

Abstract
11β-Hydroxysteroid dehydrogenase type 1 (11β-HSD1) converts inert 11keto-glucocorticoids to active 11β-hydroxy forms, thereby amplifying intracellular glucocorticoid action. Up-regulation of 11β-HSD1 in adipose tissue and liver is of pathogenic importance in metabolic syndrome. However, the mechanisms controlling 11β-HSD1 transcription are poorly understood. Glucocorticoids themselves potently increase 11β-HSD1 expression in many cells, providing a potential feed-forward system to pathology. We have investigated the molecular mechanisms by which glucocorticoids regulate transcription of 11β-HSD1, exploiting an A549 cell model system in which endogenous 11β-HSD1 is expressed and is induced by dexamethasone. We show that glucocorticoid induction of 11β-HSD1 is indirect and requires new protein synthesis. A glucocorticoid-responsive region maps to between −196 and −88 with respect to the transcription start site. This region contains two binding sites for CCAAT/enhancer-binding protein (C/EBP) that together are essential for the glucocorticoid response and that bind predominantly C/EBPβ, with C/EBPδ present in a minority of the complexes. Both C/EBPβ and C/EBPδ are rapidly induced by glucocorticoids in A549 cells, but small interfering RNA-mediated knockdown shows that only C/EBPβ reduction attenuates the glucocorticoid induction of 11β-HSD1. Chromatin immunoprecipitation studies demonstrated increased binding of C/EBPβ to the 11β-HSD1 promoter in A549 cells after glucocorticoid treatment. A similar mechanism may apply in adipose tissue in vivo where increased C/EBPβ mRNA levels after glucocorticoid treatment were associated with increased 11β-HSD1 expression. C/EBPβ is a key mediator of metabolic and inflammatory signaling. Positive regulation of 11β-HSD1 by C/EBPβ may link amplification of glucocorticoid action with metabolic and inflammatory pathways and may represent an endogenous innate host-defense mechanism.
11β-HYDROXYSTEROID DEHYDROGENASE type 1 (11β-HSD1) catalyzes the reduction of intrinsically inert 11-keto corticosteroids (cortisone and 11-dehydrocorticosterone), regenerating active glucocorticoids (cortisol and corticosterone) and increasing intracellular glucocorticoid action in cells in which it is expressed (1). In contrast, 11β-HSD2, expressed in aldosterone-target tissues such as distal nephron and colon, is a high-affinity 11β-dehydrogenase that catalyzes rapid inactivation of glucocorticoids and confers mineralocorticoid specificity upon the mineralocorticoid receptor. Both 11β-HSD isozymes, the products of distinct genes, are crucial controls of tissue glucocorticoid action.
Chronic glucocorticoid excess causes Cushing’s syndrome (obesity, hypertension, insulin resistance/type 2 diabetes, dyslipidemia, and heart disease) (2), yet plasma cortisol levels are normal in idiopathic obesity. However, in obesity/metabolic syndrome, expression and activity of 11β-HSD1 are strikingly up-regulated in adipose tissue (3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13), increasing tissue glucocorticoid levels and possibly explaining the paradoxical similarity between common metabolic syndrome/obesity spectrum disorders and rare Cushing’s syndrome (14). Understanding the basis of this tissue-specific regulation is of substantial interest.
Glucocorticoids themselves up-regulate 11β-HSD1 mRNA levels in a variety of primary cells in vitro (15, 16, 17, 18, 19), including in human adipocytes (8) and preadipocytes (20). Glucocorticoids also increase 11β-HSD1 mRNA levels in vivo (21, 22, 23, 24, 25), although the in vivo regulation is tissue specific and complex (21, 23, 24, 25). Nevertheless, 11β-HSD1 expression in mouse adipose tissue is markedly increased by corticosterone (25). The underlying mechanisms remain unknown. Most regulators of 11β-HSD1 expression are likely to act indirectly, and the only direct regulators of 11β-HSD1 gene transcription described to date comprise members of the C/EBP family of transcription factors (26, 27, 28). The 11β-HSD1 gene is transcribed from two promoters, P1 and P2 (27). Transcription in liver, brain, and adipose tissue is predominantly from P2 and is dependent upon the transcription factor CCAAT/enhancer-binding protein (C/EBPα) (26, 27). Transcription from P1 is C/EBPα independent (27). Other members of the C/EBP family also regulate hepatic 11β-HSD1 transcription (26), although their role in 11β-HSD1 regulation in other tissues remains to be determined.
Few cell lines express endogenous 11β-HSD1. Most, including HepG2 hepatoma cells, previously used to characterize the 11β-HSD1 promoter (26, 27), contain only trace amounts of endogenous 11β-HSD1 mRNA and show minimal 11β-HSD1 promoter activity in the absence of cotransfected activators (26, 27). However, the P2 promoter of 11β-HSD1 shows robust activity in transfected A549 lung epithelial cells (27), which are also glucocorticoid responsive (29, 30). Here we have investigated the critical mechanisms by which glucocorticoids themselves regulate endogenous 11β-HSD1 transcription, exploiting the A549 cell model.
RESULTS
A549 Cells Express Endogenous 11β-HSD1, Which Acts Exclusively as a Reductase
The robust activity of the 11β-HSD1 P2 promoter in A549 cells (27) suggested they may express the endogenous gene. Assay of intact A549 cells demonstrated endogenous 11β-HSD enzyme, acting as an 11keto-reductase (conversion of cortisone to cortisol; 0.23 pmol/h·mg protein), with no 11β-dehydrogenase activity detectable (Fig. 1), consistent with activity due to 11β-HSD1 but not 11β-HSD2.
Fig. 1.

Endogenous 11β-HSD1 Is Expressed in A549 Cells and Shows Exclusively Reductase Activity
Representative image of thin-layer chromatography plate showing 11keto-reductase activity (conversion of 200 nm [3H]cortisone to cortisol) but no 11β-dehydrogenase activity (conversion of 200 nm [3H]cortisol to cortisone) in intact A549 cells. Arrows indicate cortisone and cortisol. M indicates medium with [3H]steroid alone (no cells).
Glucocorticoid Induction of 11β-HSD1 mRNA in A549 Cells Is Indirect and Requires New Protein Synthesis
Addition of dexamethasone to A549 cells increased 11β-HSD1 mRNA levels within 4 h, with maximal induction at 24 h (Fig. 2A). The induction was dose dependent (Fig. 2B) and blocked by addition of RU38486 (RU486) (Fig. 2C), a glucocorticoid receptor (GR) antagonist, demonstrating its dependence upon activation of GR. The time course of 11β-HSD1 mRNA induction, with little or no effect of dexamethasone within 2 h, suggested that 11β-HSD1 is not a primary glucocorticoid response gene. Consistent with this, the induction of 11β-HSD1 mRNA was blocked by the protein synthesis inhibitor cycloheximide (Fig. 2D) and thus requires new protein synthesis.
Fig. 2.

Glucocorticoid Regulation of 11β-HSD1 in A549 Cells Is Indirect and Dependent upon New Protein Synthesis
Real-time PCR measurement of 11β-HSD1 mRNA levels, expressed as fold induction, relative to levels in vehicle-treated cells. A, 11β-HSD1 mRNA levels were increased within 4 h of addition of 10−6 m dexamethasone to A549 cells and were maximal at 24 h. B, Dexamethasone (Dex, 24 h) increased endogenous 11β-HSD1 mRNA levels in a dose-dependent manner in A549 cells. C, The GR antagonist RU38486 (RU486, 10−5 m), added 30 min before 10−6 m dexamethasone, blocked the induction of 11β-HSD1 mRNA. D, Dexamethasone induction of 11β-HSD1 mRNA in A549 cells was blocked by cycloheximide (CHX, 10−5 m) added 30 min before 10−6 m dexamethasone (4 h). Data are mean ± sem of at least three independent mRNA samples. ***, P < 0.0001.
Glucocorticoid Regulation of the 11β-HSD1 P2 Promoter Requires C/EBP-Binding Sites FP3 and FP4
The P2 promoter, but not the P1 promoter, of 11β-HSD1 is active in transiently transfected A549 cells (27) (and data not shown). Dexamethasone increased activity of a P2 promoter-reporter construct in transfected A549 cells, with a similar dose response to the endogenous gene (Fig. 3A). A deletion series of the 11β-HSD1 P2 promoter was used to localize the glucocorticoid-responsive region. Although 5′ deletion of the promoter to −196 had no effect on glucocorticoid induction, deletion from −196 to −124 abolished the response to glucocorticoid (Fig. 3B). Furthermore, internal deletions that removed this region (deletion of −1299 to −125 or deletion of −311 to −125) also abolished the dexamethasone response (Fig. 3B), demonstrating that the region between −196 and −124 is essential for glucocorticoid induction of the 11β-HSD1 P2 promoter and that sequences upstream of −311 do not contribute to the induction.
Fig. 3.

Dexamethasone Induction of the P2 Promoter of 11β-HSD1 Requires C/EBP-Binding Sites FP3 and FP4
A, In transiently transfected A549 cells, dexamethasone dose-dependently increased activity of p11β1(−1799/+49)-LUC, a luciferase reporter construct encoding −1799 to +49 of the 11β-HSD1 P2 promoter. Data are expressed as fold induction relative to vehicle-treated cells and are mean ± sem of at least two experiments, each carried out in triplicate. B, Effect of 5′ deletion or internal deletion of the 11β-HSD1 P2 promoter upon dexamethasone response. Data are expressed relative to vehicle-treated p11β1(−1799/+ 49) (arbitrarily set to 1) and are mean ± sem of at least three experiments, each carried out in triplicate. C, Effect of mutation of FP3 and/or FP4 upon the glucocorticoid response of the proximal 11β-HSD1 P2 promoter, encoded by p11β1(−196/+49)-LUC. Data are expressed relative to vehicle-treated p11β1(−196/+49)-LUC (arbitrarily set to 1) and are mean ± sem of at least three experiments, each carried out in triplicate. D, The fragment encoding −196 to −88 of the 11β-HSD1 P2 promoter in one [P(−196/−88)1], two [P(−196/−88)2], or three [P(−196/−88)3] copies confers a glucocorticoid response upon the heterologous promoter present in the vector pGL2-promoter. Data are expressed relative to vehicle-treated vector (arbitrarily set to 1) and are mean ± sem of at least three experiments, each carried out in triplicate. E, No effect of mutation of the putative GRE (indicated between FP3 and FP4 by an X) upon the glucocorticoid response of the proximal 11β-HSD1 P2 promoter, encoded by p11β1(−196/+49)-LUC, either alone or in combination with either FP3 or FP4 mutation. Data are expressed relative to vehicle-treated p11β1(−196/+49)-LUC (arbitrarily set to 1) and are mean ± sem of at least five experiments, each carried out in triplicate. In each transfection, 1 × 105 cells were seeded per well in six-well plates and transfected with 250 ng test plasmid and 250 ng pRSV-lacZ (internal control). Promoter activity is expressed as luciferase/β-galactosidase (internal control) activity. Black bars represent activity in vehicle-treated cells; white bars represent activity in dexamethasone-treated cells (10−6 m unless stated otherwise). *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
The region between −196 and +49 remained fully responsive to dexamethasone. We have shown previously that this region contains four binding sites, footprints (FP) 1–4, for members of the C/EBP family of transcription factors (26). These sites synergize, so that their combined mutation markedly attenuated or abolished C/EBPα regulation of the promoter in HepG2 hepatoma cells, but mutation individually reduced only the C/EBPα effect (26). In transiently transfected A549 cells, mutation of either FP3 or FP4 alone modestly reduced or had little effect upon dexamethasone induction of the promoter (Fig. 3C). However, mutation of both sites prevented the glucocorticoid induction (Fig. 3C), suggesting that these C/EBP- binding sites mediate the glucocorticoid response. In support of this, transfer of one, two, or three copies of the fragment encoding −196 to −88, which includes FP3 and FP4, to a heterologous promoter conferred an incremental increase in both basal promoter activity and glucocorticoid response, dependent upon copy number (Fig. 3D), confirming the presence of a glucocorticoid-responsive region between −196 and −88. Between FP3 and FP4 is a sequence that partly matches a GR consensus binding site or glucocorticoid response element (GRE) (identical at nine of 12 conserved nucleotides). However, mutation of this putative GRE did not affect the glucocorticoid response (Fig. 3E). Furthermore, the dexamethasone effect on promoters with combined mutation of the putative GRE and either FP3 or FP4 was indistinguishable from the effect on promoters with mutant FP3 or FP4 sites alone (Fig. 3E).
C/EBPβ Binds to the Glucocorticoid-Responsive Region of the 11β-HSD1 P2 Promoter
The experiments above strongly implicated members of the C/EBP family in the glucocorticoid response of the 11β-HSD1 P2 promoter. EMSA with A549 nuclear extracts showed that dexamethasone increased complex formation on an oligonucleotide encoding FP4, without changing the pattern or relative intensities of the bands (Fig. 4). Complex formation was competed by excess unlabeled oligonucleotides encoding FP4, FP3, or an oligonucleotide encoding a consensus C/EBP binding site, but not by an oligonucleotide encoding the TdT initiator sequence (Fig. 4A), demonstrating the presence of proteins with C/EBP-like binding specificity in the complexes. Supershift with C/EBP-specific antisera showed C/EBPβ present in a substantial proportion of the complexes, with C/EBPδ present in a minority (Fig. 4B). No binding by C/EBPα was observed in A549 extracts (Fig. 4B), although C/EBPα was present in protein-DNA complexes formed by liver nuclear extract (data not shown).
Fig. 4.

C/EBPβ in A549 Cells Binds to the Glucocorticoid-Responsive Region of the 11β-HSD1 P2 Promoter
A, Representative competition EMSA showing complexes formed by A549 nuclear extracts on a double-stranded oligonucleotide encoding FP4 (0.1 pmol). Lane 1 contains no added protein. Lanes 2 and 11 contain 4 μg nuclear extract from vehicle-treated (−Dex) and dexamethasone-treated (+Dex; 10−6 m, 24 h) cells, respectively. Addition of competitor oligonucleotides (5- or 50-fold molar excess) is indicated above the lanes. Oligonucleotides encoded FP4, FP3, the TdT initiator (Inr; to which C/EBP does not bind) (26 ), and a C/EBP consensus binding site (con) described previously (26 ). B, Representative EMSA showing presence of C/EBPβ and C/EBPδ, but not C/EBPα, in complexes formed by 4 μg nuclear extract from vehicle (−Dex) or dexamethasone (+Dex; 10−6 m) treated A549 cells on a oligonucleotide encoding FP4. The addition of C/EBPα (α), C/EBPβ (β), C/EBPδ (δ), or no (−) antibody is indicated above the gel. The C/EBPβ/δ supershifted complex is indicated by a black arrowhead. The first lane does not contain any added protein extract. Nonspecific complexes are indicated by asterisks.
Dexamethasone Induces C/EBPβ and C/EBPδ mRNAs as Primary Response Genes in A549 Cells
To determine whether C/EBPβ and other members of the C/EBP family are regulated by glucocorticoids in A549 cells, real-time PCR was carried out to measure C/EBPα, -β, -δ, and -ζ mRNA levels. Levels of C/EBPβ and -δ mRNA were increased after dexamethasone addition (Fig. 5A), and their induction was blocked by RU486 (Fig. 5B) but unaffected by cycloheximide (Fig. 5C). In contrast, levels of C/EBPα mRNA were decreased and C/EBPζ mRNA levels were unchanged after dexamethasone (Fig. 5A). Dexamethasone did not affect levels of GR mRNA (Fig. 5A). Western blotting of whole-cell lysates of untreated A549 cells showed C/EBPβ-immunoreactive proteins of 38, 35, and 20 kDa, corresponding to the liver-enriched activator protein (LAP) (38 and 35 kDa) and liver-enriched inhibitor protein (LIP) (20 kDa) forms of C/EBPβ (31) (Fig. 5D). After dexamethasone, intensities of all bands were increased. Cycloheximide markedly reduced the intensities of all the C/EBPβ-immunoreactive proteins, suggesting rapid turnover of the protein (Fig. 5D), consistent with reported data in myotubes (32). Similarly, levels of C/EBPδ were increased after dexamethasone but were unaffected by cycloheximide (Fig. 5D).
Fig. 5.

Dexamethasone Increases C/EBPβ and -δ Expression in A549 Cells
A–C, Real-time PCR measurement of levels of mRNAs encoding members of the C/EBP family in vehicle-treated A549 cells (black bars) or after 10−6 m dexamethasone treatment (white bars). Levels of mRNA are expressed as fold induction, relative to levels in vehicle-treated cells. Data are mean ± sem of three to 12 independent mRNA samples. *, P < 0.05; **, P < 0.001; ***, P < 0.0001. A, Levels of C/EBPβ and C/EBPδ mRNAs were induced after 24 h dexamethasone treatment. B, The induction of C/EBPβ and C/EBPδ was abolished by prior incubation with 10−5 m RU486 (RU), added 30 min before dexamethasone. Hatched bars, RU486; spotted bars, RU486 and dexamethasone. C, Cycloheximide (10−5 m), added 30 min before dexamethasone (4 h), had no effect on the induction of C/EBPβ and C/EBPδ. Hatched bars, cycloheximide; spotted bars, cycloheximide, and dexamethasone. D, Representative Western blot, probed with C/EBPβ, C/EBPδ, or tubulin antibody, of 20 μg protein extract from triplicate samples of A549 cells treated for 24 h with vehicle (−D) or 10−6 m dexamethasone (+D), with or without cycloheximide (CHX), as indicated. Sizes of protein markers are shown on the right. Immunoreactive proteins corresponding to the LAP (38 and 35 kDa) and LIP (20 kDa) isoforms of C/EBPβ are indicated.
C/EBPβ, But Not C/EBPδ, Is Required for the Dexamethasone Induction of 11β-HSD1 mRNA
Transfection of A549 cells with small interfering RNA (siRNA) specific for C/EBPβ or -δ mRNA decreased levels of the respective protein (Fig. 6). Importantly, C/EBPβ siRNA abolished the dexamethasone induction of 11 β-HSD1 mRNA (Fig. 6C), consistent with a critical role for C/EBPβ in the glucocorticoid response. In contrast, neither C/EBPδ nor scrambled siRNA had any effect on the levels of 11β-HSD1 mRNA after dexamethasone, suggesting that C/EBPδ is redundant for the response.
Fig. 6.

Dexamethasone Induction of 11β-HSD1 Is Abolished by siRNA against C/EBPβ but Is Unaffected by C/EBPδ siRNA
A, Representative Western blot (20 μg protein/lane) probed with C/EBPβ antibody showing levels of C/EBPβ protein (38-kDa LAP, 20-kDa LIP, and proteolytic products of LAP) 24 h after mock transfection (Mock) or transfection with 80 pmol C/EBPβ siRNA (C/EBPβ) or C/EBPδ siRNA (C/EBPδ) and concurrent vehicle (−) or dexamethasone (+D, 10−6 m) treatment. The blot was stripped and reprobed with tubulin antibody, as loading control. B, Western blot (20 μg protein/lane) probed with C/EBPδ antibody showing levels of C/EBPδ protein 24 h after mock transfection (Mock) or transfection with C/EBPβ siRNA (C/EBPβ) or with C/EBPδ siRNA (C/EBPδ) and concurrent vehicle (−) or dexamethasone (+D, 10−6 m) treatment. The blot was stripped and reprobed with tubulin antibody as loading control. C, Real-time PCR measurement of levels of 11β-HSD1 mRNA after mock transfection or transfection with C/EBPβ, C/EBPδ, or scrambled siRNAs. Levels of mRNA in mock-transfected vehicle-treated cells are arbitrarily set to 1. Note logarithmic scale. Data are mean ± sem of at least three independent mRNA samples. ***, P < 0.0001.
C/EBPβ Occupancy of the 11β-HSD1 Promoter Is Increased after Dexamethasone
Chromatin immunoprecipitation (ChIP) assays showed that after dexamethasone treatment of A549 cells, there was a marked increase in C/EBPβ binding to FP3 and FP4 in the endogenous 11β-HSD1 gene (Fig. 7). There was little or no association of C/EBPβ with the promoter in vehicle-treated cells, with levels of PCR product similar to those in control ChIP reactions with IgG or RNA polymerase II (not predicted to bind to the FP3/FP4 region of the 11β-HSD1 P2 promoter) (Fig. 7).
Fig. 7.

C/EBPβ Occupancy of the 11β-HSD1 Promoter Is Increased after Dexamethasone
Representative ChIP assay showing association of C/EBPβ with the endogenous 11β-HSD1 P2 promoter in A549 cells after 24 h treatment with 10−6 m dexamethasone. PCR (35 cycles) were carried out with primers spanning FP3 and FP4 on DNA after immunoprecipitation with control IgG (lanes 7 and 8), C/EBPβ antiserum (lanes 9 and 10), RNA polymerase II antiserum (RNAP; lanes 11 and 12) or after control precipitations with no antibody (No Ab, lanes 5 and 6). Lane 1 contains marker, and lane 2 a PCR carried out with water as negative control. Lanes 3 and 4 show input DNA (1%) from vehicle-treated or dexamethasone-treated cells, respectively. The assay shown is representative of two independent assays.
Overexpression of C/EBPβ in A549 Cells Increases 11β-HSD1 P2 Promoter Activity and Blocks Further Induction by Dexamethasone
To investigate whether overexpression of C/EBPβ mimics or alters the response of the 11β-HSD1 promoter to dexamethasone, A549 cells were cotransfected with 11β-HSD1 promoter-reporter plasmid and C/EBPβ, C/EBPδ, or both together. C/EBPβ increased basal expression of the promoter (Fig. 8), consistent with previous data in hepatoma cells showing C/EBPβ activates the 11β-HSD1 promoter, albeit weakly when compared with C/EBPα (26). However, it markedly attenuated the dexamethasone response (Fig. 8). In contrast, whereas there was a trend for C/EBPδ to decrease basal promoter activity, this did not achieve significance, nor did C/EBPδ affect the magnitude of the dexamethasone induction (Fig. 8). However, when C/EBPβ and C/EBPδ were cotransfected, basal activity was indistinguishable from control levels, and the dexamethasone induction was abrogated.
Fig. 8.

Overexpression of C/EBPβ Blocks the Effect of Glucocorticoid on the 11β-HSD1 P2 Promoter
Ectopic expression of C/EBPβ increased basal promoter activity and attenuated the dexamethasone response of the 11β-HSD1 P2 promoter in transiently transfected A549 cells. Ectopic expression of C/EBPδ had no effect. In each transfection, 1 × 105 cells were seeded per well in six-well plates and transfected with 250 ng 11β1(−196/+49), 250 ng pRSV-lacZ (internal control), and 50 ng C/EBP expression plasmid or empty vector. Promoter activity is expressed as luciferase/β-galactosidase (internal control) activity. Data are expressed relative to vehicle-treated 11β1(−196/+49) cotransfected with empty vector (vector; arbitrarily set to 1) and are mean ± sem of at least three experiments, each carried out in triplicate. *, Significantly different from vehicle-treated control, P < 0.05; ***, P < 0.0001. §, Significantly different from vector, P < 0.05.
Glucocorticoid Treatment in Vivo Increases 11β-HSD1 and C/EBPβ Expression in Adipose Tissue
To explore whether a similar mechanism could underlie glucocorticoid regulation of 11β-HSD1 in adipose tissue, we measured 11β-HSD1, C/EBPβ, and C/EBPδ mRNA levels in adipose tissue and liver of Pomc−/− mice (congenitally deficient in glucocorticoids) that had been treated for 10 d with corticosterone added to their drinking water. We have previously shown that this regime increased 11β-HSD1 expression in adipose tissue of Pomc−/− as well as wild-type mice (although the magnitude of the increase was lower in the latter because of higher basal levels), whereas liver 11β-HSD1 expression did not differ between genotypes or with corticosterone treatment (25). Corticosterone increased both 11β-HSD1 and C/EBPβ mRNA levels in adipose tissue of Pomc−/− mice, whereas C/EBPδ mRNA levels were unaffected. Consistent with previous data (25), glucocorticoid treatment had no effect on hepatic 11β-HSD1 expression, although C/EBPβ mRNA levels were decreased (Fig. 9B). Thus, it is possible that C/EBPβ may play a role in the tissue-specific glucocorticoid regulation of 11β-HSD1 in vivo.
Fig. 9.
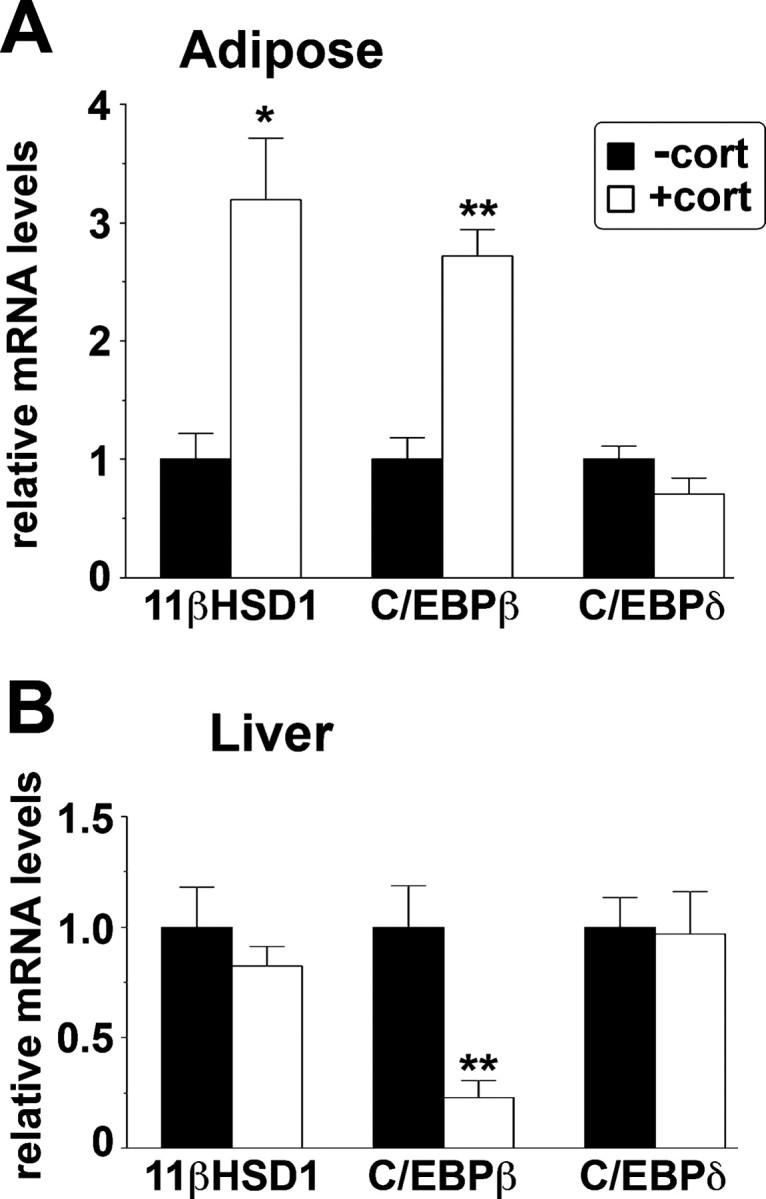
Increased C/EBPβ mRNA Levels Are Associated with Increased 11β-HSD1 mRNA Levels in Adipose Tissue of Glucocorticoid-Treated Pomc−/− Mice
Real-time PCR measurement of levels of mRNAs encoding 11β-HSD1, C/EBPβ, and C/EBPδ in retroperitoneal adipose tissue (A) or liver (B) of Pomc−/− mice (black bars) or after 10 d treatment with corticosterone in their drinking water (25 μg/ml) (white bars) as described (25 66 ). Levels of mRNA are expressed relative to levels in untreated mice, arbitrarily set to 1. Data are mean ± sem; n = 4–6 per group. *, P < 0.05; **, P < 0.001.
DISCUSSION
There are many reports of glucocorticoid induction of 11β-HSD1 mRNA levels (8, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25). Here we have shown that glucocorticoids increase endogenous expression of 11β-HSD1 in A549 cells in a GR-dependent manner, suggesting the validity of these cells as a model of this control. The time course and inhibition by cycloheximide are both consistent with indirect regulation, paralleling previous findings in primary human fibroblasts (15). Glucocorticoid regulation of the 11β-HSD1 P2 promoter required an approximately 110-bp glucocorticoid-responsive region located close to the transcription start that contains two binding sites for C/EBP transcription factors FP3 and FP4, together essential for the glucocorticoid induction of promoter activity. Mutation of the only sequence resembling a GR binding site (located between FP3 and FP4, within the glucocorticoid-responsive region) had no effect upon the glucocorticoid response. Thus, it is unlikely that DNA binding by GR to the 11β-HSD1 promoter plays any part in the regulation of 11β-HSD1 transcription by glucocorticoids.
Many studies have linked glucocorticoid action to the C/EBP family of transcription factors. In some cases, C/EBPs interact with GR upon target gene promoters, but in others, C/EBPs, primarily C/EBPβ, mediate the effect of glucocorticoids upon genes that lack GR binding sites. For example, PEPCK and TAT, classical glucocorticoid-target genes in liver, both depend upon C/EBP binding sites in addition to GR binding sites for their glucocorticoid response (33, 34, 35). Genes induced during the acute-phase response, such as that encoding α1-acid glycoprotein, typically are dependent upon C/EBP-binding sites adjacent to GR binding sites for full activation (36, 37). As well as synergizing with GR, levels of C/EBP mRNAs are regulated by glucocorticoids. C/EBPβ and C/EBPδ are induced by glucocorticoids in a variety of cell types (38, 39, 40, 41, 42, 43, 44) and mediate glucocorticoid effects. The increase in arginase expression in hepatocytes after glucocorticoid is indirect and mediated by C/EBPβ (42), and in H441 lung epithelial cells, glucocorticoid induction of Clara cell secretory protein and cytochrome P450 2B1 depends upon C/EBPβ (45 , 46). Indeed, it has been suggested that glucocorticoid enhancement of local innate host defense mechanisms are mediated via C/EBPβ, in contrast to the antiinflammatory mechanisms of glucocorticoids, which are predominantly mediated through interference with nuclear factor-κB and activator protein-1 function (47).
Here, we have shown that C/EBPβ is indispensable for the glucocorticoid regulation of 11β-HSD1. Both C/EBPβ and -δ were increased by glucocorticoid treatment of A549 cells, although C/EBPδ was not required for the glucocorticoid induction of 11β-HSD1. This is consistent with the predominant binding of C/EBPβ to the promoter in vitro, with C/EBPδ present in only a minority of complexes. Interestingly, despite the clear presence of C/EBPβ in nuclear extracts of untreated A549 cells, little or none was associated with the 11β-HSD1 promoter before dexamethasone treatment, suggesting that glucocorticoids may exert effects on C/EBPβ (e.g. regulation of posttranslational modification) additional to the increase in expression. Previous work has established the central role of C/EBPs in the regulation of 11β-HSD1 transcription (26, 27, 28). Moreover, C/EBPβ mediates the cAMP induction of 11β-HSD1 in preadipocytes, acting through the same region of the promoter (−166 to −113) as identified here (28). Indeed, other regulators of 11β-HSD1 (peroxisome proliferator-activated receptor-α and -γ and liver X receptor-α) are likely to act indirectly (48, 49, 50), possibly through members of the C/EBP family.
Exogenous C/EBPβ mimicked the effect of dexamethasone and blocked further induction by glucocorticoid. The increased basal activity of the 11β-HSD1 promoter with exogenous C/EBPβ is consistent with previous findings (26). The magnitude of the effect was less than the endogenous gene response to glucocorticoid, suggesting that exogenous C/EBP may titrate a modulator/effector of C/EBPβ action. The nature of this factor is unknown but could be a coactivator, heterodimerization partner, or posttranslational modification. C/EBPα heterodimerizes with C/EBPβ, and we have previously shown C/EBPα to be a strong activator of 11β-HSD1, including in A549 cells (27). Indeed, overexpression of C/EBPα both markedly increased basal expression of the 11β-HSD1 promoter and abolished the response to dexamethasone (unpublished observations). However, A549 cells are reported to lack endogenous C/EBPα (51), and although we detected the presence of C/EBPα mRNA in A549 cells, levels were very low (unpublished observations) and were decreased by dexamethasone, whereas C/EBPβ mRNA levels were high in these cells. Similarly, whereas C/EBPβ protein was readily detected in A549 nuclear extracts, C/EBPα was not detected.
C/EBPβ is subject to extensive posttranscriptional regulation (31). Alternate translation starts (52, 53) as well as regulated proteolysis (32, 54, 55) generate the higher molecular weight LAP and 20-kDa LIP isoforms, and the translated protein is phosphorylated on several sites, reportedly regulating its DNA-binding activity, nuclear localization, and transcriptional activity (reviewed in Ref. 31). In our experiments, all isoforms of C/EBPβ (LAP and LIP) were similarly regulated by glucocorticoids. However, because the LIP/LAP ratio depended upon the protein extraction method (unpublished observations), it is likely that the detected LIP arose at least in part from in vitro proteolysis, consistent with previous reports (55), although we cannot exclude a role for the LIP/LAP ratio in the regulation of the 11β-HSD1 promoter.
Thus, in cells in which C/EBPα is highly expressed, such as hepatocytes, it may be the dominant (positive) regulator of the P2 promoter of 11β-HSD1. This is consistent with our data in liver of Pomc−/− mice, in which glucocorticoid status had no effect on the level of 11β-HSD1 expression. In other cell types, including adipose tissue, C/EBPβ may be a dynamic regulator of 11β-HSD1 transcription in response to signaling by glucocorticoids (as shown here) or by inflammatory or innate host defense mediators that signal through C/EBPβ (56). Indeed, 11β-HSD1 may mediate some of the adverse metabolic effects of C/EBPβ. Like 11β-HSD1-deficient mice (57, 58), C/EBPβ−/− mice are insulin sensitized (59) and resist the adverse metabolic effects of a high-fat diet (60, 61). C/EBPβ deficiency also reduces obesity, fatty liver disease, and diabetes in genetically obese diabetic Leprdb/db mice (62). Moreover, it has been recently reported that C/EBPβ−/− mice show a marked reduction in pulmonary fibrosis in a bleomycin-induced mouse model of lung disease (63). It will be intriguing to determine the impact of 11β-HSD1 deficiency in this model.
The region of the 11β-HSD1 promoter that binds C/EBPβ and mediates the glucocorticoid (and cAMP) (28) response is highly conserved between the rodent and human genes (50/51 nucleotide identity), suggesting conservation of this regulation in humans. The central role of the C/EBPs in the regulation of 11β-HSD1 transcription (26, 27), and that of C/EBPβ in the hormone responsiveness of the gene (28) (this work), suggest that they may be critical in coordinating signaling from the diverse nutrient, hormonal, and inflammatory regulators of 11β-HSD1.
MATERIALS AND METHODS
Cell Culture, RNA Extraction, and Analysis
A549 cells were maintained as previously described (27). Dexamethasone (10−6 m, unless stated otherwise) treatment was for 24 h (unless stated otherwise). Cycloheximide (10−5 m) or RU38486 (RU486; 10−5 m) were added 30 min before dexamethasone or vehicle and cells harvested 4 or 24 h later. RNA was extracted after homogenization in Trizol (Invitrogen, Paisley, UK) and was resuspended in ribonuclease-free water. RNA levels were quantified by real-time PCR. Reverse transcription was carried out as previously described (64) using 2 μg RNA. PCR was carried out on a LightCycler 480, according to the manufacturer’s instructions. Mastermix and primer-probe sets for 18S, 11β-HSD1, TBP, GAPDH, C/EBPα, C/EBPβ, C/EBPδ, and C/EBPζ were purchased from Applied Biosystems (Warrington, UK). Routinely, at least two internal standards were used from among 18S, TBP, and GAPDH. Similar data were obtained with all internal standards, and none varied with dexamethasone treatment (data not shown).
11β-HSD1 Assay
11β-HSD1 was assayed in intact cells by measuring conversion of 200 nm cortisone, containing trace amounts of [3H]cortisone, to cortisol, as previously described (64).
Transfections
A549 cells were maintained and transfected as previously described (27). Briefly, cells were seeded at 1.5 × 105 cells per well of a six-well plate and transfected the after day using Lipofectamine 2000 (Invitrogen) with 250 ng test plasmid, 250 ng pRSV-LacZ (as internal control), and where appropriate, 50 ng pMSV-C/EBPβ, pMSV-C/EBPδ (gifts from S. McKnight and W.-C. Yeh), or pMSV vector. Dexamethasone (10−6 m, unless stated otherwise) or vehicle was added 1–4 h after transfection. Transfections were carried out in triplicate, and each experiment was repeated up to five times.
Reporter plasmids were as follows. A 5′ deletion series of the rat 11β-HSD1 P2 promoter has been described previously (26) and is designated 11β1(5′-endpoint/3′-endpoint). In some experiments, an identical deletion series subcloned into an alternative reporter vector, pGL3-Basic (Promega, Southampton, UK), was used. Other plasmids comprise an internal deletion series of 11β1(−1799/+49), designated Δ(5′-endpoint of deletion/3′-endpoint of deletion) and a series of fragments of the rat 11β-HSD1 promoter subcloned into pGL2-promoter, designated P(−196/−88)n, where n indicates the number of copies of the −196/−88 fragment inserted. Derivatives of 11β1(−196/+49) with mutations in FP3, FP4, or both FP3 and FP4 have been described previously (26). Mutation of the putative GRE between FP3 and FP4, changing the sequence 5′-TGGAGTAAACATTGTCCATTA-3′ (nucleotides matching the GRE consensus (65) are underlined) to 5′-TGGACCGTTCATCCAGGATTA-3′ (mutated nucleotides underlined) was carried out using a QuikChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer’s instructions; all constructs were verified by sequencing.
For siRNA transfections, 7.5 × 104 cells were seeded per well of a 12-well plate and transfected the following day with 80 pmol siRNA using Lipofectamine 2000 (Life Technologies, Paisley, UK) according to the manufacturer’s instructions. Dexamethasone (10−6 m) or vehicle was added 1–4 h after transfection and cells harvested 24 h later for RNA extraction or Western analysis. Transfections were carried out in triplicate and repeated at least twice. siRNAs were purchased from Applied Biosystems (Warrington, UK) and were scrambled siRNA (AM4611 negative control 1), C/EBPβ (ID 114495), and C/EBPδ (ID 45106).
Nuclear Extracts and EMSA
Nuclear extracts were prepared from liver or A549 cells as previously described (26). A549 cells were treated with 10−6 m dexamethasone or vehicle for 24 h before harvesting for extracts. EMSA were carried out as previously described using 0.1 pmol 32P-labeled double-stranded oligonucleotide encoding FP4 (top strand, 5′-GTTGCCTCTTACTCAATGAAATGGAGTAAA-3′; bottom strand, 5′-TTTACTCCATTTCATTGAGTAAGAGG-3′), with 5 μg added protein extract. DNA was end labeled with 32P using the Klenow fragment of DNA polymerase I as described (26). Competition assays were carried out as previously described (26) using a 5- or 50-fold molar excess of competitor oligonucleotide encoding FP4, FP3 (top, 5′-ATTGTCCATTATGAAATCCATCACGCAGG-3′; bottom, 5′-GCCTGCGTGATGGATTTCATAATGGA-3′), the TdT initiator (26) or a C/EBP consensus binding site (26). For antibody supershift assays, 1 μl C/EBPα antiserum, C/EBPβ antiserum, or C/EBPδ antiserum (Santa Cruz Biotechnology Inc., Santa Cruz, CA) was included in the preincubation before addition of 32P-labeled DNA.
Western Analysis
Cells were lysed in 0.125 m Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate, and 10% glycerol. Electrophoresis was carried out on 12% NuPage Bis-tris gels (Life Technologies) with 20 μg protein loaded. After transfer, blots were probed with antibodies to C/EBPβ or -δ at 1:500 dilution or tubulin at 1:2000 dilution (Santa Cruz Biotechnology).
ChIP Assays
ChIP assays were carried out using an Upstate EZ ChIP kit (Millipore, Billerica, MA) according to the manufacturer’s protocol. Briefly, A549 cells were grown to confluence in 10-cm dishes, then treated with 10−6 m dexamethasone or vehicle for 24 h. Proteins were cross-linked to DNA by incubation in 1% formaldehyde, and then cells were lysed in sodium dodecyl sulfate lysis buffer (provided in the kit). Chromatin was sheared by sonication using a Soniprep 150 (MSE; Beckenham, Kent, UK) with seven 10-sec pulses, at maximal amplitude. Immunoprecipitations were carried out with 5 μg C/EBP antiserum or control antibodies (rabbit IgG or anti-RNA polymerase II) supplied with the kit. One percent of the immunoprecipitation reaction was reserved as input. After reversal of cross-linking, 35 cycles of PCR (94 C for 30 sec, 56 C for 1 min, and 72 C for 1 min) were carried out with primers spanning FP3 and FP4: 5′-AGTCCTGTACAGTCATGAGCTTGG-3′ and 5′-ATTTCCCTGTCAGAGCAGCGATTG-3′.
Pomc−/− Mice and Glucocorticoid Treatment
The generation and corticosterone treatment of Pomc−/− mice has been described previously (25, 66). Mice were housed in standard conditions on a 12-h light, 12-h dark cycle (lights on 0700 h) with ad libitum access to water and chow (4.5% fat diet; Special Diet Services, Witham, UK). Eight-week-old male mice were treated for 10 d with corticosterone (25 μg/ml) in their drinking water (66). RNA was extracted from adipose tissue and liver as described previously (25). All animal protocols used in these studies were approved under the auspices of the United Kingdom Home Office Animals (Scientific Procedures) Act, 1986.
Statistics
Data were analyzed using t tests or by ANOVA followed by post hoc Fisher least significant difference. Significance was set at P < 0.05. Values are mean ± sem.
Acknowledgments
We thank members of the Endocrinology Unit, Queen’s Medical Research Institute, for helpful comments and discussions.
NURSA Molecule Pages:
Ligands: Dexamethasone | RU486;
Nuclear Receptors: GR.
Footnotes
This work was supported by a Wellcome Trust Programme grant (J.R.S. and K.E.C.) and by the European Union 6th Framework Programme DIABESITY. S.S. was the recipient of a grant from the Great Britain Sasakawa Foundation.
Disclosure Statement: S.S., C.L.E., V.K., Z.M., K.A., A.P.C., Y.N., T.O., and K.E.C. have nothing to declare. J.R.S. has consulted for Incyte, Vitae Pharmaceuticals, Ipsen, and Janssen.
First Published Online July 10, 2008
Abbreviations: C/EBPα, CCAAT/enhancer binding protein-α; ChIP, chromatin immunoprecipitation; GR, glucocorticoid receptor; GRE, glucocorticoid response element; 11β-HSD1, 11β-hydroxysteroid dehydrogenase type 1; LAP, liver-enriched activator protein; LIP, liver-enriched inhibitor protein; siRNA, small interfering RNA.
References
- 1.Seckl JR 2004. 11β-Hydroxysteroid dehydrogenases: changing glucocorticoid action. Curr Opin Pharmacol 4:597–602 [DOI] [PubMed] [Google Scholar]
- 2.Orth DN 1995. Cushing’s syndrome. N Engl J Med 332:791–803 [DOI] [PubMed] [Google Scholar]
- 3.Rask E, Olsson T, Söderberg S, Andrew R, Livingstone DE, Johnson O, Walker BR 2001. Tissue-specific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab 86:1418–1421 [DOI] [PubMed] [Google Scholar]
- 4.Paulmyer-Lacroix O, Boullu S, Oliver C, Alessi MC, Grino M 2002. Expression of the mRNA coding for 11β-hydroxysteroid dehydrogenase type 1 in adipose tissue from obese patients: an in situ hybridization study. J Clin Endocrinol Metab 87:2701–2705 [DOI] [PubMed] [Google Scholar]
- 5.Wake DJ, Rask E, Livingstone DE, Soderberg S, Olsson T, Walker BR 2003. Local and systemic impact of transcriptional up-regulation of 11β-hydroxysteroid dehydrogenase type 1 in adipose tissue in human obesity. J Clin Endocrinol Metab 88:3983–3988 [DOI] [PubMed] [Google Scholar]
- 6.Rask E, Walker BR, Soderberg S, Livingstone DE, Eliasson M, Johnson O, Andrew R, Olsson T 2002. Tissue-specific changes in peripheral cortisol metabolism in obese women: increased adipose 11β-hydroxysteroid dehydrogenase type 1 activity. J Clin Endocrinol Metab 87:3330–3336 [DOI] [PubMed] [Google Scholar]
- 7.Lindsay RS, Wake DJ, Nair S, Bunt J, Livingstone DE, Permana PA, Tataranni PA, Walker BR 2003. Subcutaneous adipose 11β-hydroxysteroid dehydrogenase type 1 activity and messenger ribonucleic acid levels are associated with adiposity and insulinemia in Pima Indians and Caucasians. J Clin Endocrinol Metab 88:2738–2744 [DOI] [PubMed] [Google Scholar]
- 8.Engeli S, Bohnke J, Feldpausch M, Gorzelniak K, Heintze U, Janke J, Luft FC, Sharma AM 2004. Regulation of 11β-HSD genes in human adipose tissue: influence of central obesity and weight loss. Obes Res 12:9–17 [DOI] [PubMed] [Google Scholar]
- 9.Kannisto K, Pietilainen KH, Ehrenborg E, Rissanen A, Kaprio J, Hamsten A, Yki-Jarvinen H 2004. Overexpression of 11β-hydroxysteroid dehydrogenase-1 in adipose tissue is associated with acquired obesity and features of insulin resistance: studies in young adult monozygotic twins. J Clin Endocrinol Metab 89:4414–4421 [DOI] [PubMed] [Google Scholar]
- 10.Desbriere R, Vuaroqueaux V, Achard V, Boullu-Ciocca S, Labuhn M, Dutour A, Grino M 2006. 11β-Hydroxysteroid dehydrogenase type 1 mRNA is increased in both visceral and subcutaneous adipose tissue of obese patients. Obesity 14:794–798 [DOI] [PubMed] [Google Scholar]
- 11.Goedecke JH, Wake DJ, Levitt NS, Lambert EV, Collins MR, Morton NM, Andrew R, Seckl JR, Walker BR 2006. Glucocorticoid metabolism within superficial subcutaneous rather than visceral adipose tissue is associated with features of the metabolic syndrome in South African women. Clin Endocrinol 65:81–87 [DOI] [PubMed] [Google Scholar]
- 12.Michailidou Z, Jensen MD, Dumesic DA, Chapman KE, Seckl JR, Walker BR, Morton NM 2007. Omental 11β-hydroxysteroid dehydrogenase 1 correlates with fat cell size independently of obesity. Obesity 15:1155–1163 [DOI] [PubMed] [Google Scholar]
- 13.Paulsen SK, Pedersen SB, Fisker S, Richelsen B 2007. 11β-HSD type 1 expression in human adipose tissue: impact of gender, obesity, and fat localization. Obesity 15:1954–1960 [DOI] [PubMed] [Google Scholar]
- 14.Seckl JR, Morton NM, Chapman KE, Walker BR 2004. Glucocorticoids and 11β-hydroxysteroid dehydrogenase in adipose tissue. Recent Prog Horm Res 59:359–393 [DOI] [PubMed] [Google Scholar]
- 15.Hammami MM, Siiteri PK 1991. Regulation of 11β- hydroxysteroid dehydrogenase activity in human skin fibroblasts: enzymatic modulation of glucocorticoid action. J Clin Endocrinol Metab 73:326–334 [DOI] [PubMed] [Google Scholar]
- 16.Jamieson PM, Chapman KE, Edwards CRW, Seckl JR 1995. 11β-hydroxysteroid dehydrogenase is an exclusive 11β-reductase in primary cultures of rat hepatocytes: effect of physicochemical and hormonal manipulations. Endocrinology 136:4754–4761 [DOI] [PubMed] [Google Scholar]
- 17.Whorwood CB, Donovan SJ, Wood PJ, Phillips DI 2001. Regulation of glucocorticoid receptor α and β isoforms and type I 11β-hydroxysteroid dehydrogenase expression in human skeletal muscle cells: a key role in the pathogenesis of insulin resistance? J Clin Endocrinol Metab 86:2296–2308 [DOI] [PubMed] [Google Scholar]
- 18.Sun K, He P, Yang K 2002. Intracrine induction of 11β-hydroxysteroid dehydrogenase type 1 expression by glucocorticoid potentiates prostaglandin production in the human chorionic trophoblast. Biol Reprod 67:1450–1455 [DOI] [PubMed] [Google Scholar]
- 19.Cooper MS, Rabbitt EH, Goddard PE, Bartlett WA, Hewison M, Stewart PM 2002. Osteoblastic 11β-hydroxysteroid dehydrogenase type 1 activity increases with age and glucocorticoid exposure. J Bone Miner Res 17:979–986 [DOI] [PubMed] [Google Scholar]
- 20.Bujalska IJ, Kumar S, Hewison M, Stewart PM 1999. Differentiation of adipose stromal cells: the roles of glucocorticoids and 11β-hydroxysteroid dehydrogenase. Endocrinology 140:3188–3196 [DOI] [PubMed] [Google Scholar]
- 21.Yang K, Berdusco ETM, Challis JRG 1994. Opposite effects of glucocorticoid on hepatic 11β-hydroxysteroid dehydrogenase mRNA and activity in fetal and adult sheep. J Endocrinol 143:121–126 [DOI] [PubMed] [Google Scholar]
- 22.Hundertmark S, Ragosch V, Schein B, Buhler H, Lorenz U, Fromm M, Weitzel HK 1994. Gestational age dependence of 11β-hydroxysteroid dehydrogenase and its relationship to the enzymes of phosphatidylcholine synthesis in lung and liver of fetal rat. Biochim Biophys Acta 1210:348–354 [DOI] [PubMed] [Google Scholar]
- 23.Jamieson PM, Chapman KE, Seckl JR 1999. Tissue- and temporal-specific regulation of 11β-hydroxysteroid dehydrogenase type 1 by glucocorticoids in vivo. J Steroid Biochem Mol Biol 68:245–250 [DOI] [PubMed] [Google Scholar]
- 24.Jellinck PH, Dhabhar FS, Sakai RR, McEwen BS 1997. Long-term corticosteroid treatment but not chronic stress affects 11β-hydroxysteroid dehydrogenase type I activity in rat brain and peripheral tissues. J Steroid Biochem Mol Biol 60:319–323 [DOI] [PubMed] [Google Scholar]
- 25.Michailidou Z, Coll AP, Kenyon CJ, Morton NM, O'Rahilly S, Seckl JR, Chapman KE 2007. Peripheral mechanisms contributing to the glucocorticoid hypersensitivity in proopiomelanocortin null mice treated with corticosterone. J Endocrinol 194:161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams LJS, Lyons V, MacLeod I, Rajan V, Darlington GJ, Poli V, Seckl JR, Chapman KE 2000. C/EBP regulates hepatic transcription of 11β-hydroxysteroid dehydrogenase type 1: a novel mechanism for cross talk between the C/EBP and glucocorticoid signalling pathways. J Biol Chem 275:30232–30239 [DOI] [PubMed] [Google Scholar]
- 27.Bruley C, Lyons V, Worsley AG, Wilde MD, Darlington GD, Morton NM, Seckl JR, Chapman KE 2006. A novel promoter for the 11β-hydroxysteroid dehydrogenase type 1 gene is active in lung and is C/EBPα independent. Endocrinology 147:2879–2885 [DOI] [PubMed] [Google Scholar]
- 28.Gout J, Tirard J, Thevenon C, Riou JP, Begeot M, Naville D 2006. CCAAT/enhancer-binding proteins (C/EBPs) regulate the basal and cAMP-induced transcription of the human 11β-hydroxysteroid dehydrogenase encoding gene in adipose cells. Biochimie 88:1115–1124 [DOI] [PubMed] [Google Scholar]
- 29.Ballard PL, Mason RJ, Douglas WH 1978. Glucocorticoid binding by isolated lung cells. Endocrinology 102:1570–1575 [DOI] [PubMed] [Google Scholar]
- 30.Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Ha C, Yamamoto KR 2004. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci USA 101:15603–15608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roesler WJ 2001. The role of C/EBP in nutrient and hormonal regulation of gene expression. Annu Rev Nutr 21:141–165 [DOI] [PubMed] [Google Scholar]
- 32.Wei W, Yang H, Cao P, Menconi M, Chamberlain C, Petkova V, Hasselgren PO 2006. Degradation of C/EBPβ in cultured myotubes is calpain-dependent. J Cell Physiol 208:386–398 [DOI] [PubMed] [Google Scholar]
- 33.Yamada K, Duong DT, Scott DK, Wang JC, Granner DK 1999. CCAAT/enhancer-binding protein β is an accessory factor for the glucocorticoid response from the cAMP response element in the rat phosphoenolpyruvate carboxykinase gene promoter. J Biol Chem 274:5880–5887 [DOI] [PubMed] [Google Scholar]
- 34.Arizmendi C, Liu S, Croniger C, Poli V, Friedman JE 1999. The transcription factor CCAAT/enhancer-binding protein β regulates gluconeogenesis and phosphoenolpyruvate carboxykinase (GTP) gene transcription during diabetes. J Biol Chem 274:13033–13040 [DOI] [PubMed] [Google Scholar]
- 35.Grange T, Roux J, Rigaud G, Pictet R 1991. Cell-type specific activity of two glucocorticoid responsive units of rat tyrosine aminotransferase gene is associated with multiple binding sites for C/EBP and a novel liver-specific nuclear factor. Nucleic Acids Res 19:131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alam T, An MR, Mifflin RC, Hsieh CC, Ge X, Papaconstantinou J 1993. trans-activation of the α1-acid glycoprotein gene acute phase responsive element by multiple isoforms of C/EBP and glucocorticoid receptor. J Biol Chem 268:15681–15688 [PubMed] [Google Scholar]
- 37.Baumann H, Gauldie J 1994. The acute phase response. Immunol Today 15:74–80 [DOI] [PubMed] [Google Scholar]
- 38.MacDougald OA, Cornelius P, Lin FT, Chen SS, Lane MD 1994. Glucocorticoids reciprocally regulate expression of the CCAAT/enhancer-binding protein α and δ genes in 3T3-L1 adipocytes and white adipose tissue. J Biol Chem 269:19041–19047 [PubMed] [Google Scholar]
- 39.Boudreau F, Blais S, Asselin C 1996. Regulation of CCAAT/enhancer binding protein isoforms by serum and glucocorticoids in the rat intestinal epithelial crypt cell line IEC-6. Exp Cell Res 222:1–9 [DOI] [PubMed] [Google Scholar]
- 40.Tomlinson JJ, Boudreau A, Wu D, Atlas E, Hache RJ 2006. Modulation of early human preadipocyte differentiation by glucocorticoids. Endocrinology 147:5284–5293 [DOI] [PubMed] [Google Scholar]
- 41.Matsuno F, Chowdhury S, Gotoh T, Iwase K, Matsuzaki H, Takatsuki K, Mori M, Takiguchi M 1996. Induction of the C/EBPβ gene by dexamethasone and glucagon in primary-cultured rat hepatocytes. J Biochem 119:524–532 [DOI] [PubMed] [Google Scholar]
- 42.Gotoh T, Chowdhury S, Takiguchi M, Mori M 1997. The glucocorticoid-responsive gene cascade. Activation of the rat arginase gene through induction of C/EBPβ. J Biol Chem 272:3694–3698 [DOI] [PubMed] [Google Scholar]
- 43.Yang H, Mammen J, Wei W, Menconi M, Evenson A, Fareed M, Petkova V, Hasselgren PO 2005. Expression and activity of C/EBPβ and δ are upregulated by dexamethasone in skeletal muscle. J Cell Physiol 204:219–226 [DOI] [PubMed] [Google Scholar]
- 44.Woltje M, Tschoke B, von Bulow V, Westenfeld R, Denecke B, Graber S, Jahnen-Dechent W 2006. CCAAT enhancer binding protein β and hepatocyte nuclear factor 3β are necessary and sufficient to mediate dexamethasone-induced up-regulation of α2HS-glycoprotein/fetuin-A gene expression. J Mol Endocrinol 36:261–277 [DOI] [PubMed] [Google Scholar]
- 45.Berg T, Didon L, Barton J, Andersson O, Nord M 2005. Glucocorticoids increase C/EBPβ activity in the lung epithelium via phosphorylation. Biochem Biophys Res Commun 334:638–645 [DOI] [PubMed] [Google Scholar]
- 46.Berg T, Cassel TN, Schwarze PE, Nord M 2002. Glucocorticoids regulate the CCSP and CYP2B1 promoters via C/EBPβ and δ in lung cells. Biochem Biophys Res Commun 293:907–912 [DOI] [PubMed] [Google Scholar]
- 47.Zhang N, Truong-Tran QA, Tancowny B, Harris KE, Schleimer RP 2007. Glucocorticoids enhance or spare innate immunity: effects in airway epithelium are mediated by CCAAT/enhancer binding proteins. J Immunol 179:578–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hermanowski-Vosatka A, Gerhold D, Mundt SS, Loving VA, Lu M, Chen Y, Elbrecht A, Wu M, Doebber T, Kelly L, Milot D, Guo Q, Wang PR, Ippolito M, Chao YS, Wright SD, Thieringer R 2000. PPARα agonists reduce 11β-hydroxysteroid dehydrogenase type 1 in the liver. Biochem Biophys Res Commun 279:330–336 [DOI] [PubMed] [Google Scholar]
- 49.Berger J, Tanen M, Elbrecht A, Hermanowski-Vosatka A, Moller DE, Wright SD, Thieringer R 2001. PPARγ ligands inhibit adipocyte 11β-hydroxysteroid dehydrogenase type 1 expression and activity. J Biol Chem 276:12629–12635 [DOI] [PubMed] [Google Scholar]
- 50.Stulnig TM, Oppermann U, Steffensen KR, Schuster GU, Gustafsson JA 2002. Liver X receptors downregulate 11β-hydroxysteroid dehydrogenase type 1 expression and activity. Diabetes 51:2426–2433 [DOI] [PubMed] [Google Scholar]
- 51.Li F, Rosenberg E, Smith CI, Notarfrancesco K, Reisher SR, Shuman H, Feinstein SI 1995. Correlation of expression of transcription factor C/EBPα and surfactant protein genes in lung cells. Am J Physiol 269:L241–L247 [DOI] [PubMed]
- 52.Descombes P, Schibler U 1991. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 67:569–579 [DOI] [PubMed] [Google Scholar]
- 53.Xiong W, Hsieh CC, Kurtz AJ, Rabek JP, Papaconstantinou J 2001. Regulation of CCAAT/enhancer-binding protein-β isoform synthesis by alternative translational initiation at multiple AUG start sites. Nucleic Acids Res 29:3087–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burgess-Beusse BL, Timchenko NA, Darlington GJ 1999. CCAAT/enhancer binding protein α (C/EBPα) is an important mediator of mouse C/EBPβ protein isoform production. Hepatology 29:597–601 [DOI] [PubMed] [Google Scholar]
- 55.Baer M, Johnson PF 2000. Generation of truncated C/EBPβ isoforms by in vitro proteolysis. J Biol Chem 275:26582–26590 [DOI] [PubMed] [Google Scholar]
- 56.Poli V 1998. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem 273:29279–29282 [DOI] [PubMed] [Google Scholar]
- 57.Morton NM, Holmes MC, Fiévet C, Staels B, Tailleux A, Mullins JJ, Seckl JR 2001. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11β-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem 276:41293–41300 [DOI] [PubMed] [Google Scholar]
- 58.Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C, Walker BR, Flier JS, Mullins JJ, Seckl JR 2004. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11β-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 53:931–938 [DOI] [PubMed] [Google Scholar]
- 59.Wang L, Shao J, Muhlenkamp P, Liu S, Klepcyk P, Ren J, Friedman JE 2000. Increased insulin receptor substrate-1 and enhanced skeletal muscle insulin sensitivity in mice lacking CCAAT/enhancer-binding protein-β. J Biol Chem 275:14173–14181 [DOI] [PubMed] [Google Scholar]
- 60.Millward CA, Heaney JD, Sinasac DS, Chu EC, Bederman IR, Gilge DA, Previs SF, Croniger CM 2007. Mice with a deletion in the gene for CCAAT/enhancer-binding protein β are protected against diet-induced obesity. Diabetes 56:161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rahman SM, Schroeder-Gloeckler JM, Janssen RC, Jiang H, Qadri I, Maclean KN, Friedman JE 2007. CCAAT/enhancing binding protein β deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology 45:1108–1117 [DOI] [PubMed] [Google Scholar]
- 62.Schroeder-Gloeckler JM, Rahman SM, Janssen RC, Qiao L, Shao J, Roper M, Fischer SJ, Lowe E, Orlicky DJ, McManaman JL, Palmer C, Gitomer WL, Huang W, O'Doherty RM, Becker TC, Klemm DJ, Jensen DR, Pulawa LK, Eckel RH, Friedman JE 2007. CCAAT/ enhancer-binding protein β deletion reduces adiposity, hepatic steatosis, and diabetes in Lepr(db/db) mice. J Biol Chem 282:15717–15729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu B, Ullenbruch MR, Jin H, Gharaee-Kermani M, Phan SH 2007. An essential role for CCAAT/enhancer binding protein β in bleomycin-induced pulmonary fibrosis. J Pathol 211:455–462 [DOI] [PubMed] [Google Scholar]
- 64.Gilmour JS, Coutinho AE, Cailhier JF, Man TY, Clay M, Thomas G, Harris HJ, Mullins JJ, Seckl JR, Savill JS, Chapman KE 2006. Local amplification of glucocorticoids by 11β-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J Immunol 176:7605–7611 [DOI] [PubMed] [Google Scholar]
- 65.Beato M 1987. Induction of transcription by steroid hormones. Biochim Biophys Acta 910:95–102 [DOI] [PubMed] [Google Scholar]
- 66.Coll AP, Challis BG, Lopez M, Piper S, Yeo GS, O'Rahilly S 2005. Proopiomelanocortin-deficient mice are hypersensitive to the adverse metabolic effects of glucocorticoids. Diabetes 54:2269–2276 [DOI] [PubMed] [Google Scholar]