

Abstract
Transduction of the insulin signal is mediated by multisite Tyr and Ser/Thr phosphorylation of the insulin receptor substrates (IRSs). Previous studies on the function of single-site phosphorylation, particularly phosphorylation of Ser-302, -307, and -318 of IRS-1, showed attenuating as well as enhancing effects on insulin action. In this study we investigated a possible cross talk of these opposedly acting serine residues in insulin-stimulated skeletal muscle cells by monitoring phosphorylation kinetics, and applying loss of function, gain of function, and combination mutants of IRS-1. The phosphorylation at Ser-302 was rapid and transient, followed first by Ser-318 phosphorylation and later by phosphorylation of Ser-307, which remained elevated for 120 min. Mutation of Ser-302 to alanine clearly reduced the subsequent protein kinase C-ζ-mediated Ser-318 phosphorylation. The Ser-307 phosphorylation was independent of Ser-302 and/or Ser-318 phosphorylation status. The functional consequences of these phosphorylation patterns were studied by the expression of IRS-1 mutants. The E302A307E318 mutant simulating the early phosphorylation pattern resulted in a significant increase in Akt and glycogen synthase kinase 3 phosphorylation. Furthermore, glucose uptake was enhanced. Because the down-regulation of the insulin signal was not affected, this phosphorylation pattern seems to be involved in the enhancement but not in the termination of the insulin signal. This enhancing effect was completely absent when Ser-302 was unphosphorylated and Ser-307 was phosphorylated as simulated by the A302E307E318 mutant. Phospho-Ser-318, sequentially phosphorylated at least by protein kinase C-ζ and a mammalian target of rapamycin/raptor-dependent kinase, was part of the positive as well as of the subsequent negative phosphorylation pattern. Thus we conclude that insulin stimulation temporally generates different phosphorylation statuses of the same residues that exert different functions in insulin signaling.
PHOSPHORYLATION OF DISTINCT residues is an important posttranslational modification to regulate the function of proteins, including enzymatic activity and creation of docking sites for other proteins, thereby transducing signals from transmembrane receptors to intracellular target molecules. The insulin receptor substrates (IRSs) are key players in the signaling network of tyrosine kinase receptors. The phosphorylation of IRS by kinases modifies their property as multidocking site proteins and consequently the diversification of the incoming signal, thereby achieving a specific and adaptive cellular response (1, 2, 3, 4, 5). The best studied IRS, IRS-1, contains 34 tyrosine residues and 244 serine/threonine residues (6). Whereas tyrosine phosphorylation of IRS-1 by the tyrosine kinase activity of the insulin receptor is the crucial event for the signal transduction to downstream targets, serine/threonine phosphorylation appears to be responsible for the precise regulation and is discussed as a major mechanism for the termination of the insulin signal (1, 2). Unbalanced chronic stimulation of IRS-1 serine kinases leads to hyperphosphorylation of IRS-1 and is a major pathophysiological mechanism in the development of insulin resistance and subsequently of type 2 diabetes (5, 7).
During the last decade, several serine/threonine residues in IRS-1 have been identified as functionally relevant phosphorylation sites, and the knowledge about the identity and regulation of the IRS-1 kinases involved is accumulating. Most of these data fit with the model of a generalized mechanism for reduced insulin action, important for the physiological determination of insulin action but also responsible for the induction of insulin resistance. These sites (corresponding to rat IRS-1 sequence) include Ser-24 (8, 9), -267 (10), -302 (11), -307 (12, 13, 14, 15, 16, 17, 18), -318 (19, 20), -332 (21), -357 (22), -408 (23), -522 (24), -612 (15, 25, 26, 27), -632 (15, 25, 28), -662 (25, 27), -731 (25), -789 (29, 30), and -1099/1100 (31, 32). The IRS-1 kinases responsible for phosphorylation of the serine residues of IRS-1 and for attenuation of insulin signaling are protein kinase C (PKC)-α (9), PKC-δ (22), PCK-θ (31, 33), c-jun-N-terminal kinase (JNK) (12, 13), inhibitor of NF κB kinase (14, 34, 35), MAPKs (15), mammalian target of rapamycin (mTOR) (15, 16, 17, 36) and its downstream kinase S6 kinase-1 (28, 32, 37), glycogen synthase kinase (GSK)-3 (21), and salt-inducible kinase-2 (30). However, serine phosphorylation of IRS-1 appears also to be involved in the positive regulation of insulin signal transduction (38). The phosphorylation of Ser-302 (39, 40, 41), -318 (42, 43), -325 (44), -629 (not present in rodents) (45), -789 (46), and -1216 (47, 48) has been associated with improved insulin signaling. Although many sites in IRS-1 have been identified, the investigation of the complex regulation and the role of IRS-1 serine phosphorylation patterns in the modulation of the activation and inhibition of insulin signal transduction are still challenging, reflected, for instance, by the inconsistent data reporting stimulatory as well as inhibitory functions for Ser-302 (11, 39, 40, 41), -318 (19, 42, 43), and -789 (29, 30, 46) in insulin signal transduction.
Very recently, capillary HPLC-electrospray tandem mass spectrometry has been introduced for global assessment of the in vivo phosphorylation pattern of IRS-1 obtained from biopsies of human vastus lateralis muscle (49, 50). Phosphorylation of 22 Ser/Thr sites from Ser-302, -307, -318 up to Ser-1223 (rat IRS-1 sequence) was detected (50). After 2 h of insulin stimulation a significant change in phosphorylation intensity could be demonstrated for several sites including a 2.6-fold increase in Ser-307 phosphorylation (50).
However, these elegant studies did not provide insight into the functional role of these phosphorylation sites either singly or in combination, nor did they reveal whether or not the phosphorylation of each site is an independent event. Therefore, it was the aim of our study to investigate whether distinct phosphorylation sites are independently phosphorylated and whether functional consequences are related to or independent of the combination of Ser phosphorylation. We focused our studies on the possible interdependence of the phosphorylation of Ser-302, -307, and -318 because the present reports describing the individual function of these residues indicated a role for Ser-307 in the attenuation of insulin signaling (12, 13, 14, 15, 16, 17, 18), whereas there was contrasting evidence on whether phospho-Ser-302 and -318 have a positive (39, 40, 41, 43) or negative effect (9, 16, 36, 37, 38, 39) on insulin signal transduction.
We found sequential and interdependent changes in the insulin-dependent phosphorylation state of Ser-302, -307, and -318 of IRS-1 in skeletal muscle cells, which was temporally mediated by different kinases. The phosphorylation of Ser-302 is an initial event and appeared to be involved in the subsequent phosphorylation of Ser-318. Phospho-Ser-302/-318 exerts a pronounced positive effect on the early insulin action followed by the attenuation of insulin action when Ser-307 and Ser-318 are phosphorylated and Ser-302 has become dephosphorylated. These novel pieces of information may also reconcile conflicting findings about the role of Ser-302 and Ser-318.
RESULTS
Ser-302, -307, and -318 Differ from Each Other in the Insulin-Induced Phosphorylation Kinetics
The phosphorylation of Ser-302, -307, and -318 at defined time points during insulin treatment has been demonstrated in cell culture, and in mice and human studies, but the phosphorylation kinetics are largely unknown. Therefore, we compared the phosphorylation states of the three sites of endogenous IRS-1 at 5, 10, 30, 60, and 120 min after insulin stimulation in L6 myotubes. We observed characteristic kinetics for each individual residue (Fig. 1, A–C): phosphorylation of Ser-302 was an early event with a maximal intensity after 10 min of insulin stimulation and a clear reduction after 30, 60, and 120 min. The phosphorylation of Ser-307 showed different kinetics with a maximum after 60 min of insulin stimulation. The early increase in the phosphorylation of Ser-318 was comparable to that of Ser-302; however, the high phosphorylation signal lasted until late phase of insulin stimulation. Thus, the data suggest a sequential phosphorylation of the three serine residues in the order Ser-302, -318, and -307 with a clear and continuous reduction of phospho-Ser-302 after 60 and 120 min far below the intensity of baseline values.
Fig. 1.

Kinetics of Insulin-Induced Serine Phosphorylation of IRS-1
A, Immunoblot analysis of phosphorylation of Ser-302, -307, -318, and IRS-1 protein in myotube cell extracts (Ser-307 and -318) and of immunoprecipitated IRS-1 (Ser-302) after stimulation with 10 nm insulin for 0–120 min as indicated. Due to the lower sensitivity of the anti-phospho-Ser-302 antibody, immunoprecipitation of IRS-1 was performed to study this phosphorylation. B, Densitometric quantification of the immunoblots (n = 4, mean ± sem). Relative phosphorylation of the indicated serine compared with maximal insulin-stimulated phosphorylation of this site set as 100%. C, Representative immunoblot of the phosphorylation of Ser-307, -318, and IRS-1 protein of immunoprecipitated IRS-1 after stimulation with 10 nm insulin for 0–120 min as indicated. D, Immunoblot analysis of tyrosine phosphorylation of immunoprecipitated IRS-1 and (E) densitometric quantification (n = 5; mean ± sem). Values of unstimulated cells were set as 1. F. Scheme of the serine phosphorylation pattern induced by insulin and the corresponding IRS-1 mutants used for the following studies. Ins, Insulin; IP, immunoprecipitation.
The insulin-induced tyrosine phosphorylation of IRS-1 showed a maximum after 5 min and was significantly decreased after 30 min with no further change after 60 and 120 min (Fig. 1, D and E). This led us to speculate that the early phosphorylated serine residues at position 302 and 318 might be involved in positive insulin signaling.
To study the interplay and potential interdependency of the opposing serine sites 302, 307, and 318, as well as their effects on signal transduction, IRS-1 mutants with glutamate (E) simulating the phosphorylated and alanine (A) imitating the unphosphorylated serine residue were generated for further experiments to simulate different phosphorylation states based on the results from the kinetic study, as shown in Fig. 1F.
Role of Phospho-Ser-302 in the Subsequent Phosphorylation of Ser-318
Next we aimed to prove the hypothesis of a sequential phosphorylation, i.e. whether the phosphorylation of Ser-302 is a prerequisite for the phosphorylation of Ser-318 and subsequently, whether phosphorylation of Ser-318 regulates the phosphorylation of Ser-307.
First we studied whether Ser-302 phosphorylation triggers phosphorylation of Ser-318. The IRS-1 A302/307 mutant simulates the absence of phospho-Ser-302 and phospho-Ser-307. We used a double mutant, because in this early phase of insulin signaling Ser-307 is not phosphorylated (Fig. 1), and expression of the IRS-1 A307 single mutant had only a marginal effect on the phosphorylation of Ser-318 (data not shown). In C2C12 cells expressing IRS-1 A302/307 mutant, no insulin-induced phosphorylation on Ser-318 was observed after 5 min of stimulation (Fig. 2A). These results suggest that the phosphorylation of Ser-302 may be involved in the subsequent rapid phosphorylation of Ser-318.
Fig. 2.

Role of Phospho-Ser-302 in the Subsequent Ser-318 Phosphorylation
A, C2C12 cells transfected with empty vector (con), IRS-1 wt, or IRS-1 A302/307 were stimulated with 10 nm insulin as indicated. A representative immunoblot for phospho-Ser-318 and the reprobe for IRS-1 are shown. In the lower part, densitometric quantification of the immunoblots is shown (n = 4; mean ± sem); *, P < 0.05 compared with IRS-1 wt transfected cells stimulated with insulin for 5 min. (B and C) C2C12 cells transfected with IRS-1 wt or IRS-1 E302A307A318 (B) and IRS-1 wt or IRS-1 A302 (C) were stimulated with 10 nm insulin for 5 min. PKC-ζ/λ was immunoprecipitated and coprecipitated IRS-1 was visualized by immunoblotting. IRS-1 protein expression in the extracts is shown in the upper panel. The lower part of the figure shows the densitometric quantification (n = 5; mean ± sem); *, P < 0.05 vs. IRS-1 wt transfected cells stimulated with insulin for 5 min; #, P < 0.05 vs. unstimulated IRS-1 wt transfected cells. con, Control; IB, immunoblot; ins, insulin; IP, immunoprecipitation; rel., relative.
Recently we have shown that phosphorylation of Ser-318 is mediated, at least in part, by PKC-ζ and that insulin induces the association of IRS-1 and PKC-ζ in a time-dependent manner (42). The IRS-1/PKC-ζ-association is found after 5 min of stimulation but is lost after 10 and 60 min (42). Therefore, we speculated that phosphorylation of Ser-302 enhances this interaction. Using C2C12 cells expressing IRS-1 E302A307A318, which simulates the basal phosphorylation state with the phosphorylation only on Ser-302 (Fig. 1F), we observed a significant increase in the association of PKC-ζ to this IRS-1 mutant after 5 min of insulin stimulation (Fig. 2B). Because we have previously shown that expression of the IRS-1 A318 single mutant does not change the association to PKC-ζ compared with IRS-1 wild-type (wt) expressing cells (42), we concluded that the imitation of a permanent Ser-302 phosphorylation could lead to an increased interaction of PKC-ζ with IRS-1. Moreover, mutation of Ser-302 to alanine reduced both basal and insulin-stimulated PKC-ζ/IRS-1 association (Fig. 2C). These data suggest a potential role of phospho-Ser-302 in the subsequent PKC-ζ-mediated phosphorylation of IRS-1.
PKC-ζ, mTOR/Raptor-Dependent Kinase, and JNK Are Involved in the Insulin-Induced, Sequential Phosphorylation of Ser-302, -307, and -318
The results shown in Fig. 2 suggest a relevant role of the initial phosphorylation of Ser-302 for the binding of PKC-ζ and the subsequent PKC-ζ-mediated phosphorylation of Ser-318. However, the ongoing phosphorylation of Ser-318 after 10 and 60 min of insulin stimulation seems not to depend on the phosphorylation at Ser-302 (Fig. 2A), which may be a hint that more than one kinase is involved in the phosphorylation of Ser-318. We followed this aspect using small interfering RNA (siRNA) approaches in C2C12 cells. First we knocked down PKC-ζ/λ (the atypical PKC isoforms expressed in mice; shortly referred to as PKC-ζ). Interestingly, the phosphorylation of Ser-318 after 5 min of insulin stimulation was completely prevented and tended to be reduced after 10 min (P = 0.06), but was not impaired after 60 min (Fig. 3A), indicating that PKC-ζ is responsible for the early, but not for the ongoing, insulin-induced phosphorylation of Ser-318. Because the lasting phosphorylation state of the studied IRS-1 region is dominated by Ser-307 and Ser-318 (Fig. 1) and the insulin-mediated phosphorylation of Ser-307 is mediated by JNK and mTOR (12, 13, 15), we first studied the potential role of these kinases in the phosphorylation of Ser-318.
Fig. 3.
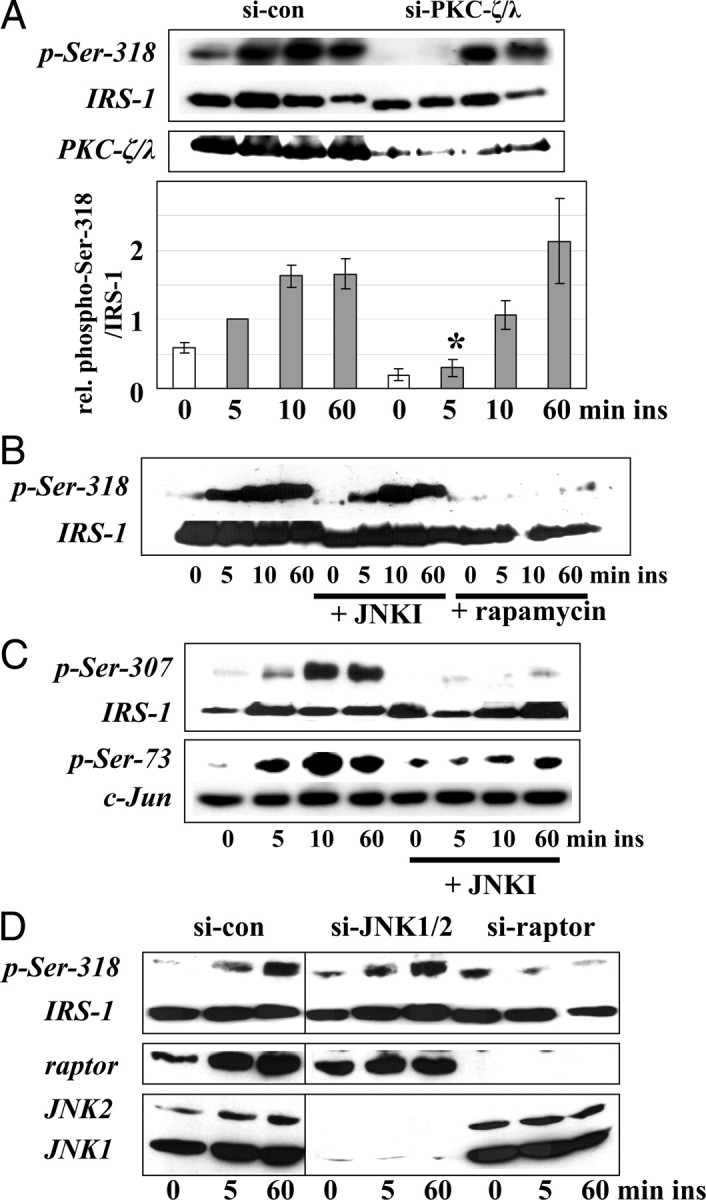
Insulin-Induced Phosphorylation of Ser-318 is First Mediated by PKC-ζ and Later on by a Raptor-Dependent Pathway
A, C2C12 cells transfected with siRNA oligonucleotides targeting PKC-ζ/λ were stimulated with 10 nm insulin as indicated. Shown are representative immunoblots for phospho-Ser-318 and PKC-ζ/λ. Membranes were reprobed for IRS-1 protein. In the lower part, densitometric quantification of the immunoblots is shown (n = 4, mean ± sem); *, P < 0.05 compared with control transfected cells stimulated with insulin for 5 min. B and C, C2C12 cells were preincubated with 10 μm JNK inhibitor (JNKI) SP 600125 (B and C) or 25 nm rapamayin (B) for 30 min before stimulation with 10 nm insulin. Representative immunoblots for phospho-Ser-318 and the reprobe for IRS-1 (B), phospho-Ser-307, phospho-Ser-73, and the corresponding reprobes for IRS-1 and c-Jun protein (C) are shown. D, C2C12 cells transfected with siRNA oligonucleotides targeting JNK1 and -2 or raptor were stimulated with 10 nm insulin as indicated. Shown are representative immunoblots for phospho-Ser-318, JNK1 and -2, and raptor. Lanes from each blot shown here are from one gel but have been loaded in a different order. con, Control; si-, small interfering; ins, insulin.
First we performed experiments using SP 600125, an inhibitor of JNK, and rapamycin, an inhibitor of the kinase mTOR (Fig. 3, B and C). We could not detect a participation of JNK in the insulin-induced Ser-318 phosphorylation in C2C12 cells at any time point studied (Fig. 3B). The effectiveness of the JNK inhibitor was indicated by the complete inhibition of the insulin-induced phosphorylation of Ser-307 and the reduction of the phosphorylation of the JNK substrate c-Jun (Fig. 3C). The use of rapamycin, shown in Fig. 3B, led to a complete inhibition of the phosphorylation of Ser-318. After that, we performed knock-down experiments of JNK1/2 as well as raptor using siRNA oligonucleotides (Fig. 3D). Knockdown of JNK1 and JNK2 could not prevent the phosphorylation of Ser-318 (Fig. 3D). Contrary to that, the knockdown of raptor prevented the phosphorylation of Ser-318 (Fig. 3D). Therefore we speculated whether a mTOR/raptor-dependent kinase is involved in the phosphorylation of Ser-318 at later time points, because both treatment of cells with rapamycin and knockdown of raptor using siRNA oligonucleotides prevented the phosphorylation of Ser-318 at all time points studied (Fig. 3, B and D). The data also suggest that a mTOR/raptor-dependent pathway may be relevant to initiate the early phosphorylation of Ser-318. Because recent reports showed a mTOR pathway-dependent phosphorylation of Ser-302 upon insulin stimulation (11, 39), we addressed this issue in our C2C12 skeletal muscle cell culture model using rapamycin to inhibit the phosphorylation of Ser-302, but no effect could be detected (data not shown).
The Subsequent Phosphorylation of Ser-307 Is Independent of the Precursory Phosphorylation of Ser-302 and -318
Because phosphorylation of Ser-302 and -318 preceded the phosphorylation of Ser-307 we investigated the potential effect of both sites on the later phosphorylation in C2C12 cells. The mutation of both serine residues had no effect on the insulin-induced phosphorylation of Ser-307 (Fig. 4A). Furthermore, based on the kinetics study showing an early decrease of phospho-Ser-302 (Fig. 1), we investigated the effect of phosphorylation of Ser-318 simulated by the E318 mutation. However, the expression of this mutant did not change the phosphorylation of Ser-307 (Fig. 4B). Because the knock-down of PKC-ζ, which prevents the early phosphorylation of Ser-318 (Fig. 2A) had also no effect on the insulin-induced phosphorylation of Ser-307 (data not shown), we suggest that the combined phosphorylation of Ser-302/-318, as well as phosphor-Ser-318 alone, are no prerequisite for the phosphorylation of Ser-307; i.e. Ser-307 phosphorylation is independently regulated.
Fig. 4.

Phosphorylation of Ser-318 Is Not Necessary for Phosphorylation of Ser-307
A and B, C2C12 cells transfected with IRS-1 wt, IRS-1 A302/318, or IRS-1 E318 were stimulated with 10 nm insulin as indicated. Shown are representative immunoblots for phospho-Ser-307 and the corresponding reprobe for IRS-1 protein and in the lower part the densitometric quantification (n = 4, mean ± sem). ins, Insulin; rel., relative.
The Early Phosphorylation of Ser-302 and Ser-318 Enhances Insulin Action
The studies on the insulin-stimulated kinetics of the phosphorylation of the three serine residues revealed an early phosphorylation pattern after 5 min of stimulation, including phosphorylation of Ser-302 and -318 (simulated with IRS-1 E302A307E318), and a late phosphorylation pattern including phosphorylation of Ser-307 and Ser-318 (simulated with IRS-1 A302E307E318) (Fig. 1F). The kinetic of the tyrosine phosphorylation of IRS-1 suggests an involvement of the early phosphorylation of Ser-302/-318 in the enhancement of insulin signaling (Fig. 1, D and E). Expression of IRS-1 E302A307E318 to investigate the function of phosphoSer-302/-318 on insulin signal transduction led to a considerable increase in the phosphorylation of Ser-473 of Akt after 10 min of insulin stimulation in C2C12 cells, indicating enhanced activation of this kinase (Fig. 5, A and B). The phosphorylation of the downstream kinase GSK-3 was also increased (Fig. 5A). Of note, the reduction of Akt phosphorylation after 60 min of insulin treatment was not significantly different from IRS-1 wt transfected cells, indicating the preceding role of phospho-Ser-302/-318 in the activation of the signaling with no detectable influence on the termination of insulin action (Fig. 5B). In contrast, C2C12 cells expressing the IRS-1 A302E307E318 mutant, which represents the phosphorylation state of this IRS-1 region in the ongoing insulin action, exhibit a slightly reduced phosphorylation of Akt and GSK-3 after 10 min of insulin stimulation. Thus, the early phosphorylation of IRS-1 on Ser-302 and Ser-318 enhances insulin signaling, and this effect is absent when Ser-307 instead of Ser-302 is phosphorylated. The effect on insulin-stimulated Akt phosphorylation was further studied using the single-alanine mutants IRS-1 A302, A307, and A318 (Fig. 5, C and D). The complete inhibition of Ser-307 phosphorylation using IRS-1 A307 led to a significant increase in Akt phosphorylation at all time points studied, underlining the important inhibitory function of this site. Expression of IRS-1 A318 reduced the Akt phosphorylation after 5 min of insulin stimulation, demonstrating the positive function of phospho-Ser-318 on acute insulin action. This has also been reported in a recent study (42). The expression of the single Ala-302 mutant had no significant effect on insulin-stimulated Akt phosphorylation.
Fig. 5.
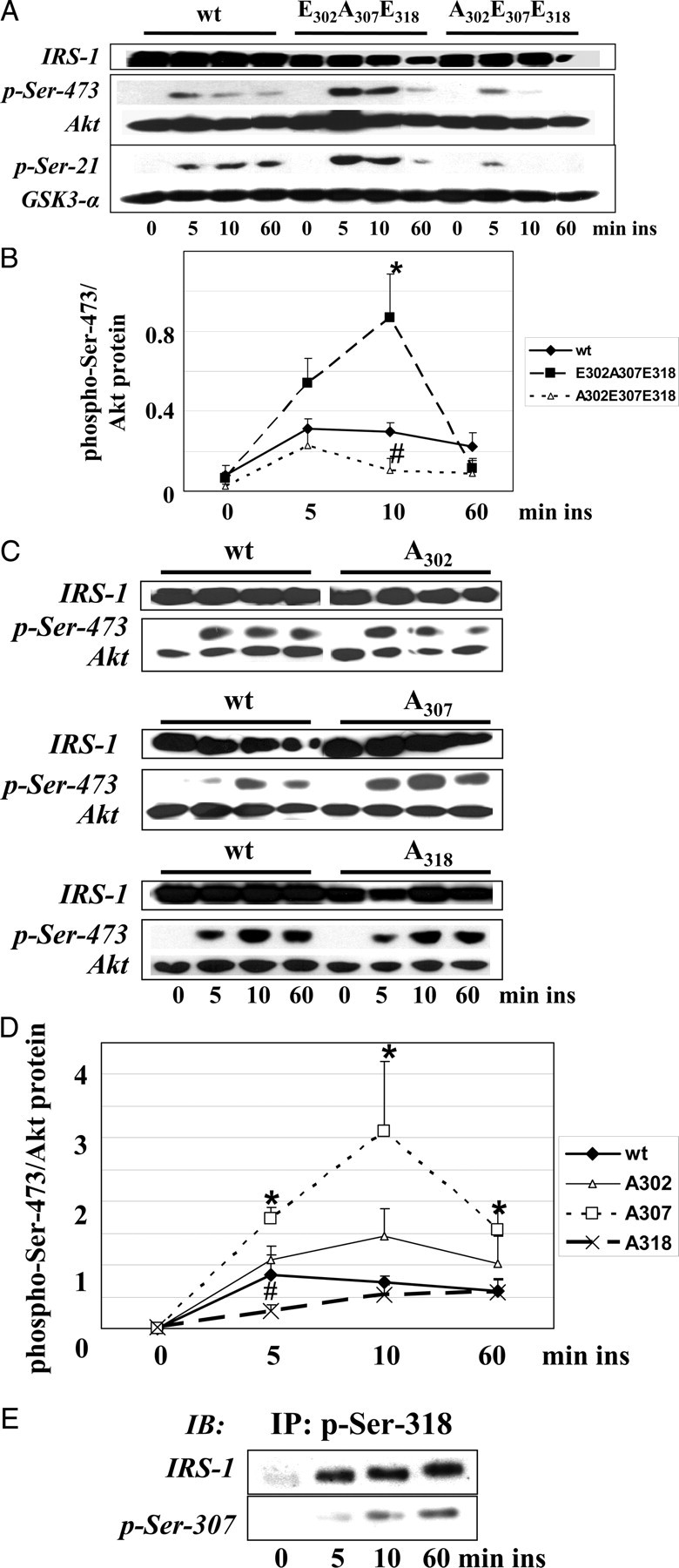
The Early Phosphorylation Pattern Enhances Insulin Action
A, C2C12 cells were transfected with IRS-1 wt, IRS-1 E302A307E318, or IRS-1 A302E307E318 and stimulated with 10 nm insulin as indicated. Cell extracts were immunoblotted for phospho-Ser-473 of Akt, phospho-Ser-9/21 of GSK-3, and IRS-1. Membranes were reprobed for Akt and GSK-3 protein. Only phosphorylation of Ser-21 of GSK-3α was detected, whereas both GSK-3α and -β were expressed (data not shown). B, Densitometric quantification of the phosphorylation of Ser-473 of Akt (n = 4, mean ± sem); *, P < 0.05 IRS-1 E302A307E318 vs. IRS-1 wt; #, P < 0.05 IRS-1 A302E307E318 vs. IRS-1 wt after 10 min of insulin stimulation. C, C2C12 cells were transfected with IRS-1 wt, IRS-1 A302, A307, or A318 and stimulated with 10 nm insulin as indicated. Cell extracts were immunoblotted for phospho-Ser-473 of Akt and IRS-1. Membranes were reprobed for Akt protein. D, Densitometric quantification of the phosphorylation of Ser-473 of Akt (n = 4, mean ± sem); *, P < 0.05 IRS-1 A307 vs. IRS-1 wt; #, P < 0.05 IRS-1 A318 vs. IRS-1 wt after 5, 10, or 60 min of insulin stimulation. E, Immunoprecipitation of IRS-1 wt transfected C2C12 cells using immunopurified anti-phospho-Ser-318 antibodies. Immunoprecipitates were immunoblotted for phospho-Ser-307 and IRS-1. IB, Immunoblot; ins, insulin; IP, immunoprecipitation.
From these data with the single-alanine mutants it was somehow unexpected that expression of the IRS-1 A302E307E318 mutant did not result in a strong down-regulation of insulin action. Earlier studies on the function of the single phosphorylation of each residue clearly implicate the involvement of Ser-307 and -318 in the attenuation of insulin action performed by loss of function studies using serine to alanine mutants (12, 13, 16, 19, 42, 51). This finding raises the question of whether both sites may occur in the phosphorylated state on the same molecule. This crucial point was investigated in a next step by the immunoprecipitation of the fraction of IRS-1, which is phosphorylated on Ser-318 upon insulin stimulation. Applying an immunopurified phospho-Ser-318 antibody we detected comparable amounts of IRS-1 after stimulation with insulin for 5, 10, and 60 min, but not in unstimulated cells (Fig. 5E), as expected from the kinetics shown in Fig. 1B. Moreover, we also found the time-dependent phosphorylation of Ser-307 in this immunoprecipitated, i.e. Ser-318 phosphorylated IRS-1 (Fig. 5E).
The Early Serine Phosphorylation of IRS-1 Enhances Glucose Uptake
The regulation of insulin action was further investigated as insulin-stimulated glucose uptake in L6 myoblasts expressing the IRS-1 triple mutants after 5 min of insulin stimulation (Fig. 6A). Simulating the early Ser-302/-318 phosphorylation by the E302A307E318 mutant led to an increased insulin-dependent glucose uptake. Of note, in the unstimulated state the glucose uptake was also significantly enhanced in cells expressing the E302A307E318 mutant. Expression of IRS-1 A302E307E318 did not reduce glucose uptake in comparison with IRS-1 wt expressing cells (Fig. 6A). This could indicate that the phosphorylation pattern has no influence on the acute insulin-stimulated glucose uptake. We also studied the effect of the single-alanine mutants IRS-1 A302, A307, and A318 (Fig. 6B). We observed a significant reduction of glucose uptake in A302 and A318 expressing cells after 5 min of insulin stimulation. These data support the positive function of phospho-Ser-302 and phospho-Ser-318 in early insulin action. Moreover, expression of IRS-1 E302 increased basal and insulin-stimulated glucose uptake (Fig. 6C), similar to the results obtained in IRS-1 E302A307E318 expressing cells (Fig. 6A). No difference in the insulin-stimulated glucose uptake was detected in IRS-1 A307 expressing cells in comparison with IRS-1 wt (Fig. 6B), despite its pronounced effect on Akt phosphorylation (Fig. 5, C and D). Because A307 represents the unphosphorylated Ser-307 residue, and phosphorylation of Ser-307 was almost absent after 5 min of insulin stimulation (Fig. 1), this result suggests that phosphorylation of Ser-307 is not involved in the early regulation of insulin-dependent glucose uptake.
Fig. 6.
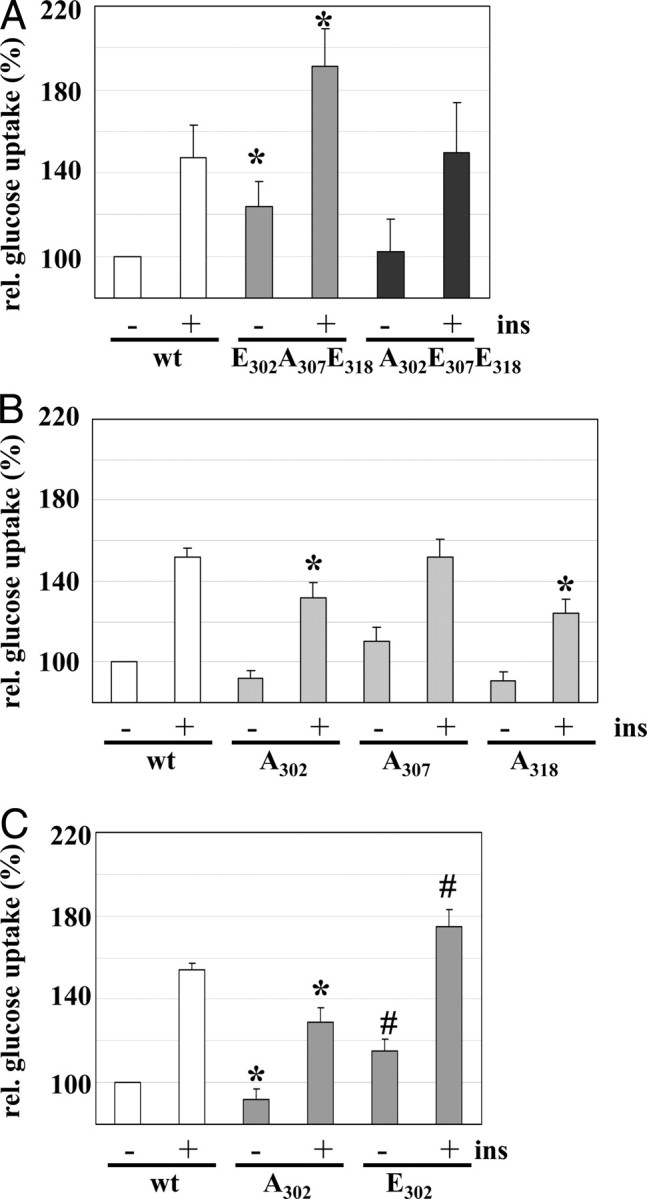
The Early Serine Phosphorylation of IRS-1 Enhances Glucose Uptake
A, Glucose uptake in L6 Glut4myc cells transfected with IRS-1 wt, IRS-1 E302A307E318, or IRS-1 A302E307E318 after 5 min of insulin stimulation. Basal glucose uptake of IRS-1 wt transfected cells was set as 100%. *, P < 0.05 vs. IRS-1 wt. B, Glucose uptake after 5 min of insulin stimulation in L6 Glut4myc cells transfected with IRS-1 wt, IRS-1 A302, IRS-1A307, and IRS-1 A318; *, P < 0.05 A302 and A318 vs. insulin-stimulated IRS-1 wt. C, Glucose uptake in L6 Glut4myc cells transfected with IRS-1 wt, IRS-1 A302, and IRS-1 E302 after 5 min of insulin stimulation. *, P < 0.05 A302 vs. basal or insulin-stimulated IRS-1 wt; #, P < 0.05 E302 vs. basal or insulin-stimulated IRS-1 wt (n = 4, mean ± sem). ins, Insulin.
DISCUSSION
During the last decade more than 20 serine/threonine residues in IRS-1 have been identified as functionally relevant phosphorylation sites. Data from 16 sites fit with the model of a generalized mechanism for reduced insulin action responsible for the induction of insulin resistance (8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32). In contrast, the phosphorylation of just three serines of IRS-1, namely Ser-325 (44), -629 (not present in rodents) (45), and -1216 (48), had recently been associated with improved insulin signaling. Of note, for an additional three sites (Ser-302, -318, and -789) inconsistent data of stimulatory as well as inhibitory functions on the insulin action have been reported (11, 19, 29, 30, 39, 40, 41, 42, 43, 46). These reports prompted us to speculate that, depending on the duration of insulin stimulation, temporally different phosphorylation statuses of the same residues in defined regions of IRS-1 might exist, which exert different functions on signal transduction. To prove this hypothesis, we investigated the cross talk of the insulin-dependent, temporal phosphorylation of potentially opposedly acting serine residues of IRS-1 in skeletal muscle cells and the implications for insulin signal transduction, by selecting Ser-307 as a negative acting site and the adjacent Ser-302 and -318.
Applying phosphorylation kinetic studies, loss of function, gain of function, and combination mutants of IRS-1, we demonstrated that each site exhibits a characteristic phosphorylation kinetics, that a sequential, interdependent phosphorylation exists, and that the interplay rather than the phosphorylation status of single-serine residues regulates insulin action. Phosphorylation of Ser-302 and -318 was found to be an early event and was accompanied by tyrosine phosphorylation of IRS-1. At later time points, coincident with reduction in tyrosine phosphorylation, phosphorylation of Ser-307 and -318 was maximal.
The controversial data on the regulation of insulin signaling by phosphorylated Ser-302 and -318, demonstrating positive (39, 40, 41, 42) as well as negative effects (11, 19, 20) could be reconciled. We found that the rapid phosphorylation of Ser-302 is an initial event and appeared to be present to some extent even without insulin stimulation. Following a maximum after 10 min of insulin stimulation, which is similar to recent reports achieved in 32D cells (39) and primary human adipocytes (52), the intensity of the Ser-302 phosphorylation decreased during the ongoing insulin action below baseline values. Moreover, the data suggest a potential role of phospho-Ser-302 in the rapid association of insulin-activated PKC-ζ with IRS-1, which, in turn, phosphorylated Ser-318. The positive function of phosphor-Ser-302/-318 on insulin signal transduction was reflected by a considerable increase in the phosphorylation of Ser-473 of Akt after 10 min of insulin action, indicating enhanced activation of this kinase paralleled by a decrease in the activation of the inhibitory downstream kinase GSK-3. The preceding role of phosphor-Ser-302/-318 in the activation of insulin signaling was underlined further by a similar reduction of Akt phosphorylation after 60 min of insulin treatment irrespectively if the cells were transfected with IRS-1 wt or the E302A307E318 mutant, which simulates the permanent phosphorylation of Ser-302 and Ser-318. The investigation of the insulin-stimulated glucose uptake resulted in additional evidence for the positive action of the combined phospho-Ser-302/-318. Of note, in the unstimulated state the glucose uptake was also significantly enhanced simulating the early Ser-302 or Ser-318 phosphorylation by the expression of the E302 or the E302A307E318 mutant in comparison with cells expressing IRS-1 wt.
Several observations implicated a possible role of Ser-302 phosphorylation in the subsequent phosphorylation of Ser-318. When the phosphorylation of Ser-302 was prevented by the substitution with alanine, the early, PKC-ζ-dependent phosphorylation of Ser-318 was blocked. Furthermore, the association of IRS-1 and PKC-ζ was increased when simulating the phosphorylation of Ser-302 by mutation to glutamate and reduced by mutation to alanine. The rapid phosphorylation of Ser-302 and Ser-318 within minutes after the addition of insulin and the activation of downstream signaling as well as glucose uptake suggested the involvement of these sites in a positive modulation of insulin action, underlining that serine phosphorylation of IRS-1 is not only a major mechanism for the attenuation of insulin action (8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32), but might also be important for a maximum positive modulation of signal transduction in addition to the well-known effect of tyrosine phosphorylation.
The participation of PKC-ζ in this early phosphorylation event provides further evidence for a predominantly positive function of atypical PKC isoforms in the physiological process of insulin signal transduction. These kinases are activated by insulin (53, 54) and are important for insulin-stimulated glucose uptake (53, 55, 56). Their positive role in insulin action is further supported by the impaired activation of atypical PKC isoforms in obese humans and type 2 diabetics (56, 57, 58, 59). However, atypical PKCs appeared also to be involved in the self-attenuation of insulin signaling via serine phosphorylation of IRS-1 (35, 60). Seven serine residues proximal to the phosphotyrosine binding domain including Ser-302, but not Ser-318 or Ser-307, could participate in this mechanism, because a seven-alanine mutant of IRS-1 confers protection from the palmitic acid-induced inhibitory effects of PKC-ζ, (23, 35). Because we have observed a dissociation of PKC-ζ and IRS-1 after 10 min of insulin stimulation (42), an indirect negative feedback loop could be responsible for the attenuation of insulin signaling, e.g. via the recently postulated PKC-ζ-dependent activation of inhibitor of NF κB kinase (35).
Whereas Ser-302 was dephosphorylated by undefined phosphatases in the ongoing insulin action, the phosphorylation of Ser-318 was maintained, but not by PKC-ζ, as clearly demonstrated in this study. Thus, one residue could be temporally and sequentially phosphorylated by different kinases during the action of the same biological stimulus. The phosphorylation of Ser-318 by insulin-stimulated kinase activity is initially mediated by PKC-ζ followed within minutes by a kinase of the mTOR/raptor-dependent pathway. The following insulin-induced JNK-mediated phosphorylation of Ser-307 appeared not dependent on the phosphorylation of Ser-318, because the phosphorylation of Ser-307 was neither prevented by knockdown of PKC-ζ, which abrogated the early phosphorylation of Ser-318, nor did the substitution with glutamate at position 318 increase the phosphorylation of Ser-307. Moreover, mutation of Ser-302 and -318 to alanine also had no effect on this phosphorylation. In a previous report it has been suggested that the phosphorylation of Ser-307 could be required for efficient phosphorylation of Ser-302 (11). Based on the findings of the present study, this interdependency appears unlikely, at least in skeletal muscle cells, during insulin stimulation, because phosphorylation of Ser-307 did not precede the phosphorylation of Ser-302.
Whereas the simultaneous early phosphorylation at Ser-302 and Ser-318 had a pronounced positive effect, the influence of the late phosphorylation at Ser-307 together with Ser-318 on insulin signaling was low, and no difference was found compared with wt IRS-1 in insulin-stimulated glucose uptake. This is somewhat surprising because both phosphorylation of Ser-307 and Ser-318 have been clearly implicated in the attenuation of insulin signaling (12, 13, 14, 15, 16, 17, 18, 19, 20). In most reports on the negative effects of single phospho-Ser-307 and phospho-Ser-318, data were obtained from studies using loss of function mutants (substitution with alanine), i.e. the simulation of an unphosphorylated state at this site resulted in the detection of a positive effect on insulin signal transduction. When we studied the effect of the single-alanine mutants, we also observed positive effects on insulin-stimulated Akt phosphorylation using IRS-1 A307. Expression of IRS-1 A318 resulted in reduced insulin action to an early time point, whereas the attenuation of insulin action after 60 min was abrogated as reported earlier (42). To rule out the possibility that the phosphorylation of Ser-307 and Ser-318, which is simulated by the A302E307E318 mutant, does not occur in living cells and this may cause the missing negative influence on signal transduction, we investigated the phosphorylation of Ser-307 in the phospho-Ser-318-immunoprecipitated fraction of IRS-1. These data clearly indicated that Ser-307 and Ser-318 could be phosphorylated on the same IRS-1 molecule in skeletal muscle cells. Another possible explanation for the weak inhibitory effect seen in this study is that in most studies reporting negative effects of these sites the cells were stimulated only with insulin, but pretreated with other activators of serine kinases such as TNF-α (12, 61), anisomycin (51), phorbol ester (19), or fatty acids (33), which clearly induces other signaling pathways and different phosphorylation patterns of signaling molecules including IRS-1 than stimulation with insulin alone. Therefore, we suggest that the phosphorylation of further serine sites of IRS-1 is required for the self-attenuation of insulin signaling.
Taken together, it is clearly the entire serine phosphorylation pattern of IRS-1 that influences insulin signaling and not the individual action of one phosphorylated serine residue. However, it can only be speculated whether the positive contribution to insulin action is mediated by only a few serine residues and the opposite negative function needs a complex pattern to down-regulate insulin signaling. Possibly, the simultaneous phosphorylation of Ser-302 and -318 leads to more significant changes in the structure of IRS-1 than combined phosphorylation of Ser-307 and -318, but this can only be a hypothetical consideration so far because the reported crystal structure of an amino-terminal IRS-1 fragment encompasses only the pleckstrin homology and phosphotyrosine binding domain (amino acid residues 4–271) (62), whereas the serine residues investigated in the present study are located distal to this region.
Taken together, our study indicates that, dependent on its duration, insulin stimulation generates different phosphorylation patterns in defined regions of IRS-1, which differentially regulate signal transduction.
MATERIALS AND METHODS
Materials
C2C12 cells were from American Type Culture Collection (ATCC, Manassas, VA). Parental L6 cells and L6 GLUT4myc cells were kindly provided from A. Klip (Toronto, Ontario, Canada). Cell culture media and supplements were from Cambrex (Verviers, Belgium). Oligonucleotides were synthesized by Life Technologies (Karlsruhe, Germany). Phosphatase inhibitors and rapamycin were obtained from Sigma (Munich, Germany). The following antibodies were used: phospho-Akt Ser-473, phospho-IRS-1 Ser-302 (Cell Signaling Technology, Frankfurt, Germany); Akt, JNK (BD Biosciences, Inc., San Diego, CA); IRS-1 (06-248), phospho-IRS-1 Ser-307 (07-247), phosphotyrosine (4G10), phospho-c-Jun Ser-73 (06-659) (Upstate Biotechnology, Inc., Lake Placid, NY); PKC-ζ/λ (sc-216), c-Jun (sc-1694) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Polyclonal anti-p-Ser-318 antiserum was raised against a synthetic peptide (SMVGGKPGpSFRVRASSD) flanking Ser-318 in IRS-1, which is conserved among mouse, rat, and human sequence. For immunoprecipitation, this antiserum was purified by a two-step immunoaffinity chromatography. In the first step, antiserum was applied to an affinity column containing the unphosphorylated peptide. In the second step, the eluate was purified using the phosphorylated peptide coupled to an affinity column.
The cytomegalovirus promoter-based expression vectors for rat IRS-1 and human PKC-ζ are described in Ref. 63 . 2-[3H (G)]-deoxy-d-glucose (SA 185–370 gBq/mmol) was from PerkinElmer (Zaventem, Belgium).
Site-Directed Mutagenesis
Mutation of serine sites of rat IRS-1 to alanine or glutamate was made by oligonucleotide-mediated mutagenesis as described elsewhere (42) or generated using the Stratagene QuikChange XL Site-Directed Mutagenesis method (Stratagene, La Jolla, CA). Positive clones were verified by sequencing.
Cell Culture
For most transfection experiments, C2C12 myoblasts were used because we observed higher expression levels, better transfection efficiencies, and no toxic effects from the transfection reagents in these cells. L6 Glut4myc myoblasts were used for determination of glucose uptake, because C2C12 cells showed almost no insulin-dependent glucose uptake. C2C12 cells were cultured in DMEM containing 25 mm glucose, 10% fetal calf serum (FCS), 2 mm glutamine; 1 × 105 cells per well of a six-well plate were transfected using the Ca3(PO4)2-DNA-coprecipitation-method (64). Forty hours after transfection, the cells were starved in DMEM (5.5 mm glucose) without FCS for 3 h and stimulated as indicated. L6 cells were cultured in α-MEM containing 5.5 mm glucose, 10% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin as described (65). For differentiation, 0.5 × 104 cells/cm2 L6 cells were seeded in differentiation medium (α-MEM containing 5.5 mm glucose, 2% FCS, 100 U/ml penicillin, and 100 μg/ml streptomycin). Myotubes were used 7–8 d after cell seeding.
Cell Lysis, Immunoprecipitation, and Western Blot Analysis
Cells were lysed with 200 μl of lysis buffer/well (50 mm HEPES, pH 7.5; 150 mm NaCl; 1.5 mm MgCl2; 1 mm EDTA; 10% glycerin; 1% Triton X-100; containing protease and phosphatase inhibitors). Total protein (400 μg) was used for immunoprecipitation. Immunoprecipitated proteins or 40 μg protein of the total extracts were separated by SDS-PAGE (7.5%), and Western blot analysis was performed as described elsewhere (43).
2-Deoxyglucose Uptake
L6 Glut4myc myoblasts seeded in 12-well plates were transfected with the IRS-1 mutants using lipofectamine 2000 (Karlsruhe, Germany). Cells were serum-starved, 48 h after transfection, for 3 h in α-MEM before the experiments. After stimulation with 100 nm insulin, cells were washed with HEPES-buffered saline (containing 140 mm NaCl; 20 mm HEPES-Na, pH 7.4; 5 mm KCl; 2.5 mm MgSO4; 1 mm CaCl2) and 300 μl/well of 2-deoxyglucose mix (HEPES-buffered saline containing 0.25 μCi/ml [3H]2-deoxyglucose and 10 μm 2-deoxyglucose) was added. After incubation at 37 C, 5% CO2 for 7 min, cells were washed three times with 0.9% NaCl and lysed with 250 μl of lysis buffer. Total amounts of cell lysates were counted by liquid scintillation counting. Protein content of lysates generally varied less than 10%.
siRNA
siRNA oligonucleotides targeting mouse PKC-λ (NM_008857), PKC-ζ (NM_008860), JNK1 (NM_016700), and JNK2 (NM_016961) were designed, synthesized, and annealed at Dharmacon Research (Lafayette, CO). siRNA oligonucleotides targeting mouse raptor (NM_028898) were from QIAGEN (Hilden, Germany). An unrelated siRNA targeting firefly luciferase was used as control in all experiments. Transfection was performed with CellPhect (Amersham Biosciences, Buckinghamshire, UK) with 50 nm siRNA according to the instructions of the manufacturer. Briefly, 1 × 105 cells per well were seeded in six-well plates and transfected in DMEM containing 25 mm glucose, 10% FCS without antibiotics. Cells were stimulated as indicated 24 h after the glycerol shock.
Statistical Analysis
Results presented are derived from at least four independent experiments. Means ± sem values were calculated, and groups of data were compared using Student′s t test. Statistical significance was set at P < 0.05.
Acknowledgments
We thank Andreas Dittmar for excellent technical support.
Footnotes
This work was supported by Grant P-LS-Prot/29 from Landesstiftung Baden-Wuerttemberg (to R.L.), Grant GRK 1302/1 from the Deutsche Forschungsgemeinschaft (to E.S.), and a grant from the Deutsche Diabetes Gesellschaft (to R.L.).
Disclosure Statement: The authors have nothing to disclose.
First Published Online October 16, 2008
Abbreviations: FCS, Fetal calf serum; GSK, glycogen synthase kinase; IRS, insulin receptor substrate; JNK, c-jun-N-terminal kinase; mTOR, mammalian target of rapamycin; PKC, protein kinase C; siRNA, small interfering RNA; wt, wild type.
References
- 1.Pirola L, Johnston AM, Van Obberghen E 2004. Modulation of insulin action. Diabetologia 47:170–184 [DOI] [PubMed] [Google Scholar]
- 2.Zick Y 2004. Uncoupling insulin signalling by serine/threonine phosphorylation: a molecular basis for insulin resistance. Biochem Soc Trans 32:812–816 [DOI] [PubMed] [Google Scholar]
- 3.Taniguchi CM, Emanuelli B, Kahn CR 2006. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96 [DOI] [PubMed] [Google Scholar]
- 4.Karlsson HK, Zierath JR 2007. Insulin signaling and glucose transport in insulin resistant human skeletal muscle. Cell Biochem Biophys 48:103–113 [DOI] [PubMed] [Google Scholar]
- 5.Fritsche L, Weigert C, Häring HU, Lehmann R 2008. How insulin receptor substrate proteins regulate the metabolic capacity of the liver—implications for health and disease. Curr Med Chem 15:1316–1329 [DOI] [PubMed] [Google Scholar]
- 6.Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, Wilden PA, Cahill DA, Goldstein BJ, White MF 1991. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 352:73–77 [DOI] [PubMed] [Google Scholar]
- 7.Bouzakri K, Koistinen HA, Zierath JR 2005. Molecular mechanisms of skeletal muscle insulin resistance in type 2 diabetes. Curr Diabetes Rev 1:167–174 [DOI] [PubMed] [Google Scholar]
- 8.Kim JA, Yeh DC, Ver M, Li Y, Carranza A, Conrads TP, Veenstra TD, Harrington MA, Quon MJ 2005. Phosphorylation of Ser24 in the pleckstrin homology domain of insulin receptor substrate-1 by Mouse Pelle-like kinase/interleukin-1 receptor-associated kinase: cross-talk between inflammatory signaling and insulin signaling that may contribute to insulin resistance. J Biol Chem 280:23173–23183 [DOI] [PubMed] [Google Scholar]
- 9.Nawaratne R, Gray A, Jorgensen CH, Downes CP, Siddle K, Sethi JK 2006. Regulation of insulin receptor substrate 1 pleckstrin homology domain by protein kinase C: role of serine 24 phosphorylation. Mol Endocrinol 20:1838–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao Z, Zuberi A, Quon MJ, Dong Z, Ye J 2003. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases. J Biol Chem 278:24944–24950 [DOI] [PubMed] [Google Scholar]
- 11.Werner ED, Lee J, Hansen L, Yuan M, Shoelson SE 2004. Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. J Biol Chem 279:35298–35305 [DOI] [PubMed] [Google Scholar]
- 12.Aguirre V, Uchida T, Yenush L, Davis R, White MF 2000. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 275:9047–9054 [DOI] [PubMed] [Google Scholar]
- 13.Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF 2001. Insulin/IGF-1 and TNF-α stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest 107:181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao Z, Hwang D, Bataille F, Lefevre M, York D, Quon MJ, Ye J 2002. Serine phosphorylation of insulin receptor substrate 1 by inhibitor κ B kinase complex. J Biol Chem 277:48115–48121 [DOI] [PubMed] [Google Scholar]
- 15.Gual P, Gremeaux T, Gonzalez T, Marchand-Brustel Y, Tanti JF 2003. MAP kinases and mTOR mediate insulin-induced phosphorylation of insulin receptor substrate-1 on serine residues 307, 612 and 632. Diabetologia 46:1532–1542 [DOI] [PubMed] [Google Scholar]
- 16.Greene MW, Sakaue H, Wang L, Alessi DR, Roth RA 2003. Modulation of insulin-stimulated degradation of human insulin receptor substrate-1 by serine 312 phosphorylation. J Biol Chem 278:8199–8211 [DOI] [PubMed] [Google Scholar]
- 17.Carlson CJ, White MF, Rondinone CM 2004. Mammalian target of rapamycin regulates IRS-1 serine 307 phosphorylation. Biochem Biophys Res Commun 316:533–539 [DOI] [PubMed] [Google Scholar]
- 18.Bouzakri K, Karlsson HK, Vestergaard H, Madsbad S, Christiansen E, Zierath JR 2006. IRS-1 serine phosphorylation and insulin resistance in skeletal muscle from pancreas transplant recipients. Diabetes 55:785–791 [DOI] [PubMed] [Google Scholar]
- 19.Moeschel K, Beck A, Weigert C, Lammers R, Kalbacher H, Voelter W, Schleicher ED, Haring HU, Lehmann R 2004. Protein kinase C-ζ-induced phosphorylation of Ser318 in insulin receptor substrate-1 (IRS-1) attenuates the interaction with the insulin receptor and the tyrosine phosphorylation of IRS-1. J Biol Chem 279:25157–25163 [DOI] [PubMed] [Google Scholar]
- 20.Hennige AM, Stefan N, Kapp K, Lehmann R, Weigert C, Beck A, Moeschel K, Mushack J, Schleicher E, Haring HU 2006. Leptin down-regulates insulin action through phosphorylation of serine-318 in insulin receptor substrate 1. FASEB J 20:1206–1208 [DOI] [PubMed] [Google Scholar]
- 21.Liberman Z, Eldar-Finkelman H 2005. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J Biol Chem 280:4422–4428 [DOI] [PubMed] [Google Scholar]
- 22.Waraich RS, Weigert C, Kalbacher H, Hennige AM, Lutz S, Häring HU, Schleicher ED, Voelter W, Lehmann R 2008. Phosphorylation of Ser-357 of rat insulin receptor substrate-1 mediates adverse effects of protein kinase C- on insulin action in skeletal muscle cells. J Biol Chem 283:11226–11233 [DOI] [PubMed] [Google Scholar]
- 23.Liu YF, Herschkovitz A, Boura-Halfon S, Ronen D, Paz K, LeRoith D, Zick Y 2004. Serine phosphorylation proximal to its phosphotyrosine binding domain inhibits insulin receptor substrate 1 function and promotes insulin resistance. Mol Cell Biol 24:9668–9681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giraud J, Haas M, Feener EP, Copps KD, Dong X, Dunn SL, White MF 2007. Phosphorylation of Irs1 at SER-522 inhibits insulin signaling. Mol Endocrinol 21:2294–2302 [DOI] [PubMed] [Google Scholar]
- 25.Mothe I, Van Obberghen E 1996. Phosphorylation of insulin receptor substrate-1 on multiple serine residues, 612, 632, 662, and 731, modulates insulin action. J Biol Chem 271:11222–11227 [DOI] [PubMed] [Google Scholar]
- 26.De Fea K, Roth RA 1997. Protein kinase C modulation of insulin receptor substrate-1 tyrosine phosphorylation requires serine 612. Biochemistry 36:12939–12947 [DOI] [PubMed] [Google Scholar]
- 27.Li J, DeFea K, Roth RA 1999. Modulation of insulin receptor substrate-1 tyrosine phosphorylation by an Akt/phosphatidylinositol 3-kinase pathway. J Biol Chem 274:9351–9356 [DOI] [PubMed] [Google Scholar]
- 28.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G 2004. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431:200–205 [DOI] [PubMed] [Google Scholar]
- 29.Qiao LY, Zhande R, Jetton TL, Zhou G, Sun XJ 2002. In vivo phosphorylation of insulin receptor substrate 1 at serine 789 by a novel serine kinase in insulin-resistant rodents. J Biol Chem 277:26530–26539 [DOI] [PubMed] [Google Scholar]
- 30.Horike N, Takemori H, Katoh Y, Doi J, Min L, Asano T, Sun XJ, Yamamoto H, Kasayama S, Muraoka M, Nonaka Y, Okamoto M 2003. Adipose-specific expression, phosphorylation of Ser794 in insulin receptor substrate-1, and activation in diabetic animals of salt-inducible kinase-2. J Biol Chem 278:18440–18447 [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Soos TJ, Li X, Wu J, Degennaro M, Sun X, Littman DR, Birnbaum MJ, Polakiewicz RD 2004. Protein kinase C θ inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J Biol Chem 279:45304–45307 [DOI] [PubMed] [Google Scholar]
- 32.Tremblay F, Brule S, Hee US, Li Y, Masuda K, Roden M, Sun XJ, Krebs M, Polakiewicz RD, Thomas G, Marette A 2007. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci USA 104:14056–14061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI 2002. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 277:50230–50236 [DOI] [PubMed] [Google Scholar]
- 34.Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, Yuan M, Li ZW, Karin M, Perret P, Shoelson SE, Shulman GI 2001. Prevention of fat-induced insulin resistance by salicylate. J Clin Invest 108:437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herschkovitz A, Liu YF, Ilan E, Ronen D, Boura-Halfon S, Zick Y 2007. Common inhibitory serine sites phosphorylated by IRS-1 kinases, triggered by insulin and inducers of insulin resistance. J Biol Chem 282:18018–18027 [DOI] [PubMed] [Google Scholar]
- 36.Tremblay F, Marette A 2001. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 276:38052–38060 [DOI] [PubMed] [Google Scholar]
- 37.Zick Y 2005. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci STKE 2005:e4 [DOI] [PubMed]
- 38.Gual P, Marchand-Brustel Y, Tanti JF 2005. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 87:99–109 [DOI] [PubMed] [Google Scholar]
- 39.Giraud J, Leshan R, Lee YH, White MF 2004. Nutrient-dependent and insulin-stimulated phosphorylation of insulin receptor substrate-1 on serine 302 correlates with increased insulin signaling. J Biol Chem 279:3447–3454 [DOI] [PubMed] [Google Scholar]
- 40.Danielsson A, Ost A, Nystrom FH, Stralfors P 2005. Attenuation of insulin-stimulated insulin receptor substrate-1 serine 307 phosphorylation in insulin resistance of type 2 diabetes. J Biol Chem 280:34389–34392 [DOI] [PubMed] [Google Scholar]
- 41.Ost A, Danielsson A, Liden M, Eriksson U, Nystrom FH, Stralfors P 2007. Retinol-binding protein-4 attenuates insulin-induced phosphorylation of IRS1 and ERK1/2 in primary human adipocytes. FASEB J 21:3696–3704 [DOI] [PubMed] [Google Scholar]
- 42.Weigert C, Hennige AM, Brischmann T, Beck A, Moeschel K, Schauble M, Brodbeck K, Haring HU, Schleicher ED, Lehmann R 2005. The phosphorylation of Ser318 of insulin receptor substrate 1 is not per se inhibitory in skeletal muscle cells but is necessary to trigger the attenuation of the insulin-stimulated signal. J Biol Chem 280:37393–37399 [DOI] [PubMed] [Google Scholar]
- 43.Weigert C, Hennige AM, Lehmann R, Brodbeck K, Baumgartner F, Schauble M, Haring HU, Schleicher ED 2006. Direct cross-talk of interleukin-6 and insulin signal transduction via insulin receptor substrate-1 in skeletal muscle cells. J Biol Chem 281:7060–7067 [DOI] [PubMed] [Google Scholar]
- 44.Paz K, Liu YF, Shorer H, Hemi R, LeRoith D, Quan M, Kanety H, Seger R, Zick Y 1999. Phosphorylation of insulin receptor substrate-1 (IRS-1) by protein kinase B positively regulates IRS-1 function. J Biol Chem 274:28816–28822 [DOI] [PubMed] [Google Scholar]
- 45.Luo M, Langlais P, Yi Z, Lefort N, De Filippis EA, Hwang H, Christ-Roberts CY, Mandarino LJ 2007. Phosphorylation of human insulin receptor substrate-1 at serine 629 plays a positive role in insulin signaling. Endocrinology 148:4895–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jakobsen SN, Hardie DG, Morrice N, Tornqvist HE 2001. 5′-AMP-activated protein kinase phosphorylates IRS-1 on Ser-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J Biol Chem 276:46912–46916 [DOI] [PubMed] [Google Scholar]
- 47.Lehmann R, Beck A, Moeschel K, Schmidt EK, Deeg M, Rapp E, Sun XJ, Kellerer M, Voelter W, Schleicher ED, Häring HU 2002. Protein kinase C-ζ phosphorylates serine/threonine residues at the C-terminal binding motif of the tyrosine phosphatase SHP-2 of insulin receptor substrate 1. Sign Transduct 1–2:40–45
- 48.Luo M, Reyna S, Wang L, Yi Z, Carroll C, Dong LQ, Langlais P, Weintraub ST, Mandarino LJ 2005. Identification of insulin receptor substrate 1 serine/threonine phosphorylation sites using mass spectrometry analysis: regulatory role of serine 1223. Endocrinology 146:4410–4416 [DOI] [PubMed] [Google Scholar]
- 49.Yi Z, Luo M, Mandarino LJ, Reyna SM, Carroll CA, Weintraub ST 2006. Quantification of phosphorylation of insulin receptor substrate-1 by HPLC-ESI-MS/MS. J Am Soc Mass Spectrom 17:562–567 [DOI] [PubMed] [Google Scholar]
- 50.Yi Z, Langlais P, De Filippis EA, Luo M, Flynn CR, Schroeder S, Weintraub ST, Mapes R, Mandarino LJ 2007. Global assessment of regulation of phosphorylation of insulin receptor substrate-1 by insulin in vivo in human muscle. Diabetes 56:1508–1516 [DOI] [PubMed] [Google Scholar]
- 51.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF 2002. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem 277:1531–1537 [DOI] [PubMed] [Google Scholar]
- 52.Danielsson A, Nystrom FH, Stralfors P 2006. Phosphorylation of IRS1 at serine 307 and serine 312 in response to insulin in human adipocytes. Biochem Biophys Res Commun 342:1183–1187 [DOI] [PubMed] [Google Scholar]
- 53.Bandyopadhyay G, Standaert ML, Galloway L, Moscat J, Farese RV 1997. Evidence for involvement of protein kinase C (PKC)-ζ and noninvolvement of diacylglycerol-sensitive PKCs in insulin-stimulated glucose transport in L6 myotubes. Endocrinology 138:4721–4731 [DOI] [PubMed] [Google Scholar]
- 54.Standaert ML, Bandyopadhyay G, Perez L, Price D, Galloway L, Poklepovic A, Sajan MP, Cenni V, Sirri A, Moscat J, Toker A, Farese RV 1999. Insulin activates protein kinases C-ζ and C-λ by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J Biol Chem 274:25308–25316 [DOI] [PubMed] [Google Scholar]
- 55.Braiman L, Alt A, Kuroki T, Ohba M, Bak A, Tennenbaum T, Sampson SR 2001. Activation of protein kinase C ζ induces serine phosphorylation of VAMP2 in the GLUT4 compartment and increases glucose transport in skeletal muscle. Mol Cell Biol 21:7852–7861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sajan MP, Standaert ML, Miura A, Bandyopadhyay G, Vollenweider P, Franklin DM, Lea-Currie R, Farese RV 2004. Impaired activation of protein kinase C-ζ by insulin and phosphatidylinositol-3,4,5-(PO4)3 in cultured preadipocyte-derived adipocytes and myotubes of obese subjects. J Clin Endocrinol Metab 89:3994–3998 [DOI] [PubMed] [Google Scholar]
- 57.Beeson M, Sajan MP, Dizon M, Grebenev D, Gomez-Daspet J, Miura A, Kanoh Y, Powe J, Bandyopadhyay G, Standaert ML, Farese RV 2003. Activation of protein kinase C-ζ by insulin and phosphatidylinositol-3,4,5-(PO4)3 is defective in muscle in type 2 diabetes and impaired glucose tolerance: amelioration by rosiglitazone and exercise. Diabetes 52:1926–1934 [DOI] [PubMed] [Google Scholar]
- 58.Kim YB, Kotani K, Ciaraldi TP, Henry RR, Kahn BB 2003. Insulin-stimulated protein kinase C λ/ζ activity is reduced in skeletal muscle of humans with obesity and type 2 diabetes: reversal with weight reduction. Diabetes 52:1935–1942 [DOI] [PubMed] [Google Scholar]
- 59.Vollenweider P, Menard B, Nicod P 2002. Insulin resistance, defective insulin receptor substrate 2-associated phosphatidylinositol-3′ kinase activation, and impaired atypical protein kinase C (ζ/λ) activation in myotubes from obese patients with impaired glucose tolerance. Diabetes 51:1052–1059 [DOI] [PubMed] [Google Scholar]
- 60.Liu YF, Paz K, Herschkovitz A, Alt A, Tennenbaum T, Sampson SR, Ohba M, Kuroki T, LeRoith D, Zick Y 2001. Insulin stimulates PKCζ-mediated phosphorylation of insulin receptor substrate-1 (IRS-1). A self-attenuated mechanism to negatively regulate the function of IRS proteins. J Biol Chem 276:14459–14465 [DOI] [PubMed] [Google Scholar]
- 61.Jiang G, Dallas-Yang Q, Liu F, Moller DE, Zhang BB 2003. Salicylic acid reverses phorbol 12-myristate-13-acetate (PMA)- and tumor necrosis factor α (TNFα)-induced insulin receptor substrate 1 (IRS1) serine 307 phosphorylation and insulin resistance in human embryonic kidney 293 (HEK293) cells. J Biol Chem 278:180–186 [DOI] [PubMed] [Google Scholar]
- 62.Dhe-Paganon S, Ottinger EA, Nolte RT, Eck MJ, Shoelson SE 1999. Crystal structure of the pleckstrin homology-phosphotyrosine binding (PH-PTB) targeting region of insulin receptor substrate 1. Proc Natl Acad Sci USA 96:8378–8383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kellerer M, Mushack J, Seffer E, Mischak H, Ullrich A, Häring HU 1998. Protein kinase C isoforms α, δ and θ require insulin receptor substrate-1 to inhibit the tyrosine kinase activity of the insulin receptor in human kidney embryonic cells (HEK 293 cells). Diabetologia 41:833–838 [DOI] [PubMed] [Google Scholar]
- 64.Chen C, Okayama H 1987. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7:2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koivisto UM, Martinez-Valdez H, Bilan PJ, Burdett E, Ramlal T, Klip A 1991. Differential regulation of the GLUT-1 and GLUT-4 glucose transport systems by glucose and insulin in L6 muscle cells in culture. J Biol Chem 266:2615–2621 [PubMed] [Google Scholar]