

Abstract
Insulin-stimulated translocation of the glucose transporter GLUT4 to the plasma membrane in muscle and fat cells depends on the phosphatidylinositide 3-kinase/Akt pathway. The downstream target AS160/TBC1D4 is phosphorylated upon insulin stimulation and contains a TBC domain (Tre-2/Bub2/Cdc16) that is present in most Rab guanosine triphosphatase-activating proteins. TBC1D4 associates with GLUT4-containing membranes under basal conditions and dissociates from membranes with insulin. Here we show that the association of TBC1D4 with membranes is required for its inhibitory action on GLUT4 translocation under basal conditions. Whereas the insulin-dependent dissociation of TBC1D4 from membranes was not required for GLUT4 translocation, its phosphorylation was essential. Many agonists that stimulate GLUT4 translocation failed to trigger TBC1D4 translocation to the cytosol, but in most cases these agonists stimulated TBC1D4 phosphorylation at T642, and their effects on GLUT4 translocation were inhibited by overexpression of the TBC1D4 phosphorylation mutant (TBC1D4-4P). We postulate that TBC1D4 acts to impede GLUT4 translocation by disarming a Rab protein found on GLUT4-containing-membranes and that phosphorylation of TBC1D4 per se is sufficient to overcome this effect, allowing GLUT4 translocation to the cell surface to proceed.
GLUCOSE UPTAKE INTO fat and muscle cells is triggered by insulin and exercise. The insulin-sensitive glucose transporter, GLUT4, is expressed in these tissues. Upon stimulation, GLUT4 translocates from intracellular vesicles to the plasma membrane (1). The Ser/Thr kinase Akt/PKB plays a key role in this process, at least in part, by phosphorylating the Rab guanosine triphosphatase-activating protein (GAP) AS160/TBC1D4 (Akt substrate of 160 kDa hereafter referred to as TBC1D4) (2, 3, 4, 5). Insulin triggers TBC1D4 phosphorylation on five sites: Ser318, Ser570, Ser588, Thr642, and Thr751. Other agonists that stimulate GLUT4 translocation, such as muscle contraction and AMP-activated protein kinase (AMPK) activators, also induce TBC1D4 phosphorylation (6, 7, 8, 9, 10, 11, 12). Phosphorylation of TBC1D4 at Thr642 induces 14-3-3 binding (13), and overexpression of a TBC1D4 mutant with four phosphorylation sites mutated to Ala (TBC1D4-4P) inhibits insulin-stimulated GLUT4 translocation to the plasma membrane in 3T3-L1 adipocytes, whereas the overexpression of the wild-type TBC1D4 (TBC1D4-wt) has no effect (4). A simultaneous mutation in the GAP domain (TBC1D4-4P,R/K) overcomes the inhibitory effect on GLUT4 translocation, indicating that GAP activity is required for this inhibition. Furthermore, the inhibitory effect of TBC1D4-4P is abolished when a constitutive 14-3-3 binding peptide sequence is inserted into the TBC1D4-4P protein, suggesting that 14-3-3 binding to Thr642 in TBC1D4 is involved in this inhibitory process (13). TBC1D4 displays in vitro GAP activity toward several Rab proteins including Rab2A, 8A, 10, and 14, which is of interest because Rabs 10, 11, and 14 have been found on purified GLUT4 vesicles (14, 15). Overexpression of constitutively active mutants encoding Rabs 8A and 14 in L6 myoblasts overcomes the inhibitory effect of the TBC1D4-4P mutant on insulin-stimulated GLUT4 translocation, whereas constitutively active Rab10 had no effect (16). Intriguingly, it has been shown (17, 18) that constitutively active Rab10 enhanced cell surface levels of GLUT4 in the absence of insulin, and knockdown of Rab10 inhibited insulin-stimulated GLUT4 translocation. Collectively these data support a model whereby TBC1D4 controls the GTP loading of a subset of Rabs that control trafficking of GLUT4 to the plasma membrane.
The question is how does insulin regulate the GAP activity of TBC1D4. Two models have been proposed. The first model is based on the observation that TBC1D4 binds to GLUT4 vesicles in the absence of insulin and dissociates in the presence of insulin (5, 14, 19). These data suggest that the GAP activity is regulated simply by localization. The second model is based on phosphorylation of TBC1D4 whereby in a manner analogous to tuberin (20), phosphorylation may regulate GAP activity per se. Distinguishing between the models is a major goal of the present study.
In this study, we investigated the requirement for the association/dissociation of TBC1D4 with GLUT4 vesicles in mediating its effects on the trafficking of GLUT4 in adipocytes. Our findings indicate that TBC1D4 association with GLUT4 vesicles is required to facilitate its inhibitory effect on GLUT4 translocation in the basal state. Intriguingly, dissociation of TBC1D4 from GLUT4 vesicles was not necessary to promote GLUT4 translocation. Moreover, many agonists that stimulate GLUT4 translocation to the cell surface [endothelin-1, IL-6, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR), adiponectin, or berberine] appear to require TBC1D4 for this action but, unlike insulin, many of these do not result in a detectable release of TBC1D4 from GLUT4 vesicles into the cytosol. These data implicate an important role for TBC1D4 phosphorylation per se in insulin action.
RESULTS
Insulin-Induced TBC1D4 Translocation to the Cytosol Is Phosphorylation Dependent
We have previously shown that TBC1D4 associates with GLUT4-containing membranes under basal conditions and dissociates from vesicles upon insulin stimulation (14). The model in which TBC1D4 inactivates the Rab, thus preventing fusion of GLUT4 vesicles with the cell surface, would require association of the GAP with the vesicles. A fundamental observation in formulating this model is that overexpression of TBC1D4-4P inhibits insulin-stimulated GLUT4 translocation in 3T3-L1 adipocytes, and mutation of the GAP activity overcomes this inhibitory effect (4). One prediction from the model is that the TBC1D4-4P mutant may remain associated with GLUT4 vesicles even in the presence of insulin, thus acting in a dominant-negative manner. Flag-tagged wild type-TBC1D4 (TBC1D4-wt) and TBC1D4-4P were expressed in 3T3-L1 adipocytes either by electroporation or retroviral transduction. Cells were then incubated in the absence or presence of insulin, followed by subcellular fractionation. The amount of TBC1D4-wt in the cytosol increased when the cells were stimulated with insulin (Fig. 1). However, TBC1D4-4P failed to translocate into the cytosol upon insulin stimulation. Whereas TBC1D4 was concentrated in the low-density microsome (LDM) fraction isolated form basal cells, we failed to observe an insulin-dependent change in TBC1D4 levels in this fraction (Fig. 1C). We have previously reported that the LDM fraction comprises both membranes and polypeptide complexes (21). Hence we next used glycerol gradient sedimentation analysis to resolve these components. As shown in Fig. 1D, the LDM fraction could be resolved into GLUT4-containing membranes (2 to 7) and free protein (10 to 12) as indicated by the use of anti-14-3-3 labeling. TBC1D4 was found in both peaks, indicating that it is localized both to membranes consistent with our previous findings (14) and to a polypeptide component. In addition, some TBC1D4 is also found at the bottom of the gradient that contains additional membrane markers such as cation-independent mannose G-phosphate receptor (data not shown). In response to insulin we observed a redistribution of TBC1D4 from membranes to the protein peak. The distribution of TBC1D4-4P was similar to TBC1D4-wt under basal conditions (Fig. 1D), although the membrane peak was considerably broader for TBC1D4-4P compared with TBC1D4-wt. Importantly, insulin had no demonstrable effect on the distribution of TBC1D4-4P in the glycerol gradient.
Fig. 1.

TBC1D4-4P Does Not Translocate to the Cytosol in Response to Insulin
A, 3T3-L1 adipocytes were transfected with either Flag-TBC1D4-wt or Flag-TBC1D4-4P by electroporation. Cells were incubated with or without 100 nm insulin for 20 min and lysed, and the cytosol fraction was obtained by differential centrifugation. A representative Western blot incubated with anti-Flag-antibody is shown. B, Quantification of seven experiments is shown (*, P < 0.05). C, Populations of 3T3-L1 adipocytes were selected for expression of GFP by FACS after retroviral transfection of the bicistronic pMIG vector alone, pMIG-Flag-TBC1D4-wt, or pMIG-Flag-TBC1D4-4P. Cells were propagated and differentiated into 3T3-L1 adipocytes. Cells were incubated in DMEM for 2 h and then incubated in the absence or presence of 100 nm insulin for 20 min. Cells were lysed and cytosol, PM, and LDM fractions were obtained by differential cell fractionation. Representative Western blots stained for TBC1D4 and GLUT4 are shown. D, The LDM fractions for TBC1D4-wt and TBC1D4-4P obtained in panel C were loaded on top of 5–25% glycerol gradients on top of a 50% sucrose cushion. Equal volumes of each fraction were subjected to SDS-PAGE and Western blotting with anti-Flag, anti-GLUT4, and anti 14-3-3 antibodies. Vec., Vector.
Targeting the TBC1D4 GAP Domain to GLUT4 Vesicles Inhibits GLUT4 Translocation
Next we wanted to determine whether association of TBC1D4 with GLUT4-containing membranes was sufficient to mediate its inhibitory effect. To investigate this we localized the TBC1D4 GAP domain to GLUT4 vesicles by fusing the GAP domain [amino acids (aa) 865–1299] to the C terminus of hemagglutinin (HA)-tagged GLUT4 (G4-1D4GAP-wt) (Fig. 2A). We chose this approach to minimize the effect of other functional domains in TBC1D4 such as insulin-regulated aminopeptidase (IRAP) binding, 14-3-3 binding, calmodulin binding, or phosphorylation sites (4, 13, 14, 19, 22). When this construct was expressed in Chinese hamster ovary (CHO)-IR-IRS-1 cells, it produced a protein product of the predicted size (Fig. 2C). The GLUT4-TBC1D4GAP-wt chimera showed considerable colocalization with IRAP similar to the GLUT4 control construct. This indicates that the fusion protein localizes to GLUT4-containing membranes, because IRAP is a well-known marker for this compartment (Fig. 3A). We next examined the insulin response of this fusion protein in adipocytes. As shown in Fig. 2, A and B, insulin caused a 7-fold increase in plasma membrane (PM) levels of the control GLUT4 construct. In contrast, insulin had no significant effect on GLUT4-TBC1D4GAP-wt. To exclude misfolding or mistargeting of the fusion protein in adipocytes, we abolished the GAP activity in this construct by mutating the catalytic arginine to an alanine (GLUT4-1D4GAP-RA). In this fusion protein the inhibitory effect was almost completely reversed, and the fusion protein was translocated to the PM when the adipocytes were stimulated with insulin (Fig. 2, A and B). These results indicate that targeting of the functional TBC1D4 GAP domain alone to GLUT4 vesicles is sufficient to inhibit GLUT4 translocation.
Fig. 2.

Fusion of TBC1D4 GAP Domain to GLUT4 Inhibits GLUT4 Translocation
A, HA-GLUT4-1D4GAP-wt, HA-GLUT4-1D4GAP-RA, HA-GLUT4-Flag control, and HA-GLUT4-1D16GAP constructs were electroporated into 3T3-L1 adipocytes. Cells were incubated with or without 200 nm insulin for 20 min and were labeled with HA antibody for surface levels (surface) and with Flag antibody for total levels (total) of the fusion construct. Images of insulin-stimulated cells are depicted, and electroporated cells are marked with an arrow. Scale bars, 30 μm. B, Quantification of three to four experiments (A) is shown (**, P < 0.005, *, P < 0.05). Gray bars represent basal conditions, and black bars represent insulin-stimulated conditions. A, CHO-IR/IRS-1 cells were transfected with vector, HA-GLUT4-1D4GAP-wt and RA (110 kDa), and HA-GLUT4-1D16GAP (104 kDa) constructs. Cells were lysed, and the fusion proteins were immunoprecipitated with Flag antibody, followed by Western blotting with GLUT4 antibody. IP, Immunoprecipitation; WB, Western blot.
Fig. 3.
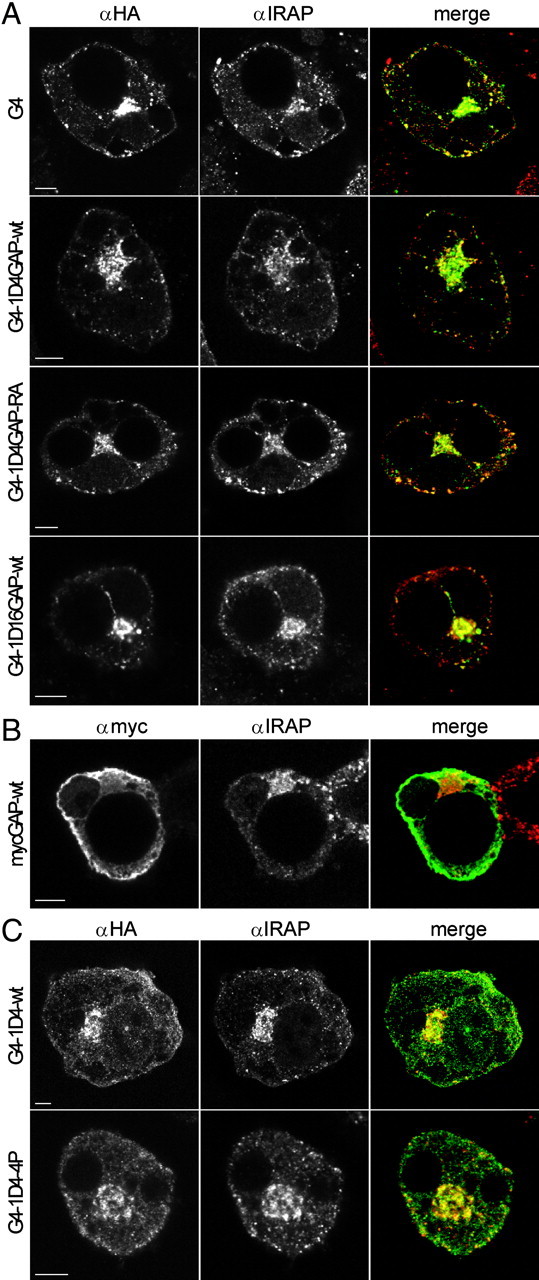
Colocalization of GLUT4-Fusion Constructs with IRAP
GLUT4 fusion constructs or myc-GAP were electroporated into 3T3-L1 adipocytes. Cells were incubated in DMEM without serum for 2 h, fixed, permeabilized, and labeled with IRAP antibody (red) and HA antibody (green) for the GLUT4 fusion construct in panels A and C or myc antibody (green) for myc-GAP-wt in panel B. Scale bar, 5 μm.
We next wanted to determine whether the observed effect was specific for the GAP domain of TBC1D4. Therefore, an unrelated GAP domain from TBC1D16 (aa 381–765) was fused to GLUT4 (G4-1D16GAP). This GAP domain displayed 27% homology with the TBC domain from TBC1D4. In contrast to that observed with GLUT4-1D4GAP, insulin stimulated the translocation of GLUT4-1D16GAP to the cell surface to a similar extent as GLUT4 (Fig. 2, A–C). This experiment shows that some of the specificity of TBC1D4 for GLUT4 trafficking is encoded in its GAP domain.
To evaluate the importance of the localization of the GAP domain to GLUT4 vesicles for its inhibition of GLUT4 translocation, we overexpressed the TBC1D4 GAP domain alone (aa 865–1299) in 3T3-L1 adipocytes. As expected, the GAP domain alone was localized to the cytosol and did not colocalize with IRAP, as it does not contain the IRAP-interacting domain (second PTB domain), which localizes TBC1D4 to GLUT4-containing membranes (Fig. 3B) (19). To measure its effect on GLUT4 translocation, the myc-tagged GAP domain was than coelectroporated with HA-GLUT4-green fluorescent protein (GFP) into adipocytes. The cells were incubated with or without insulin, fixed, and stained for surface HA to measure GLUT4 translocation. After permeabilization the cells were stained for intracellular myc to identify cells expressing the myc-GAP construct. The surface HA-GLUT4-GFP vs. total GFP (HA-GLUT4-GFP) ratio was determined in transfected cells expressing HA-GLUT4-GFP alone or in cells expressing both myc-GAP and HA-GLUT4-GFP. The GAP domain alone did not have any detectable effect on insulin-stimulated GLUT4 translocation, indicating that the association of the GAP activity of TBC1D4 with GLUT4 vesicles is required for its inhibitory role (Fig. 4).
Fig. 4.

Free TBC1D4 GAP Does Not Inhibit Insulin-Stimulated GLUT4 Translocation
Adipocytes were coelectroporated with myc-GAP-wt (GAP-wt) and HA-GLUT4-GFP (G4-GFP). Cells were incubated with or without (basal) 200 nm insulin for 20 min, fixed, and labeled with HA antibody to label surface GLUT4 only and with myc antibody after permeabilization to label cells expressing free GAP (GAP-wt). Cells expressing HA-GLUT4-GFP alone (arrows) and cells coexpressing HA-GLUT4-GFP and myc-GAP (arrowhead) were analyzed. A, Shown are representative images of four separate experiments. Scale bar, 30 μm. B, Quantification of data in panel A where the ratio of cell surface GLUT4 to total GLUT4 was quantified in cells with or without myc-GAP. Gray bars represent basal conditions, and black bars represent insulin-stimulated conditions (*, P < 0.05; **, P < 0.005).
TBC1D4 Dissociation from GLUT4 Vesicles Is Not Required for GLUT4 Translocation
The above data indicate that the localization of the TBC1D4 GAP activity to GLUT4 vesicles is required for this protein to inhibit insulin-stimulated GLUT4 translocation to the PM and that phosphorylation of TBC1D4 somehow overcomes this effect. This poses several new questions. How does phosphorylation regulate the activity of TBC1D4? What is the role of phosphorylation in the release of TBC1D4 from membranes, and does this encode the major effect of insulin on this protein? We reasoned that if the release of TBC1D4 from membranes is required for insulin action that simply fusing the TBC1D4-wt protein to GLUT4 should be sufficient to inhibit GLUT4 translocation. Hence we next produced a fusion protein comprising GLUT4 and full-length TBC1D4-wt. The GLUT4-TBC1D4-4P mutant was used as a control in this study. When these constructs were expressed in CHO-IR-IRS-1 cells, they produced protein products of the predicted size (Fig. 5A). These fusion proteins were then electroporated into 3T3-L1 adipocytes, and they colocalized to a considerable extent with IRAP (Fig. 3, A and C). Electroporated adipocytes were incubated in the presence or absence of insulin, followed by surface HA staining. The fusion of TBC1D4-4P to GLUT4 inhibited the insulin-stimulated translocation of this fusion protein to the cell surface (Fig. 5, B and C). Interestingly, the GLUT4-TBC1D4-wt fusion protein underwent insulin-dependent translocation to the PM, albeit to a lesser extent than GLUT4-GFP (Fig. 5, B and C). Both constructs showed a slight increase in PM levels under basal conditions. This may be due to some steric effects of the TBC1D4 fusion on the exposure and therefore effectiveness of the C-terminal retention signal sequences of GLUT4. These data indicate that release of TBC1D4 from membranes is not required for the insulin-dependent regulation of the protein. Hence the functional effects of insulin on TBC1D4 must be mediated via an alternate function. Based upon the dominant-negative effect of the TBC1D4-4P, but not the TBC1D4-wt protein, when fused to GLUT4, we predicted that this additional functionality is likely mediated by phosphorylation of TBC1D4 per se. We tested whether the GLUT4-TBC1D4-wt fusion protein was phosphorylated upon insulin stimulation by expressing the construct in CHO-IR/IRS-1 cells, which were incubated in the presence and absence of insulin. The fusion proteins were immunoprecipitated, subjected to SDS-PAGE, and immunoblotted with the p642 TBC1D4-specific antibody, to determine the phosphorylation at Thr642, which is the major 14-3-3 binding site in TBC1D4 (13). Insulin stimulation increased phosphorylation at the 642 site in the GLUT4-TBC1D4-wt fusion protein to a similar extent as that observed for wt TBC1D4 (Fig. 5A). Collectively, these results support a model in which phosphorylation of TBC1D4 per se is sufficient to inactivate its GAP activity, and dissociation from GLUT4 vesicles is not required for this function.
Fig. 5.
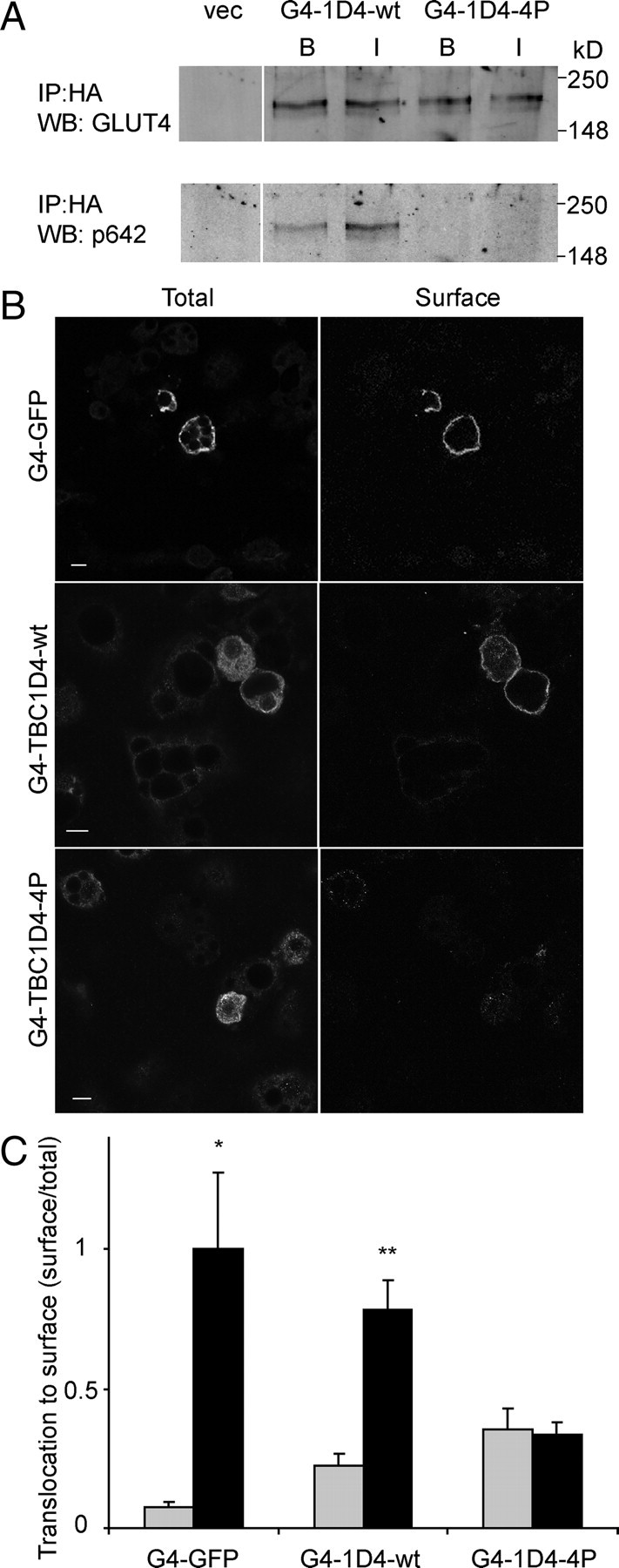
TBC1D4 Dissociation from GLUT4 Vesicles Is Not Required for GLUT4 Translocation
A, CHO-IR/IRS-1 cells were transfected with GLUT4-TBC1D4 (G4-TBC1D4-wt, -4P) constructs. Cells were incubated with (I) or without (B) 200 nm insulin for 20 min and lysed, and the fusion proteins were immunoprecipitated, followed by Western blotting with phospho-642 antibody and GLUT4 antibody. B, HA-GLUT4-GFP (G4-GFP), HA-GLUT4-TBC1D4-wt (G4-TBC1D4-wt), and HA-GLUT4-TBC1D4-4P (G4-TBC1D4-4P) were electroporated into 3T3-L1 adipocytes. Cells were labeled for surface levels (surface) and total levels (total) of the fusion construct. Shown are representative images of insulin-stimulated cells of three separate experiments. Scale bar, 10 μm. C, Quantification of data in panel B. Gray bars represent basal conditions, and black bars represent insulin-stimulated conditions (*, P < 0.05; **, P < 0.005). IP, Immunoprecipitation; vec, vector; WB, Western blot.
Alternate Agonists of GLUT4 Translocation Do Not Trigger TBC1D4 Translocation into the Cytosol in L6 Muscle Cells
Because TBC1D4 dissociation from GLUT4-containing membranes was not required for fusion, it was of interest to determine whether TBC1D4 dissociation was specific for insulin or if other agonists had the same effect. A number of other agonists have been reported to stimulate GLUT4 translocation, particularly in skeletal muscle. These include the traditional Chinese medicine berberine (23), the AMPK agonist, AICAR (9), the G protein-coupled receptor agonist endothelin-1 (24, 25), the cytokine IL-6 (26), and the adipokine adiponectin (27, 28). Intriguingly, the effects of agonists such as AICAR, berberine, endothelin-1, or IL-6 on GLUT4 translocation are mainly phosphatidylinositide 3-kinase (PI3K)-independent, and it has recently been reported that activation of the AMPK pathway also leads to phosphorylation of TBC1D4 (9, 12). We next developed a model system that would enable us to test the effects of some of these PI3K-independent agonists on TBC1D4 function. Insulin stimulated cell surface levels of GLUT4 by 5.8-fold in L6 myotubes, and this effect was almost completely inhibited by the PI3K inhibitor wortmannin, consistent with previous studies (Fig. 6A) (29). Strikingly, AICAR, adiponectin, endothelin-1, IL-6, and berberine also stimulated GLUT4 translocation by 3.3- to 5-fold. Notably whereas insulin-stimulated GLUT4 translocation was almost completely inhibited by wortmannin, this was not the case for the other agonists (Fig. 6A). In agreement with previous studies in 3T3-L1 adipocytes (5, 14), insulin stimulated the translocation of TBC1D4 into the cytosol in L6 myotubes (Fig. 5, B and C). However, other agonists tested did not trigger translocation of TBC1D4 into the cytosol (Fig. 6, B and C).
Fig. 6.
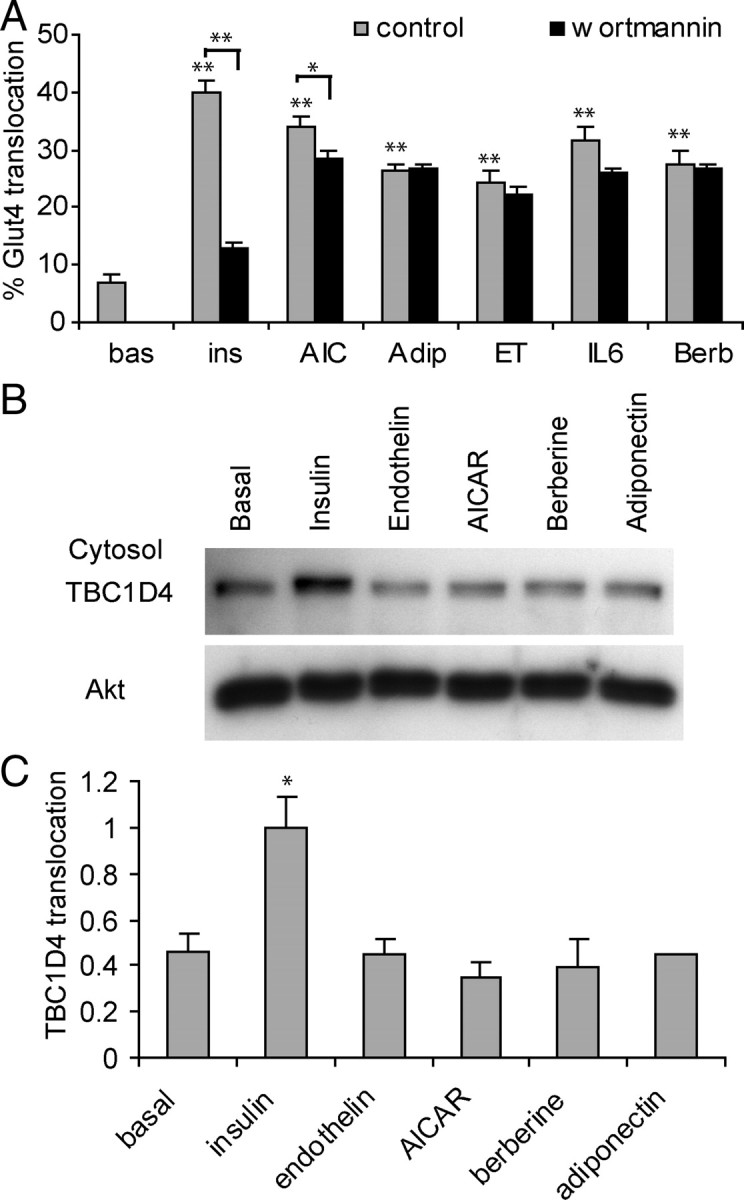
Only Insulin Causes Translocation of GLUT4 to the Cell Surface as Well as Cytosol Translocation of TBC1D4 in L6 Myotubes
A, L6 myotubes overexpressing HA-GLUT4 were incubated with 100 nm insulin, 2 mm AICAR, 2 μg/ml adiponectin, 10 nm endothelin-1, 100 ng/ml IL-6, or 10 μm berberine for 30 min after a 10-min preincubation in the absence or presence of 100 nm wortmannin. Relative HA-GLUT4 surface levels were determined by a 96-well plate fluorescence-based HA-GLUT4 translocation assay using anti-HA surface vs. total HA staining. (*, P < 0.05; **, P < 0.005; n = 3). B, L6 myotubes were incubated for 20 min with 100 nm insulin, 2 mm AICAR, 2 μg/ml adiponectin, 10 nm endothelin-1, or 10 μm berberine. Cells were lysed, and the cytosol fraction was obtained by differential cell fractionation. A representative Western blot stained for TBC1D4 is shown. C, Quantification of three experiments shown in panel B (*, P < 0.02). Adip, Adiponectin; bas, basal; Berb, berberine; ET, endothelin-1; ins, insulin; AIC, AICAR.
Most Agonists Induce Phosphorylation of the Thr642 Site of TBC1D4 in L6 Myotubes
One possible interpretation of these data (Fig. 6, B and C) is that TBC1D4 may only participate in insulin-stimulated GLUT4 translocation particularly in view of the specific effects of wortmannin (Fig. 6A). To test this we first examined phosphorylation of TBC1D4 in L6 myotubes stimulated with the alternate agonists. L6 myotubes were stimulated with or without agonists, lysed, and subjected to immunoblotting analysis with pThr642 TBC1D4 and pSer588 TBC1D4 antibodies, which specifically recognized the phosphorylation sites in TBC1D4. To enhance the sensitivity of pTBC1D4 detection, we used L6 myotubes that expressed Flag-tagged TBC1D4. A population of L6 cells that overexpressed TBC1D4 was obtained by cell sorting, and cells were differentiated into myotubes. Insulin increased the phosphorylation of TBC1D4 at Thr642 and Ser588 by 5-fold (Fig. 7, A and B). Insulin also enhanced Akt pSer473, GSK3 pSer21/9, and ERK1/2 pThr202/Tyr204 phosphorylation but had no significant effect on AMPK phosphorylation. AICAR, berberine, and adiponectin stimulated TBC1D4 phosphorylation at the 642 site but not the 588 site. Moreover, these agonists stimulated AMPK phosphorylation but had no effect on Akt and GSK3 phosphorylation. Only insulin was able to significantly stimulate ERK phosphorylation. Intriguingly, we were unable to observe any significant effect of either endothelin-1 or IL-6 on TBC1D4 phosphorylation at either the 588 or the 642 site (Fig. 7, A and B). To confirm this latter observation, we performed an additional study using a subset of agonists, but in this case we examined endogenous TBC1D4 phosphorylation using an immunoprecipitation approach. As observed in Fig. 7, insulin, AICAR, and berberine significantly increased phosphorylation at 642, whereas only insulin increased phosphorylation at 588. Endothelin-1 had no significant effect on either phosphorylation site. Interestingly berberine also had a significant effect on Ser588 phosphorylation; however, it amounted to only 20% of the effect observed with insulin.
Fig. 7.

Differential Phosphorylation of TBC1D4 on T642 and S588 by Agonists in L6 Myotubes
A, L6 myotubes overexpressing TBC1D4 at a medium level were incubated with 100 nm insulin, 2 mm AICAR, 2 μg/ml adiponectin, 10 nm endothelin-1, 100 ng/ml IL-6, or 10 μm berberine for 20 min. Cells were lysed, and 10 μg of protein per lysate was loaded onto SDS-PAGE and analyzed by Western blotting. B, Quantification of three experiments shown in panel A. C, L6 myotubes were incubated as in panel A, cells were lysed, and endogenous TBC1D4 was immunoprecipitated, and the precipitates were analyzed by Western blotting. D, Quantification of four experiments shown in panel C (*, P < 0.05; **, P < 0.005). bas, Basal; Berb, berberine; ET, endothelin-1; ins, insulin; gAd, adiponectin.
Overexpression of the TBC1D4-4P Mutant in L6 Cells Inhibits Stimulation of GLUT4 Translocation by All Agonists
TBC1D4 phosphorylation at 642 may only regulate cell surface levels of GLUT4 in response to certain agonists. In particular, TBC1D4 might not mediate the effects of endothelin-1 and IL-6 on GLUT4 translocation, because both agonists failed to stimulate TBC1D4 phosphorylation at 642. To test this we examined the effects of overexpressing the TBC1D4-4P mutant on agonist-mediated GLUT4 translocation in L6 myotubes. TBC1D4-wt and TBC1D4-4P were expressed in HA-GLUT4 expressing L6 cells using a retroviral expression plasmid that also expressed GFP as a reporter. Cells were sorted for GFP expression using fluorescence-activated cell sorting (FACS), and populations of high expressing cells were selected. These cells were stimulated with the different agonists, and GLUT4 translocation was monitored by the HA-GLUT4 translocation assay (30). Interestingly, TBC1D4-4P, but not TBC1D4-wt, inhibited the ability of all agonists to stimulate GLUT4 translocation, including the two agonists, which failed to phosphorylate TBC1D4 (Fig. 8). The degree of inhibition of the TBC1D4-4P (∼50%) was similar for each agonist, consistent with the observed impairment of contraction-stimulated glucose uptake in mouse tibialis anterior muscles electroporated with TBC1D4-4P (8). This indicates a fundamental role for TBC1D4 in many forms of agonist-stimulated GLUT4 translocation.
Fig. 8.

TBC1D4-4P Blocks Agonist-Induced GLUT4 Translocation in L6 Myotubes
Populations of L6 myoblasts were selected for high expression of GFP by FACS after retroviral transfection of the bicistronic pMIG vector alone, pMIG-TBC1D4-wt, or pMIG-TBC1D4-4P. Cells were propagated and differentiated into myotubes, and relative HA-GLUT4 translocation (surface vs. total HA-GLUT4) was measured after addition of 100 nm insulin, 2 mm AICAR, 2 μg/ml adiponectin, 10 nm endothelin-1, 100 ng/ml IL-6, or 10 μm berberine for 30 min. Results from a total of seven experiments are shown using L6 cells obtained from three independent rounds of FACS selection (*, P < 0.05; **, P < 0.005; n = 3). Adip, Adiponectin; AIC, AICAR; bas, basal; Berb, berberine; ET, endothelin; Ins, insulin.
DISCUSSION
Our laboratory has previously shown that TBC1D4 associates with GLUT4-containing membranes in the absence of insulin and dissociates in the presence of insulin (14). However, it was not known whether the association and/or dissociation of TBC1D4 was required for basal intracellular sequestration of GLUT4 and/or insulin-stimulated GLUT4 translocation, respectively. Our model, which was an adaptation of the original model proposed by Lienhard and colleagues (4) proposed that TBC1D4 is localized to GLUT4-containing membranes to maintain the Rab substrate in an inactive state, thus preventing GLUT4 translocation. Insulin-stimulated dissociation of TBC1D4 from GLUT4 membranes would then allow GTP loading and activation of the Rab, thereby facilitating fusion of GLUT4 vesicles with the PM. This model suggests that TBC1D4 localization to GLUT4 membranes is required in the absence of insulin for its role as a negative regulator of GLUT4 translocation. Overexpression of the TBC1D4-4P mutant in adipocytes inhibits insulin-stimulated GLUT4 translocation (4). We show in this study that the inhibitory TBC1D4-4P mutant failed to dissociate from GLUT4 vesicles upon insulin stimulation, supporting this hypothesis. Furthermore, we demonstrated that the TBC1D4 GAP domain fused to GLUT4 was sufficient to inhibit the insulin-stimulated translocation of this fusion construct to the PM and that this inhibitory effect was dependent on the GAP activity of TBC1D4. Interestingly, the TBC1D16 GAP domain fused to GLUT4 was unable to inhibit insulin-stimulated GLUT4 translocation to the cell surface, thus indicating direct in vivo specificity of the GAP domain itself. These data are consistent with the hypothesis that the cognate TBC1D4 Rab substrate is colocalized with GLUT4, albeit transiently, in the absence of insulin. The localization of the GAP domain to GLUT4 membranes is crucial for its inhibitory effect, because expression of the free GAP, which did not contain the IRAP-interacting domain, in adipocytes was without effect on GLUT4 translocation. It is likely that this inhibitory function may be mediated in vivo via only a small fraction of the total TBC1D4 pool in view of its enzymatic, rather than structural, role. Parenthetically, it should be noted that GLUT4 is localized to several intracellular compartments including endosomes, trans-Golgi network, and GLUT4 storage vesicles (31, 32, 33, 34), and it remains to be determined whether TBC1D4 function is confined to one of these specific locations. It was interesting in this regard that glycerol sedimentation analysis revealed that TBC1D4 maybe localized to a subcomponent in the GLUT4 vesicle peak. This is consistent with IF localization studies (14), which show incomplete overlap between TBC1D4 and GLUT4.
Insulin triggers the dissociation of TBC1D4 from GLUT4-containing membranes (5, 14). The overexpression of TBC1D4-4P inhibits GLUT4 translocation to the cell surface with insulin, whereas overexpression of the TBC1D4-wt has no effect (4). One possibility is that phosphorylation of TBC1D4 might simply release the GAP from the membrane facilitating GTP loading of its cognate Rab without necessarily modifying GAP activity per se. Our observation that direct fusion of full-length TBC1D4-4P to GLUT4 was sufficient to block GLUT4 translocation, whereas fusion of the TBC1D4-wt had no effect is not consistent with this interpretation. Interestingly, both TBC1D4-wt and -4P were targeted to membranes as shown in Fig. 1D, and TBC1D4-4P inhibited GLUT4 translocation to the PM (Fig. 1C) as described earlier by other techniques. After fusion of TBC1D4 to GLUT4, the TBC1D4 fusion constructs retained their respective effect of the nonfused TBC1D4-wt and 4P constructs on GLUT4 translocation, further indicating that TBC1D4 dissociation from membranes does not play a major role in insulin action. In light of the observation that fusion of the GAP domain alone to GLUT4 also inhibited GLUT4 translocation, it appears that additional functional features that are specific to the N terminus of TBC1D4 are required to override the GAP activity. Notably, we observed phosphorylation of the GLUT4-TBC1D4 fusion protein at the 642 site in response to insulin consistent with phosphorylation and/or 14-3-3 binding as candidates for this additional function.
Further insight into TBC1D4 function has been observed by analysis of other agonists that trigger GLUT4 translocation. Both muscle contraction and AICAR have been shown to stimulate TBC1D4 phosphorylation at the Thr642 site, possibly via AMPK (8, 9, 10). Consistent with these data we found that several agonists that activate AMPK including AICAR, berberine, and adiponectin, all stimulated TBC1D4 642 phosphorylation. Intriguingly, none of these agonists increased phosphorylation at the 588 site, nor did they trigger TBC1D4 translocation into the cytosol. The inhibition of GLUT4 translocation in response to each of these agonists after overexpression of the TBC1D4-4P mutant supports a role of TBC1D4 in each of these effects. Thus, this again supports the conclusion that the release of TBC1D4 from the membrane is not required to facilitate agonist-mediated GLUT4 translocation to the PM and nor is phosphorylation at the 588 site. One possibility is that 588 phosphorylation triggers release of TBC1D4 from membranes, and this effect may, in fact, confer a much more robust inactivation of TBC1D4 GAP activity than 642 phosphorylation alone. Consistent with this notion, we routinely observe that insulin has a more potent influence on GLUT4 translocation than other agonists (Figs. 6 and 8). This could also explain why the GLUT4-TBC1D4-wt fusion protein does not translocate to the PM to the same degree as GLUT4-GFP upon insulin stimulation (Fig. 5C). Thus, the approach described here of creating fusions between GLUT4 and the Rab GAP TBC1D4 enabled us to differentiate between the two effects of insulin on TBC1D4: phosphorylation at the 642 site vs. membrane dissociation.
Two agonists, endothelin-1 and IL-6, stimulated GLUT4 translocation in L6 myotubes, consistent with previous findings (24, 25, 26), but did not trigger TBC1D4 translocation into the cytosol, or phosphorylation of TBC1D4 at either the 588 or 642 sites. Hence, it is conceivable that these agonists might use a pathway other than TBC1D4 to stimulate GLUT4 translocation, and further analysis is required to resolve this. One possibility is that TBC1D1, a close homolog of TBC1D4, which shows induced expression when L6 myoblasts differentiate into myotubes, might mediate the effects of some of these agonists (35, 36, 37). It is also possible that these agonists phosphorylate alternate sites in TBC1D4 to facilitate inhibition of its GAP activity. Interestingly, the effect of all agonists on GLUT4 translocation to the cell surface was inhibited when the TBC1D4 phosphorylation mutant (TBC1D4-4P) was overexpressed in L6 muscle myotubes. This indicates a fundamental role for TBC1D4 in GLUT4 translocation to the cell surface for all agonists, although one possibility that we cannot exclude is that overexpression of the TBC1D4-4P might act to dominantly inhibit the actions of both endogenous TBC1D4 and TBC1D1.
In summary, these results indicate the following: 1) localization of TBC1D4 to GLUT4 membranes is required to act in its inhibitory role to allow basal intracellular sequestration of GLUT4; 2) TBC1D4 dissociation from GLUT4 membranes is not required to facilitate GLUT4 translocation to the PM; 3) the GAP domain alone of TBC1D4 possesses Rab specificity in vivo because another homologous GAP domain from TBC1D16 did not replicate the effects of TBC1D4; 3) TBC1D4 phosphorylation and/or 14-3-3 binding likely plays an essential role in inhibition of its GAP activity; and 4) many agonists that regulate GLUT4 translocation both via PI3K-dependent and -independent pathways appear to do so, at least in part, by overriding the GAP activity of TBC1D4. These findings further highlight the important role of this family of proteins in vesicle transport and, in particular, of TBC1D4 in cell surface delivery of GLUT4 and support the notion that this protein is a major convergence point for actions of many different agonists that are capable of transiently modifying the cell surface residence of molecules such as the GLUT4 transporters.
MATERIALS AND METHODS
Materials
DMEM and newborn calf serum were obtained from Invitrogen (Carlsbad, CA), myoclone-Plus fetal calf serum were from Trace Scientific (Melbourne, Australia), and antibiotics were obtained from GIBCO (Paisley, UK). Paraformaldehyde was from ProSciTech (Thuringowa, Australia). Recombinant mouse IL-6 was from BenderMed Systems (Burlingame, CA). Globular mouse adiponectin was produced and purified from BL21 cells as earlier described (38). Insulin was obtained from Calbiochem (San Diego, CA) and BSA was from Bovogen (Essendon, Australia). Bicinchoninic acid reagent, Supersignal West Pico chemiluminescent substrate, and protein G agarose beads were from Pierce Chemical Co. (Rockford, IL). Lipofectamine 2000 was from Invitrogen. Polyvinylidine difluoride membrane was from Millipore Corp. (Billerica, MA). Complete protease inhibitor cocktail tablets were from Roche (Indianapolis, IN). All other materials were obtained from Sigma Chemical Co. (St. Louis, MO). Antibodies were from Sigma [murine immunoglobulin G1-MOPC21, FLAG (F7425, F3165)], Babco [HA-peptide (16B12), Richmond, CA], and Abcam (myc, Cambridge, UK). Phosphorylation-specific antibodies against AKT, ERK, GSK3, and AMPK were from Cell Signaling Technology (Beverly, MA) and against TBC1D4 phospho-Ser588 and phospho-Thr642 were obtained from Peter Shepherd (Symansis, Auckland, New Zealand). Total TBC1D4 antibodies were from Novus (Littleton, CO) or earlier described (14). Antibodies against GLUT4 (39) have been described previously. ALEXA488-conjugated secondary antibodies and GFP antibodies (A-6455) were obtained from Molecular Probes (Leiden, The Netherlands). Cy3-, Cy5- and HRP-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA) and from Amersham (Buckinghamshire, UK). IR dye 700 or 800 conjugated secondary antibodies were from Rockland Immunochemicals (Gilbertville, PA).
DNA Constructs
TBC1D4-wt, and TBC1D4-4P in 3×FlagCMV10 constructs were a gift from Gus Lienhard (Dartmouth College, Hanover, NH) (4). TBC1D4-RA was made by site-directed mutagenesis of Arg973 to Ala. HA-GLUT4-GFP plasmid was kindly provided by Samuel Cushman (Stanford University, Stanford, CA) (40). pBabe-HA-Glut4 was described earlier (41). HA-GLUT4-TBC1D4GAP, HA-GLUT4-TBC1D4 and HA-GLUT4-TBC1D16GAP in QBI25 were constructed by PCR amplification of TBC1D4 Flag-tagged aa 865–1299, full-length Flag-tagged TBC1D4, and TBC1D16 Flag-tagged aa 381–765, respectively. The PCR products were subcloned into the KpnI and BamHI sites of HA-GLUT4-GFP in QBI25, thus replacing the GFP. The HA-GLUT4 control construct, containing a Flag-tag, was constructed by excising the full-length TBC1D4 with NotI and relegation of the remaining plasmid. Myc-GAP-wt was constructed by PCR amplification of TBC1D4 aa 865–1299, followed by subcloning into the KpnI and NotI site of pCMV-myc. pMIG Gateway was obtained from Liz Calton (Garvan Institute of Medical Research, Sydney, Australia) and was constructed by inserting the Gateway cassette into the HpaI site of pMIG (42). 3×Flag-TBC1D4-wt and 4P were amplified via PCR from 3xFlagCMV10 using primers that introduced attB1 and attB2 sites and cloned into the gateway vector pDONR221 by using BP clonase II (Invitrogen) and further cloned using LR clonase II into pMIG gateway.
Cell Culture and Transfection
3T3-L1 fibroblasts were obtained from Howard Green (Boston, MA), cultured, and differentiated into adipocytes as described elsewhere (14). CHO IR/IRS-1 cells were a gift from Morris White. L6 myoblasts were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Retrovirus for expression of pBABE and pMIG-based constructs was prepared using Plat-E cells as earlier described (41) For transient transfection, a total of 20 μg of DNA was used per 10-cm dish together with 50 μl of Lipofectamine 2000 according to the manufacturer’s protocol. 3T3-L1 fibroblasts were infected with pBabepuro-HA-GLUT4 retrovirus and selected with 2 μg/ml puromycin. For electroporation, adipocytes at d 6 after differentiation were trypsinized, washed with PBS, and washed and resuspended in cytomix (25 mm HEPES, pH 7.6; 120 mm KCl; 0.15 mm CaCl2; 10 mm K2HPO4/KH2PO4, pH 7.6; 2 mm EGTA; 5 mm MgCl2; 2 mm ATP; 5 mm reduced Glutathione). Plasmid DNA (100 μg) was mixed with the cells in a cuvette and electroporated with 200 V for 10 msec (ECM 830, BTX, San Diego, CA). Electroporated adipocytes were seeded onto gelatin-coated coverslips or cell culture dishes and used 24–48 h after electroporation. L6 myoblasts infected with pBabepuro-HA-GLUT4 retrovirus were selected with 2 μg/ml puromycin as described previously (41). 3T3-L1 fiboblasts or HA-GLUT4 expressing L6 myoblasts were infected with pMIG construct for TBC1D4-wt or TBC1D4-4P. Cells were FACS sorted 48 h after infection for medium or high GFP expression. Selected cell populations were propagated for up to 10 more passages and seeded into 10-cm dishes or 96-well plates as described before and differentiated into adipocytes or myoblasts (26, 30). L6 cells were used 4–7 d after differentiation.
Immunoprecipitation and Immunoblotting
Immunoprecipitations were carried out as previously described (13). All samples were subjected to SDS-PAGE analysis as previously described (14).
Confocal Laser Scanning Microscopy
Electroporated adipocytes were serum depleted for 2 h at 37 C, after which they were incubated in the absence or presence of 200 nm insulin for 20 min. Cells were then fixed with 3% paraformaldehyde in PBS. Fixed cells were washed with PBS, and free aldehyde groups were quenched with 50 mm glycine in PBS. The cells were then blocked in 2% BSA in PBS. For cell surface HA labeling, unpermeabilized cells were incubated with HA antibody, followed by the Cy3-conjugated secondary antibody. Subsequently or for colocalization experiments, cells were permeabilized in PBS containing 0.1% saponin and 2% BSA and labeled with one or two primary antibodies as indicated (HA, GFP, FLAG, myc), followed by incubation with Cy3 and/or ALEXA-488 conjugated secondary antibodies. Optical sections were analyzed by confocal laser scanning microscopy using a Leica TCS SP system using the ×40 lens. A slice in the middle of the adipocytes, 10–15 μm above the coverslip surface, was scanned, thus avoiding inclusion of fibroblasts in the analysis. Confocal images were scanned using exactly the same settings for each experimental condition and, in the case of double labeling experiments, each fluorophore was scanned sequentially. Fluorescence intensities were measured using the Leica confocal software. A region of interest was set around each cell, and the amount of fluorescence per unit area was determined for each channel. Surface GLUT4 staining was corrected for cell size by dividing the fluorescence intensity by the perimeter/area ratio. Surface GLUT4 staining was then calculated for each cell as a proportion of total GLUT4 expression per cell.
Cell Fractionation
Cell fractionation of L6 myotubes and 3T3-L1 adipocytes was performed as earlier described (14). Glycerol gradients were performed as in Ref. 43 .
HA-GLUT4 Translocation Assay
HA-GLUT4 translocation in L6 myotubes was measured in a 96-well plate assay as earlier described (26, 30). The GFP signal due to infection with pMIG, pMIG-TBC1D4-wt, or -4P retrovirus was included in the background subtraction and remained constant under different conditions.
Statistics
All data are expressed as average ± sem. Significance was determined by t test (two-sided, unpaired, equal variance).
Acknowledgments
We thank Drs. Gus Lienhard and Samuel Cushman for providing us with reagents. We thank Peter Shepherd (Symansis, Auckland, New Zealand) for providing us with the phospho-642 and phospho-588 antibodies. We thank Dr. Will Hughes for advice in microscopy and Dr. Tilman Brummer and Dr. Bronwyn Hegarty for critically reading the manuscript.
Footnotes
This work was supported in part by grants from the National Health and Medical Research Council of Australia and Diabetes Australia Research Trust (to D.E.J.). J.S. was supported by fellowships from the Swiss National Foundation and the Novartis Stiftung.
Author Disclosure: We declare that there is no conflict of interest that would prejudice the impartiality of this work.
First Published Online September 18, 2008
Abbreviations: aa, Amino acids; AICAR, 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside; AMPK, AMP-activated protein kinase; CHO, Chinese hamster ovary; FACS, fluorescence-activated cell sorting; GAP, guanosine triphosphatase-activating protein; GLUT4, glucose transporter 4; GSV, GLUT4 storage vesicles; HA, hemagglutinin; IRAP, insulin-regulated aminopeptidase; LDM, low density microsome; PI3K, phosphatidylinositide 3-kinase; PM, plasma membrane; TBC1D4, Tre2/Bub2/Cdc16 domain family member 4 (also called AS160, Akt substrate of 160 kDa); TBC1D4-4P, TBC1D4 mutant with four phosphorylation sites; TBC1D4-wt, wild-type TBC1D4; TBC1D16, Tre2/Bub2/Cdc16 domain family member 16.
References
- 1.Bryant NJ, Govers R, James DE 2002. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 3:267–277 [DOI] [PubMed] [Google Scholar]
- 2.Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE 2005. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab 2:263–272 [DOI] [PubMed] [Google Scholar]
- 3.Zeigerer A, McBrayer MK, McGraw TE 2004. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell 15:4406–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE 2003. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278:14599–14602 [DOI] [PubMed] [Google Scholar]
- 5.Kane S, Sano H, Liu SC, Asara JM, Lane WS, Garner CC, Lienhard GE 2002. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem 277:22115–22118 [DOI] [PubMed] [Google Scholar]
- 6.Deshmukh A, Coffey VG, Zhong Z, Chibalin AV, Hawley JA, Zierath JR 2006. Exercise-induced phosphorylation of the novel Akt substrates AS160 and filamin A in human skeletal muscle. Diabetes 55:1776–1782 [DOI] [PubMed] [Google Scholar]
- 7.Bruss MD, Arias EB, Lienhard GE, Cartee GD 2005. Increased phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle in response to insulin or contractile activity. Diabetes 54:41–50 [DOI] [PubMed] [Google Scholar]
- 8.Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ 2006. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem 281:31478–31485 [DOI] [PubMed] [Google Scholar]
- 9.Thong FS, Bilan PJ, Klip A 2007. The Rab GTPase-activating protein AS160 integrates Akt, protein kinase C, and AMP-activated protein kinase signals regulating GLUT4 traffic. Diabetes 56:414–423 [DOI] [PubMed] [Google Scholar]
- 10.Treebak JT, Glund S, Deshmukh A, Klein DK, Long YC, Jensen TE, Jorgensen SB, Viollet B, Andersson L, Neumann D, Wallimann T, Richter EA, Chibalin AV, Zierath JR, Wojtaszewski JF 2006. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes 55:2051–2058 [DOI] [PubMed] [Google Scholar]
- 11.Cartee GD, Wojtaszewski JF 2007. Role of Akt substrate of 160 kDa in insulin-stimulated and contraction-stimulated glucose transport. Appl Physiol Nutr Metab 32:557–566 [DOI] [PubMed] [Google Scholar]
- 12.Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, Sakamoto K, Hirshman MF, Goodyear LJ 2006. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 55:2067–2076 [DOI] [PubMed] [Google Scholar]
- 13.Ramm G, Larance M, Guilhaus M, James DE 2006. A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem 281:29174–29180 [DOI] [PubMed] [Google Scholar]
- 14.Larance M, Ramm G, Stöckli J, van Dam EM, Winata S, Wasinger V, Simpson F, Graham M, Junutula JR, Guilhaus M, James DE 2005. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem 280:37803- 37813 [DOI] [PubMed] [Google Scholar]
- 15.Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS, Lienhard GE 2005. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J 391:87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishikura S, Bilan PJ, Klip A 2007. Rabs 8A and 14 are targets of the insulin-regulated Rab-GAP AS160 regulating GLUT4 traffic in muscle cells. Biochem Biophys Res Commun 353:1074–1079 [DOI] [PubMed] [Google Scholar]
- 17.Sano H, Eguez L, Teruel MN, Fukuda M, Chuang TD, Chavez JA, Lienhard GE, McGraw TE 2007. Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab 5:293–303 [DOI] [PubMed] [Google Scholar]
- 18.Sano H, Roach WG, Peck GR, Fukuda M, Lienhard GE 2008. Rab10 in insulin-stimulated GLUT4 translocation. Biochem J 411:89–95 [DOI] [PubMed] [Google Scholar]
- 19.Peck GR, Ye S, Pham V, Fernando RN, Macaulay SL, Chai SY, Albiston AL 2006. Interaction of the Akt substrate, AS160, with the glucose transporter 4 vesicle marker protein, insulin-regulated aminopeptidase. Mol Endocrinol 20:2576–2583 [DOI] [PubMed] [Google Scholar]
- 20.Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J, Guo R, Johnson CL, Kiguchi K, Walker CL 2006. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J Cell Biol 173:279–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark SF, Martin S, Carozzi AJ, Hill MM, James DE 1998. Intracellular localization of phosphatidylinositide 3-kinase and insulin receptor substrate-1 in adipocytes: potential involvement of a membrane skeleton. J Cell Biol 140:1211–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kane S, Lienhard GE 2005. Calmodulin binds to the Rab GTPase activating protein required for insulin-stimulated GLUT4 translocation. Biochem Biophys Res Commun 335:175–180 [DOI] [PubMed] [Google Scholar]
- 23.Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, Ye JM, Lee CH, Oh WK, Kim CT, Hohnen-Behrens C, Gosby A, Kraegen EW, James DE, Kim JB 2006. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 55:2256–2264 [DOI] [PubMed] [Google Scholar]
- 24.Imamura T, Ishibashi K, Dalle S, Ugi S, Olefsky JM 1999. Endothelin-1-induced GLUT4 translocation is mediated via Gα(q/11) protein and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. J Biol Chem 274:33691–33695 [DOI] [PubMed] [Google Scholar]
- 25.Wu-Wong JR, Berg CE, Wang J, Chiou WJ, Fissel B 1999. Endothelin stimulates glucose uptake and GLUT4 translocation via activation of endothelin ETA receptor in 3T3-L1 adipocytes. J Biol Chem 274:8103–8110 [DOI] [PubMed] [Google Scholar]
- 26.Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen-Behrens C, Watt MJ, James DE, Kemp BE, Pedersen BK, Febbraio MA 2006. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 55:2688–2697 [DOI] [PubMed] [Google Scholar]
- 27.Ceddia RB, Somwar R, Maida A, Fang X, Bikopoulos G, Sweeney G 2005. Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia 48:132–139 [DOI] [PubMed] [Google Scholar]
- 28.Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, Liu F, Dong LQ 2006. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol 8:516–523 [DOI] [PubMed] [Google Scholar]
- 29.Somwar R, Niu W, Kim DY, Sweeney G, Randhawa VK, Huang C, Ramlal T, Klip A 2001. Differential effects of phosphatidylinositol 3-kinase inhibition on intracellular signals regulating GLUT4 translocation and glucose transport. J Biol Chem 276:46079–46087 [DOI] [PubMed] [Google Scholar]
- 30.Govers R, Coster AC, James DE 2004. Insulin increases cell surface GLUT4 levels by dose dependently discharging GLUT4 into a cell surface recycling pathway. Mol Cell Biol 24:6456–6466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE 1991. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J Cell Biol 113:123–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin S, Millar CA, Lyttle CT, Meerloo T, Marsh BJ, Gould GW, James DE 2000. Effects of insulin on intracellular GLUT4 vesicles in adipocytes: evidence for a secretory mode of regulation. J Cell Sci 113:3427–3438 [DOI] [PubMed] [Google Scholar]
- 33.Ramm G, Slot JW, James DE, Stoorvogel W 2000. Insulin recruits GLUT4 from specialized VAMP2-carrying vesicles as well as from the dynamic endosomal/trans-Golgi network in rat adipocytes. Mol Biol Cell 11:4079–4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larance M, Ramm G, James DE 2008. The GLUT4 code. Mol Endocrinol 22:226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roach WG, Chavez JA, Miinea CP, Lienhard GE 2007. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. Biochem J 403:353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S, Murphy J, Toth R, Campbell DG, Morrice NA, Mackintosh C 2008. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem J 409:449–459 [DOI] [PubMed] [Google Scholar]
- 37.Chavez JA, Roach WG, Keller SR, Lane WS, Lienhard GE 2008. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J Biol Chem 283:9187–9195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Xu A, Knight C, Xu LY, Cooper GJ 2002. Hydroxylation and glycosylation of the four conserved lysine residues in the collagenous domain of adiponectin. Potential role in the modulation of its insulin-sensitizing activity. J Biol Chem 277:19521–19529 [DOI] [PubMed] [Google Scholar]
- 39.James DE, Brown R, Navarro J, Pilch PF 1988. Insulin-regulatable tissues express a unique insulin-sensitive glucose-transport protein. Nature 333:183–185 [DOI] [PubMed] [Google Scholar]
- 40.Malide D, Yewdell JW, Bennink JR, Cushman SW 2001. The export of major histocompatibility complex class I molecules from the endoplasmic reticulum of rat brown adipose cells is acutely stimulated by insulin. Mol Biol Cell 12:101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shewan AM, van Dam EM, Martin S, Luen TB, Hong W, Bryant NJ, James DE 2003. GLUT4 recycles via a trans-Golgi network (TGN) subdomain enriched in Syntaxins 6 and 16 but not TGN38: involvement of an acidic targeting motif. Mol Biol Cell 14:973–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Parijs L, Refaeli Y, Lord JD, Nelson BH, Abbas AK, Baltimore D 1999. Uncoupling IL-2 signals that regulate T cell proliferation, survival, and Fas-mediated activation-induced cell death. Immunity 11:281–288 [DOI] [PubMed] [Google Scholar]
- 43.Wei ML, Bonzelius F, Scully RM, Kelly RB, Herman GA 1998. GLUT4 and transferrin receptor are differentially sorted along the endocytic pathway in CHO cells. J Cell Biol 140:565–575 [DOI] [PMC free article] [PubMed] [Google Scholar]