

Abstract
Tumorigenesis is a multistep process involving dysregulated cell growth and metastasis. Considerable evidence implicates a mitogenic action of estrogen in early ovarian carcinogenesis. In contrast, its influence in the metastatic cascade of ovarian tumor cells remains obscure. In the present study, we showed that 17β-estradiol (E2) increased the metastatic potential of human epithelial ovarian cancer cell lines. E2 treatment led to clear morphological changes characteristic of epithelial-mesenchymal transition (EMT) and an enhanced cell migratory propensity. These morphological and functional alterations were associated with changes in the abundance of EMT-related genes. Upon E2 stimulation, expression and promoter activity of the epithelial marker E-cadherin were strikingly suppressed, whereas EMT-associated transcription factors, Snail and Slug, were significantly up-regulated. This up-regulation was attributed to the increase in gene transcription activated by E2. Depletion of endogenous Snail or Slug using small interfering RNA (siRNA) attenuated E2-mediated decrease in E-cadherin. In addition, E2-induced cell migration was also neutralized by the siRNAs, suggesting that both transcription factors are indispensable for the prometastatic actions of E2. More importantly, by using selective estrogen receptor (ER) agonists, forced expression, and siRNA approaches, we identified that E2 triggered the metastatic behaviors exclusively through an ERα-dependent pathway. We also showed that ERβ had an opposing action on ERα because the presence of ERβ completely inhibited the EMT and down-regulation of E-cadherin induced by ERα. Collectively, this study provides a compelling argument that estrogen can potentiate tumor progression by EMT induction and highlights the crucial role of ERα in ovarian tumorigenesis.
OVARIAN CANCER is the leading cause of death from gynecological tumors and is the fourth most frequent cause of death from cancers in females. The disease is highly lethal because of its aggressive metastasis within the peritoneal cavity and the fact that it is often diagnosed at an advanced stage. Epithelial ovarian cancers originating from the ovarian surface epithelium constitute approximately 90% of human ovarian cancers (1).
Epithelial-mesenchymal transition (EMT) is a developmental process of crucial importance in cell differentiation, morphogenesis, and growth. This process allows cells to dissociate from the epithelial tissue and adopt a motile phenotype. During this process, epithelial cells become elongated, lose their polarity and cellular junctions, and show mesenchymal, fibroblast-like properties. Accumulating evidence has also proposed that similar processes occur during cancer progression (2). Because of its clinical importance, intensive research has been undertaken to identify reliable molecular markers and key mediators of EMT. They include proteins that actively control cell junction formation, cytoskeletal remodeling, and cell motility such as cadherins, matrix metalloproteinases, and several transcription factors (3, 4).
Among all members in the cadherin superfamily, E-cadherin represents one of the best-characterized markers of EMT and it is regarded as a tumor suppressor. It mediates calcium-dependent intercellular junctions, and abolishment of its function induces mesenchymal phenotypes. It also influences intracellular signaling pathways through its association with catenins, which are cytoplasmic counterparts of cadherins and are implicated in numerous pathological states including malignancy (5). Mutation or decreased expression of E-cadherin is often correlated with advanced-stage carcinomas (6, 7, 8, 9). E-cadherin can be silenced or functionally inactivated by several mechanisms, including down-regulation of gene expression, gene mutations, and posttranslational regulation. Down-regulation of gene expression can occur by promoter hypermethylation, histone deacetylation, and transcriptional regulation (10, 11, 12).
Transcriptional repression is emerging as a predominant mechanism controlling the expression of E-cadherin in most carcinomas. Several transcriptional repressors, which include Snail (13), Slug (14), SIP-1/ZEB-2 (15), E12/E47 (16), and Twist (17), have been characterized. They are found to interact with the proximal E-boxes of the E-cadherin promoter. Snail and Slug are zinc finger proteins that belong to the Snail superfamily (18). An inverse correlation between E-cadherin and Snail or Slug has been described in many different cell systems (13, 19, 20). Additionally, Snail and Slug have been detected at sites of EMT in vertebrates (18). Ectopic expression of Snail and Slug in epithelial cells caused EMT by acquisition of migratory and invasive behaviors, which were also evident in ovarian cancer (13, 21). Thus, these two transcription factors are considered as promising markers for malignancy, and identifying their upstream regulators is necessary.
Risk factors associating with ovarian cancers have been proposed in many epidemiological studies. Among them, age, family history, and infertility are known to increase the risk of ovarian cancer, whereas parity, oral contraceptive use, breast feeding, hysterectomy, or tubal ligation decreases the risk (22, 23). Although it is still under debate, there is a growing body of evidence that supports the contribution of estrogen in some of these risk factors. First, ovary is a main source and target tissue of estrogen. Second, estrogen receptors are found in human ovarian surface epithelium and more than 60% of ovarian cancer cells (24). Finally, a positive correlation between ovarian cancer incidence and estrogen has been established by many experimental and epidemiological studies (25, 26, 27, 28, 29, 30). Recent meta-analysis of cohort studies, case-control studies, randomized controlled trials and cancer registry studies has shown that estrogen therapy is a critical risk factor for ovarian cancer (31). The mitogenic effect of estrogen may provide one possible explanation for the increased risk. However, whether estrogen may play an additional role in ovarian cancer cell metastasis is poorly understood.
Here, our findings demonstrated that 17β-estradiol (E2) exposure led to acquisition of both phenotypic and molecular attributes typical of EMT in ovarian epithelial cancer cells BG-1. E2 promoted a spindle-like and mesenchymal phenotype and significantly enhanced cell motility. These pronounced changes were associated with inhibition of E-cadherin expression. Furthermore, we identified estrogen receptor (ER)α as a critical mechanistic link in mediating the promotion of EMT, whereas in the presence of ERβ, the pro-metastatic effects of ERα were inhibited. This is the first demonstration of the distinct roles of ERα and ERβ in ovarian tumor metastasis through the differential regulation of EMT-related genes.
RESULTS
E2 Stimulates EMT and Migration of BG-1 Cells
First, we aimed to elucidate the functional role of E2 in EMT of BG-1 cells, an estrogen-responsive ovarian cancer cell line. BG-1 cells underwent apparent changes in morphology compatible with EMT after 48 h of E2 (10−7 m) treatment (Fig. 1A). The phenotypic changes observed include conversion from cuboidal, epithelial morphology to spindle-shaped morphology, increased intercellular separation, and scattered and increased formation of pseudopodia. Epithelial cells that undergo EMT are characterized by increased motility; we therefore asked whether E2 could increase migratory ability of BG-1 by using a wound healing assay. E2 was administered 24 h before the wound was generated, and the rate of cell migration was subsequently measured at 0, 4, and 24 h after treatment. The wounded area of BG-1 cell monolayers healed slowly in vehicle-treated cells (Fig. 1B). On the contrary, in the presence of E2, the closure of the wounded gap was significantly accelerated at 24 h (Fig. 1B). The percentage of cell-free area at the indicated time points compared with that at time zero was determined (Fig. 1B, lower panel). In E2-treated cells, cell-free area was decreased by more than 2-fold compared with control cells at 24 h (P < 0.05). During the incubation period, cells moved forward and closed the gap independently of cell division. There was no significant increase in cell growth within 48 h under the same treatment conditions as revealed by cell proliferation assay (data not shown) and was consistent with previous studies (32, 33). We conclude that E2 promotes EMT, which leads to increased migration of BG-1 cells.
Fig. 1.
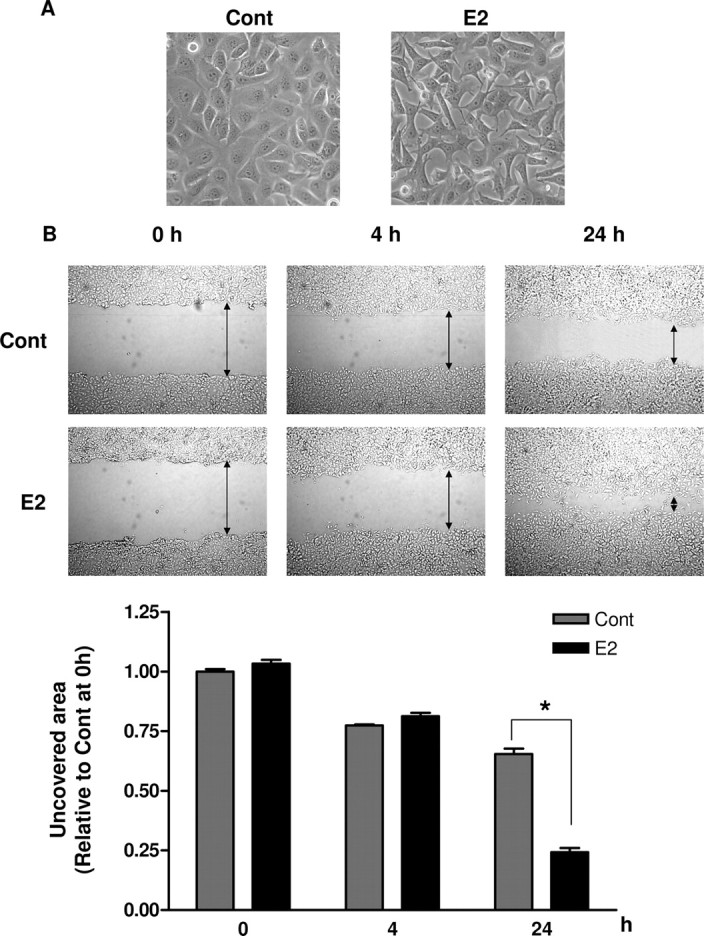
BG-1 Cells Are Induced by E2 to Undergo EMT
A, BG-1 cells were treated with vehicle (Cont) or 10−7 m E2 for 48 h. Morphological changes were observed by phase-contrast microscope, and representative photographs of at least three independent experiments are shown. Original magnification, ×200. B, Confluent monolayers of BG-1 cells were wounded with a uniform scratch, washed to remove cell debris, and cultured for indicated times in the presence of vehicle (Cont) or 10−7 m E2. Images of cell cultures were captured at 0, 4, and 24 h after scratching; representative pictures are shown in upper panel. The arrow indicates wound edge. The amount of wound repair was expressed as uncovered area at the indicated time compared with initial uncovered area of vehicle-treated control at time zero (lower panel). Values are the mean ± sd of three separate experiments. *, P < 0.05 compared with control. Cont, Control.
E2 Suppresses E-Cadherin Expression in Dose- and Time-Dependent Manner
Decrease of E-cadherin, a widely accepted characteristic associated with EMT, is often correlated to higher mobility of tumor cells (34). Given the role of E2 in controlling EMT as demonstrated in Fig. 1, we investigated the effect of E2 on the expression of this epithelial marker by Western blot analysis. Dose-response studies showed that lower concentrations of E2 (10−11 m to 10−15 m) did not affect E-cadherin expression. The down-regulation effect became apparent with doses of 10−9 m and 10−7 m of E2 and was maximal at 10−7 m (Fig. 2A) (P < 0.05). Densitometric analyses of three independent experiments confirmed this finding (Fig. 2A, lower panel). Cells were also treated with 10−7 m E2 for different time courses. As shown in a representative blot in Fig. 2B, protein levels of E-cadherin were significantly decreased after E2 treatment. Maximal inhibition was seen at 48 h (a 70% decline) (P < 0.05), and the effect was still detectable after 72 h. To determine whether ER activation was involved, the effect of E2 was examined in the presence of the pure ER antagonist, ICI 182,780 (ICI). As shown in Fig. 2B, the down-regulation was prevented by 10−7 m ICI. These results suggest ER is a mediator of the suppression effects on E-cadherin by E2.
Fig. 2.

E2 Down-Regulates E-Cadherin Expression
A, BG-1 cells were treated with vehicle (Cont) or increasing concentrations of E2 (10−15 to 10−7 m) for 48 h. B, BG-1 cells were treated with either vehicle (Cont), 10−7 m E2 or in combination with 10−7 m ICI for 24, 48, and 72 h. Whole-cell lysates was extracted, and E-cadherin protein (E-Cad) was detected by Western blot. β-Actin was used as a loading control. The immunoblot shown is representative of three independent experiments. Cumulative results for quantitative densitometry of the three experiments are shown in lower panels. Mean ± sd values are depicted for protein abundance expressed as percentage in control cells. *, P < 0.05 compared with control. +, P < 0.05 compared with the corresponding time-matched vehicle control. Cont, Control.
Transcriptional Repression of E-Cadherin by E2
To assess whether the reduction in E-cadherin expression was due to transcriptional regulation, E-cadherin mRNA levels after E2 (10−7 m) treatment were evaluated by real-time PCR. E-cadherin mRNA expression was decreased by approximately 50% as early as 12 h after E2 administration, with sustained decreases observed thereafter (Fig. 3A) (P < 0.05). This suggests that E-cadherin expression is suppressed in BG-1 cells at the transcriptional level. To confirm this, cells were transfected with reporter construct cloned with the E-cadherin proximal promoter and treated with E2 for periods up to 72 h. Concurrent with the loss of mRNA expression, treatment with E2 inhibited E-cadherin promoter activity (Fig. 3B). The promoter activity was significantly decreased by 25% after 24 h. This inhibitory effect persisted after 48–72 h, producing up to 50% decrease (P < 0.05). ICI was able to overcome this inhibitory effect because E2 and ICI in combination increased the transcription activity compared with E2-treated cells (Fig. 3B).
Fig. 3.

E2 Reduces E-Cadherin mRNA Synthesis through Transcriptional Repression
A, BG-1 cells were cultured in the presence of vehicle (Cont) or 10−7 m E2 for various time periods indicated. Total RNA was isolated and used for real-time PCR analysis of E-cadherin mRNA expression with gene-specific primers. GAPDH primers were used to normalize data, and results are expressed as fold change relative to time zero. B, BG-1 cells were transfected with 1 μg reporter construct of E-cadherin promoter. All cells were cotransfected with 0.5 μg pSV-βGal as an internal control for transcription efficiency. Cells were treated 8 h after transfection with vehicle (Cont), 10−7 m E2, or in combination with 10−7 m ICI, for an additional 24, 48, or 72 h. The luciferase activities were calculated relative to the promoterless vector (pGL3-Basic) and expressed as fold change relative to vehicle control at the corresponding time point. The data are shown as mean ± sd of three repeated experiments. *, P < 0.05 compared with the corresponding time-matched vehicle control. Cont, Control; E-Cad, E-cadherin.
E2 Enhances Expression and Promoter Activities of Snail and Slug
Transcription of E-cadherin is known to be regulated by the Snail and Slug repressors (13). To elucidate the mechanisms through which E2 suppresses E-cadherin, the protein levels of Snail and Slug after E2 treatment were investigated by Western blot. As shown in Fig. 4A, both Snail and Slug were significantly elevated in dose-dependent manner after 8 h of E2 treatment (P < 0.05). In comparison, analysis of Twist, which is another known E-cadherin repressor (17), showed no change in protein level with the same treatment (data not shown).
Fig. 4.

E2 Regulates Expression and Gene Promoter Activities of Snail and Slug
A, BG-1 cells were treated with vehicle (Cont) or increasing concentrations of E2 (10−15 to 10−7 m) for 8 h. Whole-cell lysates were extracted, and Snail and Slug were detected by Western blot. β-Actin was used as a loading control. The immunoblot shown is representative of three independent experiments. Cumulative results for quantitative densitometry of the three experiments are shown in left panel. B, BG-1 cells were cultured in the presence of vehicle (Cont) or 10−7 m E2 for the various time periods indicated and harvested for RNA extraction. C, Cells were treated with 10−7 m E2 or vehicle (Cont) for 1 h. Then, at time 0 h, the transcription inhibitor actinomycin D (5 μg/ml) was added to the medium and cells were harvested at the indicated time points. Total RNA was isolated and used for real-time PCR analysis of Snail or Slug mRNA expression with gene-specific primers. GAPDH primers were used to normalize data, and results are expressed as fold change relative to time zero. D, BG-1 cells were transfected with either human Snail or Slug promoter-reporter gene construct and pSV-βGal for normalization. The luciferase activities were determined in cell lysates 2–8 h after initiation of E2 exposure. The luciferase activities were calculated relative to the promoterless vector (pGL3-Basic) and expressed as fold change relative to vehicle control at corresponding time point. The data are shown as mean ± sd of three repeated experiments. *, P < 0.05 compared with control. +, P < 0.05 compared with the corresponding time-matched vehicle control. Cont, Control.
The time course effects of E2 on Snail and Slug mRNA levels were determined by real-time PCR as shown in Fig. 4B. E2 caused a marked induction of Snail mRNA at 1 h (∼2-fold increase), with significant increased mRNA levels being maintained within 1–4 h. Similarly, E2 treatment elevated Slug mRNA with a maximal stimulation reached in 1 h. However, the induction was transient, and the level of Slug mRNA returned to basal level at 4 h. Having established a correlation between E2 and Snail or Slug expression, we asked whether E2 stabilized their mRNA transcripts by actinomycin D chase experiment. The apparent half-lives of both Snail and Slug mRNA transcripts were approximately 1 h in control cells and were not changed upon E2 treatment (Fig. 4C). Thus, the induction of Snail and Slug in response to E2 was not due to an effect on message stability. We next investigated whether E2 can regulate their transcriptions. To this end, we performed luciferase promoter assays and examined the ability of E2 to transactivate luciferase reporter plasmids containing the human Snail or Slug promoter. Figure 4D showed that both Snail and Slug promoters were strongly activated by E2. These activations were already evident after 2 h of E2 treatment (P < 0.05). Induction of the activities peaked at 4 h (2- to 3-fold) and started to decrease at 8 h.
Snail and Slug Are Responsible for E2-Induced Repression of E-Cadherin and Cell Migration
The ability of E2 to increase Snail and Slug expressions implicates that these two repressors might be involved in transcriptional repression of E-cadherin in BG-1 cells. To confirm this, we used small interfering RNAs (siRNAs) to knock down Snail and Slug expressions in BG-1 cells. Depletion of endogenous Snail and Slug with siRNAs (sequence 1) was verified by Western blot (Fig. 5A). We also showed that knockdown of either Snail or Slug significantly interrupted the E2-mediated down-regulation of E-cadherin (Fig. 5B), whereas nonspecific siRNA had no observable effect. Another set of siRNAs with different sequences to Snail and Slug (sequence 2) was used to rule out the off-target effects, and the same results were obtained (supplemental Fig. S1 published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). Concomitantly, suppression of E-cadherin promoter by E2 was significantly abolished by transfection with either Snail or Slug siRNA, but not with nonspecific siRNA (Fig. 5C).
Fig. 5.

Snail and Slug Mediate the Regulation of E-Cadherin and Cell Migration by E2
A, BG-1 cells were transfected with either 250 nm of Snail siRNA (sequence no. 1), Slug siRNA (sequence no. 1) or nonspecific siRNA (NS siRNA) for 48 h. Efficacy of the siRNAs was determined by Western blot analysis. B, Eight hours after transfection with the siRNAs, BG-1 cells were treated with vehicle (Cont) or 10−7 m E2 for an additional 48 h. Whole-cell lysates was extracted and E-cadherin protein (E-Cad) was detected by Western blot. β-Actin was used as a loading control. C, BG-1 cells were transfected with the human E-cadherin promoter luciferase construct and pSV-βGal in combination with either one of the siRNAs. Eight hours after transfection, cells were incubated with vehicle (Cont) or 10−7 m E2 for an additional 48 h. The luciferase activities were calculated relative to the promoterless vector (pGL3-Basic) and expressed as fold change relative to vehicle control. The data are shown as mean ± sd of three repeated experiments. D, Scratch was made by scraping the monolayer 24 h after transfection of the siRNAs, and cells were incubated with medium containing vehicle (Cont) or 10−7 m E2. Wound closure was photographed after 24 h of cell migration. The amount of wound repair was expressed as uncovered area at the indicated time compared with initial uncovered area of vehicle-treated control at time zero. Values are the mean ± sd of three separate experiments. *, P < 0.05 compared with control. Cont, Control.
To further investigate the potential contribution of Snail and Slug in E2-induced cell migration, wound healing assay was performed with their corresponding siRNAs. As shown in Fig. 5D, cells treated with E2 efficiently migrated into the wound compared with control BG-1 cells or cells treated with control siRNA. In contrast, cells transfected with Snail or Slug siRNA reversed E2-induced migration into the wound. Together these findings indicate that Snail and Slug play pivotal roles in mediating EMT induction by E2.
ERα, But Not ERβ, Mediates E2-Induced EMT
To dissect which ER subtype plays a dominant role in the pro-metastatic effect of E2, BG-1 cells were treated with selective ER subtype ligands. The ERα-specific agonist, propyl pyrazole triol (PPT), is 410-fold more potent in binding to ERα than ERβ (35), whereas diarylpropionitrile (DPN) is an ERβ agonist with more than 70-fold higher binding affinity for ERβ than ERα (36). BG-1 cells were treated with either 10−7/10−8 m PPT or 10−7/10−8 m DPN for 48 h and harvested for E-cadherin detection by Western blot. PPT induced a comparable reduction in E-cadherin protein expression as E2 (Fig. 6A). In contrast, DPN induced an approximately 50% increase in the expression. Next, we confirmed this result by abrogating endogenous ERs in the cells. The efficiency of the siRNAs was confirmed by Western blot (supplemental Fig. S2A). Selective knockout of ERα dramatically reversed the down-regulation of E-cadherin expression and its promoter activity upon E2 administration (Fig. 6, B and C). Neither nonspecific siRNA nor ERβ-specific siRNA had any effect. Consistently, depletion of ERα also significantly abolished the cell morphological changes and cell migration induced by E2 (Fig. 6, D and E).
Fig. 6.
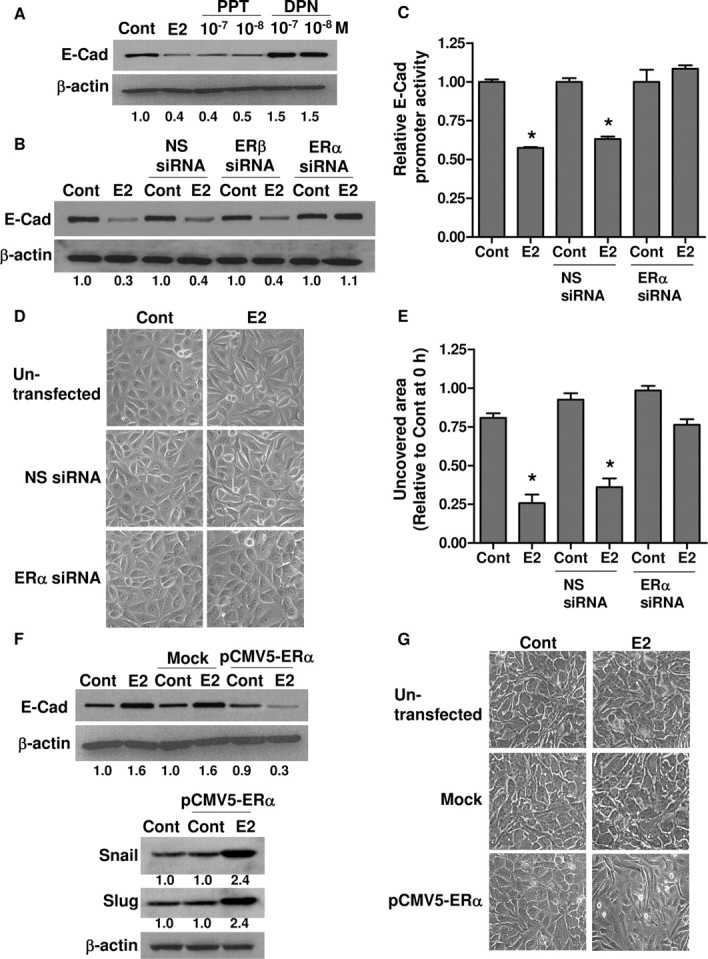
ERα Is Required for EMT Induction by E2
A, BG-1 cells were treated with either vehicle (Cont), 10−7 m E2, 10−7/10−8 m DPN, or PPT for 48 h. B, BG-1 cells were transfected with nonspecific siRNA (NS siRNA), ERα siRNA, or ERβ siRNA and subsequently treated with 10−7 m E2 for 48 h. E-cadherin protein (E-Cad) and β-actin were detected by Western blot. C, Cells were transfected with E-cadherin promoter construct and pSV-βGal in combination with the indicated siRNAs. Eight hours after transfection, cells were incubated with vehicle (Cont) or 10−7 m E2 for an additional 48 h. The luciferase activities were calculated relative to the promoterless vector (pGL3-Basic) and expressed as fold change relative to vehicle control. D, Morphological changes of cells were observed by phase-contrast microscope and representative photographs are shown. Original magnification, ×200. E, Scratch was made by scraping the monolayer 24 h after siRNA transfection, and cells were incubated with medium containing vehicle (Cont) or 10−7 m E2. Wound closure was photographed after 24 h. The amount of wound repair was expressed as uncovered area compared with initial uncovered area of vehicle-treated control at time zero. Values are the mean ± sd of three separate experiments. F, SKOV-3 cells were transfected with pCMV5-ERα or without plasmid DNA (mock) for 8 h and subsequently treated with 10−7 m E2 or vehicle (Cont) alone for another 48 h. E-cadherin (E-Cad), Snail, Slug, and β-actin were detected by Western blot. G, Morphological changes of SKOV-3 cells under the same treatment were also observed by phase-contrast microscope, and representative photographs are shown. Original magnification, ×200. The experiments were done in triplicate and repeated thrice. *, P < 0.05 compared with control. Numerical values below each lane of the immunoblots represent quantification of the relative protein level by densitometry (normalized to β-actin protein level). Cont, Control.
To further address the involvement of the ER subtypes, we restored ERα in SKOV-3 cells by transient transfection of full-length ERα (pCMV5-ERα). This ovarian cancer cell line is ERβ positive but ERα negative due to inactivating mutation, which renders the cells insensitive to estrogen in terms of cell proliferation and gene induction (24, 37). Overexpression of ERα was confirmed by Western blot, and it was comparable to that detected in ERα-expressing BG-1, indicating that the expression was within normal levels (supplemental Fig. S2B). The receptor functionality was verified by profound activation of an estrogen-responsive reporter (thymidine kinase-ERE-Luc) in the presence of E2 or PPT (data not shown). Mock-transfected cells were used as control. As shown, untransfected or mock-transfected SKOV-3 cells showed significant elevation of E-cadherin protein expression in response to E2 (Fig. 6F). In marked contrast, in ERα-overexpressing SKOV-3 cells, E2 decreased E-cadherin expression with parallel increases in both Snail and Slug expressions (Fig. 6F). In line with this, we also observed pronounced morphological changes consistent with EMT (Fig. 6G). These data, together with those obtained from BG-1 cells (Fig. 6, A–E), further illustrate that the acquisition of metastatic properties is ERα dependent.
ERβ Opposes the Pro-Metastasis Effects of ERα
The findings in Fig. 6, A and F, suggest that ERβ may have an opposite effect to ERα. To further examine the role of ERβ in the metastatic activities of ovarian cancer cells, we used siRNA to knock down ERβ in SKOV-3 cells. Figure 7A shows that ERβ siRNA specifically inhibited the expression of ERβ, but not ERα. More importantly, ERβ siRNA abolished the up-regulation of E-cadherin by E2 (Fig. 7B). The E2-mediated increase of cell-free area could also be efficiently blocked by ERβ siRNA (Fig. 7C).
Fig. 7.
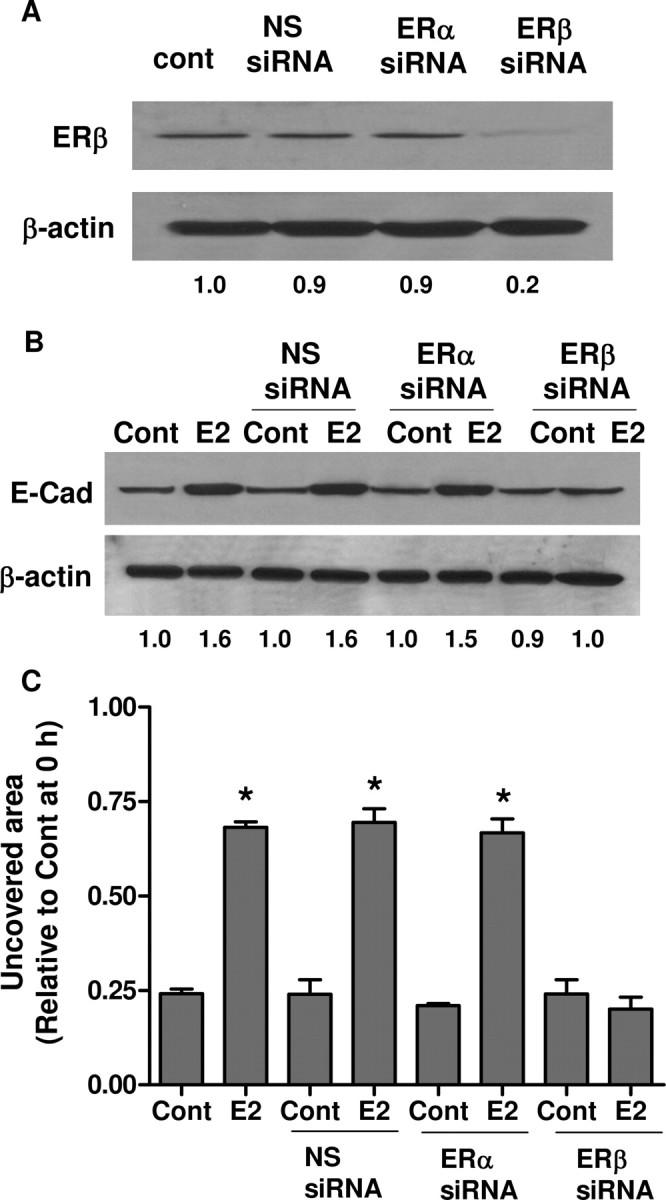
ERβ Up-Regulates E-Cadherin and Inhibits Cell Migration
A, SKOV-3 cells were transfected with nonspecific siRNA (NS siRNA), ERα siRNA or ERβ siRNA. Efficiencies of the siRNA were confirmed by Western blot with ERβ antibody. B, SKOV-3 cells were transfected with the siRNAs and subsequently treated with 10−7 m E2 for 48 h. E-cadherin (E-Cad) and β-actin were detected by Western blot. C, Scratch was made by scraping the monolayer 24 h after siRNA transfection, and cells were incubated with medium containing vehicle (Cont) or 10−7 m E2. Wound closure was photographed after 24 h. The amount of wound repair was expressed as uncovered area compared with initial uncovered area of vehicle-treated control at time zero. *, P < 0.05 compared with control. Numerical values below each lane of the immunoblots represent quantification of the relative protein level by densitometry (normalized to β-actin protein level). Cont, Control.
As an alternative approach, we introduced ERβ (pRST7-ERβ) into BG-1 cells by transient transfection. The overexpression of ERβ was verified by Western blotting (Fig. 8A). Figure 8, B and C, shows that the expression of ERβ significantly inhibited the E2-mediated down-regulation of E-cadherin protein and gene transcription. As expected, reduced Snail and Slug protein expressions could be observed in the ERβ-transfected cells (Fig. 8B). ERβ overexpression also inhibited the EMT and migration induced by E2 (Fig. 8, D and E). These results suggest that ERβ may function as a negative modulator of ERα in ovarian cancer cell metastasis.
Fig. 8.

ERβ Opposes ERα-Induced EMT
A, BG-1 cells were transfected with pRST7-ERβ or without plasmid DNA (mock). Cell lysates were extracted for ERβ detection by Western blot. B, BG-1 cells were transfected for 8 h and subsequently treated with 10−7 m E2 or vehicle (Cont) for an additional 48 h. E-cadherin (E-Cad), Snail, Slug, and β-actin were detected by Western blot. C, Cells were transfected with pRST7-ERβ, E-cadherin promoter construct, and pSV-βGal. Eight hours after transfection, cells were incubated with vehicle (Cont) or 10−7 m E2 for an additional 48 h. The luciferase activities were calculated relative to the promoterless vector (pGL3-Basic) and expressed as fold change relative to vehicle control. D, Morphological changes of BG-1 cells transfected with pRST7-ERβ or without plasmid DNA (mock) were also observed by phase-contrast microscope, and representative photographs are shown. Original magnification, ×200. E, Scratch was made by scraping the monolayer 24 h after transfection, and cells were incubated with medium containing vehicle (Cont) or 10−7 m E2. Wound closure was photographed after 24 h. The amount of wound repair was expressed as uncovered area compared with initial uncovered area of vehicle-treated control at time zero. Values are the mean ± sd of three separate experiments. *, P < 0.05 compared with control. Numerical values below each lane of the immunoblots represent quantification of the relative protein level by densitometry (normalized to β-actin protein level). Cont, Control.
DISCUSSION
The role of estrogen in ovarian tumorigenesis remains unclear and controversial. However, compelling evidence associates estrogen with increased ovarian cancer incidence (27, 28, 31). Clinical and epidemiological studies have shown that long-term use of estrogen replacement therapy increases the risk of ovarian cancer (38). Breastfeeding and use of oral contraceptives, which appear protective in a number of studies, are associated with reduced serum concentration of estradiol (22, 23). Experimental results also implicate a role of estrogen in ovarian carcinogenesis. Cell survival and proliferation could be enhanced by estrogen both in vivo and in vitro (25, 26, 29, 30). Given the importance of estrogen demonstrated by these studies, understanding the mechanistic basis through which it controls ovarian tumorigenesis will have profound biological and medical implications. In fact, the proliferative effects of estrogen were found to be mediated through regulation of genes involved in growth control, such as c-myc, bcl-2 and ezrin (25, 30, 39). In contrast to the role in regulating cell proliferation, very little is known about whether and how estrogen promotes cell metastasis. Moreover, previous data on the effects of cell invasiveness and motility by estrogen were inconsistent (30, 40). In the present study, our findings extend the understanding of the involvement of estrogen in EMT and regulation of EMT-associated proteins. We provided supporting evidence, to our knowledge for the first time, that E2 can induce EMT of ovarian cancer cells. We also demonstrated that E2 enhanced cellmigratory ability through transcriptional regulation of E-cadherin and its repressors, Snail and Slug. Moreover, our results clearly established ERα as an essential mechanistic link in mediating the induction of EMT by E2. Of importance, our data indicated that ERβ counteracts ERα signaling in mediating the metastatic responses to E2.
EMT is a prerequisite for tumor progression by which epithelial cancer cells modulate their phenotypes and acquire metastatic capabilities. The E2-induced phenotypic changes demonstrated in our data are consistent with changes seen during EMT (2). Changes in cell morphology, from a cobblestone-like appearance to more elongated shape, cell dissociation with reduced cell-cell contacts, and pseudopodia formation were evident. Exactly how E2 promotes EMT and cell motility remains to be elucidated. In this regard, our data implied that the E2-induced EMT is, at least in part, mediated through repression of E-cadherin. Reduced expression of E-cadherin has been found in advanced-stage ovarian tumors and ectopic expression of E-cadherin inhibited cell invasion in vitro (41, 42, 43). Interestingly, although unlike other types of epithelial malignancy, E-cadherin expression is frequently acquired in primary ovarian carcinomas for epithelial differentiation, it is repressed in late-stage tumors for metastasis (44, 45). However, the cause for this subsequent loss of expression remains unclear. In ovarian cancer cells, E-cadherin gene mutation is infrequent (11). Rather, gene methylation and up-regulation of E-cadherin transcriptional repressors have been reported (46, 47). From our present data, estrogen might represent one of the mechanisms that silence E-cadherin in ERα-positive ovarian cancer cells. In agreement with this, previous studies also suggest a role for estrogen in regulating E-cadherin expression, but the underlying mechanism has not been fully investigated (48, 49). Because numerous transcription repressors have been characterized in controlling E-cadherin transcription, it is likely that the repressors involved are dependent on cell type and cell context. Our data clearly revealed that two transcription factors, Snail and Slug, act in concert in the suppression of E-cadherin promoter activity by E2.
The Snail gene family of transcription factors is best known for its ability to trigger EMT through repression of E-cadherin (13, 14, 50). Snail and Slug are also involved in the regulation of tight junction proteins (51, 52), matrix metalloproteinases (53, 54), and cytokeratins (55). High expression of Snail and Slug is often found in metastatic ovarian tumor cells compared with primary tumors (56). Consistent with these observations, we demonstrated the importance of Snail and Slug in E2-induced cell migration by inhibiting transcription of E-cadherin in ovarian cancer cells. The identification of Snail and Slug as novel estrogen-responsive genes in this report helps to confirm the role of estrogen in ovarian carcinogenesis. According to Elloul et al. (56), it has been suggested that Snail is regulated at the posttranscriptional level in ovarian carcinoma, whereas Slug is probably regulated transcriptionally. Here we found that the up-regulation of both Snail and Slug by E2 was predominantly mediated through activation of their promoter activities. The ERs elicit gene transcription by interacting with the classical estrogen response element (ERE) or non-ERE elements that bind heterologous transcription factors, including activator protein 1 sites (57), Sp1 sites (58) and cAMP-response elements (59). Of particular note, putative ERE sites can be found in the Snail promoter region (60), but not in the Slug promoter. By sequence homology search, we found several potential Sp-1, activator protein 1, and cAMP-response element sites within the 5′-flanking regions of Snail and Slug. In addition, ER might also be recruited to the 3′-enhancer downstream of the coding region, constituting a combinatorial network for transcription regulation (61). Whether E2 activates the two promoters through 5′-ERE, non-ERE sites, or 3′-regulatory region has yet to be determined.
In metastatic ovarian cancer cells, only ERα is present and ERβ expression is often lost (62, 63, 64, 65). This implicates that the ER subtypes may play distinct roles in ovarian carcinogenesis. However, their functions and the molecular mechanisms in ovarian carcinogenesis have just begun to be unveiled. ERα promotes ovarian cancer cell growth, and several potential ERα-regulated genes have been identified by cDNA microarray (66). These genes include junctional and extracellular matrix proteins (e.g. cadherin 6, fibronectin, and vimentin), metastasis-related proteinases (e.g. matrix metalloproteineses and urokinase-type plasminogen activator), and regulators of cell cycle (e.g. cyclin B1) (66). Consistently, it has been shown in other cancer types that ERα promotes carcinogenesis through activation of genes linked to cell proliferation and invasion, such as cyclin D1 (67), c-myc (68), and fibulin-1 C (69). In contrast, overexpression of ERβ exerts a protective role against cancer development by inducing apoptosis and inhibiting migration/invasion (68, 70, 71). Therefore, overexpression of ERα may represent a more metastatic behavior of cancer cells. Accordingly, we provided several lines of evidence that induction of EMT by E2 is mediated specifically by ERα, but not by ERβ. First, ERα is predominant whereas ERβ is barely detectable in BG-1 cells (our unpublished data and Ref. 65), implicating ERα as the major mediator of estrogen signaling in the cells. Second, the involvement of ERα was confirmed by pharmacological agonists and siRNAs. Third, the transforming capabilities of E2 could be observed in SKOV-3 cells with ERα restored by overexpression. This cell line is ERα negative but ERβ positive due to mutation in the ERα transcript and is insensitive to estrogen with respect to cell growth (24, 37). In contrast to our observations, a previous study suggested an ERα-independent pathway in regulation of E-cadherin and Snail by E2 in ovarian cancer cells (48). There is no definite explanation for this discrepancy yet, and more extensive experiments are needed to confirm the mechanisms underlying E2 effects in those cells. It is also possible that, in some cell types, regulation of these genes by E2 might depend on an alternative pathway. ER-independent, nongenomic signaling of E2 has been proposed to occur through membrane-localized G protein-coupled receptor GPR30 (72, 73). Whether this signaling cascade is involved warrants further investigation. Intriguingly, another remarkable finding in this study is the demonstration of the tumor suppressor activity of ERβ by counteracting ERα signaling. ERβ induced E-cadherin expression and inhibited migration of the ERα-negative SKOV-3 cells. Moreover, introduction of ERβ potently blocked the pro-metastatic effects of E2 in BG-1 cells. It is interesting to note that ERβ negatively regulates the decrease of E-cadherin by ERα, and this counteracting effect of ERβ was mediated at the promoter level. Indeed, it has been proposed that modulation of ERα-targeted genes by ERβ may contribute to the contrasting actions between the two ER subtypes (67, 74). Our data also suggest that this might be the case in our paradigm because ERα is responsible for E2-induced Snail and Slug expression, which could be reversed by ERβ. The molecular mechanism underlying this differential regulation by each ER subtype is currently under investigation in our laboratory.
Given the indispensable role of EMT in promoting progression of many carcinomas, it is important to unravel the mechanisms that govern this process. Our findings herein showed that E2 can enhance ovarian tumor cell motility by triggering EMT through ERα. The induction of cell transformation in vitro and the identification of E-cadherin, Snail, and Slug as novel transcriptional targets of estrogen signaling confirm the carcinogenicity of the hormone. Our discovery that ERα and ERβ differentially mediate the EMT process provides a possible explanation for the observed increase in the ERα:ERβ expression ratio during ovarian carcinogenesis. It also carries clinical implications for selective targeting of the ERs in therapeutic and prevention strategies against ovarian cancer.
MATERIALS AND METHODS
Cell Culture
The human ovarian adenocarcinoma cell line BG-1 was kindly provided by Dr. K. S. Korach (National Institute of Environmental Health Sciences, NIH, Research Triangle Park, NC) (75). The SKOV-3 ovarian adenocarcinoma cell line was obtained from American Type Culture Collection (Manassas, VA). The cells were maintained in DMEM/F12 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 mg/ml streptomycin (Life Technologies, Inc., Gaithersburg, MD) in a humidified atmosphere of 5% CO2 at 37 C.
Treatments
Cells were stripped from endogenous steroids by changing the medium to phenol red-free DMEM/F12 containing 10% charcoal-dextran stripped fetal bovine serum (HyClone Laboratories, Inc., Logan, UT) 48 h before treatment. Cells were then incubated in fresh medium with E2 (Sigma, St. Louis, MO), ICI (ER antagonist) (Tocris, Ballwin, MO), alone or in combination for indicated times. In some cases, cells were treated with either ERα agonist PPT or ERβ agonist DPN for 48 h (Tocris). Control cultures received the same volume of ethanol. To study mRNA stability, cells were treated with ethanol or E2 for 1 h. Actinomycin D (5 μg/ml) (Sigma) was then added to the cultures, and total RNA was prepared at the times indicated for up to 8 h. All treatments were performed in duplicate or triplicate in each experiment.
Transient Transfection
Snail- and Slug-specific siRNA oligos (predesignated siRNA) were purchased from QIAGEN (Valencia, CA). Sense sequence of Snail no. 1: 5′-GCG AGC UGC AGG ACU CUA A-3′; Snail no. 2: 5′-GGU GUG ACU AAC UAU GCA A-3′; Slug no. 1: 5′-GGA CCA CAG UGG CUC AGA A-3′; and Slug no. 2: 5′-CUC CGA AGC CAA AUG ACA A-3′. ERα- and ERβ-specific siRNAs were purchased from Invitrogen (Burlington, Ontario, Canada). Sense sequence of ERα: 5′-GGG CUC UAC UUC AUC GCA U-3′; ERβ: 5′-GCA GAC CAC AAG CCC AAA U-3′. Cells cultured in six-well plates were transfected with siRNA duplexes using Lipofectamine 2000 (Invitrogen). Nonspecific siRNA duplexes were used as negative controls (QIAGEN). Cells were treated for 48 h to allow maximum knockdown.
Full-length human ERα (pCMV5-ERα) and ERβ (pRST7-ERβ) expression plasmids were kindly provided by Dr. B. S. Katzenellenbogen (Department of Molecular and Integrative Physiology, University of Illinois at Urbana Champaign, IL) and Dr. D. P. McDonnell (Department of Pharmacology and Cancer Biology, Duke University Medical Center, Durham, NC), respectively.
Reverse Transcription and Real-Time Quantitative PCR
Total RNA was isolated using TRIzol reagent (Invitrogen). Single-stranded cDNA was synthesized from 2 μg of total RNA (Amersham Biosciences, Baie d'Urfé, Quebec, Canada). Quantitative real-time PCR was carried out using ABI Prism 7300 Sequence Detector System (Applied Biosystems, Foster City, CA) and SYBR green PCR master mix (Applied Biosystems). Gene-specific primers were designed using Applied Biosystems’ Primer Express 3.0 software. The following oligonucleotides (Invitrogen) were used for PCR amplification: E-cadherin, sense, 5′-ACA GCC CCG CCT TAT GAT T-3′; and antisense, 5′-TCG GAA CCG CTT CCT TCA-3′; Snail, sense, 5′-CCC CAA TCG GAA GCC TAA CT-3′; and antisense, 5′-GCT GGA AGG TAA ACT CTG GAT TAG A-3′; and Slug, sense, 5′-TTC GGA CCC ACA CAT TAC CT-3′; and antisense, 5′-GCA GTG AGG GCA AGA AAA AG-3′. The relative mRNA level was normalized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels with the following specific primers: 5′-CCT CCC GCT TCG CTC TCT-3′ (forward), 5′-TGG CGA CGC AAA AGA AGA T-3′ (reversed). The thermal profile consisted of 2 min at 50 C, 10 min at 95 C, followed by 40 cycles of 15 sec at 95 C and 1 min at 60 C. The melting temperature profiles of amplicons were determined to show the specificity of amplification.
Immunoblot Analysis
Cells were harvested in lysis buffer (1% Triton-X, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate) with protease inhibitors cocktail (Sigma) and protein concentrations determined by the Bradford protein assay (Bio-Rad Laboratories, Inc., Hercules, CA). Protein (20 μg) was subjected to SDS-PAGE followed by electrotransfer onto nitrocellulose membrane. Membranes were incubated overnight at 4 C with mouse monoclonal human E-cadherin antibody (1:2000) (BD Transduction Laboratories, Heidelberg, Germany), human Snail antibody (1:1000) (Abgent, San Diego, CA), human Slug antibody (1:1000) (Abgent), human ERα antibody (1:1000), and ERβ antibody (1:1000) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Blots were reprobed with anti-β-actin antibody (1:2000) (Santa Cruz Biotechnology) to confirm equal loading. The immunocomplex was detected with appropriate horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences) and visualized using enhanced chemiluminescence detection system (ECL) (Amersham Biosciences). All blots were repeated in at least three different experiments. Quantitative densitometry was performed using Scion Image software (Scion Corp., Frederick, MD).
Reporter Gene Assay
Luciferase reporter vector containing human E-cadherin promoter (−178 to +66) was kindly provided by Dr. Yoshito Ihara (Nagasaki University, Japan) (76). Plasmid containing the human Slug promoter (−3371/+1) was a kind gift of Dr. Kaname Kawajiri (Research Institute for Clinical Oncology, Saitama, Japan) (77). The human Snail promoter reporter plasmid (−869/+59) was a generous gift of Dr. Antonio Garcia de Herreros (Universitat Pompeu Fabra, Barcelona, Spain) (78). All plasmids were cloned in the promoterless pGL3-Basic vector. Cells were transfected with 1 μg of each firefly luciferase reporter construct together with 0.5 μg pSV-βGal plasmid as normalization reference for transfection efficiency. Transfections were performed with Lipofectamine 2000 (Invitrogen). Cells were treated with E2, ICI, or vehicle 6 h after transfection. Where indicated, cells were also cotransfected with Snail or Slug siRNA. Cells transfected with promoterless vector (pGL3-Basic) were used as control for background luciferase activity. Cells were then harvested in reporter lysis buffer (Promega Corp., Madison, WI) at designated time points. Luciferase activity and β-galactosidase enzyme activity were determined with luciferase reagent and the β-galactosidase enzyme assay system (Promega). Luciferase units were calculated as luciferase activity/β-galactosidase activity and are presented as the mean ± sd of three individual experiments with triplication. The fold change was calculated by comparison with the promoterless pGL3-Basic.
Scratch Assay
Cells were grown to confluence and formed a monolayer covering the surface of the entire plate. Cells were kept in medium with appropriate treatments for 24 h before scratching. A linear scratch (∼0.7 mm wide) was made gently with sterile pipette tip across the diameter of the well and rinsed with PBS to remove debris. Fresh media for continued drug treatment were then added. For each well, at least six pictures were taken with microscope at a magnification of ×10 at three time points, 0, 4, and 24 h after scratch. The percentage of nonrecovered wound area was calculated by dividing the nonrecovered area after treatments by the initial wound area at time zero.
Statistics
All assays were repeated at least three times in duplicate or triplicate, and the graphic data were presented as the mean ± sd. Statistical analysis was performed where appropriate using the Student’s t test or ANOVA followed by Tukey’s post hoc test (GraphPad Software, San Diego, CA). Differences with P < 0.05 were considered to be statistically significant.
Acknowledgments
We thank Dr. Nelly Auersperg for valuable comments on the manuscript and Dr. Takayo Ota for technical assistance. We thank Dr. Yoshito Ihara for the human E-cadherin promoter, Dr. Kaname Kawajiri for human Slug promoter, Dr. Antonio Garcia de Herreros for human Snail promoter, Dr. Benita S. Katzenellenbogen for pCMV5-ERα overexpression plasmid, Dr. Donald P. McDonnell for pRST7-ERβ overexpression plasmid, and Dr. J. Larry Jameson for ERE luciferase vector (thymidine kinase-ERE-Luc).
NURSA Molecule Pages:
Ligands: 17β-Estradiol | Fulvestrant;
Nuclear Receptors: ERα | ERβ.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research (to P.C.K.L.) and the Hong Kong Research Grants Council (to A.S.T.W.).
Disclosure Statement: The authors have nothing to disclose.
First Published Online June 11, 2008
Abbreviations: DPN, Diarylpropionitrile; E2, 17β-estradiol; EMT, epithelial-mesenchymal transition; ER, estrogen receptor; ERE, estrogen response element; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ICI, ICI 182,780; PPT, propyl pyrazole triol; siRNA, small interfering RNA.
References
- 1.Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC 2001. Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev 22:255–288 [DOI] [PubMed] [Google Scholar]
- 2.Thiery JP 2002. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2:442–454 [DOI] [PubMed] [Google Scholar]
- 3.Lee JM, Dedhar S, Kalluri R, Thompson EW 2006. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 172:973–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moreno-Bueno G, Cubillo E, Sarrio D, Peinado H, Rodriguez-Pinilla SM, Villa S, Bolos V, Jorda M, Fabra A, Portillo F, Palacios J, Cano A 2006. Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer Res 66:9543–9556 [DOI] [PubMed] [Google Scholar]
- 5.Behrens J 2000. Control of β-catenin signaling in tumor development. Ann NY Acad Sci 910:21–33; discussion 33–25 [DOI] [PubMed] [Google Scholar]
- 6.Hu XC, Loo WT, Chow LW 2002. E-cadherin promoter methylation can regulate its expression in invasive ductal breast cancer tissue in Chinese woman. Life Sci 71:1397–1404 [DOI] [PubMed] [Google Scholar]
- 7.Mitselou A, Ioachim E, Peschos D, Charalabopoulos K, Michael M, Agnantis NJ, Vougiouklakis T 2007. E-cadherin adhesion molecule and syndecan-1 expression in various thyroid pathologies. Exp Oncol 29:54–60 [PubMed] [Google Scholar]
- 8.Hirata K, Ajiki T, Okazaki T, Horiuchi H, Fujita T, Kuroda Y 2006. Frequent occurrence of abnormal E-cadherin/β-catenin protein expression in advanced gallbladder cancers and its association with decreased apoptosis. Oncology 71:102–110 [DOI] [PubMed] [Google Scholar]
- 9.Roviello F, Corso G, Pedrazzani C, Marrelli D, De Falco G, Berardi A, Garosi L, Suriano G, Vindigni C, De Stefano A, Leoncini L, Seruca R, Pinto E 2007. Hereditary diffuse gastric cancer and E-cadherin: description of the first germline mutation in an Italian family. Eur J Surg Oncol 33:448–451 [DOI] [PubMed] [Google Scholar]
- 10.Hennig G, Lowrick O, Birchmeier W, Behrens J 1996. Mechanisms identified in the transcriptional control of epithelial gene expression. J Biol Chem 271:595–602 [DOI] [PubMed] [Google Scholar]
- 11.Risinger JI, Berchuck A, Kohler MF, Boyd J 1994. Mutations of the E-cadherin gene in human gynecologic cancers. Nat Genet 7:98–102 [DOI] [PubMed] [Google Scholar]
- 12.Tamura G, Yin J, Wang S, Fleisher AS, Zou T, Abraham JM, Kong D, Smolinski KN, Wilson KT, James SP, Silverberg SG, Nishizuka S, Terashima M, Motoyama T, Meltzer SJ 2000. E-Cadherin gene promoter hypermethylation in primary human gastric carcinomas. J Natl Cancer Inst 92:569–573 [DOI] [PubMed] [Google Scholar]
- 13.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA 2000. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2:76–83 [DOI] [PubMed] [Google Scholar]
- 14.Bolos V, Peinado H, Perez-Moreno MA, Fraga MF, Esteller M, Cano A 2003. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 116:499–511 [DOI] [PubMed] [Google Scholar]
- 15.Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F 2001. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7:1267–1278 [DOI] [PubMed] [Google Scholar]
- 16.Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A 2001. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem 276:27424–27431 [DOI] [PubMed] [Google Scholar]
- 17.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA 2004. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117:927–939 [DOI] [PubMed] [Google Scholar]
- 18.Nieto MA 2002. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol 3:155–166 [DOI] [PubMed] [Google Scholar]
- 19.Hajra KM, Chen DY, Fearon ER 2002. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res 62:1613–1618 [PubMed] [Google Scholar]
- 20.Jiao W, Miyazaki K, Kitajima Y 2002. Inverse correlation between E-cadherin and Snail expression in hepatocellular carcinoma cell lines in vitro and in vivo. Br J Cancer 86:98–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurrey NK, K A, Bapat SA 2005. Snail and Slug are major determinants of ovarian cancer invasiveness at the transcription level. Gynecol Oncol 97:155–165 [DOI] [PubMed] [Google Scholar]
- 22.Brekelmans CT 2003. Risk factors and risk reduction of breast and ovarian cancer. Curr Opin Obstet Gynecol 15:63–68 [DOI] [PubMed] [Google Scholar]
- 23.Runnebaum IB, Stickeler E 2001. Epidemiological and molecular aspects of ovarian cancer risk. J Cancer Res Clin Oncol 127:73–79 [DOI] [PubMed] [Google Scholar]
- 24.Lau KM, Mok SC, Ho SM 1999. Expression of human estrogen receptor-α and -β, progesterone receptor, and androgen receptor mRNA in normal and malignant ovarian epithelial cells. Proc Natl Acad Sci USA 96:5722–5727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi KC, Kang SK, Tai CJ, Auersperg N, Leung PC 2001. Estradiol up-regulates antiapoptotic Bcl-2 messenger ribonucleic acid and protein in tumorigenic ovarian surface epithelium cells. Endocrinology 142:2351–2360 [DOI] [PubMed] [Google Scholar]
- 26.Bai W, Oliveros-Saunders B, Wang Q, Acevedo-Duncan ME, Nicosia SV 2000. Estrogen stimulation of ovarian surface epithelial cell proliferation. In Vitro Cell Dev Biol Anim 36:657–666 [DOI] [PubMed] [Google Scholar]
- 27.Gambacciani M, Monteleone P, Sacco A, Genazzani AR 2003. Hormone replacement therapy and endometrial, ovarian and colorectal cancer. Best Pract Res Clin Endocrinol Metab 17:139–147 [DOI] [PubMed] [Google Scholar]
- 28.Rao BR, Slotman BJ 1991. Endocrine factors in common epithelial ovarian cancer. Endocr Rev 12:14–26 [DOI] [PubMed] [Google Scholar]
- 29.Silva EG, Tornos C, Deavers M, Kaisman K, Gray K, Gershenson D 1998. Induction of epithelial neoplasms in the ovaries of guinea pigs by estrogenic stimulation. Gynecol Oncol 71:240–246 [DOI] [PubMed] [Google Scholar]
- 30.Song J, Fadiel A, Edusa V, Chen Z, So J, Sakamoto H, Fishman DA, Naftolin F 2005. Estradiol-induced ezrin overexpression in ovarian cancer: a new signaling domain for estrogen. Cancer Lett 220:57–65 [DOI] [PubMed] [Google Scholar]
- 31.Greiser CM, Greiser EM, Doren M 2007. Menopausal hormone therapy and risk of ovarian cancer: systematic review and meta-analysis. Hum Reprod Update 13:453–463 [DOI] [PubMed] [Google Scholar]
- 32.Giacalone PL, Daures JP, Ouafik L, Martin PM, Laffargue F, Maudelonde T 2003. Steroids and adrenomedullin growth patterns in human ovarian cancer cells: estrogenic-regulation assay. Gynecol Oncol 91:651–656 [DOI] [PubMed] [Google Scholar]
- 33.Stopper H, Schmitt E, Gregor C, Mueller SO, Fischer WH 2003. Increased cell proliferation is associated with genomic instability: elevated micronuclei frequencies in estradiol-treated human ovarian cancer cells. Mutagenesis 18:243–247 [DOI] [PubMed] [Google Scholar]
- 34.Hirohashi S, Kanai Y 2003. Cell adhesion system and human cancer morphogenesis. Cancer Sci 94:575–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun J, Meyers MJ, Fink BE, Rajendran R, Katzenellenbogen JA, Katzenellenbogen BS 1999. Novel ligands that function as selective estrogens or antiestrogens for estrogen receptor-α or estrogen receptor-β. Endocrinology 140:800–804 [DOI] [PubMed] [Google Scholar]
- 36.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA 2001. Estrogen receptor-β potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem 44:4230–4251 [DOI] [PubMed] [Google Scholar]
- 37.Hua W, Christianson T, Rougeot C, Rochefort H, Clinton GM 1995. SKOV3 ovarian carcinoma cells have functional estrogen receptor but are growth-resistant to estrogen and antiestrogens. J Steroid Biochem Mol Biol 55:279–289 [DOI] [PubMed] [Google Scholar]
- 38.Lacey JV, Jr., Mink PJ, Lubin JH, Sherman ME, Troisi R, Hartge P, Schatzkin A, Schairer C 2002. Menopausal hormone replacement therapy and risk of ovarian cancer. JAMA 288:334–341 [DOI] [PubMed] [Google Scholar]
- 39.Chien CH, Wang FF, Hamilton TC 1994. Transcriptional activation of c-myc proto-oncogene by estrogen in human ovarian cancer cells. Mol Cell Endocrinol 99:11–19 [DOI] [PubMed] [Google Scholar]
- 40.Hayashido Y, Lucas A, Rougeot C, Godyna S, Argraves WS, Rochefort H 1998. Estradiol and fibulin-1 inhibit motility of human ovarian- and breast-cancer cells induced by fibronectin. Int J Cancer 75:654–658 [DOI] [PubMed] [Google Scholar]
- 41.Voutilainen KA, Anttila MA, Sillanpaa SM, Ropponen KM, Saarikoski SV, Juhola MT, Kosma VM 2006. Prognostic significance of E-cadherin-catenin complex in epithelial ovarian cancer. J Clin Pathol 59:460–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Darai E, Scoazec JY, Walker-Combrouze F, Mlika-Cabanne N, Feldmann G, Madelenat P, Potet F 1997. Expression of cadherins in benign, borderline, and malignant ovarian epithelial tumors: a clinicopathologic study of 60 cases. Hum Pathol 28:922–928 [DOI] [PubMed] [Google Scholar]
- 43.Yuecheng Y, Hongmei L, Xiaoyan X 2006. Clinical evaluation of E-cadherin expression and its regulation mechanism in epithelial ovarian cancer. Clin Exp Metastasis 23:65–74 [DOI] [PubMed] [Google Scholar]
- 44.Maines-Bandiera SL, Auersperg N 1997. Increased E-cadherin expression in ovarian surface epithelium: an early step in metaplasia and dysplasia? Int J Gynecol Pathol 16:250–255 [DOI] [PubMed] [Google Scholar]
- 45.Veatch AL, Carson LF, Ramakrishnan S 1994. Differential expression of the cell-cell adhesion molecule E-cadherin in ascites and solid human ovarian tumor cells. Int J Cancer 58:393–399 [DOI] [PubMed] [Google Scholar]
- 46.Imai T, Horiuchi A, Wang C, Oka K, Ohira S, Nikaido T, Konishi I 2003. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am J Pathol 163:1437–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rathi A, Virmani AK, Schorge JO, Elias KJ, Maruyama R, Minna JD, Mok SC, Girard L, Fishman DA, Gazdar AF 2002. Methylation profiles of sporadic ovarian tumors and nonmalignant ovaries from high-risk women. Clin Cancer Res 8:3324–3331 [PubMed] [Google Scholar]
- 48.Ding JX, Feng YJ, Yao LQ, Yu M, Jin HY, Yin LH 2006. The reinforcement of invasion in epithelial ovarian cancer cells by 17 β-estradiol is associated with up-regulation of Snail. Gynecol Oncol 103:623–630 [DOI] [PubMed] [Google Scholar]
- 49.Oesterreich S, Deng W, Jiang S, Cui X, Ivanova M, Schiff R, Kang K, Hadsell DL, Behrens J, Lee AV 2003. Estrogen-mediated down-regulation of E-cadherin in breast cancer cells. Cancer Res 63:5203–5208 [PubMed] [Google Scholar]
- 50.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A 2000. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2:84–89 [DOI] [PubMed] [Google Scholar]
- 51.Ikenouchi J, Matsuda M, Furuse M, Tsukita S 2003. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci 116:1959–1967 [DOI] [PubMed] [Google Scholar]
- 52.Ohkubo T, Ozawa M 2004. The transcription factor Snail downregulates the tight junction components independently of E-cadherin downregulation. J Cell Sci 117:1675–1685 [DOI] [PubMed] [Google Scholar]
- 53.Jorda M, Olmeda D, Vinyals A, Valero E, Cubillo E, Llorens A, Cano A, Fabra A 2005. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J Cell Sci 118:3371–3385 [DOI] [PubMed] [Google Scholar]
- 54.Yokoyama K, Kamata N, Fujimoto R, Tsutsumi S, Tomonari M, Taki M, Hosokawa H, Nagayama M 2003. Increased invasion and matrix metalloproteinase-2 expression by Snail-induced mesenchymal transition in squamous cell carcinomas. Int J Oncol 22:891–898 [PubMed] [Google Scholar]
- 55.Tripathi MK, Misra S, Chaudhuri G 2005. Negative regulation of the expressions of cytokeratins 8 and 19 by SLUG repressor protein in human breast cells. Biochem Biophys Res Commun 329:508–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elloul S, Silins I, Trope CG, Benshushan A, Davidson B, Reich R 2006. Expression of E-cadherin transcriptional regulators in ovarian carcinoma. Virchows Arch 449:520–528 [DOI] [PubMed] [Google Scholar]
- 57.Webb P, Lopez GN, Uht RM, Kushner PJ 1995. Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol 9:443–456 [DOI] [PubMed] [Google Scholar]
- 58.Porter W, Saville B, Hoivik D, Safe S 1997. Functional synergy between the transcription factor Sp1 and the estrogen receptor. Mol Endocrinol 11:1569–1580 [DOI] [PubMed] [Google Scholar]
- 59.Sabbah M, Courilleau D, Mester J, Redeuilh G 1999. Estrogen induction of the cyclin D1 promoter: involvement of a cAMP response-like element. Proc Natl Acad Sci USA 96:11217–11222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moggs JG, Murphy TC, Lim FL, Moore DJ, Stuckey R, Antrobus K, Kimber I, Orphanides G 2005. Anti-proliferative effect of estrogen in breast cancer cells that re-express ERα is mediated by aberrant regulation of cell cycle genes. J Mol Endocrinol 34:535–551 [DOI] [PubMed] [Google Scholar]
- 61.Eeckhoute J, Carroll JS, Geistlinger TR, Torres-Arzayus MI, Brown M 2006. A cell-type-specific transcriptional network required for estrogen regulation of cyclin D1 and cell cycle progression in breast cancer. Genes Dev 20:2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rutherford T, Brown WD, Sapi E, Aschkenazi S, Munoz A, Mor G 2000. Absence of estrogen receptor-β expression in metastatic ovarian cancer. Obstet Gynecol 96:417–421 [DOI] [PubMed] [Google Scholar]
- 63.Chan KK, Wei N, Liu SS, Xiao-Yun L, Cheung AN, Ngan HY 2008. Estrogen receptor subtypes in ovarian cancer: a clinical correlation. Obstet Gynecol 111:144–151 [DOI] [PubMed] [Google Scholar]
- 64.Brandenberger AW, Tee MK, Jaffe RB 1998. Estrogen receptor α (ER-α) and β (ER-β) mRNAs in normal ovary, ovarian serous cystadenocarcinoma and ovarian cancer cell lines: down-regulation of ER-β in neoplastic tissues. J Clin Endocrinol Metab 83:1025–1028 [DOI] [PubMed] [Google Scholar]
- 65.Pujol P, Rey JM, Nirde P, Roger P, Gastaldi M, Laffargue F, Rochefort H, Maudelonde T 1998. Differential expression of estrogen receptor-α and -β messenger RNAs as a potential marker of ovarian carcinogenesis. Cancer Res 58:5367–5373 [PubMed] [Google Scholar]
- 66.O'Donnell AJ, Macleod KG, Burns DJ, Smyth JF, Langdon SP 2005. Estrogen receptor-α mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocr Relat Cancer 12:851–866 [DOI] [PubMed] [Google Scholar]
- 67.Liu MM, Albanese C, Anderson CM, Hilty K, Webb P, Uht RM, Price Jr RH, Pestell RG, Kushner PJ 2002. Opposing action of estrogen receptors α and β on cyclin D1 gene expression. J Biol Chem 277:24353–24360 [DOI] [PubMed] [Google Scholar]
- 68.Lazennec G, Bresson D, Lucas A, Chauveau C, Vignon F 2001. ER β inhibits proliferation and invasion of breast cancer cells. Endocrinology 142:4120–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moll F, Katsaros D, Lazennec G, Hellio N, Roger P, Giacalone PL, Chalbos D, Maudelonde T, Rochefort H, Pujol P 2002. Estrogen induction and overexpression of fibulin-1C mRNA in ovarian cancer cells. Oncogene 21:1097–1107 [DOI] [PubMed] [Google Scholar]
- 70.Treeck O, Pfeiler G, Mitter D, Lattrich C, Piendl G, Ortmann O 2007. Estrogen receptor β1 exerts antitumoral effects on SK-OV-3 ovarian cancer cells. J Endocrinol 193:421–433 [DOI] [PubMed] [Google Scholar]
- 71.Cheng J, Lee EJ, Madison LD, Lazennec G 2004. Expression of estrogen receptor β in prostate carcinoma cells inhibits invasion and proliferation and triggers apoptosis. FEBS Lett 566:169–172 [DOI] [PubMed] [Google Scholar]
- 72.Filardo EJ, Quinn JA, Frackelton Jr AR, Bland KI 2002. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol 16:70–84 [DOI] [PubMed] [Google Scholar]
- 73.Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Ando S, Maggiolini M 2007. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17β-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res 67:1859–1866 [DOI] [PubMed] [Google Scholar]
- 74.Lindberg MK, Moverare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, Ohlsson C 2003. Estrogen receptor (ER)-β reduces ERα-regulated gene transcription, supporting a “ying yang” relationship between ERα and ERβ in mice. Mol Endocrinol 17:203–208 [DOI] [PubMed] [Google Scholar]
- 75.Geisinger KR, Kute TE, Pettenati MJ, Welander CE, Dennard Y, Collins LA, Berens ME 1989. Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors. Cancer 63:280–288 [DOI] [PubMed] [Google Scholar]
- 76.Hayashida Y, Urata Y, Muroi E, Kono T, Miyata Y, Nomata K, Kanetake H, Kondo T, Ihara Y 2006. Calreticulin represses E-cadherin gene expression in Madin-Darby canine kidney cells via Slug. J Biol Chem 281:32469–32484 [DOI] [PubMed] [Google Scholar]
- 77.Ikuta T, Kawajiri K 2006. Zinc finger transcription factor Slug is a novel target gene of aryl hydrocarbon receptor. Exp Cell Res 312:3585–3594 [DOI] [PubMed] [Google Scholar]
- 78.Barbera MJ, Puig I, Dominguez D, Julien-Grille S, Guaita-Esteruelas S, Peiro S, Baulida J, Franci C, Dedhar S, Larue L, Garcia de Herreros A 2004. Regulation of Snail transcription during epithelial to mesenchymal transition of tumor cells. Oncogene 23:7345–7354 [DOI] [PubMed] [Google Scholar]