManus MT2004. MicroRNA-responsive ‘sensor’ transgenes uncover Hox-like and other developmentally regulated patterns of vertebrate microRNA expression. Nat Genet 36:1079–1083 [DOI] [PubMed] [Google Scholar]
- 39.Sethupathy P, Megraw M, Hatzigeorgiou AG2006. A guide through present computational approaches for the identification of mammalian microRNA targets. Nat Methods 3:881–886 [DOI] [PubMed] [Google Scholar]
- 40.Cui Q, Yu Z, Purisima EO, Wang E2006. Principles of microRNA regulation of a human cellular signaling network. Mol Syst Biol 2:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM2005. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell 123:1133–1146 [DOI] [PubMed] [Google Scholar]
- 42.Shazand K, Baban S, Prive C, Malette B, Croteau P, Lagace M, Racine JB, Hugo P2004. FOXO1 and c-jun transcription factors mRNA are modulated in endometriosis. Mol Hum Reprod 10:871–877 [DOI] [PubMed] [Google Scholar]
- 43.Cicatiello L, Ambrosino C, Coletta B, Scalona M, Sica V, Bresciani F, Weisz A1992. Transcriptional activation of jun and actin genes by estrogen during mitogenic stimulation of rat uterine cells. J Steroid Biochem Mol Biol 41:523–528 [DOI] [PubMed] [Google Scholar]
- 44.Hyder SM, Nawaz Z, Chiappetta C, Yokoyama K, Stancel GM1995. The protooncogene c-jun contains an unusual estrogen-inducible enhancer within the coding sequence. J Biol Chem 270:8506–8513 [DOI] [PubMed] [Google Scholar]
- 45.Salmi A, Heikkila P, Lintula S, Rutanen EM1998. Cellular localization of c-jun messenger ribonucleic acid and protein and their relation to the proliferation marker Ki-67 in the human endometrium. J Clin Endocrinol Metab 83:1788–1796 [DOI] [PubMed] [Google Scholar]
- 46.Niwa K, Hashimoto M, Morishita S, Yokoyama Y, Lian Z, Tagami K, Mori H, Tamaya T2000. Preventive effects of danazol on endometrial carcinogenesis in mice. Cancer Lett 158:133–139 [DOI] [PubMed] [Google Scholar]
- 47.Pan Q, Luo X, Toloubeydokhti T, Chegini N2007. The expression profile of micro-RNA in endometrium and endometriosis and the influence of ovarian steroids on their expression. Mol Hum Reprod 13:797–806 [DOI] [PubMed] [Google Scholar]
- 48.Noyes R, Hertig AT, Rock J1975. Dating the endometrial biopsy. Am J Obstet Gynecol 122:262–263 [DOI] [PubMed] [Google Scholar]
- 49.Thomson JM, Parker J, Perou CM, Hammond SM2004. A custom microarray platform for analysis of microRNA gene expression. Nat Methods 1:47–53 [DOI] [PubMed] [Google Scholar]
- 50.Churchill GA2002. Fundamentals of experimental design for cDNA microarrays. Nat Genet 32(Suppl):490–495 [DOI] [PubMed]
- 51.Cahill S, Smyth P, Finn SP, Denning K, Flavin R, O'Regan EM, Li J, Potratz A, Guenther SM, Henfrey R, O'Leary JJ, Sheils O2006. Effect of ret/PTC 1 rearrangement on transcription and post-transcriptional regulation in a papillary thyroid carcinoma model. Mol Cancer 5:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burmistrova OA, Goltsov AY, Abramova LI, Kaleda VG, Orlova VA, Rogaev EI2007. MicroRNA in schizophrenia: genetic and expression analysis of miR-130b (22q11). Biochemistry (Mosc) 72:578–582 [DOI] [PubMed] [Google Scholar]
- 53.Megraw M, Sethupathy P, Corda B, Hatzigeorgiou AG2007. miRGen: a database for the study of animal microRNA genomic organization and function. Nucleic Acids Res 35:D149–D155 [DOI] [PMC free article] [PubMed]
- 54.Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, Volinia S, Guler G, Morrison CD, Chan KK, Marcucci G, Calin GA, Huebner K, Croce CM2007. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci USA 104:15805–15810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Croxtall JD, Jamil A, Ayub M, Colletta AA, White JO1992. TGF-β stimulation of endometrial and breast-cancer cell growth. Int J Cancer 50:822–827 [DOI] [PubMed] [Google Scholar]
- 56.Hammond MG, Oh ST, Anners J, Surrey ES, Halme J1993. The effect of growth factors on the proliferation of human endometrial stromal cells in culture. Am J Obstet Gynecol 168:1131–1136; discussion 1136–1138 [DOI] [PubMed] [Google Scholar]
- 57.Oosterlynck DJ, Meuleman C, Lacquet FA, Waer M, Koninckx PR1994. Flow cytometry analysis of lymphocyte subpopulations in peritoneal fluid of women with endometriosis. Am J Reprod Immunol 31:25–31 [DOI] [PubMed] [Google Scholar]
- 58.Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ2008. A Double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 68:7846–7854 [DOI] [PubMed] [Google Scholar]
- 59.Christoffersen NR, Silahtaroglu A, Orom UA, Kauppinen S, Lund AH2007. miR-200b mediates post-transcriptional repression of ZFHX1B. RNA 13:1172–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu Z, Wang C, Wang M, Li Z, Casimiro MC, Liu M, Wu K, Whittle J, Ju X, Hyslop T, McCue P, Pestell RG2008. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol 182:509–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sylvestre Y, De Guire V, Querido E, Mukhopadhyay UK, Bourdeau V, Major F, Ferbeyre G, Chartrand P2007. An E2F/miR-20a autoregulatory feedback loop. J Biol Chem 282:2135–2143 [DOI] [PubMed] [Google Scholar]
- 62.Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, Croce CM, Brunetti E, Grignani F, Peschle C2007. MicroRNAs 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat Cell Biol 9:775–787 [DOI] [PubMed] [Google Scholar]
- 63.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M2007. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 100:416–424 [DOI] [PubMed] [Google Scholar]
- 64.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A2006. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet 38:1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu C, Lu Y, Pan Z, Chu W, Luo X, Lin H, Xiao J, Shan H, Wang Z, Yang B2007. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci 120:3045–3052 [DOI] [PubMed] [Google Scholar]