

Abstract
Glucocorticoids have major antiinflammatory effects. Because COX-2 is the rate-limiting enzyme for proinflammatory prostaglandins, this study investigated the combinatorial inhibitory role of glucocorticoid receptor (GR) in COX-2 gene induction in macrophages and sought to identify the molecular mechanisms for that inhibition. Glucocorticoid-activated GR repressed COX-2 gene induction by lipopolysaccharide (LPS). Activated GR inhibited LPS-induced activator protein 1 activity, which in turn decreased activating transcription factor 2/c-Jun phosphorylation. The inhibition of MAPK-dependent activating transcription factor 2/c-Jun phosphorylation by GR in COX-2 repression was a result of MAPK phosphatase-1 (MKP-1) induction. Although GR did not inhibit LPS-induced p65 phosphorylation or nuclear factor-κB DNA binding activity, deletion of the nuclear factor-κB binding site in the COX-2 gene suppressed the ability of glucocorticoid to attenuate COX-2 induction. Chromatin immunoprecipitation and transfection assays revealed that a p65 DNA complex involving GR-bound GR-interacting protein 1 (GRIP1) also contributed to COX-2 repression. Additional knockdown and transfection assays identified other inflammatory genes coordinately regulated by MKP-1 and GRIP1. In summary, activated GR was found to antagonize the LPS-dependent induction of the COX-2 gene via a novel combinatorial mechanism involving MKP-1-mediated activator protein 1 inhibition and GR/GRIP1 recruitment to the p65 DNA complex; moreover, this work facilitated the identification of other GR-responding MKP-1/GRIP1-regulated genes.
Glucocorticoid receptor (GR) antagonizes induction of inflammatory genes via a novel combinatorial mechanism involving MKP-1-mediated AP-1 inhibition and GR/GRIP1 recruitment to the p65 DNA complex.
Macrophages play a central role in the inflammatory process by producing a group of primary mediators that includes prostanoid lipid metabolites, NO, and cytokines. Of these, the production of prostaglandins (PGs) represents an important step in the mediation of the inflammatory process (1). Because COX-2 is the rate-limiting enzyme for PG production, transcriptional activation of this gene leads to a large production of PGs (2, 3).
The glucocorticoids are a group of compounds that greatly affect the inflammatory process: they have potent antiinflammatory and immunosuppressive activities, which render glucocorticoids indispensable for the management of inflammatory, autoimmune, and lymphoproliferative diseases (4, 5, 6, 7). Glucocorticoids influence gene expression via binding of the glucocorticoid receptor (GR) to the glucocorticoid response element (GRE) of a target gene (8). The tethering model proposes that activated GR may be recruited to the target gene through protein-protein interaction with transcription factors [e.g. activator protein 1 (AP-1), nuclear factor-κB (NF-κB), and signal transducer and activator of transcription] (9, 10, 11). Additionally, studies indicate that GR/GRE binding may affect adjacent regulatory elements through the composite mode. Glucocorticoids inhibit inflammatory genes by means of multiple mechanisms including perturbation of signaling pathways, sequestration of transcription factors, corepressor recruitment, and inhibition of transcriptional initiation machinery. Most of the previous studies on the role of glucocorticoids in inflammatory gene regulation have focused on the role of single transcription factor modulation by glucocorticoids.
One approach to work out these regulatory mechanisms is to study the modulating effect of glucocorticoid treatment on the action of a known COX-2 inducer. Lipopolysaccharide (LPS) is a COX-2 inducer that mainly stimulates toll-like receptor (TLR)-4 in macrophages. As a result of LPS binding, the TLR cytosolic domain then recruits adaptors such as MYD88, toll-IL-1 receptor domain-containing adaptor protein/MaL, toll-IL-1 receptor domain-containing adaptor-inducing interferon β (TRIF), and TRIF-related adaptor molecule. This interaction with the adaptors leads to activation of the MAPKs and/or the NF-κB pathways for COX-2 gene induction (12, 13). In addition, gene regulation of COX-2 involves the cooperative action of transcription factors, such as AP-1, NF-κB, cAMP response element binding protein (CREB), and CCAAT/enhancer binding protein (C/EBP) (1, 14, 15, 16).
The interaction between ligand-bound TLRs and MYD88 leads to activation of the MAPKs that, in turn, activate transcription factors (e.g. AP-1, CREB, and C/EBPs) through phosphorylation cascades (17, 18). Because TLRs regulate diverse signaling pathways, the concentration levels of activated effector molecules must be balanced by negative regulators. Among those regulators, MAPK phosphatase-1 (MKP-1) preferentially dephosphorylates activated MAPKs; therefore, MKP-1 inactivates the transcription factors required for the induction of inflammatory genes (19, 20, 21). The MKP-1 gene is induced by inflammatory stimuli (22). Glucocorticoids can also induce MKP-1 via GR (23, 24). However, the role of MKP-1 induction by glucocorticoids in antiinflammatory action is still under debate (20, 25, 26). Moreover, the downstream network responsible for COX-2 repression has not been fully understood, although it has been recognized that MKP-1 induction by GR partly contributes to COX-2 gene inhibition (20).
As previously stated, TLRs regulate the NF-κB pathway. Glucocorticoids also affect this pathway. GR represses the NF-κB-dependent induction of the IL-8 and TNF-α genes (7, 27). One plausible explanation for this effect is that GR might induce IκBα for the inhibition of the p65 subunit. Alternatively, GR may be recruited to the target gene via protein-protein interaction (11, 27). However, IκBα sequestration of p65 and the possible interaction between GR and p65 may be gene specific (7, 28). Moreover, the inhibitory effect of GR on the NF-κB-mediated induction of the COX-2 gene and the molecular mechanism for this effect remain to be clarified.
This study investigated the GR-mediated attenuation of COX-2 induction by LPS in macrophages, in terms of identifying the responsible molecules and the signaling cascade. Here, we report that GR represses LPS induction of COX-2 by means of a combinatorial pathway that involves both GR-dependent MKP-1 induction and interaction between activated p65 and GR-bound GR interacting protein-1 (GRIP1). Moreover, the novel GR-inhibitory mechanism elucidated during the course of this study allows us to identify other inflammatory genes regulated by the same pathway.
Results
GR attenuation of the LPS induction of COX-2
To assess the effects of GR on the LPS induction of COX-2, Raw264.7 cells were incubated with LPS alone or in combination with dexamethasone (LPS+dexamethasone, dexamethasone pretreatment for 30 min). LPS treatment (24 h) induced COX-2 levels, whereas glucocorticoid treatment attenuated that response in a concentration-dependent manner (Fig. 1A, left). A time course experiment showed that dexamethasone (0.1 μm) inhibited the induction of COX-2 by LPS at all time points examined (ranging between 6 h and 48 h) (Fig. 1A, right). Real-time RT-PCR analysis indicated that COX-2 repression by dexamethasone was accompanied by a significant decrease in the amount of COX-2 mRNA (Fig. 1B, left). COX-2 mRNA was not detected in cells treated with either vehicle or dexamethasone alone (data not shown).
Fig. 1.
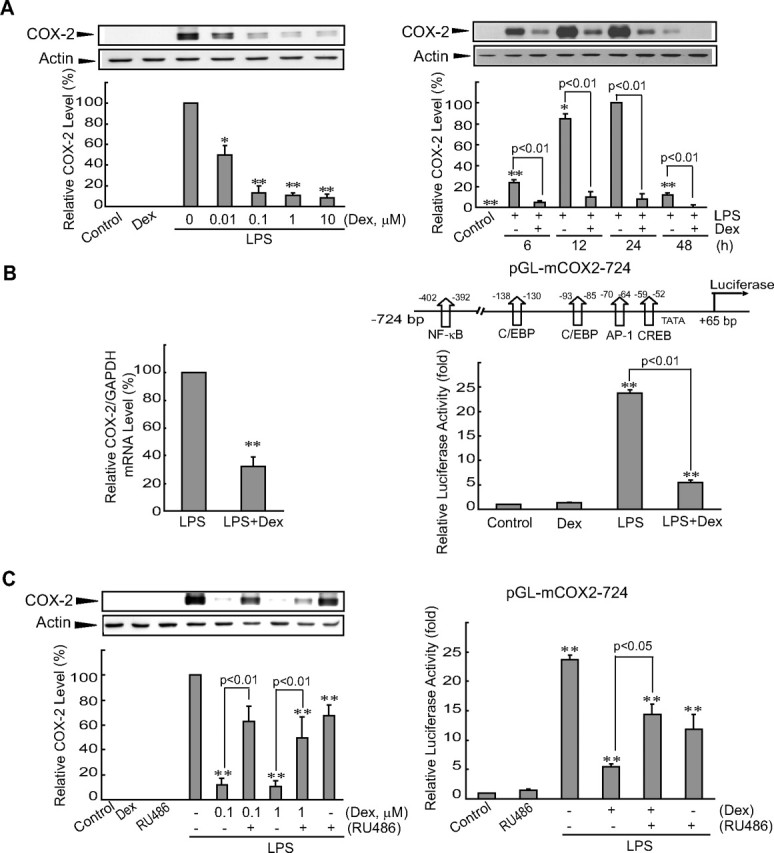
Activated GR inhibits LPS induction of COX-2. A, COX-2 immunoblots. Raw264.7 cells were incubated with varying concentrations of dexamethasone (Dex) for 30 min and were continuously exposed to 0.3 μg/ml LPS for 24 h (left). Time course effects were assessed in cells treated with 0.1 μm Dex (right). Each lane was loaded with 10 μg of cell lysate. Data represent the mean ± se of three replicates; each relative value was normalized with respect to the COX-2 level in cells treated with LPS for 24 h. B, Transcriptional inhibition by Dex of the LPS induction of the COX-2 gene. COX-2 mRNA levels were measured by real-time RT-PCR assays in cells treated with LPS for 6 h after 0.1 μm Dex pretreatment for 30 min (LPS+Dex). Data represent mean ± se of six replicates, with the COX-2 mRNA level in LPS-treated cells used as the normalization standard (left). COX-2 reporter activity was measured in cells that had been transfected with pGL-mCOX2-724 and pCMV-LacZ, and subsequently treated with LPS or LPS+Dex for 24 h. Data represent mean ± se of four replicates (right). C, Reversal by RU486 of the Dex-inhibitory effect on COX-2 induction. Cells were treated as described above with or without 0.1 μm RU468 treatment. In the left graph, data represent relative expression of each treatment group compared with that of LPS. For all graphs, the statistical significance of differences between each treatment group and the normalization standard/control was determined (*, P < 0.05; **, P < 0.01).
To determine whether glucocorticoid treatment inhibits COX-2 gene transcription, reporter gene assays were performed in cells that had been transfected with pGL-mCOX-2-724, which contained the luciferase structural gene downstream of the COX-2 promoter (−724 bp). Treatment of the transfected cells with LPS caused a 25-fold increase in luciferase activity compared with control (Fig. 1B, right). Exposure of the cells to LPS+dexamethasone resulted in a substantial decrease in luciferase induction compared with cells treated only with LPS. Next, we examined the effect of RU486, a GR antagonist, on dexamethasone’s repression of COX-2 induction by LPS. Pretreatment of the cells with RU486 (0.1 μm, 30 min) diminished the ability of dexamethasone to repress both COX-2 protein and luciferase induction by LPS (Fig. 1C). A slight decrease by RU486 in LPS induction of COX-2 might be due to its partial agonist effect for GR (29). Overall, these results showed that GR inhibits the activation of COX-2 gene transcription by LPS in macrophages.
GR inhibition of c-Jun N-terminal kinase (JNK)/p38 and activation of activating transcription factor 2 (ATF2)/c-Jun
To investigate whether GR inhibits major transcription factors, we first asked whether GR activation affects MAPK phosphorylation, a requirement for AP-1-mediated COX-2 induction (16, 30). Indeed, LPS increased phosphorylation of all three MAPKs, whereas glucocorticoid treatment inhibited the phosphorylation of JNK and p38 (Fig. 2A). The level of ERK1/2 phosphorylation by LPS was not changed by dexamethasone treatment. Because ATF2 and c-Jun are well-recognized substrates of JNK and p38 (31, 32, 33), the effect of glucocorticoid on their phosphorylation was examined. Immunoblot analyses confirmed phosphorylation of ATF2 and c-Jun after LPS treatment. Glucocorticoid treatment reduces the ability of LPS to promote the phosphorylation of ATF2 and c-Jun (Fig. 2B).
Fig. 2.
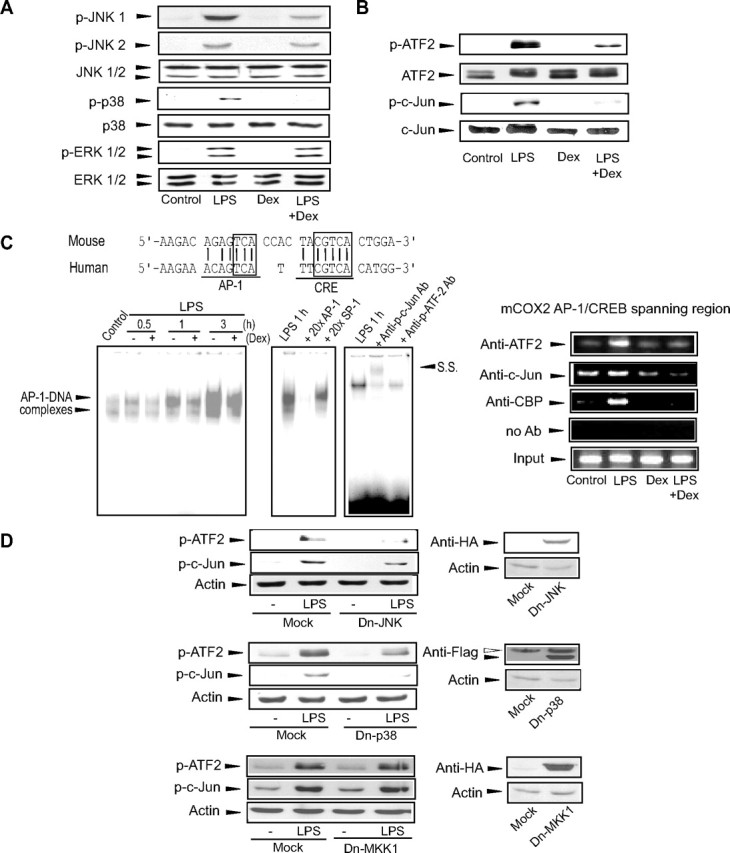
Inhibition of MAPK phosphorylation by GR diminishes the ability of LPS to promote ATF2/c-Jun DNA binding. A, Inhibition of JNK1/2 and p38 phosphorylation by GR. Phosphorylated or total MAPKs were immunoblotted from the lysates of cells treated with LPS or LPS+Dex for 30 min. B, Inhibition of ATF2 and c-Jun phosphorylation by GR. Phosphorylated or total ATF2 and c-Jun were immunoblotted from the lysates of cells treated as indicated above for 1 h. C, AP-1 gel shift and ChIP assays. The depicted sequences show alignment of the AP-1 binding sites in the murine and human COX-2 genes (upper). The boxes indicate core nucleotide sequences. Gel shift assays were performed with the nuclear extracts of cells treated with LPS or LPS+Dex (left). Arrowheads indicate the region showing AP-1 DNA binding. The specificity of AP-1 DNA binding was assessed by excess probe competition, antibody supershift, or immunocompetition assays (S.S., supershift). For ChIP assays, DNA-protein complexes immunoprecipitated with antibody were PCR amplified using the primers flanking the AP-1/CREB binding site. The input control was set as one tenth of the cross-linked lysates. D, Inhibition of ATF2 and c-Jun phosphorylation by Dn mutants of JNK or p38. Phosphorylated ATF2 or c-Jun was immunoblotted from the lysates of transfected cells (with the plasmid encoding Dn-JNK, Dn-p38, or Dn-MKK1) that were subsequently treated with LPS for 1 h. pCDNA3.1(+) was used as a mock transfection control. Immunoblots for tagged epitopes verified overexpression of Dn-MAPKs (open arrowhead, nonspecific band). The results were confirmed by at least three replicates. Ab, Antibody; CRE, cAMP response element; Dex, dexamethasone; HA, hemagglutinin.
Activated ATF2 and c-Jun both bind to the AP-1 binding element for the transactivation of inflammatory genes (33, 34, 35). We next explored whether GR inhibits the binding between AP-1 and DNA that is required for COX-2 induction. Murine and human COX-2 genes share the conserved AP-1 binding site (Fig. 2C). Gel shift analyses of protein binding to the murine AP-1 binding site indicated that dexamethasone inhibited LPS-activated AP-1 DNA binding (Fig. 2C, left), which is consistent with additional data showing that dexamethasone treatment caused a 50% decrease in LPS-induced AP-1 reporter activity (data not shown). The addition of a 20-fold excess of unlabeled AP-1 oligonucleotide to nuclear extracts abolished AP-1 DNA binding, whereas excess unlabeled specificity protein 1 oligonucleotide failed to do so (Fig. 2C, left). Supershift and immunoinhibition experiments confirmed that the phosphorylated forms of ATF2 and c-Jun were involved in formation of the AP-1 DNA complex (Fig. 2C, left). To determine whether ATF2 or c-Jun associates with the CREB-binding protein (CBP) on the COX-2 gene, ChIP assays were performed using the antibodies against those proteins (Fig. 2C, right). Immunoprecipitated DNA complexes were PCR amplified using the primers flanking the proximal and distal regions of the AP-1/CREB binding site. The intensities of PCR products were all distinctly higher in LPS-treated cells than vehicle-treated ones. It is worth noting that dexamethasone almost completely reversed the induction effect of LPS on the band intensities of ATF2, c-Jun, and CBP (Fig. 2C, right).
In addition, the effect of a histone deacetylase inhibitor [trichostatin A (TSA)] on GR repression of the formation of the CBP-transcription factor complex was assessed. TSA treatment somewhat antagonized the ability of glucocorticoid to repress COX-2 induction (Fig. S1A published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org), suggesting that GR recruits histone deacetylase. Similarly, TSA treatment partially restored the ability of LPS to induce luciferase from pGL-mCOX-2-724 containing the AP-1 binding site (supplemental Fig. S1B). ChIP analysis using antiacetylated histone H3 antibody supported the supposition that dexamethasone decreases chromatin decondensation for the COX-2 gene (Fig. S1C). These results provide evidence that GR inhibits not only the phosphorylation of ATF2 and c-Jun, but also their DNA binding and CBP recruitment.
To understand the mechanism underlying ATF2 and c-Jun phosphorylation, we examined the effect of dominant-negative (Dn) mutant of JNK or p38 on the phosphorylation of those proteins. Transfection of either Dn-JNK or Dn-p38 in macrophages decreased the LPS-induced phosphorylation of ATF2 and c-Jun (Fig. 2D), whereas transfection with Dn-MKK1 (Dn-MEK1) had no effect. Therefore, ATF2 and c-Jun phosphorylation depends on LPS activation of JNK1 and p38.
Role of GR-induced MKP-1 in ATF2/c-Jun dephosphorylation and COX-2 repression
Glucocorticoids stimulate MKP-1 induction through its GRE (23, 24). To confirm the role of GR in MKP-1 induction, the effect of dexamethasone on MKP-1 expression was determined. Dexamethasone effectively induced MKP-1 in a dose-dependent manner via transactivation (pGL-MKP1–1716) (supplemental Fig. S2A). RU486 reversed the ability of dexamethasone to induce MKP-1, confirming GR dependency. Moreover, GR enhanced the ability of LPS to induce MKP-1 (Fig. S2B). These results demonstrate that GR activation leads to MKP-1 induction, which synergizes the action of LPS.
Next, the effect of MKP-1 overexpression on the phosphorylation of ATF2, c-Jun, and MAPKs was assessed to determine the effect of glucocorticoid-induced MKP-1 on COX-2 gene expression. LPS-induced phosphorylation of JNK and p38 was inhibited by the ectopic expression of MKP-1 using pSG5-mMKP1, as well as that of ATF2 and c-Jun (Fig. 3A). Phosphorylation of ERK1/2 was not altered by MKP-1 expression. MKP-1 overexpression abrogated the ability of LPS to induce luciferase production from pGL-mCOX-2-724 (Fig. 3B, left). Moreover, small interfering RNA (siRNA) knockdown of murine MKP-1 reversed COX-2 repression by GR. Overexpression of human MKP-1, the transcript of which is insensitive to murine MKP-1 siRNA, inhibited the LPS induction of COX-2 luciferase expression, but was not knocked down by the siRNA (Fig. 3B, middle). Immunoblottings confirmed the species specificity of murine MKP-1 siRNA (Fig. 3B, right).
Fig. 3.
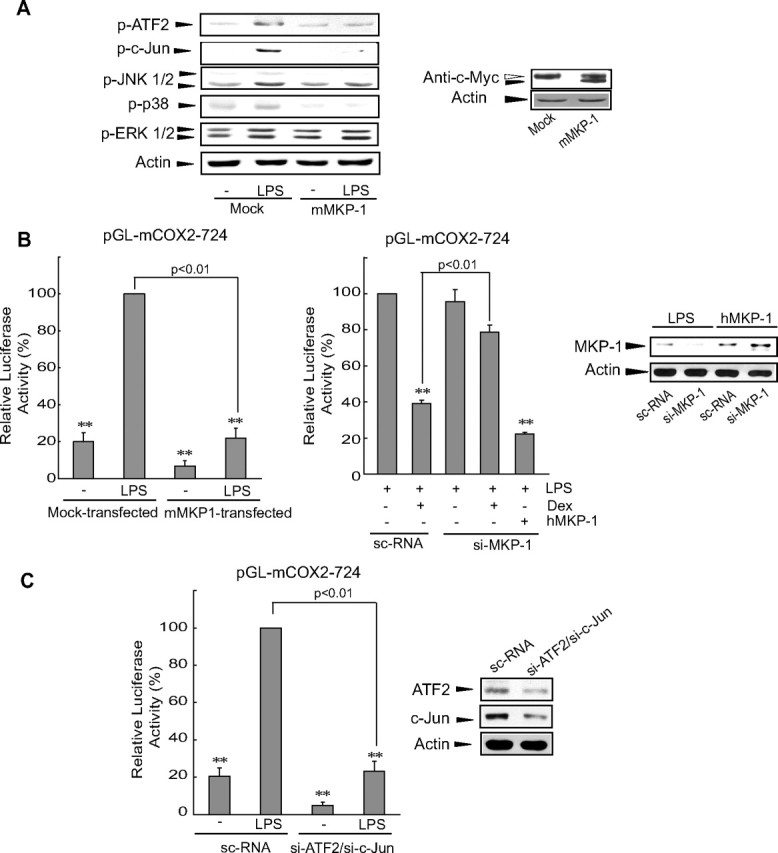
MKP-1 induction by GR leads to COX-2 gene repression through the dephosphorylation of MAPK or ATF2/c-Jun. A, Inhibition of JNK- and p38-dependent ATF2/c-Jun phosphorylation by MKP-1. Phosphorylated MAPK and ATF2/c-Jun were immunoblotted from the lysates of cells that had been transfected with either empty vector (pSG5) or pSG5-mMKP1 encoding murine MKP-1 and that were subsequently treated with LPS (for 30 min and 1 h for MAPK and ATF2/c-Jun assays, respectively). B, The role of MKP-1 in GR repression of COX-2 induction. Either pSG5-mMKP1 or pSG5-hMKP1 in combination with murine MKP-1 siRNA (si-MKP-1) was cotransfected with pGL-mCOX2-724. COX-2 luciferase activities were measured 24 h after treatment. Relative activity represents the level of luciferase compared with that of LPS-treated, mock- [/or scrambled control siRNA (sc-RNA-)] transfected cells. MKP-1 immunoblotting verifies the species specificity of si-MKP-1 (right). C, Inhibition of LPS induction of the COX-2 gene by an ATF2/c-Jun knockdown. Luciferase activity from pGL-mCOX2-724 was measured in cells that had been transfected with ATF2 and c-Jun siRNAs and had been subsequently treated with LPS for 24 h. Immunoblots confirmed protein silencing. Data were normalized in comparison with the levels shown in the sc-RNA-transfected, LPS-treated cells. For all graphs, the statistical significance of differences between each treatment groups and the normalization standard/control was determined (**, P < 0.01). Dex, Dexamethasone; si-ATF2, ATF2 siRNA; si-c-Jun, c-Jun siRNA.
We then examined the effects of ATF2/c-Jun knockdown on luciferase induction. LPS-induced luciferase expression in cells containing pGL-mCOX-2-724 was almost completely inhibited by cotransfection of siRNAs directed against ATF2 and c-Jun (Fig. 3C). In light of these results, we conclude that GR-induced MKP-1 antagonizes JNK- and p38-dependent phosphorylation of ATF2 and c-Jun. This antagonistic action thereby inhibits AP-1-dependent COX-2 gene induction by LPS.
Effects of GR on NF-κB and CREB activation by LPS
In addition to AP-1, other transcription factors such as NF-κB and CREB play roles in the induction of COX-2 by inflammatory stimuli (1). IκBα phosphorylation and proteasomal degradation precede NF-κB activation; activated NF-κB consisting of p65 and p50 translocates into the nucleus after being relieved from its IκBα binding. Separately, CREB is phosphorylated and primed for target gene transactivation through multiple pathways involving MAPKs. We observed that LPS enhanced the binding of both NF-κB and CREB to DNA; the level of LPS-mediated enhancement failed to be inhibited by glucocorticoid treatment (Fig. 4A). Chromatin immunoprecipitation (ChIP) assays confirmed that inhibition in NF-κB or CREB DNA binding could not be elicited by dexamethasone (Fig. 4B): multiple analyses showed that the band intensities of LPS+dexamethasone samples for p65 and CREB were 110 ± 13% and 121 ± 12% of LPS samples. In addition, immunoblot analyses were performed to explore whether COX-2 gene repression by dexamethasone involves IκBα phosphorylation. Because dexamethasone treatment did not affect IκBα and p65 phosphorylation levels, activated GR did not change the promotion of p65 activation by LPS (Fig. 4C). Similarly, dexamethasone treatment failed to inhibit the increase in CREB phosphorylation caused by LPS. Overall, GR activation does not change either the phosphorylation status or the DNA binding activity of NF-κB or CREB.
Fig. 4.

Activated GR does not change either NF-κB or CREB activation. A, Gel shift assays for NF-κB or CREB DNA binding. Nuclear extracts were incubated with NF-κB or CREB binding oligonucleotides. The binding specificity was confirmed by excess probe competition or supershift assay. B, ChIP assays. DNA-protein complexes were precipitated with anti-p65 or anti-CREB antibody and were subjected to PCR amplification using the flanking primers for either the NF-κB or the AP-1/CREB binding site. One tenth of cross-linked lysates served as the input control. C, Effects of GR on IκBα, p65, or CREB phosphorylation. The phosphorylated forms of IκBα, p65, or CREB were immunoblotted from the lysates of cells treated as indicated above for 1 h. The levels of the unphosphorylated forms verify equal protein loading. The results were confirmed by at least three replicates. Dex, Dexamethasone.
GR recruitment to the p65 DNA complex in COX-2 repression
Because NF-κB is known to be tethered by GR (7, 11), an assessment of whether COX-2 repression resulted from a GR-tethered alteration in NF-κB activity was performed. The region comprising the NF-κB binding site of the COX-2 gene was amplified by PCR in samples of genomic DNA-protein complex that had been immunoprecipitated with GR. Among the samples examined, LPS+dexamethasone treatment notably increased the intensity of PCR product (Fig. 5A). By contrast, GR failed to bind to the AP-1/CREB region of the binding site (Fig. 5A). Moreover, immunoprecipitation and immunoblot assays identified direct binding of GR with p65 after LPS+dexamethasone treatment (Fig. 5B).
Fig. 5.

Activated GR is recruited to p65 bound to the NF-κB binding site of the COX-2 gene. A, ChIP assays showing interaction of GR with activated p65 bound to the NF-κB binding site (upper). Genomic DNA-protein complex was precipitated with anti-GR antibody, and the immunoprecipitate was subjected to PCR amplification using the flanking primers for either the NF-κB or AP-1 binding site. Data represent mean ± se of three replicates, with the relative band intensity of NF-κB binding site in vehicle-treated cells used as the normalization standard (*, P < 0.05)(lower). B, Immunoprecipitation-immunoblot assays. GR immunoprecipitate was immunoblotted with anti-p65 antibody to assess the presence of an interaction between GR and p65 (upper). Immunoprecipitate using preimmune IgG was used as a negative control. GR immunoblotting was performed as a loading control. The statistical significance of differences between each treatment group and the normalization control was determined from three replicates (*, P < 0.05) (lower) (IP, immunoprecipitation; IB, immunoblot). C, Inhibition of COX-2 gene induction by mutation of the NF-κB binding site. The relative luciferase activity was determined in cells that had been transfected with either pGL-mCOX2-724 or pGL-mCOX2(mNF-κB)-724 and had been subsequently treated as indicated above for 24 h. Data represent mean ± se of three replicates normalized against the vehicle control. Statistical significance of differences between treatments groups compared with vehicle control in cells transfected with pGL-mCOX2(mNF-κB)-724 was determined (**, P < 0.01; N.S., not significant). Ab, Antibody; Dex, dexamethasone.
Given the important role of NF-κB in COX-2 expression, we next examined whether mutation of the NF-κB binding site affects the GR repression of LPS-induced COX-2 gene expression. Mutation of the NF-κB binding site in pGL-mCOX-2-724 [i.e. pGL-mCOX2-(mNF-κB)-724] decreased the ability of LPS to induce COX-2 reporter activity (Fig. 5C). However, activated GR did not further suppress luciferase induction by LPS in cells with pGL-mCOX2-(mNF-κB)-724, indicating that deletion of the NF-κB binding site removed the GR-dependent effect. The inhibitory effect of GR on NF-κB was confirmed by the NF-κB-driven promoter activity (data not shown). These data verify that GR has a role in NF-κB-dependent COX-2 gene induction. Taken together, our results provide evidence that the specific binding of GR to activated p65 recruited to the COX-2 promoter contributes to COX-2 repression.
Binding of GRIP1 to the GR-NF-κB DNA complex
The observation that GR binds to p65 recruited to the NF-κB binding site prompted us to search for a negative regulator of the COX-2 gene. Other work has shown that GR may recruit silencing mediators for retinoid and thyroid hormone receptors (36, 37) or nuclear receptor corepressors (38). Transfection experiments using the constructs encoding retinoid and thyroid hormone receptor or nuclear receptor corepressor indicated that those corepressors might not be responsible for COX-2 repression (data not shown). In a continuing effort to find the negative regulator, the interaction between GRIP1 and GR was examined because GRIP1 functions as a coactivator or a corepressor (10, 11, 39). ChIP assays revealed that LPS+dexamethasone treatment promoted binding of GRIP1 to the NF-κB binding site (Fig. 6A). Immunoprecipitation and immunoblot analysis supported the notion that GRIP1 binds to GR in cells treated with either dexamethasone or LPS+dexamethasone (Fig. 6B). These results indicate that GRIP1-bound GR is recruited to p65 DNA complex.
Fig. 6.
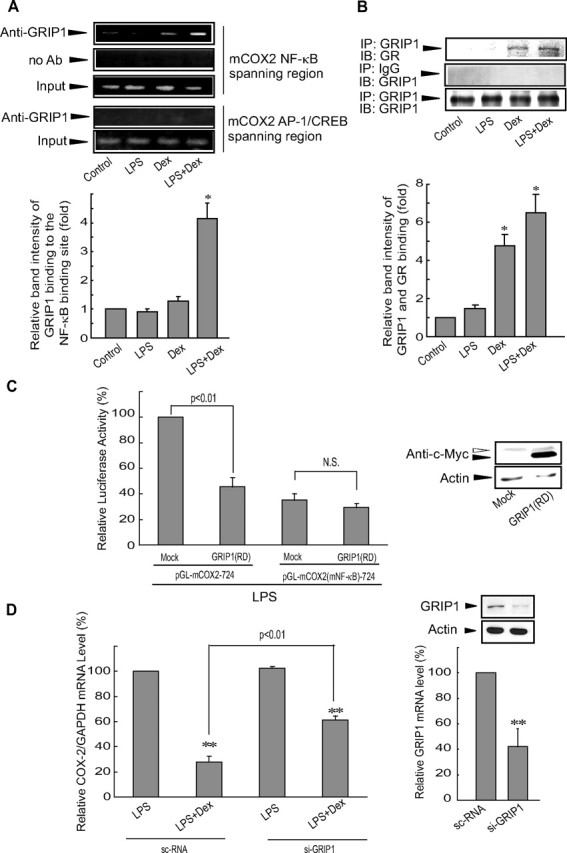
FIG. 6.
GR-bound GRIP1 is recruited to p65 DNA complex for COX-2 repression. A, Interaction between GR-bound GRIP1 and activated p65. DNA-protein complex was precipitated with anti-GRIP1 antibody, and the immunoprecipitate was subjected to PCR amplification (upper). Data represent mean ± se of three replicates, with the relative band intensity of NF-κB binding site in vehicle-treated cells used as the normalization standard (*, P < 0.05) (lower). B, GRIP1 immunoprecipitate was immunoblotted for GR (upper). The statistical significance of differences between each treatment group and the normalization control was determined from three replicates (*, P < 0.05) (lower) (IP, immunoprecipitation; IB, immunoblot). C, GRIP1-RD inhibition of NF-κB-dependent COX-2 induction. Luciferase activity from either pGL-mCOX2-724 or pGL-mCOX2(mNF-κB)-724 was measured in the lysates of cells that had been cotransfected with a mock vector or GRIP1-RD and had been subsequently treated with LPS for 24 h. Immunoblotting for c-Myc confirmed overexpression of myc-tagged GRIP1-RD. Data represent the mean ± se of three replicates (N.S., nonsignificance of mean difference). D, Reversal by si-GRIP1 of GR repression of COX-2 induction. The relative COX-2 mRNA levels to those of GAPDH were assessed by real-time RT-PCR assays in cells treated with LPS or LPS+Dex for 6 h after either scrambled control siRNA (sc-RNA) or mGRIP1 siRNA transfection (left). Knockdown of GRIP1 was verified by a decrease in GRIP1 protein (right upper) or mRNA (right lower). Data represent the mean ± se of three separate replicates with data normalized against the level of COX-2 mRNA in sc-RNA-transfected cells treated with LPS. The statistical significance of differences between each treatment group and the normalization standard was determined (**, P < 0.01). Ab, Antibody; Dex, dexamethasone; si-GRIP1, GRIP1 siRNA.
To further study the interaction between GRIP1 and activated GR, the possible effect of the repressor domain (RD) of GRIP1 (GRIP1-RD) on luciferase expression from either pGL-mCOX2-724 or pGL-mCOX2-(mNF-κB)-724 (Fig. 6C) was explored. In this functional study, we used the expanded domain of GRIP1 comprising both nuclear receptor interacting domain (NID) and RD due to stability questions concerning the use of the RD alone (11, 41). In LPS-treated cells, GRIP1-RD transfection significantly inhibited luciferase induction from pGL-mCOX2-724. However, GRIP1-RD had no effect on luciferase induction from pGL-mCOX2-(mNF-κB)-724. Moreover, siRNA knockdown of GRIP1 reversed COX-2 repression by GR (Fig. 6D). Collectively, these results demonstrate that the interaction of GRIP1 with activated GR inhibits NF-κB-mediated COX-2 gene induction.
GR regulation of other MKP-1/GRIP1-responsive genes
To better understand the extent of gene regulation by the combinatorial mechanism involving both the MKP-1 regulation by GR and the GRIP1-GR-NF-κB interaction, we tried to identify other inflammatory genes regulated by this mechanism. In silico analysis of microarray data obtained from LPS-treated interferon regulatory factor (IRF3)-, MYD88-, or MKP-1-knockout cells suggested that the COX-2, IL-6, CXCL-5, REL, CCL4, FOS1, DTR, CXCL1, CCL3, and F3 genes might be regulated by the MYD88 and MKP-1 pathway independently of IRF3 (supplemental Fig. S3). GRIP1 siRNA knockdown and Dn-MKP-1 transfection experiments verified that GR repression of the COX-2, IL-6, or CXCL-5 gene was obviously reversed by GRIP1 knockdown and to much greater extents by GRIP1 knockdown plus Dn-MKP-1 transfection (Fig. 7A). These findings are consistent with the results of in silico analysis, demonstrating that the genes are regulated by the GR-mediated MKP-1 and GRIP1-dependent pathway (Fig. 7B).
Fig. 7.
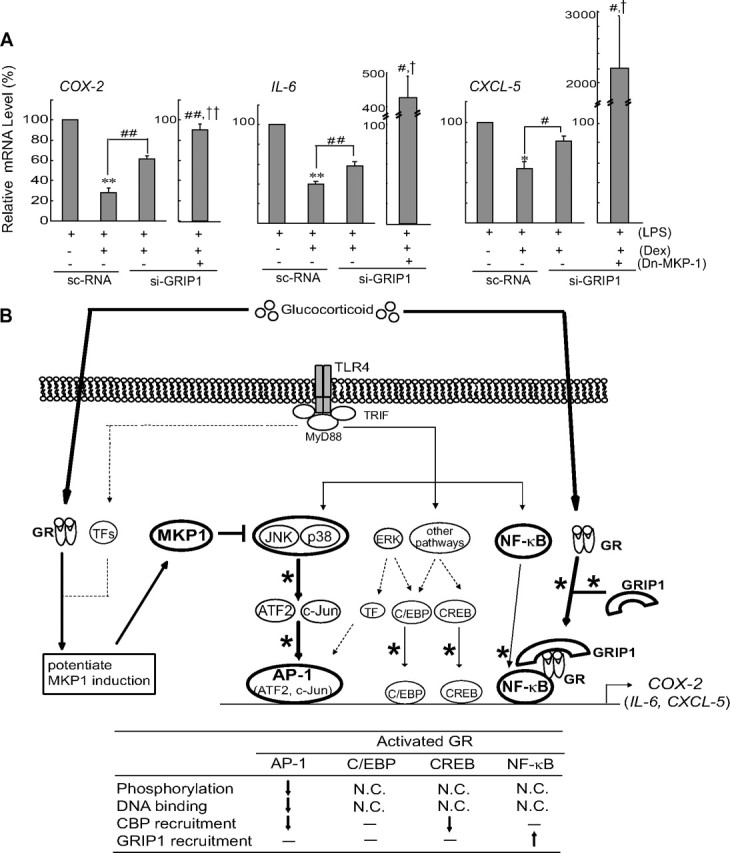
Activated GR inhibits the induction of other MKP-1/GRIP1-regulated genes. A, RT-PCR assays. COX-2, IL-6, and CXCL-5 transcripts were measured by real-time RT-PCR analyses in cells that had been treated with LPS or LPS+Dex after transfection of si-GRIP1 and/or pSG5-mMKP1-CS (Dn-MKP-1). Mock transfection represents transfection of both scrambled control siRNA (sc-RNA) and pSG5. Data represent the mean ± se of three replicates; the relative response level of each treatment was normalized against the response to LPS treatment in the mock/sc-RNA control (tests of significance of mean differences compared with LPS: **, P < 0.01; *, P < 0.05; tests of significance of mean differences compared with sc-RNA-transfected cells treated with LPS+Dex: ##, P < 0.01; #, P < 0.05; and tests of significance of mean differences compared with mock/si-GRIP1 and Dn-MKP-1/si-GRIP1-transfected cells treated with LPS+Dex: ††, P < 0.01; †, P < 0.05). B, A schematic diagram illustrating the combinatorial inhibitory mechanism (bold lines) regulated by GR. GR inhibits TLR4-dependent COX-2 gene induction in macrophages via a novel inhibitory mechanism that involves MKP-1-mediated AP-1 inhibition and GR-GRIP1 recruitment to p65 DNA complex. Dex, Dexamethasone; N.C., not changed; siGRIP1, GRIP1 siRNA; TF, transcription factor.
Discussion
COX-2 gene induction by LPS is regulated by multiple transcription factors (e.g. AP-1, NF-κB, CREB, and C/EBP), and the formation of the activated transcription factor-DNA complex recruits either coactivators or corepressors for gene regulation. Because mutation of each transcription factor’s binding site partly impairs the induction of COX-2 gene, a set of transcription factors must be necessary for full responsiveness (3, 14, 16, 30). In the present study, we confirmed that GR effectively inhibits LPS induction of COX-2 at the transcriptional level. Numerous studies have shown that glucocorticoids inhibit inflammatory genes by mechanisms including simple, composite, or tethering modes. Nevertheless, the regulatory mechanism of GR seems to be more complex and gene specific than can be explained using any single mode. Here, we reveal a novel MKP-1/GRIP1-dependent combinatorial mechanism for COX-2 gene repression by GR. Moreover, our work identifies IL-6 and CXCL-5 as other genes regulated by this mechanism.
In this study, we identified that the transcription factor tethered by activated GR is NF-κB, but not AP-1 (Fig. 5A). Among the AP-1 components (e.g. ATF2, Jun, Fos, and Fra), ATF2 and c-Jun bind to the AP-1 response element to form an activating DNA complex that is required for gene induction by inflammatory or genotoxic stimuli (33, 34, 35). Glucocorticoid treatment inhibits LPS-induced AP-1 activity as a consequence of the inhibition of ATF2 and c-Jun phosphorylation; therefore, AP-1-mediated COX-2 induction by LPS is counterbalanced by GR activation. Studies have shown that ATF2 and c-Jun are the substrates of JNK and p38 (31, 32, 33). Two experimental observations strongly support an inhibitory role of GR in MAPK-mediated ATF2/c-Jun activation: 1) GR repression of COX-2 was accompanied by JNK and p38 dephosphorylation and 2) the Dn mutants of JNK and p38 inhibited ATF2 and c-Jun phosphorylation. Our data shown in Fig. 2C and supplemental Fig. S1C indicated that GR activation allows the ATF2/c-Jun complex to dissociate from the COX-2 gene, which prevents CBP recruitment and histone acetylation. In addition, COX-2 repression by glucocorticoid may result, at least in part, from the decreased recruitment of ATF2/c-Jun to the AP-1 DNA element and the consequent decrease in CBP coactivator binding. In this study, dexamethasone treatment did not completely inhibit AP reporter activity. The remaining AP-1 activity might be due to involvement of other AP-1 members such as Fos and Fra, both of which are regulated by ERK (40, 41).
MKP-1 serves as a feedback regulator of immune responses (21, 22, 25), as evidenced by the increased susceptibility of MKP-1-deficient mice to anaphylaxis. However, this regulatory effect is reversed by glucocorticoid treatment (26). Moreover, COX-2 repression by glucocorticoid has been shown to be partly restored by MKP-1 deficiency (20), suggesting a reciprocal relationship between MKP-1 and MAPK activation. In the present study, we reveal the role of MKP-1, induced by GR, in inhibiting JNK and p38. This finding is consistent with previous reports showing that MKP-1 dephosphorylates MAPKs at threonine and tyrosine residues (22, 42). Although the murine MKP-1 gene contains GREs in its promoter region (23, 43), and thus may have the potential to respond to glucocorticoids, the precise GRE location within the promoter has not been functionally characterized. First, in silico analysis revealed that the MKP-1 gene contains one full GRE and two half-sites (MGI no. 105120). Subsequent promoter deletion analysis of the MKP-1 gene enabled us to verify the location of the functional GRE site within the promoter region between −1499 and −1481 bp (Cho, I. J. and S. G. Kim, unpublished data). Next, we found that dexamethasone treatment enhanced LPS-dependent MKP-1 induction. The first important finding of this study is that the enhanced MKP-1 induction by LPS+dexamethasone causes JNK and p38 inactivation, which in turn decreases the level of ATF2 and c-Jun phosphorylation. Therefore, COX-2 repression by GR may result from the inhibition of MAPK-dependent ATF2/c-Jun phosphorylation by way of MKP-1 induction. This conclusion is further strengthened by the observation that MKP-1 overexpression attenuates the ability of LPS to phosphorylate ATF2/c-Jun and to transactivate the COX-2 gene. The results of this study concur with previous reports which found that MKP-1 deficiency causes sustained activation of JNK and p38, thus increasing the production of proinflammatory cytokines (20, 22, 25, 44).
The TLR signaling pathway regulates other transcription factors via MAPKs, including CREB and C/EBP. Several cellular kinases (e.g. p90RSK, calmodulin-dependent kinase IV, protein kinase C, and protein kinase A) phosphorylate CREB (45). In particular, p38 regulates CREB phosphorylation via MSK1 (45). Despite the observed decrease in levels of p38 phosphorylation associated with GR treatment, LPS-induced CREB phosphorylation and DNA binding activity were unaffected by glucocorticoid in this study. Previously, we reported that C/EBPβ plays a role in COX-2 induction in macrophages treated with the combination of LPS and ceramide (46). Past work in this laboratory indicated that GR treatment did not change chemical-induced C/EBPβ activation in hepatocytes (36). In addition to the present data showing that GR activation does not impact ERK1/2 phosphorylation (which is responsible for C/EBPβ activation) (47), gel shift results confirmed that C/EBPβ does not contribute to COX-2 repression by GR (data not shown). Collectively, these results suggest that COX-2 repression by GR is not due to alterations in CREB or C/EBPβ activation.
The second important finding of this work is the identification of NF-κB as the transcription factor associated with COX-2 gene repression by GR. Activated GR directly binds to p65 recruited to the NF-κB binding site, demonstrating for the first time that p65 is the transcription factor tethered by activated GR on the COX-2 gene. This result is consistent with previous findings showing that GR might be recruited to target genes via interactions with transcription factors such as NF-κB, AP-1, or signal transducer and activator of transcription (9, 10, 11) and that the zinc binding region of GR interacts with the RHD domain of p65 for IL-8 repression (27). GR enhances the synthesis of IκBα in immune cells (5, 48), which opens the possibility of NF-κB inhibition by GR. However, sequestration of p65 by IκBα induction appears to depend on cell type (7). Our finding that GR activation does not change either NF-κB DNA binding or IκBα and p65 phosphorylation excludes the possibility that glucocorticoid inhibits NF-κB activity via IκBα induction. Some researchers have proposed that GR is recruited to the AP-1 DNA complex (10, 11). In contrast, our ChIP analysis clearly shows that GR does not bind to the AP-1 region of the COX-2 gene. Granted, it is possible that GR inhibits the level of IRF3 binding to activated p65 (28). However, the observation that IRF3 activation by LPS minimally affected COX-2 induction (15) rules out a possible interaction of GR with IRF3 playing a role in COX-2 repression.
GRIP1 dually functions as a corepressor or coactivator by binding to nuclear receptors (11, 39, 49, 50). In the present study, we elucidated the interactions among the GRIP1, GR, and p65 components involved in COX-2 repression. This concept is corroborated by two observations: 1) the NID-RD domain of GRIP1 inhibits COX-2 induction by LPS and 2) COX-2 repression by GR is abrogated by either mutating the NF-κB region or GRIP1 knockdown. GR interacts with GRIP1 without altering NF-κB activity. Previous studies have shown that p38 phosphorylated GRIP1 and potentiated coactivator activity (51). Thus, p38 inhibition as a result of GR-dependent MKP-1 induction might cause GRIP1 to present its repressor domain. GRIP1 is also involved in IL-8 gene regulation (11), matching with our finding that GRIP1 bound with GR contributes to the inhibition of NF-κB-driven activity in the COX-2 gene.
Finally, Ogawa et al. (28) attempted to elucidate the GR-mediated inhibitory mechanisms of inflammatory genes through the use of in silico and microarray analyses, claiming that inflammatory gene regulation depends on the inhibition of NF-κB binding with IRF3. Alternatively, other inflammatory genes may be independently regulated by IRF3. To determine whether our model for the mechanism of COX-2 repression can be extended to other genes, we analyzed microarray data from MKP-1-, MYD88-, and IRF3-deficient cells treated with LPS, which enabled us to predict if these genes are regulated in a similar manner (supplemental Fig. S3). One important experimental observation, which substantiates this concept, concerns the reversal by GRIP1 siRNA (si-GRIP1)/Dn-MKP-1 transfection of GR-induced IL-6 and CXCL-5 repression, thus identifying a GR-responding/IRF3-independent regulatory mechanism in macrophages.
Based on this present work, we conclude that GR antagonizes the induction of certain inflammatory genes via a novel combinatorial mechanism, which involves MKP-1-mediated AP-1 inhibition and GRIP1-GR recruitment to the p65 DNA complex (Fig. 7B). Our findings may offer insight into the GR signaling network for inflammatory gene regulation.
Materials and Methods
Materials
Anti-COX-2 antibody was obtained from Cayman Chemical Co. (Ann Arbor, MI). Antibodies directed against phospho-ATF2, ATF2, phospho-c-Jun, phospho-CREB, CREB, phospho-IκBα, phospho-p65, phospho-p38, p38, phospho-JNK1/2, JNK1/2, phospho-ERK1/2, and ERK1/2 were supplied by Cell Signaling Technology (Beverly, MA). Antibodies directed against c-myc, hemagglutinin, c-Jun, CBP, GRIP1, GR, IκBα, and p65 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). LPS (Escherichia coli 026:B6), dexamethasone, RU486, antiflag antibody, antiactin antibody, and other reagents were supplied by Sigma-Aldrich (St. Louis, MO).
Cell culture and treatment
Raw264.7 cells, a murine macrophage cell line, were purchased from American Type Culture Collection (Manassas, VA) and were cultured in DMEM containing 10% fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. The cells were maintained at 37 C in a humidified atmosphere containing 5% CO2. For all experiments, cells were grown to 80–90% confluency, and the cell population was subjected to no more than 20 cell passages. After 24 h serum starvation, the cells were incubated with dexamethasone or other agents for 30 min and then were continuously exposed to LPS for the indicated time period.
Immunoblot analysis
The cells were scraped, transferred to microtubes, and allowed to swell by adding lysis buffer. Total lysates were prepared as previously described (14, 52). Immunoblot analyses were performed according to the established procedures (14, 52). The proteins of interest in cell lysates were resolved by SDS-PAGE and were transferred to nitrocellulose membrane. The protein bands were visualized using an ECL chemiluminescence system (Amersham, Buckinghamshire, UK). Equal loading of samples was verified by immunoblotting for actin. Relative protein levels were determined by scanning densitometry. At least three replicates were performed for each experiment.
Real-time RT-PCR
Total RNA was isolated from cells using an RNeasy mini kit (QIAGEN, Valencia, CA). The RNA (2 μg each) isolate was reverse transcribed using oligo-d(T)16 primers to obtain cDNA. Real-time PCR was carried out according to the manufacturer’s instructions (Light-Cycler 2.0; Roche, Mannheim, Germany). The relative levels of COX-2 (sense, 5′-TCTCCAACCTCTCCTACTAC-3′; antisense, 5′-GCACGTAGTCTTCGATCACT-3′, 624 bp); IL-6 (sense, 5′-TTCCATCCAGTTGCCTTCTT-3′; antisense, 5′-ATTTCCACGATTTCCCAGAG-3′, 170 bp); and CXCL-5 (sense, 5′-GCTGCCCCTTCCTCAGTCAT-3′; antisense, 5′-CACCGTAGGGCACTGTGGAC-3′, 129 bp) were normalized based on the level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH; sense, 5′-TCGTGGAGTCTACTGGCGT-3′; antisense, 5′-GCCTGCTTCACCACCTTCT-3′, 510 bp) using Lightcycler software 4.0 (Roche). After PCR amplification, a melting curve of each amplicon was determined to verify its accuracy.
Plasmid construction, transfection, and luciferase assays
pGL-mCOX2-724 (COX-2-luciferase construct) and its NF-κB mutant construct were generated as previously described (53). Briefly, Raw264.7 cells were cotransfected with pCMV-LacZ and the COX-2 luciferase construct using Lipofectamine (Invitrogen, Carlsbad, CA). The transfected cells were incubated in MEM for 12 h and then were exposed to 0.3 μg/ml LPS with or without dexamethasone (0.1 μm, for 24 h). To normalize transfection efficiency, β-galactosidase activity was measured at 420 nm by using o-nitrophenyl-β-d-galactopyranoside as a substrate.
For some experiments, the cells were cotransfected with pSG5-mMKP1 [or pTag3A-GRIP1(RD)] in combination with pGL-mCOX2-724. pSG5 (or pCMV-Tag3A) was used for mock transfection. pSG5-mMKP1, which encodes the murine MKP-1 gene, and its Dn plasmid, pSG5-mMKP1-CS, were supplied by Dr. Tonk (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). pTag3A-GRIP1(RD), which encodes the Myc-tagged NID and RD domains of GRIP1, was generated by PCR amplification (from 648 to 1007 amino acids) of the murine GRIP1 gene using mouse cDNA as a template. The amplified DNA was cloned into pCMV-Tag3A (Stratagene, La Jolla, CA). DNA sequencing (ABI 7700 DNA cycle sequencer; Applied Biosystems, Foster, CA) verified authenticity of the constructs.
The JNK1 Dn mutant (KmJNK1), the Dn p38α mutant (pCMV5-Dn-p38α), and the Dn MKK1 (MEK1) mutant (MKK1-8E) were provided by Dr. Dhanasekaran (Temple University, Philadelphia, PA), Dr. Moon (Duksung Woman’s University, Seoul, Korea), and Dr. Ahn (Howard Hughes Medical Institute, University of Colorado, Boulder, CO), respectively.
Gel retardation assays
Nuclear extracts were prepared as previously described (14, 52). Double-stranded, end-labeled DNA probes for the consensus sequences of AP-1 (5′-CGCTTGATGAGTCAGCCGGAA-3′), NF-κB (5′-AGTTGAGGGGACTTTCCCAGGC-3′), and CREB (5′-AGAGATTGCCTGACGTCAG AGAGCTAG-3′) were used for gel-shift assays. The binding specificities of proteins to DNA were confirmed by both competition and supershift (or immunoinhibition) reactions. For the competition assays, a 20-fold molar excess of unlabeled oligonucleotide was added to each reaction mixture before addition of the radiolabeled probe. A specificity protein 1 oligonucleotide (5′-ATTCGATCGGGGCGGGGCGAGC-3′) was used as a negative control. In other assays, the reaction mixture was incubated with antibody (2 μg each, for 20 min) after the addition of the labeled probe, and the reaction was continued for another hour at 25 C for supershift or immunoinhibition of protein-DNA complex. Samples were loaded onto 4% polyacrylamide gels at 100 V. The gels were removed, fixed, and dried, followed by autoradiography.
ChIP analysis
The cells treated as specified were cross linked by adding formaldehyde to a 1% final concentration. ChIP assays were conducted, as described previously (36, 53). PCR was performed with the primers flanking the AP-1/CREB (sense, 5′-TGCGCAACTCACTGAAGCAG-3′; antisense, 5′-TGACTGACTCCTGAAGTCTTA-3′, 169 bp) or the NF-κB (sense, 5′-ATGTGGACCCTGACAGAGGA-3′; antisense, 5′-TCTCCGGTTTCCTCCCAGTC-3′, 222 bp) binding sites of murine COX-2 gene. Precipitates obtained from the reaction mixture without antibody and 10% of cross-linked lysates were used as negative and input controls, respectively.
Immunoprecipitation
To assess the interaction between p65 and GR (or GR-bound GRIP1), cell lysates (500 μg/ml each) were incubated with anti-GR (or anti-GRIP1) antibody or preimmune IgG overnight at 4 C. The antigen-antibody complex was immunoprecipitated after incubation for 2 h at 4 C with protein G-agarose. Immune complexes were solubilized in 2× Laemmli buffer. Protein samples were resolved and were immunoblotted with anti-p65 (or anti-GR) antibody.
siRNA knockdown
Scrambled control siRNA and siRNAs of murine MKP-1, ATF2, c-Jun, and GRIP1 were supplied by Dharmacon (Lafayette, CO). Cells were cotransfected with pGL-mCOX2-724 and siRNA (100 pmol) using Lipofectamine2000 according to the manufacturer’s instructions, and luciferase activity was subsequently measured. After transfection for 24 h, the cells were preincubated with dexamethasone for 30 min and then continuously exposed to LPS for 24 h. For some experiments, cells were cotransfected with the siRNA specific for murine MKP-1 with a construct encoding human MKP-1 (pSG5-hMKP-1). Human MKP-1 expression construct was kindly provided by Dr. Keyse (Ninewells Hospital, Dundee, Scotland, UK) (54). After transfection of siRNA directed against murine GRIP1 (100 pmol, 24 h), the cells were incubated in MEM for 6 h and then were exposed to LPS or LPS+dexamethasone for 6 h. The relative mRNA levels of COX-2, IL-6, and CXCL-5 were determined by real-time RT-PCR assays. Immunoblot or real-time RT-PCR assays were performed to confirm the silencing of MKP-1, ATF2, c-Jun, or GRIP1.
Statistical analysis
One-way ANOVA procedures were used to assess significant differences among treatment groups. For each significant effect of treatment, the Newman-Keuls test was used for comparisons of multiple group means.
Acknowledgments
We thank Dr. J. S. Brooks for helpful discussion and English editing. The kind donation of pSG5-mMKP1 and pSG5-mMKP1-CS from Dr. Tonk, pSG5-hMKP1 from Dr. Keyse, pGL2-MKP1-1716 from Dr. Cato, KmJNK1 from Dr. Dhanasekaran, pCMV5-Dn-p38α from Dr. Moon, and MKK1-8E from Dr. Ahn is gratefully acknowledged.
NURSA Molecule Pages:
Coregulators: GRIP1;
Ligands: Dexamethasone | RU486;
Nuclear Receptors: GR.
Footnotes
This work was supported by Grant R11-2007-107-01001-0 from the Korea Science and Engineering Foundation funded by the Korea government (Ministry of Education, Science and Technology).
Disclosure Statement: The authors have nothing to disclose.
First Published Online October 22, 2008
Abbreviations: ATF2, Activating transcription factor 2; AP-1, activator protein 1; CBP, CREB-binding protein; C/EBP, CCAAT/enhancer binding protein; ChIP, chromatin immunoprecipitation; CREB, cAMP response element binding protein; Dn, dominant negative; GR, glucocorticoid receptor; GRE, glucocorticoid response element; GRIP1, GR-interacting protein 1; IRF3, interferon regulatory factor 3; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharide; MKP-1, MAPK phosphatase-1; NF-κB, nuclear factor-κB; NID, nuclear receptor-interacting domain; PG, prostaglandin; RD, repressor domain; si-GRIP1, GRIP1 siRNA; siRNA, small interfering RNA; TLR, toll-like receptor; TSA, trichostatin A.
References
- 1.Wadleigh DJ, Redddy ST, Kopp E, Ghosh S, Herschman HR2000. Transcriptional activation of the cyclooxygenase-2 gene in endotoxin-treated Raw264.7 macrophages. J Biol Chem 275:6259–6266 [DOI] [PubMed] [Google Scholar]
- 2.Lee SH, Soyoola E, Chanmugam P, Hart S, Sun W, Zhong H, Liou S, Simmons D, Hwang D1992. Selective expression of mitogen-inducible cyclooxygenase in macrophages stimulated with lipopolysaccharide. J Biol Chem 267:25934–25938 [PubMed] [Google Scholar]
- 3.Reddy ST, Herschman HR1994. Ligand-induced prostaglandin synthase requires expression of the TIS10/PGS-2 prostaglandin synthase gene in murine fibroblast and macrophages. J Biol Chem 269:15473–15480 [PubMed] [Google Scholar]
- 4.Almawi WY, Hess DA, Reider MJ1998. Multiplicity of glucocorticoids action in inhibiting allograft rejection. Cell Transplant 7:511–523 [DOI] [PubMed] [Google Scholar]
- 5.Barnes PJ1998. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond) 94:557–572 [DOI] [PubMed] [Google Scholar]
- 6.Almawi WY, Melemedjian OK2002. Negative regulation of nuclear factor-κB activation and function by glucocorticoid. J Mol Endocrinol 28:69–78 [DOI] [PubMed] [Google Scholar]
- 7.De Bosscher K, Vanden Berghe W, Haegeman G2003. The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 24:488–522 [DOI] [PubMed] [Google Scholar]
- 8.Tsai MJ, O'Malley BW1994. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 63:45–86 [DOI] [PubMed] [Google Scholar]
- 9.Takeda T, Kurachi H, Yamamoto T, Nishio Y, Morishige K, Miyake A, Murata Y 1998 Crosstalk between the interleukin-6 (IL-6)-JAK-STAT and the glucocorticoid-nuclear receptor pathway: synergistic activation of IL-6 response element by IL-6 and glucocorticoid. J Endocrinol 159:323–330 [DOI] [PubMed] [Google Scholar]
- 10.Rogatsky I, Zarember KA, Yamamoto KR2001. Factor recruitment and TIF2/GRIP1 corepressor activity at a collagenase-3 response element that mediates regulation by phorbol esters and hormones. EMBO J 20:6071–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rogatsky I, Luecke HF, Leitman DC, Yamamoto KR2002. Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc Natl Acad Sci USA 99:16701–16706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barton GM, Medizhitov R2003. Toll-like receptor signaling pathways. Science 300:1524–1525 [DOI] [PubMed] [Google Scholar]
- 13.Miller SI, Ernst RK, Bader MW2005. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol 3:36–46 [DOI] [PubMed] [Google Scholar]
- 14.Cho MK, Cho YH, Lee GH, Kim SG2004. Induction of cyclooxygenase-2 by bovine type I collagen in macrophages via C/EBP and CREB activation by multiple cell signaling pathways. Biochem Pharmacol 67:2239–2250 [DOI] [PubMed] [Google Scholar]
- 15.Vila-del Sol V, Fresno M2005. Involvement of TNF and NF-κ B in the transcriptional control of cyclooxygenase-2 expression by IFN-γ in macrophages. J Immunol 174:2825–2833 [DOI] [PubMed] [Google Scholar]
- 16.Kang YJ, Wingerd BA, Arakawa T, Smith WL2006. Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. J Immunol 177:8111–8122 [DOI] [PubMed] [Google Scholar]
- 17.Hazzalin CA, Mahadevan LC2002. MAPK-regulated transcription: a continuously variable gene switch? Nat Rev Mol Cell Biol 3:30–40 [DOI] [PubMed] [Google Scholar]
- 18.Johnson GL, Lapadat R2002. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298:1911–1912 [DOI] [PubMed] [Google Scholar]
- 19.Camps M, Nichols A, Arkinstall S2000. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J 14:6–16 [PubMed] [Google Scholar]
- 20.Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, Saklatvala J, Clark AR2006. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med 203:1883–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T2006. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol 176:1899–1907 [DOI] [PubMed] [Google Scholar]
- 22.Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA2006. Dynamic regulation of pro- and antiinflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci USA 103:2274–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC2001. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J 20:7108–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark AR2003. MAP kinase phosphatase 1: a novel mediator of biological effects of glucocorticoids? J Endocrinol 178:5–12 [DOI] [PubMed] [Google Scholar]
- 25.Zhao Q, Wang X, Nelin LD, Yao Y, Matta R, Manson ME, Baliga RS, Meng X, Smith CV, Bauer JA, Chang CH, Liu Y2006. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med 203:131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maier JV, Brema S, Tuckermann J, Herzer U, Klein M, Stassen M, Moorthy A, Cato AC2007. Dual specificity phosphatase 1 knockout mice show enhanced susceptibility to anaphylaxis but are sensitive to glucocorticoids. Mol Endocrinol 21:2663–2671 [DOI] [PubMed] [Google Scholar]
- 27.Nissen RM, Yamamoto KR2000. The glucocorticoid receptor inhibits NFκB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 14:2314–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK2005. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell 122:707–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chivers JE, Cambridge LM, Catley MC, Mak JC, Donnelly LE, Barnes PJ, Newton R2004. Differential effects of RU486 reveal distinct mechanisms for glucocorticoid repression of prostaglandin E release. Eur J Biochem 271:4042–4052 [DOI] [PubMed] [Google Scholar]
- 30.Chang YJ, Wu MS, Lin JT, Chen CC2005Helicobacter pylori-induced invasion and angiogenesis of gastric cells is mediated by cyclooxygenase-2 induction through TLR2/TLR9 and promoter regulation. J Immunol 175:8242–8252 [DOI] [PubMed] [Google Scholar]
- 31.Gupta S, Campbell D, Derijard B, Davis RJ1995. Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267:389–393 [DOI] [PubMed] [Google Scholar]
- 32.Fukunaga R, Hunter T1997. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J 16:1921–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karin M, Gallagher E2005. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 57:283–295 [DOI] [PubMed] [Google Scholar]
- 34.Angel P, Hattori K, Smeal T, Karin M1988. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell 55:875–885 [DOI] [PubMed] [Google Scholar]
- 35.Hayakawa J, Mittal S, Wang Y, Korkmaz KS, Adamson E, English C, Omichi M, McClelland M, Mercola D2004. Identification of promoters bound by c-Jun/ATF2 during rapid large-scale gene activation following genotoxic stress. Mol Cell 16:521–535 [DOI] [PubMed] [Google Scholar]
- 36.Ki SH, Cho IJ, Choi DW, Kim SG2005. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPβ TAD and Nrf2 Neh4/5: role of SMRT recruited to GR in GSTA2 gene repression. Mol Cell Biol 25:4150–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szapary D, Huang Y, Simons Jr SS1999. Opposing effects of corepressor and coactivators in determining the dose-response curve of agonists, and residual agonist activity of antagonists, for glucocorticoid receptor-regulated gene expression. Mol Endocrinol 13:2108–2121 [DOI] [PubMed] [Google Scholar]
- 38.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, Rosenfeld MG1995. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 37:397–404 [DOI] [PubMed] [Google Scholar]
- 39.Reily MM, Pantoja C, Hu X, Chinenov Y, Rogatsky I2006. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J 25:108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hurd TW, Culbert AA, Webster KJ, Tavaré JM2002. Dual role for mitogen-activated protein kinase (Erk) in insulin-dependent regulation of Fra-1 (fos-related antigen-1) transcription and phosphorylation. Biochem J 368:573–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chalmers CJ, Gilley R, March HN, Balmanno K, Cook SJ2007. The duration of ERK1/2 activity determines the activation of c-Fos and Fra-1 and the composition and quantitative transcriptional output of AP-1. Cell Signal 19:695–704 [DOI] [PubMed] [Google Scholar]
- 42.Franklin CC, Kraft AS1997. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem 272:16917–16923 [DOI] [PubMed] [Google Scholar]
- 43.Noguchi T, Metz R, Chen L, Mattei MG, Carrasco D, Bravo R1993. Structure, mapping, and expression of erp, a growth factor-inducible gene encoding a nontransmembrane protein tyrosine phosphatase, and effect of ERP on cell growth. Mol Cell Biol 13:5195–5205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shepherd EG, Zhao Q, Welty SE, Hansen TN, Smith CV, Liu Y2004. The function of mitogen-activated protein kinase phosphatase-1 in peptidoglycan-stimulated macrophages. J Biol Chem 279:54023–54031 [DOI] [PubMed] [Google Scholar]
- 45.Johannessen M, Delghandi MP, Moens U2004. What turns CREB on? Cell Signal 16:1211–1227 [DOI] [PubMed] [Google Scholar]
- 46.Cho YH, Lee CH, Kim SG2003. Potentiation of lipopolysaccharide-inducible cyclooxygenase 2 expression by C2-ceramide via c-Jun N-terminal kinase-mediated activation of CCAAT/enhancer binding protein β in macrophages. Mol Pharmacol 63:512–523 [DOI] [PubMed] [Google Scholar]
- 47.Nakajima T, Kinoshita S, Sasagawa T, Sasaki K, Naruto M, Kishimoto T, Akira S1993. Phosphorylation at threonine-235 by a ras-dependent mitogen-activated protein kinase cascade is essential for transcription factor NF-IL6. Proc Natl Acad Sci USA 90:2207–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scheinman R, Cogswell PC, Lofquist A, Baldwin Jr AS1995. Role of transcriptional activation of IκΒα in mediation of immunosuppression by glucocorticoids. Science 270:283–286 [DOI] [PubMed] [Google Scholar]
- 49.Lefstin JA, Yamamoto KR1998. Allosteric effects of DNA on transcriptional regulators. Nature 392:885–888 [DOI] [PubMed] [Google Scholar]
- 50.Rosenfeld MG, Lunyak VV, Glass CK2006. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 20:1405–1428 [DOI] [PubMed] [Google Scholar]
- 51.Frigo DE, Basu A, Nierth-Simpson EN, Weldon CB, Dugan CM, Elliott S, Collins-Burow BM, Salvo VA, Zhu Y, Melnik LI, Lopez GN, Kushner PJ, Curiel TJ, Rowan BG, McLachlan JA, Burow ME2006. p38 mitogen-activated protein kinase stimulates estrogen-mediated transcription and proliferation through the phosphorylation and potentiation of the p160 coactivator glucocorticoid receptor-interacting protein 1. Mol Endocrinol 20:971–983 [DOI] [PubMed] [Google Scholar]
- 52.Kang KW, Choi SY, Cho MK, Lee CH, Kim SG2003. Thrombin induces nitric-oxide synthase via Gα12/13-coupled protein kinase C-dependent I-κBα phosphorylation and JNK-mediated IκΒα degradation. J Biol Chem 278:17368–17378 [DOI] [PubMed] [Google Scholar]
- 53.Ki SH, Choi MJ, Lee CH, Kim SG2007. Gα12 specifically regulates COX-2 induction by sphingosine-1-phosphate: role for JNK-dependent ubiquitinylation and degradation of IκB. J Biol Chem 282:1938- 1947 [DOI] [PubMed] [Google Scholar]
- 54.Groom LA, Sneddon AA, Alessi DR, Dowd S, Keyse SM1996. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. EMBO J 15:3621–3632 [PMC free article] [PubMed] [Google Scholar]