

Abstract
Rapid progress in mapping nuclear receptor binding sites, referred to as “location analysis,” has recently been achieved through the use of chromatin immunoprecipitation approaches. Location analysis can be performed on a single locus or cover a complete genome, and the resulting datasets can be probed to identify direct target genes and/or investigate the molecular mechanisms by which nuclear receptors control gene expression. In addition, when coupled with other genetic and functional genomics investigative methods, location analysis has proven to be a powerful tool with which to identify novel biological functions of nuclear receptors and build transcriptional regulatory networks. Thus, the knowledge gained from several recent chromatin immunoprecipitation-based studies has challenged basic concepts of nuclear receptor action, offered new insights into gene-regulatory mechanisms, and led to the identification of nuclear receptor-controlled biological functions.
NUCLEAR RECEPTORS constitute a large family of transcription factors that regulate developmental and physiological processes through direct control of gene expression. In addition, deregulation of nuclear receptor activity has been associated with a wide range of human diseases, including cancer. Consequently, the identification of the full complement of the target genes specific to each member of the family is crucial to our understanding of their biological roles. Moreover, knowledge about the location and composition of genomic regulatory regions encompassing nuclear receptor binding sites would facilitate the study of the molecular determinants and mechanisms governing the specificity of action of each receptor in response to physiological cues, natural ligands, or therapeutically relevant drugs. Until recently, the identification of genes and the characterization of the regulatory elements suspected of being targeted by nuclear receptors were mainly accomplished on a gene-by-gene basis, a process that was both inefficient and labor intensive. In addition, these studies were mostly limited to the proximal promoter regions of regulated genes.
The concurrent advent of chromatin immunoprecipitation (ChIP) assays, DNA microarray-based technologies (gene chip), and whole-genome/high-throughput sequencing has recently led to the development of new approaches to identify the location of bound transcription factors and other chromatin-associated proteins on a genome-wide scale. Herein, we will review how these new techniques were applied to the study of individual nuclear receptors and discuss how the results of these studies introduced novel paradigms of nuclear receptor action and led to the recognition that nuclear receptors orchestrate comprehensive transcriptional regulatory networks linking gene regulation and physiology.
NUCLEAR RECEPTOR-DNA INTERACTIONS
Like other transcription factors, nuclear receptors control the expression of their target genes mostly through association with specific DNA-regulatory elements. Nuclear receptor binding sites are generally referred to as hormone response elements (HREs) that are composed of one (for monomer binding) or two (for homo- and heterodimer binding) short 6-bp sequences known as core half-sites. The primary sequence of the core half-sites is largely limited to two distinct motifs, AGGTCA and AGAACA; thus binding specificity is mainly dictated by the spatial organization of the two half-sites required for binding by nuclear receptor dimers. Receptors binding DNA as monomers recognize a single half-site flanked at the 5′-end by A/T-rich motifs of 3 to 6 bp in length. Despite the apparent specificity of the HRE-receptor interaction, nuclear receptor binding sites often deviate considerably from the defined consensus half-site sequences. In addition, nuclear receptors can also bind to DNA through tethering to other transcription factors. These binding properties make the recognition of nuclear receptor binding sites by visually scanning regulatory sequences of target genes or even through more sophisticated bioinformatics efforts, a difficult and imprecise task. In addition, the occurrence of HRE-like motifs throughout the genome seems to be much more prevalent than the actual functional usage of these sites in vivo (1, 2). In agreement with this observation, functional HREs are not lonesome entities, but rather are surrounded by other transcription factor binding sites that, in combination, can impart gene- and cell-specific regulation (3). Therefore, rigorous location analyses of nuclear receptor binding sites in genomes require direct physical measurements of their interaction with DNA.
ChIP-BASED APPROACHES
The development of the ChIP assay was a defining event in the study of protein-DNA interactions in vivo (4, 5). The technique allows the identification of DNA elements bound by proteins such as histones or transcription factors under a specific cellular context and is thus well suited for the study of nuclear receptors. Briefly, the protein-DNA interactions are fixed with formaldehyde, a cross-linking reagent, the chromatin is subsequently sheared to an average length of 500-1000 bp, and the protein of interest is immunoprecipitated along with its bound DNA using a specific antibody, a strict and often limiting requirement as ChIP-grade antibodies are difficult to find for most nuclear receptors. After reverse cross-link, the enriched DNA is purified and the level of enrichment is evaluated by quantitative PCR (qPCR) using specific primers for the bound regions. Normalization is achieved by comparing to an IgG-background sample and controlled by monitoring negative genomic regions where no binding of the protein is expected. The use of the ChIP-qPCR technique is now considered the gold standard to confirm the presence of a nuclear receptor at a putative regulatory region. ChIP-qPCR assays have been particularly useful in studying the molecular mechanisms involved in the cyclical recruitment of coregulatory proteins by nuclear receptors at specific enhancers and promoters after hormonal stimulation of a target gene (6, 7, 8, 9, 10, 11). Although knowledge of the precise location of the transcription factor’s binding site in the genomic region under study is required to perform a standard ChIP experiment, the end result of the immunoprecipitation assay consists of a pool of enriched genomic targets bound by the factor. This led to the development of genome-wide ChIP-based approaches that proved extremely useful in large-scale identification of genomic targets of numerous transcription factors (12, 13, 14, 15, 16, 17), monitoring the presence of components of the preinitiation complex at active promoters (18) and mapping specific modifications of histones residing in transcriptionally active regions of the genome (19, 20, 21). The ChIP-based techniques that were used to map nuclear receptor binding sites include ChIP-cloning, ChIP-paired end-tag (PET), ChIP coupled to a DNA selection and ligation (ChIP-DSL), and several variations of ChIP-on-chip (Table 1).
Table 1.
ChIP-Based Techniques Used to Study Nuclear Receptor Genomic Localization
| Technique | Procedure | Assay | Identification of Genomic Regions | Binging Site Resolution | Extensive Sequencing Required | Bias Introduced by the Technique | Repeat Regions Masked | Need for Data Normalization |
|---|---|---|---|---|---|---|---|---|
| ChIP-cloning | Linker ligation; entire ChIP fragment cloned | Sequencing | Unbiased | Low | Yes | No | No | No |
| ChIP-PET | Concatenate and clone PET fragments | Sequencing | Unbiased | High | Yes | No | No | Yes |
| ChIP-chip (PCR-based array) | Linker ligation; Amplification; labeling with fluorescent dye | Microarray hybridization | Biased | Low | No | Amplification | Yes | Yes |
| ChIP-chip (tilled array) | Linker ligation; Amplification; Labeling with fluorescent dye | Microarray hybridization | Array dependent | High | No | Amplification | Yes | Yes |
| ChIP-DSL | Select and anneal oligos; Labeling with fluorescent dye | Microarray hybridization | Biased | Low | No | Amplification; DSL oligo pool | Yes | Yes |
LOCATION ANALYSIS OF NUCLEAR RECEPTOR BINDING SITES
To date, ChIP-based approaches were used to investigate the binding of seven different nuclear receptors to genomic DNA in human and mouse tissues or cell lines. The results of these studies are summarized below and in Table 2.
Table 2.
Nuclear Receptors Target Discovery Using ChIP-Based Approaches
| Factor | Technique1 | Tissue/Cell Line | Bound Regions | Genomic Distribution |
|---|---|---|---|---|
| HNF4α | 2 | Human liver | 1555 | Promoters |
| HNF4α | 2 | Human pancreatic islets | 1423 | Promoters |
| ERα | 1 | MCF-7 | 12 | 8/12 <10 kb of TSS; 4/12 <2 kb of TSS |
| ERα | 1 | MCF-7 | 32 | n/a |
| ERα | 1 | MCF-7 | 94 | Intronic (45.7%); <10 kb upstream (12.8%); >50 kb upstream (17%) |
| ERα | 1 | U2OS | 173 | Intronic (38%); <10 kb upstream (24%); >10 kb upstream (38%) |
| ERα | 1 | Mouse uterus | n/a | n/a |
| ERα | 2 | MCF-7 | 153 | Promoters |
| ERα | 3 | MCF-7 | 57 | Mostly distal |
| ERα | 4 | MCF-7 | 3665 | Distal (96%) |
| ERα | 5 | MCF-7 | 1234 | Distal (95%) |
| ERα | 6 | MCF-7 | 578 | Promoters |
| ERα | 7 | MCF-7 | 47 | Promoters (79%) |
| ERα | 8 | MCF-7 | 92 | Promoters |
| ERα | 4 | Mouse liver | 5568 | >10 kb (60%) |
| GR | 9 | A549 | 8 | Promoters |
| GR | 10 | A549 | 73 | <5 kb from TSS of nearest responsive gene (31%) |
| AR | 3 | LNCaP | 90 | <500 kb from TSS of nearest responsive gene (38%) |
| AR | 11 | HPr-1AR | 524 | <10 kb from TSS of nearest responsive gene (33%) |
| AR | 2 | LNCaP | 1532 | Promoters |
| ERRα and ERRγ | 2 | Mouse heart | 736 | Promoters |
| ERRα | 2 | Mouse bone-derived macrophages | 215 | Promoters |
| VDR | 12 | ST2 | 3 (VDR) 5 (RANKL) | Intronic (VDR) >15 kb upstream TSS (RANKL) |
Table 2A.
Cont.
| Motif Discovery | Correlation with Regulated Expression | PolII Occupancy or Histone Modifications | Reference |
|---|---|---|---|
| n/a | n/a | 50% of PolII occupred promoters bound by HNF4α | |
| n/a | 10% of 133 HNF4α-dependent genes showed binding (23 ) | 50% of PolII occupied promoters bound by HNF4α | 22 |
| ERE; half-sites | 6/12 of genes regulated by E2 | n/a | 25 |
| n/a | n/a | n/a | 28 |
| n/a | 43% of the genes with proximal binding are regulated by E2 | PolII recruited to all tested ERα-bound regions (enhanced) upon E2 treatment | |
| ERE (11%) | n/a | n/a | 30 |
| n/a | n/a | n/a | 31 |
| ERE (70%) | n/a | PolII recruited to all tested ERα-bound regions (enhanced upon E2 treatment) | 32 |
| ERE (49%); forkhead | 11/12 E2-regulated genes on chr.21–22 were bound by ERα at promoter | PolII recruited to all tested ERα-bound regions | 2 |
| ERE; AP-1; OCT1; C/EBP; forkhead | E2-up-regulated and late -down-regulated genes associated with ERα binding within 50 kb of TSS (33%) | ERα-bound regions dispersed throughout genome, whereas polII binding correlates with TSS | |
| ERE (71%) | E2-upregulated genes associated with ERα binding within 50 kb of TSS | n/a | 35 |
| ERE (44%) | 9% of ERα-bound promoters associated with E2 - responsive genes | PolII recruited to 43% of all promoters along with AcH3K9 and Me2H3K4 modifications | 36 |
| ERE and AP-1 | ERα-bound promoters correlate with E2- responsiveness | 47% ERα-bound promoters showed E2-dependent PolII recruitment, which correlated with AcH3K9 | 3 |
| n/a | n/a | 40/92 showed AcH3K9; 28/92 showed H3K9Me2; PolII recruitment correlates with ERα recruitment | 38 |
| ERE; ETS; forkhead | 22/209 E2-responsive genes bound by ERα at promoter | n/a | 39 |
| GRE | 8/11 GR-bound promoters correlate with glucocorticoid-regulated genes | n/a | 43 |
| GRE (68%) | 88% of GR-bound regions associated with glucocorticoid responsiveness | n/a | 44 |
| ARE (10%); noncanonical ARE (68%); forkhead; GATA; Oct | 38% of AR-bound regions located within 500 kb of androgen-regulated genes | n/a | 45 |
| ARE (69%) | 84% of the androgen-responsive genes represented on array are bound by AR | n/a | 46 |
| ARE (27%); ETS | 6% of AR-bound promoters showed altered expression upon androgen treatment | n/a | 47 |
| ERRE, NRF-1, CREB, STAT3 | n/a | n/a | 48 49 |
| n/a | High correlation for mitochondrial respiration pathway | n/a | 50 |
| VDRE | Binding correlates with VDR or RNAKL expression | PolII recruitment correlates with histone acetylation | 52 53 |
1, ChIP-cloning; 2, ChIP-on-chip on promoter array (1 kb PCR-generated fragments); 3, ChIP-on-chip on chromosome 21–22 tilled array; 4, ChIP-on-chip on whole-genome tilled array; 5, ChIP-PET; 6, ChIP-DSL (40-oligomer promoter array); 7, ChIP-on-chip on array consisting of ∼900 regions (1 kb PCR-generated fragments): ∼600 E2-responsive promoters, ∼250 control promoters, and ∼50 nonpromoter regions; 8, ChIP-on-chip on CpG islands; 9, ChIP scanning of 11 glucocorticoid-responsive gene promoters; 10, ChIP-on-chip on oligonucleotide tilled array covering large regions around TSS of 548 glucocorticoid-responsive genes; 11, ChIP-on-chip on oligonucleotide tilled array covering large regions around TSS of 205 androgen-responsive genes; 12, ChIP-on-chip on oligonucleotide tiled array covering large regions around the TSS of VDR and RANKL genes. n/a, nonapplicable; ERRE, ERR response element; GRE, glucocorticoid response element; VDRE, vitamin D response element.
Hepatocyte Nuclear Factor (HNF)4-α
The orphan nuclear receptor HNF4-α (nuclear receptor 2A1) was the first member of the family to be analyzed for the localization of its binding sites employing a ChIP-on-chip approach (22). Using an approximately 13,000 human promoter array, this study identified 1,555 and 1,423 promoters bound by HNF4-α in human liver and pancreatic islets, respectively. The number of bound promoters identified in both tissues was surprisingly large, especially considering that the array was relatively limited both in terms of coverage, approximately 1 kb per promoter region, and number of genes on the array. HNF4-α was also found to bind to almost half of the actively transcribed genes in these tissues as measured via promoter occupancy by RNA polymerase II (PolII), suggesting that HNF-4α acts as a master regulator of the liver and pancreatic islet transcriptomes. This assertion was recently challenged by the results of expression profiling in HNF-4α-deficient pancreatic β-cells (23). Of 133 genes the expression of which was dependent on the presence of HNF-4α, only 13 were in the list of HNF-4α-bound promoters, indicating that HNF-4α is probably not a controller of the global transcriptional program in these cells (23). However, the study by Odom et al. (22) showed that HNF-4α, through binding to the promoter of HNF-1α, participates in a multicomponent-regulatory network that may have a broader role in liver and pancreatic islets development and function.
Estrogen Receptor-α (ERα)
The location of ERα (nuclear receptor 3A1) binding sites has been extensively investigated using all types of ChIP-based approaches and array designs. Together, the results of these experiments provided not only mechanistic insights into how ERα works as a transcription factor but, because most studies were performed with the MCF-7 human breast cancer cell line, also allows for an easier comparison of the technical advantages and disadvantages of each approach.
The first study reporting the isolation of ERα-bound regulatory modules was based on ChIP cloning (24, 25). Twelve ERα-bound genomic fragments were isolated from estradiol (E2)-treated MCF-7 cells and functionally characterized for transcriptional activity, modulation of cognate target gene expression, and corecruitment of coactivators upon E2 treatment. In particular, this study identified an ERα-bound genomic fragment located in the first intron of the gene, 4 kb downstream from the first promoter, encoding retinoic acid receptor-α (RARA). The RARA gene was known to be regulated by E2, but classic functional characterization of the promoter had shown only a limited response to the hormone (26, 27). In contrast, the intronic fragment was shown to be a cross-species conserved regulatory region essential for the hormonal regulation of the gene. Despite its limited scope, this initial study established the concept that ChIP-based approaches would be essential to identify regulatory elements involved in long-range control of gene expression by nuclear receptors. The ChIP-cloning technique was subsequently used to identify a number of ERα-bound genomic fragments from chromatin isolated from MCF-7 (28, 29) and U2OS (30) cells as well as mouse uteri (31), and some of these fragments were linked to E2-regulated genes such as WNT11 (40 kb upstream), AQP5 (promoter), and BARD1 (intronic, 74 kb downstream).
The development of high-throughput ChIP-on-chip technologies enabled the identification and localization of ERα-bound genomic loci on a larger scale. Initially, promoter arrays consisting of 1 kb PCR products encompassing promoter regions of annotated genes were used to map ERα-bound promoters throughout the human genome (32). This study identified 153 promoters bound by ERα upon E2 treatment in MCF-7 cells, including the promoter of the gene encoding the pioneer factor forkhead box A1 (FOXA1), the expression of which correlates with the presence of ERα in breast tumors (33). Unbiased analysis of the genomic ERα-bound fragments showed enrichment of the canonical estrogen response element (ERE). In addition, motif-finding analysis using the consensus FOXA1 binding site revealed that a subset (∼12%) of the ERα-bound promoters contained FOXA1 recognition sites. Concurrently, the utilization of the whole-chromosome 21 and 22 tiled arrays led to the identification of 57 ERα-bound sites within 32 discrete clusters (2). This study was the first to demonstrate that binding of ERα occurs largely at regions located far away from the promoters of genes. As observed with ERα-bound promoters, several distal ERα binding sites were shown to include FOXA1 recognition motifs. Both groups demonstrated that ablation of FOXA1 expression in MCF-7 cells suppressed ERα binding to the TIFF (pS2) promoter and hindered the induction of TIFF expression by E2. These findings, coupled with the observation that FOXA1 was required for E2-induced reentry into the cell cycle, led to the suggestion that FOXA1 and functionally related factors could be used to define subdomains of the estrogen response (32).
Carroll et al. (34) subsequently extended their initial chromosome-wide ChIP-on-chip analysis to the entire human genome. In this study, they identified 3665 high- confidence ERα-bound genomic loci in MCF-7 cells treated with E2. This unique resource, referred to as the ERα cistrome, provided the first comprehensive map of ERα binding sites in human breast cancer cells. Of particular interest, mapping of the ERα-bound regions to the nearest gene revealed that only 4% of the identified segments were located in the vicinity (800 bp upstream to 200 downstream) of the transcription start site (TSS) of annotated genes. Nonetheless, correlation of ERα binding with early E2-regulated genes showed that one third were associated with ERα binding within 50 kb of TSSs, demonstrating the presence of ERα-bound regions at E2-regulated genes. Bioinformatics analysis of the segments revealed enrichment not only for the ERE motif, but also for Forkhead, activator protein 1 (AP-1), OCT1, and CCAAT enhancer binding protein (C/EBP) motifs, indicating a universal role for cooperating factors in ERα-dependent transcription. Subsequent analysis (74) revealed that distal ER binding sites associates with FOXA1 which induces changes to chromatin structure at enhancers.
The ChIP-PET technique, a high-throughput variation of the ChIP-cloning approach, was also used to map the genomic binding targets of ERα in MCF-7 cells (35). Identification of 1234 high-confidence ERα-bound regions after E2 treatment was achieved, and the fragments were found to be highly enriched (71%) for the presence of ERE-like motifs. Coupling to expression profiling of E2-treated MCF-7 cells revealed that 287 E2-regulated genes were associated with ERα-bound fragments located within 50 kb of their TSS. A unique facet of this study was the finding that ERα-bound fragments seem to be poorly conserved between primates and other mammalian species because only approximately 17% of the sites appeared to be under positive selection. Although the majority of ERα binding sites can be found far away from TSSs, a nonnegligible number of promoters have also been identified as genuine ERα targets.
A variant of the ChIP-promoter array, ChIP-DSL, identified 578 high-confidence ERα-bound promoters in MCF-7 cells (36). Although a strong correlation between ERα and PolII recruitment at these sites was observed, only 54 of 879 E2-induced genes displayed ERα binding at their promoters. These data once again suggest that most E2-induced genes are indirect targets or that their transcriptional regulation by ERα is mediated via distal cis-regulatory regions.
Kininis et al. (37) used antibodies against ERα, steroid receptor coactivator-1, RNA PolII, and acetylated histones (AcH3K9) to perform ChIP-on-chip analyses and monitor variation in recruitment of these factors upon E2 treatment in MCF-7 cells. The custom-made promoter array included the promoter region of approximately 600 known estrogen-regulated genes, about 50 distal nonpromoter regions previously shown to be bound by ERα as well as approximately 250 control promoters. The experiment identified 47 ERα-bound genomic regions, 22 of which showed E2-dependent changes in recruitment of PolII. A strong correlation was also observed between ERα and steroid receptor coactivator-1 colocalization and between the recruitment of PolII and AcH3K9 upon E2 treatment. These observations suggested that nuclear receptor signaling is involved in the regulation of the chromatin state. A CpG island/promoter array was also used to identify 92 ERα-bound promoter regions in MCF-7 cells (38). Computational modeling showed that 13 of these promoters had c-MYC binding sites in close proximity (13–214 bp) of a consensus ERE. Treatment with E2 was shown to enhance the interaction between ERα and c-MYC.
Although most reports on the identification ERα genomic targets were carried out in MCF-7 cells, other tissues have also been used in ChIP-based experiments. As previously mentioned, Kobayashi et al. (31) used the ChIP-cloning technique to identify ERα-bound fragments in E2-treated mouse uteri. ChIP-on-chip using chromatin from mouse liver and a whole-genome tiled array identified 5568 ERα binding regions (39). Whereas most ERα-bound regions were localized distal to TSS (60% >10 kb upstream), a significant number of binding sites were found at promoters. Indeed, the authors also identified 948 ERα-bound regions using a 25K promoter array. Remarkably, 811 (85%) of those promoter regions were also identified by the whole-genome tiled array. The ERα-bound promoters were found to be enriched for genes involved in energy metabolism and wound response.
Finally, a combination of computational prediction of transcriptional regulatory modules and ChIP-based methodologies were also used to identify ERα targets (40, 41, 42). Of interest, predicted cis-regulatory modules, a subset of them containing an ERE consensus motif, showed an enrichment near the 3′-end of genes and in regions distal from genes (41).
Glucocorticoid Receptor (GR)
The first attempt to identify primary GR (nuclear receptor 3C1) targets in A549 human lung adenocarcinoma cells involved the use of a ChIP-scanning assay, where multiple standard ChIPs of the GR were performed with a series of primer sets covering 3 kb upstream of the TSSs of known glucocorticoid-regulated genes (43). Eight GR-bound regions were identified using this approach providing a first mean to identify direct GR target genes. More recently, the same group used a tiled array covering a 100-kb region centered on the TSS of 548 potentially glucocorticoid-induced genes to study GR occupancy in A549 cells (44). The study identified 73 GR-bound regions of which 64 were associated with GR-responsive genes, indicating that GR occupancy is a primary determinant of glucocorticoid responsiveness in these cells. The GR-bound regions were evenly distributed between upstream and downstream regions of TSS; 31% of the sites were located proximal to promoters, whereas 63% were distal. This suggests that GR, as it was observed for ERα, can act at long distances to regulate target genes, but also that the occurrence of GR binding sites at promoters may be more frequent than for ERα. Further analysis of the GR-bound regions revealed strong sequence interspecies conservation, especially at the core GR binding sites. Computational approaches allowed the identification of a series of imperfect palindromes similar to the previously established GR core binding motif as the most prominent motif found (68% of the segments). Analysis of the sequence also revealed enrichment for other factors such as AP-1, ETS (v-ets erythroblastosis virus E26 oncogene homolog), Sp1, C/EBP, and HNF4 confirming that GR binding sites are embedded within complex composite elements. Subsequent study (75) used the Chip-chip approach to study the interaction of GR with the chromatin landscape and showed that binding occurs at nuclease accessible sites and mediates H2AZ histone variant exchange upon hormone activation.
Androgen Receptor (AR)
Recently, three groups identified genomic loci bound by the AR (nuclear receptor 3C4). First, tiled oligonucleotide microarrays were used to map AR binding sites throughout human chromosomes 21 and 22 (45). The ChIP-on-chip was performed with chromatin isolated from the prostate cancer cell line LNCaP, and 90 AR-bound regions were identified on the two chromosomes. Only 38% of the AR-bound loci were located within 500 kb from the TSS of androgen-regulated genes. Although the canonical androgen response element (ARE) was poorly recognized (10%) using an unbiased analysis of the AR-bound sequences, 68% had a noncanonical ARE composed of either a single ARE half-site or two half-sites arranged in different orientations and various spacing. This observation suggests that AR rarely binds genomic DNA via a canonical ARE, which may explain in part its specificity of action in vivo vis-à-vis other receptors such as GR and the progesterone receptor that recognize the same consensus motif in vitro. This study also reported the enrichment of many other DNA-binding motifs associated with the AR-bound regions such as Forkhead, GATA, and Oct. The factors FOXA1, GATA2, and Oct1 were shown to act in concert with AR on chromatin at androgen-regulated loci.
Second, Bolton et al. (46) used an array consisting of tiled 100-kb genomic regions centered on the TSS of 205 AR-responsive genes. The study identified 524 AR-bound regions in HPr-IAR cells, 33% of which were located within 10 kb of the TSS of the nearest responsive genes. Remarkably, and in contrast to the data of Wang et al. (45), it was found that 84% of the 205 androgen-regulated genes that were represented on the array were associated with one or more AR-binding regions, and that a canonical ARE occurred in 69% of the AR-bound fragments.
Third, PCR-generated promoter arrays have also been used in ChIP-on-chip to identify AR target promoters in LNCaP cells (47). This study identified 1532 potential AR-bound promoters, of which 92 of the associated genes showed altered expression in androgen-treated LNCaP cells. This result once again contrasted with the report from Wang et al. (45), which showed recruitment of AR mainly to regions distal to AR-regulated genes as well as extremely low recruitment at promoters. Again, only 27% of the AR-bound regions identified contained a canonical ARE. On the other hand, the ARE half-site was highly enriched in the target promoters (79%) as were the binding sites for members of the ETS family of transcription factors (70% of the promoters had co-occurrence of an ARE half-site and an ETS site). The binding of ETS1 was shown to be androgen dependent at a subset of AR-bound promoters, and, conversely, the presence of ETS1 was shown to be required for androgen regulation of these genes.
Estrogen-Related Receptors (ERRs)
A custom-made mouse 19K promoter array was used to identify ERRα- (nuclear receptor 3B1), and/or ERRγ- (nuclear receptor 3B3) bound regions in mouse adult and newborn heart tissues as well as in bone-derived macrophages (48, 49, 50). Together, these studies identified more than 700 distinct promoters bound by one or two of the ERR isoforms. Cross-validation by ChIP-qPCR showed that all the promoters identified in the adult heart ChIP-on-chip experiments were, in fact, bound by either or both ERRα and ERRγ, demonstrating for the first time functional redundancy between isoforms of the same subfamily of nuclear receptors at the genomic level. Analysis of the bound promoters showed them to drive the expression of a network of genes involved in the control of energy metabolism and specific heart functions such as calcium handling and contractile work. The biological relevance of the regulatory pathway identified by ChIP-on-chip was validated by demonstrating that ERRα is essential for the bioenergetic and functional adaptation to cardiac pressure overload in the mouse (51). Motif-finding algorithms indicated that estrogen-related receptor response element (ERR) target promoters were enriched for ERRE, Nuclear respiratory factor 1 (NRF-1), cAMP response element-binding protein (CREB), and signal transducer and activator of transcription (STAT)3 binding sites whereas functional studies showed cooperative activation of target promoters by ERRα, STAT3, and CREB. Similarly, the ChIP-on-chip study using chromatin isolated from the hearts of newborn mice identified ERRγ target genes essential for the transition from glycolytic to oxidative metabolism at birth (49). The third study, using chromatin from bone-derived macrophages, identified a large number of ERRα target genes encoding mitochondrial respiratory chain machinery, many of which were induced upon treatment with interferon γ (50). Consequently, the loss of ERRα results in decreased mitochondrial gene expression, intracellular reactive oxygen species level, and bacterial clearance in interferon γ-activated macrophages. Through the utilization of a combination of ChIP-on-chip experiments, expression profiling, and phenotypic analyses of knock-out mice, these studies demonstrate the usefulness of promoter arrays to identify novel biological functions regulated by nuclear receptors.
Vitamin D Receptor (VDR)/Retinoid X Receptors
Pike and co-workers (52, 53) used a ChIP-on-chip approach to identify VDR (nuclear receptor 1I1) binding sites across the entire mouse VDR and RANKL loci. Three VDR-bound regions were identified distal to the TSS of the VDR gene and correlated with the localization of retinoid X receptor (nuclear receptor 2B1), acetylated histone (AcH), and PolII recruitment. Likewise, five VDR-bound regions were identified upstream of the TSS of the RANKL gene, the farthest located more than 70 kb from the promoter. This far upstream enhancer was shown to be able to confer vitamin D inducibility to a heterologous promoter. Together, these locus-specific studies demonstrated the relevance of distal enhancers in VDR transcriptional regulation.
OVERLAP BETWEEN STUDIES
One observation emerging from the ChIP-based datasets is the variable degree of overlap between the targets identified in different studies for the same receptor under similar experimental conditions. As described above, several different approaches have been used to generate lists of genomic targets of the ERα in MCF-7 cells upon E2 treatment. Comparison of the ERα target lists obtained by ChIP-PET (35) and the whole genome ChIP-on-chip using Affymetrix tiled arrays (34) reveals an overlap of approximately 50%. Likewise, of the 153 promoters identified by Laganière et al. (32) using a promoter array, 76 (48%) and 60 (39%) of the promoters could be found in the lists generated by the whole genome tiled array (34) and the ChIP-DSL (36) studies, respectively. However, 101 (66%) of the 153 target promoters could be found in the combined lists. One exception is the 85% overlap observed between ERα-bound sites in mouse liver on a whole-genome tiled array vs. the ERα-bound sites on a promoter array (39). Because most target lists were extensively validated by standard ChIP-qPCR and thus presumed legitimate, each list generated to date must be incomplete.
Several factors could account for the variation observed between the different studies, the first one being the technical approach used. Each approach is capable of identifying targets not found by the other technique. Each experimental approach appears to be complementary, and thus merging results from several studies may be required to obtain the full cistrome for a given receptor. Disparity could also be caused by the genetic variability of the cell lines, especially for cancer cells, the antibody used, and amplification and labeling methods. The type of platform used for ChIP-on-chip (tiled vs. PCR-generated, whole genome vs. promoters, probe design, exclusion of genomic regions containing repetitive sequences, etc.) as well as the different algorithms employed for data analysis can also account for variability of the results. The fact that a group using the same conditions (chromatin, antibody, tiled arrays) obtained an 85% overlap in ERα target lists in two distinct experiments indicates that all these factors are important for the reproducibility of the data sets (39).
It is also probable that nuclear receptors are recruited to some sites in a more transient manner, leading to unstable interaction with DNA or to transitory looping interactions and thus making binding more difficult to detect by some ChIP-based techniques. This feature probably accounts for the strong overlap observed for the highest ranked targets whereas less overlap is seen for the low ranking targets. However, although each specific experimental condition may yield a validated set of target genes, it is important to keep in mind that the targets obtained from these lists must be assessed for biological activity in the proper context.
CORRELATION BETWEEN BINDING AND GENE EXPRESSION
Another important point highlighted by these studies of nuclear receptor target identification is the nominal level of correlation between receptor binding and hormone-dependent gene expression. So et al. (44) and Bolton et al. (46) reported a relatively high level of correlation between binding and gene expression for GR and AR, respectively. It should be pointed out that these two studies were performed using arrays containing mostly loci known to be hormonally regulated. As reviewed above, the correlation level between binding and hormone-regulated expression drops considerably when unbiased approaches are used. All studies tend to assign the closest gene as the target gene of a bound region, a method that may not reflect the actual mechanism of gene regulation. It is striking to observe that when comparing the total of induced genes to the ones that show recruitment of the receptor at their target promoters or even within 50 kb of their TSSs, the fraction of regulated genes with receptor binding is relatively low (34, 36, 45). However, both Carroll et al. (34) and Kwon et al. (36) observed PolII binding at the promoters of most E2-regulated genes. These observations imply that hormonal regulation of most target genes is indirect and/or that occupancy of a site does not routinely entail regulation of the nearest gene in the cell or tissue under study. On the other hand, as emerging data have suggested, PolII might be preloaded or paused at the promoters of many genes (54, 55). Thus, the presence of PolII at promoters may not necessarily indicate transcriptional regulation of genes because the actual regulation can occur subsequently to its binding (37).
As previously mentioned, it is also possible that the low level of overlap between binding and expression is caused, in part, by the fact that the individual lists of targets generated from these studies are incomplete. Moreover, the binding of a nuclear receptor to a specific site does not necessarily correlate with the functionality of the element. Nonetheless, such observations underscore the need to reevaluate the mode of action of nuclear receptors. The classical model of regulation of target genes through direct binding to proximal promoters or even to distant enhancers could represent only a minority of the transcriptional activity generated in response to the activation of some receptors. In this context, it will be important to map and characterize the complete transcriptional regulatory networks involved in hormonal control of gene expression (56, 57).
PROXIMAL PROMOTERS
Although hundreds of target promoters have been identified by location analyses, one of the common themes that emerge from most studies is that nuclear receptors frequently bind to genomic regions located far from annotated TSSs. One explanation for these results is that several enhancers that currently map to regions hundreds of kilobases away from the nearest annotated gene could, in fact, be positioned close to a real but, as yet unrecognized, gene or an alternative TSS of a known but distant gene. This concept is supported by the observation that PolII was shown to be recruited to some distant binding sites (34, 37). Findings generated by several recent whole-genome studies that underline the issue of mammalian genome complexity provide additional support to this notion. In particular, the definition of the core promoter, which represents the minimal region required for initiation of basal transcription, has evolved significantly over the last few years. First, it was recently suggested that more than half of the human genes commonly utilize several differentially regulated alternative promoters (18, 58). This leads to the generation of many alternative amino-terminal transcripts for the same gene, often linked to tissue- and cell-specific promoter use. This structural attribute may be one of the main driving forces behind the complexity of the proteome in higher eukaryotes. Second, some genes could use the promoter(s) of other neighboring genes in specific cells and developmental stages, as reported for Drosophila (58). Third, many TSSs have also been associated with exons and 3′-untranslated regions of other genes (58, 59). This gives rise to the concept of broad promoters, which is of great importance when studying gene regulation (60). Such a high level of intricacy in promoter assignment also renders more complex the distribution of nuclear receptor-bound loci to either promoters or distal elements (Fig. 1). Consequently, ChIP-based studies probably overlook many regulated genes due to mis-annotation of broad promoters. Assignment of a targeted locus to a gene must be done cautiously because promoter usage may vary according to context. Whereas more and more knowledge is gained on the architecture of the mammalian genome, the regulatory networks that take place within it are understood to be more complex. Therefore, due to the complexity of the transcriptome, unequivocal association of a nuclear receptor-bound locus with a corresponding regulated gene will remain an imprecise process in the near future.
Fig. 1.
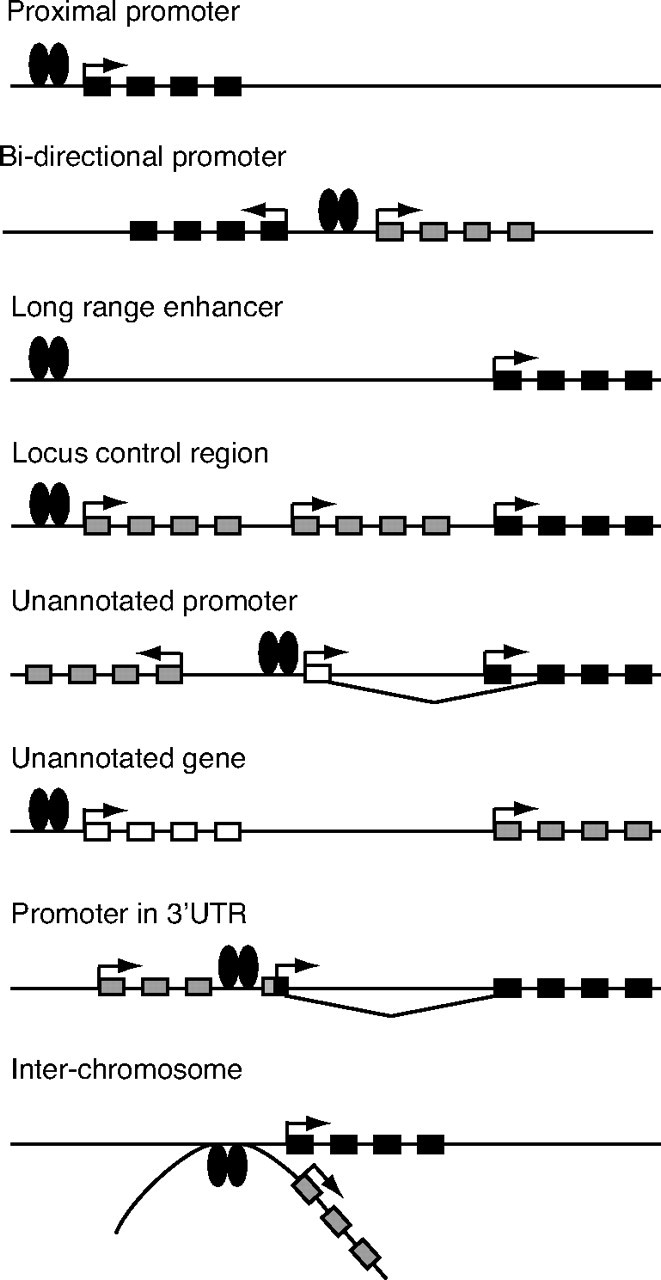
Transcriptome Complexity and Assignment of Binding Sites to Target Genes
Binding sites for nuclear receptors identified by ChIP-based experiments are usually assigned to the nearest annotated gene. Binding sites located in proximal promoter regions can be assigned with high confidence to the associated gene, although specific regulation of one gene over another in transcriptional units sharing a bidirectional promoter is possible. Unambiguous assignment of a target gene to a binding site located far away from its TSS requires the use of the 3C technology. A locus control region can regulate the expression of an individual gene within a cluster in a spatiotemporal manner. Binding sites located far away from a known gene could actually be close to an unannotated promoter or in the vicinity of an unknown gene. A significant number of promoters have been found in the 3′-untranslated region of another gene. Finally, a binding site located on one chromosome can be used to bring a transcription factor close to a gene located on another chromosome. A gene in black represents the actual target of the nuclear receptor bound to a proximal or distal site; a gene in gray indicates the annotated gene nearest to a binding site but may not be the actual target of the receptor; a gene in white denotes the presence of an unannotated gene. UTR, Untranslated region.
ACTING AT A DISTANCE
Given the fact that many, if not most, remote genomic loci bound by nuclear receptors will not be masked promoters, their assignment to target genes represents an even greater challenge. Numerous studies have addressed the question of how remote activators interact with their target genes (61, 62, 63, 64). Several models of distal regulation have been proposed (65, 66). These different models all suggest that the enhancer is brought in close proximity to its cognate promoter. Recent data indicate that enhancers mediate contact with their cognate gene promoters through the formation of nuclear domains of histone modification and of intra- and interchromosomal loops (65). These domains might rearrange genes within a three-dimensional nuclear region that favors transcription, also termed “hyperaccessible active chromatin hub” (67). Taken together, these features represent a major challenge for the study of gene regulation by remote elements with the key test residing in the unequivocal assignment of distal enhancers to specific target genes. Indeed, distal enhancers were shown to routinely regulate not the nearest gene but genes often located hundreds of kilobases from the remote enhancers as well as genes located on a different chromosome (68).
Chromosome capture conformation (3C) is a technique that is now commonly used to assess the recruitment of a distal enhancer, when bound by a transcription factor, to a specific promoter (69). Detection of such looping events by the 3C technique has been achieved for nuclear receptors (2, 11). Wang et al. (11, 45) showed that whereas AR binds mainly to regions distal to AR-regulated genes, looping between these distal regions and promoters occurs and allows PolII to track from the enhancer to the promoter and to initiate transcription. Further development of high-throughput 3C-based methods to capture DNA interactions like the 3C-on-chip (70), 3C-carbon copy, and ChIP-loop assays (71, 72) will be essential to facilitate nuclear receptor target discovery and improve our understanding of the role of DNA folding in gene regulation (73).
CONCLUDING REMARKS
ChIP-based studies have now been performed with several nuclear receptors and common themes, and new concepts have emerged from the analysis of the vast amount of information generated by these experiments. First is the realization that most nuclear receptor binding sites are located distal from the TSS of annotated genes. This regulatory feature represents a considerable challenge for assigning a binding site to a specific gene, especially when the complexity of the transcriptome is taken into account. Because this observation was made in studies exploring the functions of steroid hormone receptors such as ERα, AR, and GR, it is not known whether this mode of action will be valid for all members of the superfamily of nuclear receptors. For example, studies with the orphan nuclear receptors HNF-4α and ERRα and γ have identified several hundred targeted promoters. Second is the finding that binding to DNA via tethering with another transcription factor is a relatively common occurrence. In general, bioinformatics analysis of nuclear receptor binding regions have shown that approximately 70% contain a recognizable cognate HRE, inferring that about 30% of binding events involve tethering to another DNA-binding factor. Third is the observation that nuclear receptor binding regions are enriched for binding sites for specific classes of transcription factors such as AP-1, Oct, Forkhead, ETS, STAT, and CREB. Functional studies have shown that nuclear receptor action at certain target genes is often dependent on the presence of specific factors. This phenomenon was put in evidence by the original discovery that FOXA1 is required for ERα binding at promoters and enhancers harboring FOXA1 binding sites (2, 32). Likewise, ETS1 regulates the transcriptional activity of AR (47). These observations thus reinforce the concept that nuclear receptor binding sites are only one part of complex multicomponent enhancers. Fourth is the somewhat disconcerting finding that there is relatively little overlap between receptor binding and changes in gene expression. Although the expression of hundreds of genes may be modulated by the presence of a hormone or a drug, only a small number of these genes can be associated with a nuclear receptor binding site identified by ChIP-based techniques. The reverse is also true, i.e. the expression of only a small number of identified targets usually changes in response to a specific stimulus. The lack of more extensive overlaps may indicate that ChIP-based techniques are not as well developed and sensitive as the expression-profiling techniques but also may reflect the difficulty of accurately assigning a binding site to a targeted gene. It is likely that many hormonally regulated genes are induced as secondary effects and that the induction of a small number of direct targets has considerable influence on subsequent changes in gene expression. Thus, our complete understanding of nuclear receptors’ action will depend on the construction of regulatory networks. Whereas several different approaches are required to build complex regulatory networks and connect them with biological functions (Fig. 2), the application of ChIP-based approaches in combination with systematic functional analysis of the data will be crucial to achieve this goal. The study of nuclear receptors action has always been instrumental to improving our understanding of the molecular mechanisms of gene regulation and its direct links with physiology and disease. As the work reviewed here shows, nuclear receptors will continue to be at the forefront of new discoveries in this field.
Fig. 2.
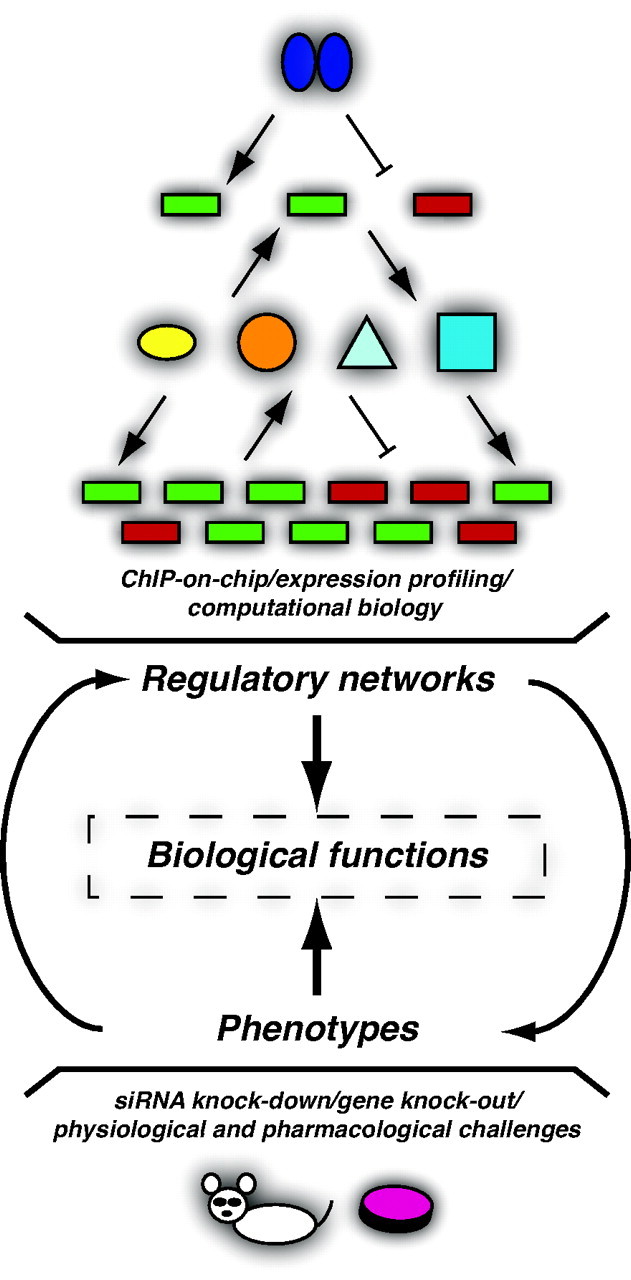
Regulatory Networks, Phenotypic Analyses, and Biological Functions of Nuclear Receptors
Nuclear receptors control the expression of a limited set of primary target genes. Some of these genes encode transcription factors, coregulator proteins, micro-RNAs, and posttranslational modifiers of proteins that will both influence the original primary response and lead to a broad secondary response. ChIP-based, expression profiling, and computational biology approaches are considered a useful set of tools with which to build regulatory networks (top). In parallel, phenotypic analyses using small interfering RNA knockdown, gene knock-out and overexpression, as well as physiological and pharmacological challenges in cell-based and animal models are used to study the overall functions of a particular nuclear receptor (bottom). When integrated, the two complementary approaches can lead to the identification of nuclear receptor-dependent biological functions for which we have a clear understanding of the molecular mechanisms underlying the observed phenotypes (middle). A nuclear receptor is represented by two blue ovals; genes are represented by green and red boxes for up- and down-regulated genes, respectively; gene products are represented by diverse shapes and colors. Arrows indicate activation; blunt arrows represent repression. siRNA, small interfering RNA.
Acknowledgments
We thank members of the Giguère laboratory for critical reading of the manuscript.
NURSA Molecule Pages:
Coregulators: SRC-1;
Ligands: 17β-Estradiol;
Nuclear Receptors: AR | ERα | ERRα | ERRγ | GR | HNF4α | RXRα | VDR.
Footnotes
This work was supported by the Canadian Institutes of Health Research.
Disclosure Statement: The authors have nothing to disclose.
First Published Online February 21, 2008
Abbreviations: AcH, Acetylated histone; AP-1, activator protein 1; AR, androgen receptor; ARE, androgen response element; 3C, chromosome conformation capture; C/EBP, CAAT enhancer binding protein; ChIP, chromatin immunoprecipitation; ChIP-DSL, ChIP coupled to a DNA selection and ligation; ChIP-PET, ChIP-paired end tag; CREB, cAMP response element-binding protein; ER, estrogen receptor; ERE, estrogen response element; ERR, estrogen related receptor; ETS1, v-ets erythroblastosis virus E26 oncogene homolog 1; FOXA1, forkhead box A1; GR, glucocorticoid receptor; HNF, hepatocyte nuclear factor; HRE, hormone response element; PolII, polymerase II; qPCR, quantitative PCR; STAT, signal transducer and activator of transcription; TSS, transcription start site; VDR, vitamin D receptor.
References
- 1.Bourdeau V, Deschenes J, Metivier R, Nagai Y, Nguyen D, Bretschneider N, Gannon F, White JH, Mader S 2004. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol Endocrinol 18:1411–1427 [DOI] [PubMed] [Google Scholar]
- 2.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M 2005. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122:33–43 [DOI] [PubMed] [Google Scholar]
- 3.Lefstin JA, Yamamoto KR 1998. Allosteric effects of DNA on transcriptional regulators. Nature 392:885–888 [DOI] [PubMed] [Google Scholar]
- 4.Solomon MJ, Larsen PL, Varshavsky A 1988. Mapping protein-DNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell 53:937–947 [DOI] [PubMed] [Google Scholar]
- 5.Lee TI, Johnstone SE, Young RA 2006. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc 1:729–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burakov D, Crofts LA, Chang CP, Freedman LP 2002. Reciprocal recruitment of DRIP/Mediator and p160 coactivator complexes in vivo by estrogen receptor. J Biol Chem 277:14359–14362 [DOI] [PubMed] [Google Scholar]
- 7.Shang Y, Myers M, Brown M 2002. Formation of the androgen receptor transcription complex. Mol Cell 9:601–610 [DOI] [PubMed] [Google Scholar]
- 8.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M 2000. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103:843–852 [DOI] [PubMed] [Google Scholar]
- 9.Metivier R, Penot G, Carmouche RP, Hubner MR, Reid G, Denger S, Manu D, Brand H, Kos M, Benes V, Gannon F 2004. Transcriptional complexes engaged by apo-estrogen receptor-α isoforms have divergent outcomes. EMBO J 23:3653–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F 2003. Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115:751–763 [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Carroll JS, Brown M 2005. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell 19:631–642 [DOI] [PubMed] [Google Scholar]
- 12.Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, Liu J, Zhao XD, Chew JL, Lee YL, Kuznetsov VA, Sung WK, Miller LD, Lim B, Liu ET, Yu Q, Ng HH, Ruan Y 2006. A global map of p53 transcription-factor binding sites in the human genome. Cell 124:207–219 [DOI] [PubMed] [Google Scholar]
- 13.Cam H, Balciunaite E, Blais A, Spektor A, Scarpulla RC, Young R, Kluger Y, Dynlacht BD 2004. A common set of gene regulatory networks links metabolism and growth inhibition. Mol Cell 16:399–411 [DOI] [PubMed] [Google Scholar]
- 14.Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD 2002. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 16:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA 2000. Genome-wide location and function of DNA binding proteins. Science 290:2306–2309 [DOI] [PubMed] [Google Scholar]
- 16.Weinmann AS, Yan PS, Oberley MJ, Huang TH, Farnham PJ 2002. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev 16:235–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bieda M, Xu X, Singer MA, Green R, Farnham PJ 2006. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res 16:595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B 2005. A high-resolution map of active promoters in the human genome. Nature 436:876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA 2007. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130:77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39:311–318 [DOI] [PubMed] [Google Scholar]
- 21.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Odom DT, Zizlsperger N, Gordon DB, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe PA, Gifford DK, Fraenkel E, Bell GI, Young RA 2004. Control of pancreas and liver gene expression by HNF transcription factors. Science 303:1378–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta RK, Gao N, Gorski RK, White P, Hardy OT, Rafiq K, Brestelli JE, Chen G, Stoeckert Jr CJ, Kaestner KH 2007. Expansion of adult β-cell mass in response to increased metabolic demand is dependent on HNF-4α. Genes Dev 21:756–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laganière J, Deblois G, Giguère V 2003. Nuclear receptor target gene discovery using high throughput chromatin immunoprecipitation. In: Russell DW, Mangelsdorf DJ, eds. Methods in enzymology. San Diego: Academic Press; 339–350 [DOI] [PubMed]
- 25.Laganière J, Deblois G, Giguère V 2005. Functional genomics identifies a mechanism for estrogen activation of the retinoic acid receptor α1 gene in breast cancer cells. Mol Endocrinol 19:1584–1592 [DOI] [PubMed] [Google Scholar]
- 26.Rishi AK, Shao ZM, Baumann RG, Li XS, Sheikh MS, Kimura S, Bashirelahi N, Fontana JA 1995. Estradiol regulation of the human retinoic acid receptor α gene in human breast carcinoma cells is mediated via an imperfect half-palindromic estrogen response element and Sp1 motifs. Cancer Res 55:4999–5006 [PubMed] [Google Scholar]
- 27.Sun G, Porter W, Safe S 1998. Estrogen-induced retinoic acid receptor α 1 gene expression: role of estrogen receptor-Sp1 complex. Mol Endocrinol 12:882–890 [DOI] [PubMed] [Google Scholar]
- 28.Creekmore AL, Ziegler YS, Boney JL, Nardulli AM 2007. Estrogen receptor α regulates expression of the breast cancer 1 associated ring domain 1 (BARD1) gene through intronic DNA sequence. Mol Cell Endocrinol 267:106–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin Z, Reierstad S, Huang CC, Bulun SE 2007. Novel estrogen receptor-α binding sites and estradiol target genes identified by chromatin immunoprecipitation cloning in breast cancer. Cancer Res 67:5017–5024 [DOI] [PubMed] [Google Scholar]
- 30.Levy N, Tatomer D, Herber CB, Zhao X, Tang H, Sargeant T, Ball LJ, Summers J, Speed TP, Leitman DC 2008. Differential regulation of native estrogen receptor regulatory elements by estradiol, tamoxifen, and raloxifene. Mol Endocrinol 22:287–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi M, Takahashi E, Miyagawa S, Watanabe H, Iguchi T 2006. Chromatin immunoprecipitation-mediated target identification proved aquaporin 5 is regulated directly by estrogen in the uterus. Genes Cells 11:1133–1143 [DOI] [PubMed] [Google Scholar]
- 32.Laganière J, Deblois G, Lefebvre C, Bataille AR, Robert F, Giguère V 2005. Location analysis of estrogen receptor α target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc Natl Acad Sci USA 102:11651–11656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415:530–536 [DOI] [PubMed] [Google Scholar]
- 34.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M 2006. Genome-wide analysis of estrogen receptor binding sites. Nat Genet 38:1289–1297 [DOI] [PubMed] [Google Scholar]
- 35.Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, Chiu KP, Lipovich L, Barnett DH, Stossi F, Yeo A, George J, Kuznetsov VA, Lee YK, Charn TH, Palanisamy N, Miller LD, Cheung E, Katzenellenbogen BS, Ruan Y, Bourque G, Wei CL, Liu ET 2007. Whole-genome cartography of estrogen receptor α binding sites. PLoS Genet 3:e87 [DOI] [PMC free article] [PubMed]
- 36.Kwon YS, Garcia-Bassets I, Hutt KR, Cheng CS, Jin M, Liu D, Benner C, Wang D, Ye Z, Bibikova M, Fan JB, Duan L, Glass CK, Rosenfeld MG, Fu XD 2007. Sensitive ChIP-DSL technology reveals an extensive estrogen receptor α-binding program on human gene promoters. Proc Natl Acad Sci USA 104:4852–4857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kininis M, Chen BS, Diehl AG, Isaacs GD, Zhang T, Siepel AC, Clark AG, Kraus WL 2007. Genomic analyses of transcription factor binding, histone acetylation, and gene expression reveal mechanistically distinct classes of estrogen-regulated promoters. Mol Cell Biol 27:5090–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng AS, Jin VX, Fan M, Smith LT, Liyanarachchi S, Yan PS, Leu YW, Chan MW, Plass C, Nephew KP, Davuluri RV, Huang TH 2006. Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor-α responsive promoters. Mol Cell 21:393–404 [DOI] [PubMed] [Google Scholar]
- 39.Gao H, Falt S, Sandelin A, Gustafsson JA, Dahlman-Wright K 2008. Genome-wide identification of estrogen receptor α binding sites in mouse liver. Mol Endocrinol 22:10–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin CY, Strom A, Vega VB, Kong SL, Yeo AL, Thomsen JS, Chan WC, Doray B, Bangarusamy DK, Ramasamy A, Vergara LA, Tang S, Chong A, Bajic VB, Miller LD, Gustafsson JA, Liu ET 2004. Discovery of estrogen receptor a target genes and response elements in breast tumor cells. Genome Biol 5:R66 [DOI] [PMC free article] [PubMed]
- 41.Blanchette M, Bataille AR, Chen X, Poitras C, Laganière J, Lefebvre C, Deblois G, Giguère V, Ferretti V, Bergeron D, Coulombe B, Robert F 2006. Genome-wide computational prediction of transcriptional regulatory modules reveals new insights into human gene expression. Genome Res 16:656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin VX, Leu YW, Liyanarachchi S, Sun H, Fan M, Nephew KP, Huang TH, Davuluri RV 2004. Identifying estrogen receptor α target genes using integrated computational genomics and chromatin immunoprecipitation microarray. Nucleic Acids Res 32:6627–6635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Ha C, Yamamoto KR 2004. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci USA 101:15603–15608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.So AY, Chaivorapol C, Bolton EC, Li H, Yamamoto KR 2007. Determinants of cell- and gene-specific transcriptional regulation by the glucocorticoid receptor. PLoS Genet 3:e94 [DOI] [PMC free article] [PubMed]
- 45.Wang Q, Li W, Liu XS, Carroll JS, Janne OA, Keeton EK, Chinnaiyan AM, Pienta KJ, Brown M 2007. A hierarchical network of transcription factors governs androgen receptor-dependent prostate cancer growth. Mol Cell 27:380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolton EC, So AY, Chaivorapol C, Ha CM, Li H, Yamamoto KR 2007. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev 21:2005–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Massie CE, Adryan B, Barbosa-Morais NL, Lynch AG, Tran MG, Neal DE, Mills IG 2007. New androgen receptor genomic targets show an interaction with the ETS1 transcription factor. EMBO Rep 8:871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguère V 2007. Genone-wide orchestration of cardiac functions by orphan nucler receptors ERRα and γ. Cell Metab 5:345–356 [DOI] [PubMed] [Google Scholar]
- 49.Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker JW, Giles W, Naviaux RK, Giguère V, Evans RM 2007. ERRγ directs and maintains the transition to oxidative metabolism in the post-natal heart. Cell Metab 6:16–24 [DOI] [PubMed] [Google Scholar]
- 50.Sonoda J, Laganière J, Mehl IR, Barish GD, Chong LW, Li X, Scheffler IE, Mock DC, Bataille AR, Robert F, Lee C-H, Giguère V, Evans RM 2007. Nuclear receptor ERRα and coactivator PGC-1β are effectors of IFN-γ induced host defense. Genes Dev 21:1909–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huss JM, Imahashi K-I, Dufour C, Weinheimer CJ, Courtois M, Kovacs A, Giguère V, Murphy E, Kelly DP 2007. The nuclear receptor ERRα is required for the bioenergetic and functional adaption to cardiac pressure overload. Cell Metab 6:25–37 [DOI] [PubMed] [Google Scholar]
- 52.Zella LA, Kim S, Shevde NK, Pike JW 2006. Enhancers located within two introns of the vitamin D receptor gene mediate transcriptional autoregulation by 1,25-dihydroxyvitamin D3 Mol Endocrinol 20:1231–1247 [DOI] [PubMed] [Google Scholar]
- 53.Kim S, Yamazaki M, Zella LA, Meyer MB, Fretz JA, Shevde NK, Pike JW 2007. Multiple enhancer regions located at significant distances upstream of the transcriptional start site mediate RANKL gene expression in response to 1,25-dihydroxyvitamin D3 J Steroid Biochem Mol Biol 103:430–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, Adelman K 2007. RNA polymerase is poised for activation across the genome. Nat Genet 39:1507–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, Young RA 2007. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet 39:1512–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blais A, Dynlacht BD 2005. Constructing transcriptional regulatory networks. Genes Dev 19:1499–1511 [DOI] [PubMed] [Google Scholar]
- 57.Walhout AJ 2006. Unraveling transcription regulatory networks by protein-DNA and protein-protein interaction mapping. Genome Res 16:1445–1454 [DOI] [PubMed] [Google Scholar]
- 58.Denoeud F, Kapranov P, Ucla C, Frankish A, Castelo R, Drenkow J, Lagarde J, Alioto T, Manzano C, Chrast J, Dike S, Wyss C, Henrichsen CN, Holroyd N, Dickson MC, Taylor R, Hance Z, Foissac S, Myers RM, Rogers J, Hubbard T, Harrow J, Guigo R, Gingeras TR, Antonarakis SE, Reymond A 2007. Prominent use of distal 5′ transcription start sites and discovery of a large number of additional exons in ENCODE regions. Genome Res 17:746–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trinklein ND, Karaoz U, Wu J, Halees A, Force Aldred S, Collins PJ, Zheng D, Zhang ZD, Gerstein MB, Snyder M, Myers RM, Weng Z 2007. Integrated analysis of experimental data sets reveals many novel promoters in 1% of the human genome. Genome Res 17:720–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sandelin A, Carninci P, Lenhard B, Ponjavic J, Hayashizaki Y, Hume DA 2007. Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat Rev Genet 8:424–436 [DOI] [PubMed] [Google Scholar]
- 61.Wang JC, Giaever GN 1988. Action at a distance along a DNA. Science 240:300–304 [DOI] [PubMed] [Google Scholar]
- 62.Rippe K, von Hippel PH, Langowski J 1995. Action at a distance: DNA-looping and initiation of transcription. Trends Biochem Sci 20:500–506 [DOI] [PubMed] [Google Scholar]
- 63.Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P 2002. Long-range chromatin regulatory interactions in vivo. Nat Genet 32:623–626 [DOI] [PubMed] [Google Scholar]
- 64.Ptashne M, Gann A 1997. Transcriptional activation by recruitment. Nature 386:569–577 [DOI] [PubMed] [Google Scholar]
- 65.Dean A 2006. On a chromosome far, far away: LCRs and gene expression. Trends Genet 22:38–45 [DOI] [PubMed] [Google Scholar]
- 66.Li Q, Barkess G, Qian H 2006. Chromatin looping and the probability of transcription. Trends Genet 22:197–202 [DOI] [PubMed] [Google Scholar]
- 67.de Laat W, Grosveld F 2003. Spatial organization of gene expression: the active chromatin hub. Chromosome Res 11:447–459 [DOI] [PubMed] [Google Scholar]
- 68.Lomvardas S, Barnea G, Pisapia DJ, Mendelsohn M, Kirkland J, Axel R 2006. Interchromosomal interactions and olfactory receptor choice. Cell 126:403–413 [DOI] [PubMed] [Google Scholar]
- 69.Dekker J, Rippe K, Dekker M, Kleckner N 2002. Capturing chromosome conformation. Science 295:1306–1311 [DOI] [PubMed] [Google Scholar]
- 70.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W 2006. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet 38:1348–1354 [DOI] [PubMed] [Google Scholar]
- 71.Dostie J, Dekker J 2007. Mapping networks of physical interactions between genomic elements using 5C technology. Nat Protoc 2:988–1002 [DOI] [PubMed] [Google Scholar]
- 72.Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD, Dekker J 2006. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 16:1299–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Simonis M, Kooren J, de Laat W 2007. An evaluation of 3C-based methods to capture DNA interactions. Nat Methods 4:895–901 [DOI] [PubMed] [Google Scholar]
- 74.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M 2008. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 132:958–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.John S, Sabo PJ, Johnson TA, Sung MH, Biddie SC, Lightman SL, Voss TC, Davis SR, Meltzer PS, Stamatoyannopoulos JA, Hager GL 2008. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol Cell 29:611–624 [DOI] [PubMed] [Google Scholar]