

Abstract
The farnesoid X receptor (FXR) is a key metabolic regulator in the liver by maintaining the homeostasis of liver metabolites. Recent findings suggest that FXR may have a much broader function in liver physiology and pathology. In the present work, we identify a novel role of FXR in protecting liver cell from apoptosis induced by nutritional withdrawal including serum deprivation in vitro or starvation in vivo. Two FXR ligands, chenodeoxycholic acid (CDCA) and GW4064, rescued HepG2 cells from serum deprivation-induced apoptosis in a dose-dependent manner. This effect of FXR on apoptotic suppression was compromised when FXR was knocked down by short interfering RNA. Similarly, the effects of both CDCA and GW4064 were abolished after inhibition of the MAPK pathway by a specific inhibitor of MAPK kinase 1/2. Immunoblotting results indicated that FXR activation by CDCA and GW4064 induced ERK1/2 phosphorylation, which was attenuated by serum deprivation. In vivo, FXR−/− mice exhibited an exacerbated liver apoptosis and lower levels of phosphorylated-ERK1/2 compared to wild-type mice after starvation. In conclusion, our results suggest a novel role of FXR in modulating liver cell apoptosis.
HEPATOCYTE APOPTOSIS IS an important part of the tightly controlled homeostatic mechanism regulating liver function and is regulated by many signaling pathways (1). Disruption of the balance between cell growth and apoptosis may result in liver diseases such as cholestasis, cirrhosis, and liver cancer (2, 3).
Farnesoid X receptor (FXR, NR1H4) is a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors. Recent studies demonstrate that FXR is a central metabolic regulator in bile acid (BA) homeostasis as well as lipid and glucose metabolism (4, 5, 6). FXR coordinates the expression of genes involved in BA production, efflux, influx, and detoxification in the liver. Moreover, FXR activation alleviates the hepatotoxicity of hepatotoxins (7, 8, 9) and protects against cholestatic liver injury in rats (10, 11), which suggests that a key function of FXR is for hepatoprotection. In addition to controlling the levels of BAs in liver, FXR also helps accelerate normal liver regeneration in response to increased BA stress after 70% hepatectomy (9). The hepatoprotective role of FXR is essential for normal liver physiology. For example, FXR null mice spontaneously develop liver tumors due to chronic liver injury as they age (12).
Nutrition is essential for cell proliferation and survival in vitro and in vivo (13, 14, 15). For example, serum deprivation is a potent stimulus for the induction of apoptosis in different cell lines, including human hepatoma HepG2 cells (16). This study aimed to determine whether FXR plays a protective role in liver cell apoptosis induced by nutritional withdrawal both in vitro and in vivo. We asked three fundamental questions: 1) Does FXR activation by its ligands, chenodeoxycholic acid (CDCA) and GW4064, inhibit serum deprivation-induced apoptosis? 2) Is there more severe apoptosis in FXR−/− liver in vivo after starvation? 3) Which signal transduction pathway may mediate the effect of FXR in apoptosis suppression? The results indicate that FXR activation suppresses serum deprivation-induced apoptosis of HepG2 cells by activating the ERK1/2 MAPK pathway and FXR also inhibits liver cell apoptosis induced by starvation in animals.
RESULTS
CDCA and GW4064 Attenuated the Serum Deprivation-Induced Apoptosis of HepG2 Cells
Serum deprivation was used to induce cell apoptosis in HepG2 cells. Serum withdrawal for 72 h strongly induced cell death, as shown by the increased number of detached cells with a round shape (supplemental Fig. S1A published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). However, incubation of the cells with FXR ligands, CDCA (50 μmol/liter) and GW4064 (2 μmol/liter), inhibited cell death in the serum free condition, and the cells had similar morphologies to those incubated with serum (supplemental Fig. S1A). We also compared the number of cells that remained attached to the dish by counting the cell number using a Coulter particle counter. Serum deprivation decreased the number of attached cells, but cell attachment was significantly induced by both CDCA and GW4064 treatments (supplemental Fig. S1B), suggesting that FXR ligands can rescue serum deprivation-induced cell death. To confirm that the observed cell death phenotype after serum deprivation was due to apoptosis, we collected both the detached cells and the cells that remained attached to the bottom of the dishes and subjected them to a DNA ladder assay. Only the detached cells showed DNA laddering, indicative of apoptosis (supplemental Fig. S2).
To quantitatively measure the effects of CDCA and GW4064 in rescuing cell death, we used a live cell propidium iodide (PI) staining method, described by Zamai et al. (17). This method identifies three distinguishable populations of cells (healthy, dead, or in the process of apoptosis) according to the intensity of PI staining. Serum withdrawal caused most of the cells to shift into the process of apoptosis and increased the number of apoptotic cells (Fig. 1A). Treatment with CDCA (50 μmol/liter) and GW4064 (2 μmol/liter) reversed the effect of serum deprivation by causing the majority of cells to shift back to the healthy condition. The results were confirmed by the PI/Annexin V double staining (supplemental Fig. S3). To test whether CDCA and GW4064 also inhibit HepG2 cell apoptosis induced by a DNA-damaging agent, we treated the cells with 2 μg/ml bleomycin and then incubated the cells with CDCA (50 μmol/liter) and GW4064 (2 μmol/liter). However, both ligands failed to rescue the bleomycin-induced apoptosis (supplemental Fig. S4), suggesting that the antiapoptotic effect of FXR is not applicable to all inducers.
Fig. 1.
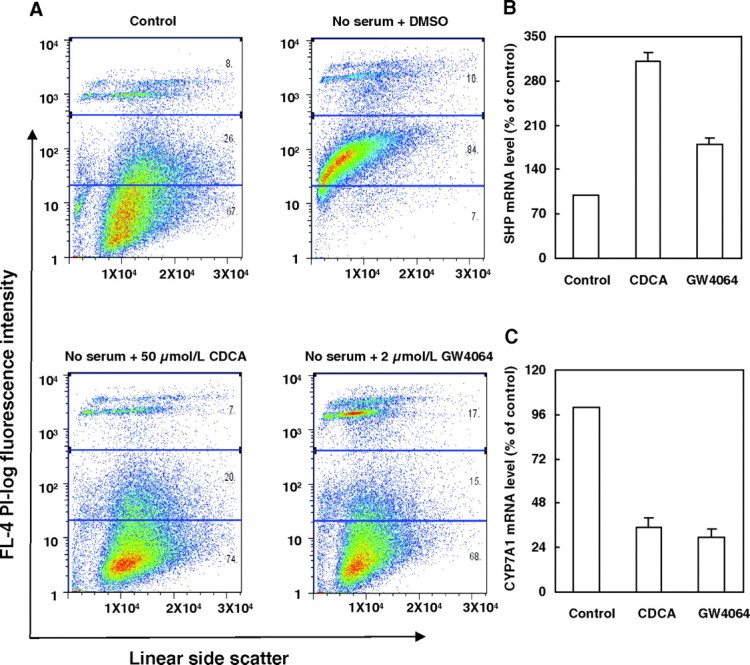
CDCA or GW4064 Attenuates Serum Deprivation-Induced Apoptosis of HepG2 Cells
A, Representative fluorescence-activated cell sorting analysis of serum-deprived HepG2 cells treated with vehicle (DMSO), CDCA (50 μmol/liter), or GW4064 (2 μmol/liter) for 48 h, and then harvested and stained with PI. Cells cultured in complete culture medium were used as controls (n = 3). B and C, FXR activation by CDCA (50 μmol/liter) or GW4064 (2 μmol/liter) was assessed by measuring SHP and CYP7A1 expression in vehicle (DMSO)-treated and CDCA- or GW4064-treated HepG2 cells in serum-free medium. HepG2 cells were treated with vehicle (DMSO) as controls. Expression was measured by real-time PCR and normalized as a ratio using β-actin mRNA as a control gene. The mRNA levels of SHP (B) and CYP7A1 (C) are represented as the percent transcript abundance in vehicle-, CDCA-, or GW4064-treated HepG2 cells vs. the corresponding control groups. Data are the means of three independent experiments ± sem. L, Liter.
Both CDCA and GW4064 have been shown to activate FXR and to change the expression of FXR target genes in HepG2 cells (18). Therefore, we measured the mRNA levels of two previously reported FXR-target genes, short heterodimer partner (SHP) (19) and cholesterol 7 α-hydroxylase (CYP7A1) (20), in serum-deprived HepG2 cells incubated with 50 μmol/liter CDCA or 2 μmol/liter GW4064. Incubation with either CDCA or GW4064 increased SHP mRNA levels (Fig. 1B). As expected, CYP7A1 mRNA was suppressed by these two FXR ligands (Fig. 1C). These results suggest that CDCA and GW4064 protect against serum deprivation-induced HepG2 cell apoptosis concomitant with FXR activation.
To further demonstrate the specific effects of CDCA and GW4064 on suppressing serum deprivation-induced cell apoptosis, we performed a dose response experiment. As shown in Fig. 2, A–D, both CDCA and GW4064 rescued HepG2 cell death in a remarkably dose-dependent manner. GW4064 began to show an effect on cell death at concentrations as low as 0.1 μmol/liter, which is consistent with its potent agonist effect on FXR activation (21). However, when we treated the cells with 100 μmol/liter CDCA, the number of dead cells did not decrease compared with that of treatment with 50 μmol/liter CDCA or even increased compared with the control, indicating that 100 μmol/liter CDCA may be cytotoxic to HepG2 cells. Therefore, in later experiments, we regularly used CDCA at a concentration of 50 μmol/liter and GW4064 at concentration of 2 μmol/liter.
Fig. 2.
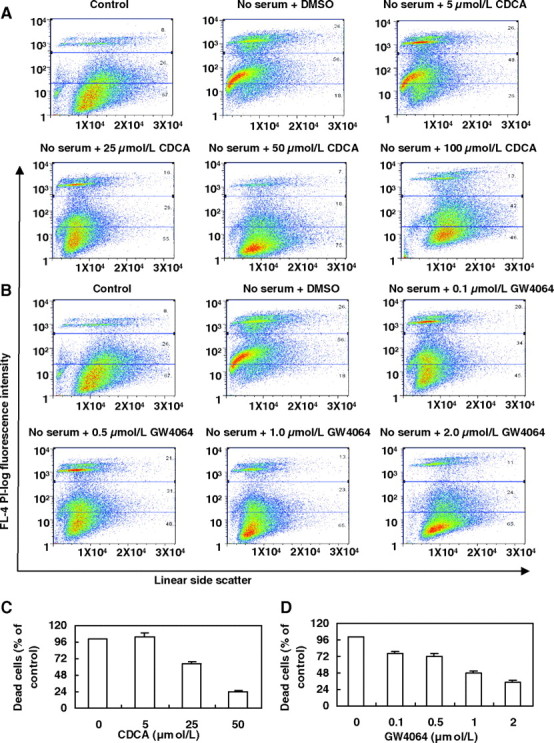
CDCA and GW4064 Attenuate Serum Deprivation-Induced Apoptosis of HepG2 Cells in a Dose-Dependent Manner
A and B, Representative fluorescence-activated cell sorting analysis of serum-deprived HepG2 cells treated with vehicle (DMSO), different concentrations of CDCA (5, 25, 50, 100 μmol/liter), or GW4064 (0.1, 0.5, 1.0, 2.0 μmol/liter) for 48 h. Cells were then harvested and stained with PI. HepG2 cells incubated in complete culture medium for 72 h were used as controls (n = 3). C and D, HepG2 cells were treated as described above. Dead cells were counted as a percentage of the total number of cells using fluorescence-activated cell sorting and are represented as the percent dead cells in the different concentrations of CDCA or GW4064 vs. the control groups (no CDCA or GW4064 treatment). Data are the means of three independent experiments ± sem. L, Liter.
FXR Is Required for Mediating the Effects of CDCA and GW4064 on Suppression of HepG2 Cell Apoptosis
To test whether FXR is required to mediate the effects of CDCA and GW4064 on suppression of HepG2 cell apoptosis, we examined the effects of these two ligands on apoptosis when FXR mRNA was reduced by FXR-specific short interfering RNA (siRNA) duplexes. Compared with the mock and control siRNA-transfected cells, transfection of FXR-specific siRNA in HepG2 cells reduced FXR mRNA levels by approximately 65% (100% or 93.7 ± 4.7% vs. 35.0 ± 1.8%) (Fig. 3A). To investigate the effect of FXR knockdown, mRNA levels of the known FXR target genes SHP and bile salt export pump (BSEP) were measured in the same samples. As expected, FXR ligand treatment significantly increased SHP and BSEP gene expression in mock or control siRNA-transfected cells, but no effect was seen in cells receiving FXR siRNA treatment (Fig. 3, B and C), suggesting that the FXR siRNA was effective at reducing FXR activity. As expected, after FXR siRNA transfection, both CDCA and GW4064 failed to suppress the serum deprivation-induced apoptosis of HepG2 cells (Fig. 3D). These data strongly support the idea that sufficient level of FXR expression is required to mediate the effects of CDCA and GW4064 on suppression of cell apoptosis.
Fig. 3.

FXR Is Required to Mediate the Effects of CDCA and GW4064 on Suppressing Serum Deprivation-Induced Apoptosis of HepG2 Cells
A–C, HepG2 cells were transfected with siRNA specific for FXR (FXR siRNA), a siRNA negative control (c-siRNA), or no siRNA (Mock). After 8 h of transfection, cells were switched to serum-free medium and treated with vehicle control (DMSO), CDCA (50 μmol/liter), or GW4064 (2 μmol/liter) for 24 h. The mRNA levels of FXR (A) were measured by quantitative real-time PCR and are presented as the percent transcript abundance in c-siRNA or FXR siRNA transfected cells vs. the Mock control groups. The mRNA levels of SHP (B) and BSEP (C) are represented as the percent transcript abundance in vehicle-, CDCA-, or GW4064-treated HepG2 cells vs. the corresponding vehicle control groups. *, P < 0.05 less than the corresponding values in Mock or c-siRNA group. D, After 8 h of transfection with FXR siRNA, c-siRNA, or no siRNA, cells were switched to serum-free medium and treated with vehicle control (DMSO), CDCA (50 μmol/liter), or GW4064 (2 μmol/liter) for 48 h. Cells were harvested and stained with PI. Dead cells were counted as a percentage of the total number of cells using fluorescence-activated cell sorting and represented as the percent dead cells in vehicle-, CDCA-, or GW4064-treated HepG2 cells vs. the corresponding vehicle control groups. Data are the means of three independent experiments ± sem. *, P < 0.01 greater than the corresponding values in Mock or c-siRNA group.
FXR Activation by CDCA and GW4064 Activates the MAPK Pathway in Serum-Deprived HepG2 Cells
Previous studies indicate that liver cell apoptosis is modulated by several different signaling pathways, such as the protein kinase A (PKA), MAPK, and phosphoinositol-3-OH kinase (PI3K) signaling pathways (22). We investigated whether FXR activation by CDCA or GW4064 activated these signaling pathways to inhibit serum deprivation-induced apoptosis of HepG2 cells. We cotreated the HepG2 cells with specific pathway inhibitors (MAPK inhibitor, UO126; PI3K inhibitor, Ly294002; PKA inhibitor, H-89; Jun N-terminal kinase (JNK) inhibitor peptide II; MAPK p38 inhibitor, SB203580) and assessed the impact on HepG2 cell survival. Treatment with H-89, JNK inhibitor peptide II, and SB203580 resulted in no significant effects of CDCA and GW4064 on cell viability (Fig. 4A). In contrast, addition of the MAPK kinase (MEK)1/2 inhibitor, UO126, increased the ratio of apoptotic cells from approximately 26% to about 92% in the CDCA group and from approximately 41% to about 85% in the GW4064 group. However, the PI3K inhibitor, Ly294002, showed a differentiated effect on CDCA and GW4064; it partially inhibited the effect of CDCA, but completely abolished the effect of GW4064, on the suppression of apoptosis, which may be caused by the fact that CDCA is a nonspecific ligand of FXR and has activities on targets other than FXR. In summary, these results demonstrate that the MAPK signaling pathway is required for mediating the effects of both CDCA and GW4064 on rescuing apoptosis of HepG2 cells induced by serum deprivation.
Fig. 4.
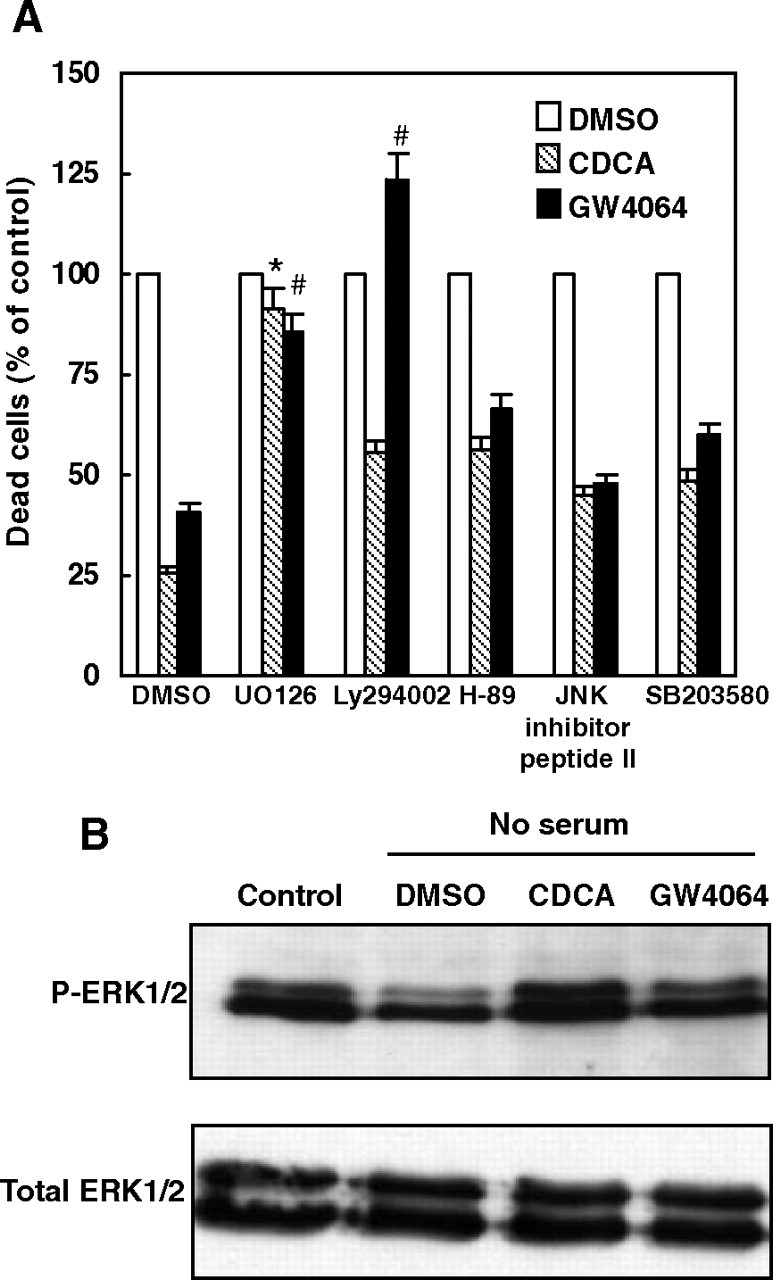
Effects of Inhibitors of Different Signaling Pathways on HepG2 Cell Apoptosis Induced by Serum Deprivation
A, Serum-deprived HepG2 cells were pretreated with vehicle (DMSO), UO126 (10 μmol/liter), Ly294002 (20 μmol/liter), H89 (6 μmol/liter), JNK inhibitor peptide II (10 μmol/liter), or SB203580 (10 μmol/liter) for 30 min before exposure to vehicle control, CDCA (50 μmol/liter) or GW4064 (2 μmol/liter) for 48 h. Cells were then harvested and stained with PI. Dead cells were counted as a percentage of the total number of cells using fluorescence-activated cell sorting and are represented as the percent dead cells in vehicle-, CDCA-, or GW4064-treated HepG2 cells vs. the corresponding vehicle control groups. Data are the means of three independent experiments ± sem. *, P < 0.05 greater than vehicle DMSO plus CDCA-treated value; #, P < 0.05 greater than vehicle DMSO plus GW4064-treated value. B, CDCA and GW4064 treatments increased the phosphorylation of ERK1/2 in serum-deprived HepG2 cells. Serum-deprived HepG2 cells were treated with vehicle control (DMSO), CDCA (50 μmol/liter), or GW4064 (2 μmol/liter) for 30 min. Cells cultured in complete culture medium were used as controls. Cells were lysed and total cell lysates were analyzed by Western blot using anti-p-ERK1/2 or anti-ERK1/2 antibodies. P-ERK1/2, Phosphorylated ERK1/2.
To further confirm the activation of the MAPK pathway by CDCA and GW4064, we tested whether CDCA or GW4064 treatment increased the phosphorylation of ERK1/2, downstream targets of MEK1/2, in serum-deprived HepG2 cells. As shown in Fig. 4B, ERK1/2 phosphorylation was strongly reduced by serum deprivation in HepG2 cells, which is consistent with the work of Das et al. (23). However, ERK1/2 phosphorylation was returned to normal levels by CDCA or GW4064 treatments. In contrast, the expression of total ERK1/2 protein, which served as a loading control, was not changed by different treatments.
Enhanced Liver Apoptosis in Starved FXR−/− Mice
It has been reported that food restriction induces hepatic apoptosis in vivo probably due to the decreased growth factor signaling (14). To further address whether that FXR functions to suppress apoptosis in vivo, we compared the apoptosis in wild-type and FXR−/− mice fasted for 4 d. The levels of alanine transaminase (ALT), a common marker for liver damage, were significantly increased by starvation in FXR−/− mice compared with the wild-type mice, the ALT levels of which were not significantly affected by fasting (Fig. 5A). The liver apoptosis induced by food withdrawal was confirmed by terminal deoxy-nucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assays. As shown in Fig. 5, B and C, significant TUNEL-positive staining was detected in the liver of FXR−/− mice compared with the wild-type mice after fasting. Hematoxylin and eosin (H&E) staining showed that there was no hepatic necrosis in these mice (Fig. 5D) and, as stated previously (12), there were many vacuoles in FXR−/− groups due to lipid deposits. The results of TUNEL and H&E staining suggest that FXR−/− mice have an enhanced apoptosis in response to food withdrawal. Because the data in vitro show that ERK activation plays an important role in FXR-mediated suppression of apoptosis induced by serum deprivation in HepG2 cells, we thus tested ERK activation in vivo after fasting mice for 4 d. The total ERK protein expression was not changed, but phosphorylated ERK expression was suppressed by loss of FXR (supplemental Fig. S5 and Fig. 5E). Food withdrawal did not change the levels of total ERK but decreased phosphorylated ERK in both wild-type and FXR−/− mice. The extent of decrease of ERK activation in response to food withdrawal was higher in FXR−/− mice (Fig. 5E). These results suggest that, similar to in vitro serum withdrawal-induced apoptosis in HepG2 cells, ERK activation may be important in mediating the effects of FXR on suppression of liver apoptosis induced by food withdrawal.
Fig. 5.

Enhanced Liver Apoptosis and Decreased ERK Activation in FXR−/− Mice after Starvation
A, ALT levels in wild-type and FXR−/− mice. *, P < 0.05 greater than the control group of FXR−/−. B, The representative TUNEL staining of liver sections from wild-type and FXR−/− livers (×20). C, Statistical analysis of the number of TUNEL-positive cells per total number of cells. The number of cells in at least 20 microscopic fields was counted. *, P < 0.05 greater than the control group of FXR−/−. D, Representative H&E staining of liver sections from wild-type and FXR−/− livers (×200) E, Immunoblot analysis for phosphorylated ERK1/2, total ERK1/2, and β-actin from total protein pools of wild-type and FXR−/− individual livers (n = 4). The expression levels of P-ERK and total ERK were normalized by total ERK and β-actin, respectively. FXRKO, FXR knockout; WT, wild type.
DISCUSSION
FXR is a key regulator of BA homeostasis. In addition, FXR plays an important role in hepatoprotection to prevent the toxic effect of BAs on liver when they reach abnormally high levels. For example, in animal models of cholestasis, GW4064 provides protection against liver injury via FXR activation (11). Moreover, administration of GW4064 to mice fed a lithogenic diet prevents the development of cholesterol gallstone disease. These protective effects for FXR have been attributed to the transcriptional regulation of genes involved in BA metabolism and transportation in hepatocytes. We recently demonstrated that FXR−/− mice developed spontaneous liver tumors, and we observed strong cell apoptosis in liver, suggesting that FXR may also protect liver cells from apoptosis (12). In the current study, we first examined whether FXR could directly modulate cell apoptosis in a human hepatoma cell line, HepG2. The use of serum deprivation to induce apoptosis in HepG2 cells is well established and is widely used by numerous laboratories (13, 16, 24, 25). Our results clearly show that FXR activation strongly suppresses serum deprivation-induced apoptosis in HepG2 cells, suggesting a novel function of FXR in cytoprotection by suppressing cell apoptosis. These effects were further confirmed by showing that enhanced apoptosis was induced by starvation in FXR−/− mice compared with wild-type mice. This antiapoptosis function, combined with FXR’s effect on lowering BAs in the liver, may constitute a powerful hepatoprotection pathway, which is essential for maintaining normal liver physiology.
The effects of FXR on suppressing cell apoptosis are previously uncharacterized in liver cells. However, De Gottardi et al. (26) have reported that FXR may suppress apoptosis in a Barrett’s esophagus-derived cell line. In contrast, Swales et al. (27) showed that activation of FXR induced apoptosis in breast cancer cells; however, the high concentrations of GW4064 (50 μmol/liter) used in their study may cause FXR-independent effect. The difference in observations between Swales et al. and our group may also result from tissue-specific differences between liver and breast cells. On the other hand, our results are consistent with another observation by Silva et al. (28). They showed that the apoptosis of MDA-MB-231 breast cancer cells was suppressed by low doses of deoxycholic acid or CDCA at 10 μmol/liter. The role of FXR in breast cancer needs to be confirmed in vitro and determined in vivo. It will be interesting to study whether FXR also suppresses apoptosis in other tissues such as esophagus in vivo.
Our results show that FXR activation increases ERK phosphorylation, which is consistent with results showing that ERK activation protects various cell types from apoptosis (29, 30, 31, 32, 33). Activation of ERK constitutes a major antiapoptotic pathway activated by a variety of extracellular agonists such as growth factors or hormones (34). ERK targets a number of antiapoptotic regulators, including proteins of the Bcl-2 family, caspases, or inhibitor of apoptosis proteins (35, 36, 37, 38). In HepG2 cells, we observed an enhanced effect on serum deprivation-induced apoptosis by inhibiting the MAPK/ERK1/2 pathway, regardless of the presence of CDCA or GW4064. FXR activation increased ERK phosphorylation. Moreover, wild-type mice showed resistance to apoptosis induced by starvation and a higher level of phosphorylated ERK compared with FXR−/− mice, suggesting that ERK signaling may also be important for suppression of apoptosis during starvation in a FXR-dependent manner in vivo. Therefore, we conclude that ERK1/2 pathway is at least one of the signaling pathways that may mediate the effect of FXR in suppressing liver cell apoptosis.
The enhanced ERK activation by other nuclear receptors has been demonstrated previously. For example, peroxisome proliferator-activated receptor-γ has been shown to crosstalk with ERK pathways (39, 40, 41). Papageorgiou et al. (42) reported that peroxisome proliferator-activated receptor-γ ligands induced a rapid phosphorylation of ERK1/2 in many cell lines. Narayanan et al. (43) reported that the vitamin D receptor interacts with ERK. Ligand activation of vitamin D receptor results in a rapid activation of ERK in bone cell lines. Estrogen receptor has been shown to interact with PI3K leading to activation of ERK1/2 (44). Whether FXR has a similar effect on ERK pathway remains to be tested. It will be interesting to determine whether FXR directly interacts with ERK, which contributes to the repression of liver cell apoptosis.
It should be noted that serum deprivation-induced apoptosis is complicated by removing the majority of growth factors and hormones. It will be interesting to know whether FXR suppresses apoptosis induced by other specific pathways. One potentially similar role of FXR may apply to BA-induced liver cell apoptosis. Both in vitro and in vivo experiments indicate that high levels of BAs can directly induce apoptosis in hepatocytes by activating the Fas pathway in a ligand-independent manner, possibly by membrane perturbation (45, 46, 47). BAs also simultaneously activate an intrinsic antiapoptotic mechanism to ameliorate their proapoptotic effects. BAs activate multiple cell-signaling cascades, such as the ERK1/2, JNK, and PI3K pathways (48, 49), inside hepatocytes to increase cell survival and proliferation (22, 50, 51). However, the mechanisms by which BAs activate signal transduction pathways to suppress cell death processes in hepatocytes are unclear. Because FXR is the primary BA sensor and provides hepatoprotection against BA-induced liver injury (11, 52), we hypothesize that FXR may similarly reduce the toxic effect of BAs by directly modulating cell apoptosis.
A similar role in suppression of liver apoptosis was also reported for the xenobiotic receptors, constitutive androstane receptor (53) and pregnane X receptor (54, 55). Activation of constitutive androstane receptor by its ligand, 1,4-bis [2-(3,5-dichloropyridyloxy)] benzene, strongly suppressed Fas-activated liver apoptosis (53). The similarity among the roles of members in NR1 group in suppressing liver apoptosis raises an intriguing possibility that the NR1I and NR1H groups of nuclear receptors, which share the most homologous sequence, may process the same cell-protective function. It will be interesting to determine whether other receptors, such as liver X receptor, in this group of nuclear hormone receptors have similar functions in regulating cell apoptosis.
In conclusion, this study reveals that FXR activation by its ligands rescues the serum deprivation-induced apoptosis in HepG2 cells by activating the MAPK/ERK1/2 pathway and FXR also suppress apoptosis induced by starvation in vivo. Our findings indicate that the previously suggested beneficial effects of FXR activation may result from both the benefit of maintaining BA homeostasis and of suppressing cell death by FXR in liver. Further understanding of these mechanisms will facilitate the development of FXR agonists as therapeutic drugs to reduce liver damage in certain human liver diseases.
MATERIALS AND METHODS
Material
CDCA and PI were purchased from Sigma Chemical Co. (St. Louis, MO). Phospho-/total-ERK1/2 antibodies were purchased from Cell Signaling Technologies (Worcester, MA). β-Actin antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Bleomycin antibiotic, PI3K inhibitor (LY294002), MEK inhibitor (U0126), JNK inhibitor II, PKA inhibitor (H-89), and p38 inhibitor (SB203580) were purchased from Calbiochem (San Diego, CA) as powder, then dissolved in sterile dimethylsulfoxide (DMSO), and stored in the dark at −80 C.
Cell Culture and Experimental Treatments
Human hepatoblastoma cells (HepG2) were grown in complete culture medium [high glucose DMEM (with l-glutamine) supplied with 10% (vol/vol) inactivated fetal calf serum and 1% (vol/vol) antibiotics-antimycotics]. Cultures were fed with fresh medium twice weekly. For experiments, 6 × 105 HepG2 cells were seeded in 60-mm culture dishes with complete culture medium. The following day, cells were switched to medium without serum. After 24 h, CDCA or GW4064 was added at the specified concentrations (CDCA: 5, 25, 50, or 100 μmol/liter; GW4064: 0.1, 0.5, 1.0, or 2.0 μmol/liter), and cells were collected after a 48-h incubation. For inhibitor experiments, cells were pretreated with inhibitors for 30 min, after which either 50 μmol/liter of CDCA or 2 μmol/liter of GW4064 was added. Cells were collected after a 48-h incubation.
Cell Counting
After treatment with CDCA or GW4064 for the indicated time, the floating cells were aspirated away, and the attached cells were trypsinized and collected. Cells were counted using a Coulter particle counter (Coulter Corp., Miami, FL) according to the manufacturer’s instructions.
DNA Laddering Assay
HepG2 cells were cultured with complete culture medium or medium without fetal bovine serum (FBS) for 72 h. Genomic DNA isolation was performed according to the manufacturer’s instructions using the ZR Genomic DNA II kit (Zymo Research Corp., Orange, CA). Isolated DNA was run on 1% agarose gels, visualized by ethidium bromide staining, and photographed using an AlphaImager 2000 Documentation and Analysis System (Alpha Innotech Corp., San Leandro, CA).
Flow Cytometric Assay for Detection of Apoptotic Cells
After treatment with indicated reagents, suspended and attached HepG2 cells (∼1 × 106 cells) were harvested by trypsinization and washed twice with PBS. Cells were stained with PI (20 μg/ml in PBS) and/or with PI/Annexin V according to the manufacture’s instructions (BD Pharmingen Annexin V:FITC Apoptosis Detection Kit I). Flow cytometric analysis was performed with a CyAn-ADP flow cytometer DakoCytomation, Fort Collins, CO).
RNA Isolation and Quantitative Real-Time PCR
Total RNA was extracted from HepG2 cells using Tri-Reagent (Molecular Research Center, Inc., Cincinnati, OH). Quantitative real-time PCR was performed using the Power SYBR Green PCR Master Mix protocol (Applied Biosystems, Foster City, CA). Amplification of β-actin was used as an internal reference. β-Actin primers were obtained from Ambion, Inc. (Austin, TX). Quantitative PCR analysis was conducted using the ABI 7300 Sequence Detection System. Sequences of the primers used for real-time PCR are as follows. FXR: forward, 5′-GATGCCTGTAACAAAGAAGCCCC-3′; reverse, 5′-CACACAGTTGCCCCCGTTTTTAC-3′; SHP: forward, 5′-GTGGCTTCAATGCTGTCTGGAG-3′; reverse, 5′-CAGGCTGGTCGGAATGGACTT G-3′; CYP7A1: forward, 5′-CATCACAAATCCC TTGTCATACC-3′; reverse, 5′-ATACATCCCTTCTGTCACCCAGG-3′; BSEP: forward, 5′-CCAGGGAAATCAA GCTCTTAA-3′; reverse, 5′-ACTTATGATCTACAACAGCT-3′.
FXR siRNA
FXR siRNA SMART pool and control siRNA were purchased from Dharmacon (Lafayette, CO) (sequence information is patented and protected by Dharmacon) and transfected into HepG2 cells using DharmaFECT 1 reagent (Dharmacon). After an 8-h transfection, the cells were transferred into serum-free medium and treated with vehicle (DMSO), CDCA, or GW4064.
Animals
The wild-type and FXR−/− mice that have been extensively crossed to C57BL/6 background (56) were held in a pathogen-free animal facility under standard 12-h light,12-h dark cycle. Mice were fed standard rodent chow and water ad libitum. Male wild-type and FXR−/− mice at 8 wk of age were fasted for 4 d after which mice were killed and blood and livers were taken for further analysis. All procedures followed the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Analysis of Alanine Transaminase (ALT) Activity
Liver injury was quantified by measurement of plasma enzyme activities of serum alanine aminotransferase (ALT) measured at the City of Hope Helford Research Hospital.
Liver Histology
When experiments terminated, livers were removed, and small pieces were fixed in 4% formaldehyde-PBS solution, embedded in paraffin, sectioned at 5 μm, and used for TUNEL staining by a TUNEL kit (Roche, Indianapolis, IN) and/or used for H&E staining (12).
Immunoblot Analysis
At indicated time points after treatment, HepG2 cells were lysed for 15 min with lysis buffer and centrifuged at 12,000 × g at 4 C for 15 min. Liver tissue extracts were prepared by homogenation in lysis buffer as described previously (57). The samples were resolved by 10% SDS-PAGE, transferred to nitrocellulose membranes, and blotted using anti-phospho-ERK1/2, anti-total-ERK1/2 or anti-β-actin antibodies. The membranes were washed with Tris-buffered saline-Tween 20 and then incubated with antirabbit secondary antibody conjugated to horseradish peroxidase (1:5000) (Amersham Biosciences, Buckinghamshire, UK). Bands on blots were visualized using an enhanced chemiluminescence detection system (PerkinElmer Life Sciences, Wellesley, MA) and quantified with a computerized digital imaging system using AlphaImager 2000 software (Alpha Innotech).
Statistics
All data represent at least three independent experiments and are expressed as the mean ± sem. Student’s t test was used to calculate P values. P < 0.05 was considered significant.
Acknowledgments
We thank Kris Justus and Keely Walker for the proofreading.
NURSA Molecule Pages:
Ligands: GW4064;
Nuclear Receptors: FXRα.
Footnotes
This work was supported by Sidney Kimmel Foundation for Cancer Research and the Margaret E. Early Medical Research Trust.
Present address for X.H.: Department of Pathology, Fujian Medical University, 88 Jiaotong Road, Fuzhou, Fujian 350004, China.
Disclosure Statement: The authors have nothing to disclose.
First Published Online April 24, 2008
Y.-D.W. and F.Y. contributed equally to this work.
Abbreviations: ALT, Alanine transaminase; BA, bile acid; BSEP, bile salt export pump; CDCA, chenodeoxycholic acid; CYP7A1, cholesterol 7 α-hydroxylase; DMSO, dimethylsulfoxide; FXR, farnesoid X receptor; H&E, hematoxylin and eosin; JNK, Jun N-terminal kinase; MEK, MAPK kinase; PI, propidium iodide; PI3K, phosphoinositol-3-OH kinase; PKA, protein kinase A; SHP, short heterodimer partner; siRNA, short interfering RNA; TUNEL, terminal deoxy-nucleotidyl transferase-mediated dUTP nick end-labeling.
References
- 1.Kren BT, Trembley JH, Fan G, Steer CJ 1997. Molecular regulation of liver regeneration. Ann NY Acad Sci 831:361–381 [DOI] [PubMed] [Google Scholar]
- 2.Chen W, Woodruff TK, Mayo KE 2000. Activin A-induced HepG2 liver cell apoptosis: involvement of activin receptors and Smad proteins. Endocrinology 141:1263–1272 [DOI] [PubMed] [Google Scholar]
- 3.Farinati F, Cardin R, Fiorentino M, D'Errico A, Grigioni W, Cecchetto A, Naccarato R 2001. Imbalance between cytoproliferation and apoptosis in hepatitis C virus related chronic liver disease. J Viral Hepat 8:34–40 [DOI] [PubMed] [Google Scholar]
- 4.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J 2004. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest 113:1408–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, Caron S, Torpier G, Fruchart JC, Gonzalez FJ, Kuipers F, Staels B 2006. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem 281:11039–11049 [DOI] [PubMed] [Google Scholar]
- 6.Wang YD, Chen WD, Huang W 2008. FXR, a target for different diseases. Histol Histopathol 23:621–627 [DOI] [PubMed] [Google Scholar]
- 7.Stedman C, Liddle C, Coulter S, Sonoda J, Alvarez JG, Evans RM, Downes M 2006. Benefit of farnesoid X receptor inhibition in obstructive cholestasis. Proc Natl Acad Sci USA 103:11323–11328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma K, Saha PK, Chan L, Moore DD 2006. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest 116:1102–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, Dong B, Huang X, Moore DD 2006. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 312:233–236 [DOI] [PubMed] [Google Scholar]
- 10.Guo GL, Lambert G, Negishi M, Ward JM, Brewer Jr HB, Kliewer SA, Gonzalez FJ, Sinal CJ 2003. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem 278:45062–45071 [DOI] [PubMed] [Google Scholar]
- 11.Liu Y, Binz J, Numerick MJ, Dennis S, Luo G, Desai B, MacKenzie KI, Mansfield TA, Kliewer SA, Goodwin B, Jones SA 2003. Hepatoprotection by the farnesoid X receptor agonist GW4064 in rat models of intra- and extrahepatic cholestasis. J Clin Invest 112:1678–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W 2007. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res 67:863–867 [DOI] [PubMed] [Google Scholar]
- 13.Bai J, Cederbaum AI 2006. Cycloheximide protects HepG2 cells from serum withdrawal-induced apoptosis by decreasing p53 and phosphorylated p53 levels. J Pharmacol Exp Ther 319:1435–1443 [DOI] [PubMed] [Google Scholar]
- 14.Grasl-Kraupp B, Bursch W, Ruttkay-Nedecky B, Wagner A, Lauer B, Schulte-Hermann R 1994. Food restriction eliminates preneoplastic cells through apoptosis and antagonizes carcinogenesis in rat liver. Proc Natl Acad Sci USA 91:9995–9999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tessitore L, Tomasi C, Greco M 1999. Fasting-induced apoptosis in rat liver is blocked by cycloheximide. Eur J Cell Biol 78:573–579 [DOI] [PubMed] [Google Scholar]
- 16.Zhuge J, Cederbaum AI 2006. Serum deprivation-induced HepG2 cell death is potentiated by CYP2E1. Free Radic Biol Med 40:63–74 [DOI] [PubMed] [Google Scholar]
- 17.Zamai L, Canonico B, Luchetti F, Ferri P, Melloni E, Guidotti L, Cappellini A, Cutroneo G, Vitale M, Papa S 2001. Supravital exposure to propidium iodide identifies apoptosis on adherent cells. Cytometry 44:57–64 [PubMed] [Google Scholar]
- 18.Barbier O, Torra IP, Sirvent A, Claudel T, Blanquart C, Duran-Sandoval D, Kuipers F, Kosykh V, Fruchart JC, Staels B 2003. FXR induces the UGT2B4 enzyme in hepatocytes: a potential mechanism of negative feedback control of FXR activity. Gastroenterology 124:1926–1940 [DOI] [PubMed] [Google Scholar]
- 19.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA 2000. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6:517–526 [DOI] [PubMed] [Google Scholar]
- 20.Xu G, Li H, Pan LX, Shang Q, Honda A, Ananthanarayanan M, Erickson SK, Shneider BL, Shefer S, Bollineni J, Forman BM, Matsuzaki Y, Suchy FJ, Tint GS, Salen G 2003. FXR-mediated down-regulation of CYP7A1 dominates LXRα in long-term cholesterol-fed NZW rabbits. J Lipid Res 44:1956–1962 [DOI] [PubMed] [Google Scholar]
- 21.Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, Plunket KD, Creech KL, Moore LB, Wilson JG, Lewis MC, Jones SA, Willson TM 2000. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem 43:2971–2974 [DOI] [PubMed] [Google Scholar]
- 22.Schoemaker MH, Conde de la Rosa L, Buist-Homan M, Vrenken TE, Havinga R, Poelstra K, Haisma HJ, Jansen PL, Moshage H 2004. Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology 39:1563–1573 [DOI] [PubMed] [Google Scholar]
- 23.Das B, Yeger H, Tsuchida R, Torkin R, Gee MF, Thorner PS, Shibuya M, Malkin D, Baruchel S 2005. A hypoxia-driven vascular endothelial growth factor/Flt1 autocrine loop interacts with hypoxia-inducible factor-1α through mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 pathway in neuroblastoma. Cancer Res 65:7267–7275 [DOI] [PubMed] [Google Scholar]
- 24.Baek JH, Jang JE, Kang CM, Chung HY, Kim ND, Kim KW 2000. Hypoxia-induced VEGF enhances tumor survivability via suppression of serum deprivation-induced apoptosis. Oncogene 19:4621–4631 [DOI] [PubMed] [Google Scholar]
- 25.Mitsui H, Takuwa N, Maruyama T, Maekawa H, Hirayama M, Sawatari T, Hashimoto N, Takuwa Y, Kimura S 2001. The MEK1-ERK map kinase pathway and the PI 3-kinase-Akt pathway independently mediate anti-apoptotic signals in HepG2 liver cancer cells. Int J Cancer 92:55–62 [PubMed] [Google Scholar]
- 26.De Gottardi A, Dumonceau JM, Bruttin F, Vonlaufen A, Morard I, Spahr L, Rubbia-Brandt L, Frossard JL, Dinjens WN, Rabinovitch PS, Hadengue 2006. Expression of the bile acid receptor FXR in Barrett's esophagus and enhancement of apoptosis by guggulsterone in vitro. Mol Cancer 5:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swales KE, Korbonits M, Carpenter R, Walsh DT, Warner TD, Bishop-Bailey D 2006. The farnesoid X receptor is expressed in breast cancer and regulates apoptosis and aromatase expression. Cancer Res 66:10120–10126 [DOI] [PubMed] [Google Scholar]
- 28.Silva J, Dasgupta S, Wang G, Krishnamurthy K, Ritter E, Bieberich E 2006. Lipids isolated from bone induce the migration of human breast cancer cells. J Lipid Res 47:724–733 [DOI] [PubMed] [Google Scholar]
- 29.Namgaladze D, Kollas A, Brune B 2008. Oxidized LDL attenuates apoptosis in monocytic cells by activating ERK signaling. J Lipid Res 49:58–65 [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Wezeman M, Zhang X, Lin P, Wang M, Qian J, Wan B, Kwak LW, Yu L, Yi Q 2007. Human C-reactive protein binds activating Fcγ receptors and protects myeloma tumor cells from apoptosis. Cancer Cell 12:252–265 [DOI] [PubMed] [Google Scholar]
- 31.Allen TR, Krueger KD, Hunter III WJ, Agrawal DK 2005. Evidence that insulin-like growth factor-1 requires protein kinase C-ε, PI3-kinase and mitogen-activated protein kinase pathways to protect human vascular smooth muscle cells from apoptosis. Immunol Cell Biol 83:651–667 [DOI] [PubMed] [Google Scholar]
- 32.Boucher MJ, Morisset J, Vachon PH, Reed JC, Laine J, Rivard N 2000. MEK/ERK signaling pathway regulates the expression of Bcl-2, Bcl-X(L), and Mcl-1 and promotes survival of human pancreatic cancer cells. J Cell Biochem 79:355–369 [PubMed] [Google Scholar]
- 33.Gardai SJ, Whitlock BB, Xiao YQ, Bratton DB, Henson PM 2004. Oxidants inhibit ERK/MAPK and prevent its ability to delay neutrophil apoptosis downstream of mitochondrial changes and at the level of XIAP. J Biol Chem 279:44695–44703 [DOI] [PubMed] [Google Scholar]
- 34.Ballif BA, Blenis J 2001. Molecular mechanisms mediating mammalian mitogen-activated protein kinase (MAPK) kinase (MEK)-MAPK cell survival signals. Cell Growth Differ 12:397–408 [PubMed] [Google Scholar]
- 35.Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ 2003. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem 278:18811–18816 [DOI] [PubMed] [Google Scholar]
- 36.Scheid MP, Schubert KM, Duronio V 1999. Regulation of bad phosphorylation and association with Bcl-x(L) by the MAPK/Erk kinase. J Biol Chem 274:31108–31113 [DOI] [PubMed] [Google Scholar]
- 37.Derouet M, Thomas L, Cross A, Moots RJ, Edwards SW 2004. Granulocyte macrophage colony-stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl-1. J Biol Chem 279:26915–26921 [DOI] [PubMed] [Google Scholar]
- 38.Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR 2003. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol 5:647–654 [DOI] [PubMed] [Google Scholar]
- 39.Burgermeister E, Seger R 2007. MAPK kinases as nucleo-cytoplasmic shuttles for PPARγ. Cell Cycle 6:1539–1548 [DOI] [PubMed] [Google Scholar]
- 40.Vicent GP, Ballare C, Nacht AS, Clausell J, Subtil-Rodriguez A, Quiles I, Jordan A, Beato M 2006. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell 24:367–381 [DOI] [PubMed] [Google Scholar]
- 41.Burgermeister E, Chuderland D, Hanoch T, Meyer M, Liscovitch M, Seger R 2007. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor γ. Mol Cell Biol 27:803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papageorgiou E, Pitulis N, Msaouel P, Lembessis P, Koutsilieris M 2007. The non-genomic crosstalk between PPAR-γ ligands and ERK1/2 in cancer cell lines. Expert Opin Ther Targets 11:1071–1085 [DOI] [PubMed] [Google Scholar]
- 43.Narayanan R, Sepulveda VA, Falzon M, Weigel NL 2004. The functional consequences of cross-talk between the vitamin D receptor and ERK signaling pathways are cell-specific. J Biol Chem 279:47298–47310 [DOI] [PubMed] [Google Scholar]
- 44.Mannella P, Brinton RD 2006. Estrogen receptor protein interaction with phosphatidylinositol 3-kinase leads to activation of phosphorylated Akt and extracellular signal-regulated kinase 1/2 in the same population of cortical neurons: a unified mechanism of estrogen action. J Neurosci 26:9439–9447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qiao L, McKinstry R, Gupta S, Gilfor D, Windle JJ, Hylemon PB, Grant S, Fisher PB, Dent P 2002. Cyclin kinase inhibitor p21 potentiates bile acid-induced apoptosis in hepatocytes that is dependent on p53. Hepatology 36:39–48 [DOI] [PubMed] [Google Scholar]
- 46.Kwo P, Patel T, Bronk SF, Gores GJ 1995. Nuclear serine protease activity contributes to bile acid-induced apoptosis in hepatocytes. Am J Physiol 268:G613–G621 [DOI] [PubMed]
- 47.Higuchi H, Yoon JH, Grambihler A, Werneburg N, Bronk SF, Gores GJ 2003. Bile acids stimulate cFLIP phosphorylation enhancing TRAIL-mediated apoptosis. J Biol Chem 278:454–461 [DOI] [PubMed] [Google Scholar]
- 48.Rao YP, Studer EJ, Stravitz RT, Gupta S, Qiao L, Dent P, Hylemon PB 2002. Activation of the Raf-1/MEK/ERK cascade by bile acids occurs via the epidermal growth factor receptor in primary rat hepatocytes. Hepatology 35:307–314 [DOI] [PubMed] [Google Scholar]
- 49.Qiao L, Yacoub A, Studer E, Gupta S, Pei XY, Grant S, Hylemon PB, Dent P 2002. Inhibition of the MAPK and PI3K pathways enhances UDCA-induced apoptosis in primary rodent hepatocytes. Hepatology 35:779–789 [DOI] [PubMed] [Google Scholar]
- 50.Han SI, Studer E, Gupta S, Fang Y, Qiao L, Li W, Grant S, Hylemon PB, Dent P 2004. Bile acids enhance the activity of the insulin receptor and glycogen synthase in primary rodent hepatocytes. Hepatology 39:456–463 [DOI] [PubMed] [Google Scholar]
- 51.Dent P, Fang Y, Gupta S, Studer E, Mitchell C, Spiegel S, Hylemon PB 2005. Conjugated bile acids promote ERK1/2 and AKT activation via a pertussis toxin-sensitive mechanism in murine and human hepatocytes. Hepatology 42:1291–1299 [DOI] [PubMed] [Google Scholar]
- 52.Kast HR, Goodwin B, Tarr PT, Jones SA, Anisfeld AM, Stoltz CM, Tontonoz P, Kliewer S, Willson TM, Edwards PA 2002. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem 277:2908–2915 [DOI] [PubMed] [Google Scholar]
- 53.Baskin-Bey ES, Huang W, Ishimura N, Isomoto H, Bronk SF, Braley K, Craig RW, Moore DD, Gores GJ 2006. Constitutive androstane receptor (CAR) ligand, TCPOBOP, attenuates Fas-induced murine liver injury by altering Bcl-2 proteins. Hepatology 44:252–262 [DOI] [PubMed] [Google Scholar]
- 54.Zhou J, Liu M, Zhai Y, Xie W 2008. The anti-apoptotic role of pregnane X receptor in human colon cancer cells. Mol Endocrinol 22:860–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zucchini N, de Sousa G, Bailly-Maitre B, Gugenheim J, Bars R, Lemaire G, Rahmani R 2005. Regulation of Bcl-2 and Bcl-xL anti-apoptotic protein expression by nuclear receptor PXR in primary cultures of human and rat hepatocytes. Biochim Biophys Acta 1745:48–58 [DOI] [PubMed] [Google Scholar]
- 56.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ 2000. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102:731–744 [DOI] [PubMed] [Google Scholar]
- 57.Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, Chua SS, Wei P, Heyman RA, Karin M, Moore DD 2002. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell 2:721–731 [DOI] [PubMed] [Google Scholar]