

Abstract
Cholesterol transport in the mitochondrial membrane, an essential step of steroid biosynthesis, is mediated by a protein complex containing the steroidogenic acute regulatory (StAR) protein. The importance of this transporter is underscored by mutations in the human StAR gene that cause lipoid congenital adrenal hyperplasia, male pseudohermaphroditism, and adrenal insufficiency. StAR transcription in steroidogenic cells is hormonally regulated and involves several transcription factors. The nuclear receptor NUR77 is present in steroidogenic cells, and its expression is induced by hormones known to activate StAR expression. We have now established that StAR transcription in cAMP-stimulated Leydig cells requires de novo protein synthesis and involves NUR77. We found that cAMP-induced NUR77 expression precedes that of StAR both at the mRNA and protein levels in Leydig cells. In these cells, small interfering RNA-mediated NUR77 knockdown reduces cAMP-induced StAR expression. Chromatin immunoprecipitation assays revealed a cAMP-dependent increase in NUR77 recruitment to the proximal StAR promoter, whereas transient transfections in MA-10 Leydig cells confirmed that NUR77 can activate the StAR promoter and that this requires an element located at −95 bp. cAMP-induced StAR and NUR77 expression in Leydig cells was found to require a Ca2+/calmodulin-dependent protein kinase (CaMK)-dependent signaling pathway. Consistent with this, we show that within the testis, CaMKI is specifically expressed in Leydig cells. Finally, we report that CaMKI transcriptionally cooperates with NUR77, but not steroidogenic factor 1, to further enhance StAR promoter activity in Leydig cells. All together, our results implicate NUR77 as a mediator of cAMP action on StAR transcription in steroidogenic Leydig cells and identify a role for CaMKI in this process.
THE STEROIDOGENIC ACUTE regulatory (StAR) protein gene encodes a protein implicated in the shuttling of cholesterol from the outer to the inner mitochondrial membrane, an essential step for the initiation of steroidogenesis. This crucial role of the StAR gene in humans is underscored by mutations leading to lipoid congenital adrenal hyperplasia accompanied by a loss in steroid synthesis in the gonads and adrenals (1). If no treatment is undertaken, this leads to death within days to weeks after birth due to deficiency in mineralocorticoid and glucocorticoid production (2). Moreover, because StAR is also required for testosterone production in testicular Leydig cells, StAR−/− male mice are pseudohermaphrodites exhibiting female genitalia (3, 4).
Chronologically, the hormonally mediated increase in steroid hormone biosynthesis occurs into two sequential steps: first, the acute response (within minutes) results in increased mobilization and delivery of cholesterol precursors to the inner mitochondrial membrane, and second, the chronic response (within hours) involves increased transcription and translation of genes encoding essential components of steroidogenesis (5, 6). Although the StAR gene is regulated by various hormones and locally produced paracrine/autocrine factors in Leydig cells (reviewed in Ref. 7), the main pathway regulating StAR expression, and consequently steroidogenesis in these cells, involves the pituitary gonadotropin LH, known to act primarily through the cAMP/protein kinase A (PKA) pathway (8). Activation of this pathway results in posttranslational modifications and/or de novo synthesis of transcription factors involved in StAR promoter activation. Other signaling pathways involved in the up-regulation of StAR expression include arachidonic acid metabolites (9, 10, 11), calcium-regulated signal transduction (12), chloride ion channels (13, 14), MAPK/ERKs (15, 16), and protein kinase C (17).
Much like the different signaling pathways, several transcription factors have also been shown to regulate StAR promoter activity. These include steroidogenic factor 1 (SF-1), GATA4, CCAAT/enhancer binding protein-β (C/EBPβ), sterol regulatory element-binding protein (SREBP), specificity protein 1 (SP1), cAMP response element (CRE)-binding protein (CREB)/CRE modulator (CREM), members of the activation protein 1 (AP-1) family (c-FOS and c-JUN), and DAX-1 (dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1; also known as NR0B1; reviewed in Refs. 6 , 18 , and 19). More recently, distal-less homeobox 5 (DLX5) has been reported to play a role in StAR gene expression by interacting with GATA4 (20). The involvement of newly synthesized transcription factors in hormone-induced StAR transcription remains a matter of debate. There have been reports suggesting that the initial period of LH/cAMP-induced StAR promoter activity does not require de novo protein synthesis (21, 22). On the other hand, other studies have concluded that the cAMP-mediated increase in StAR transcription is significantly reduced by inhibitors of protein translation (23, 24), suggesting that maximal induction of StAR transcription in response to hormonal stimulation might indeed require de novo synthesis of transcription factors.
None of the transcription factors identified so far as regulators of StAR transcription, however, is rapidly and strongly induced at the protein level in response to hormonal stimulation in Leydig cells. Rather, their activation appears to rely on posttranslational modifications. NUR77 [nerve growth factor induced-B (NGFI-B), TR3, nuclear receptor 4A1 (NR4A1)] is a member of the NUR77 family of orphan neural receptors, which also includes NURR1 (NR4A2) and neural orphan receptor 1 [NOR1 (NR4A3)]. Members of this family are characterized as immediate-early response genes, in that their expression is rapidly and strongly induced by various stimuli in numerous tissues (25, 26, 27), including hormonally stimulated steroidogenic cells (28, 29, 30, 31, 32, 33). Moreover, changes in NUR77 intracellular localization in response to certain stimuli (34) also modulate its transactivation potential. NUR77 is known to bind as a monomer to a regulatory element similar to that recognized by the nuclear receptor SF-1/NR5A1, which is known to regulate expression of many steroidogenic genes (35). This suggests that these two nuclear receptors might regulate a common set of genes. Indeed, NUR77 activates the promoter of several genes involved in testosterone biosynthesis in Leydig cells that are also known to be regulated by SF-1, including the human HSD3B2 (29, 36, 37), rat P450c17 (38), and mouse Hsd3b1 promoters (39). Because Nur77 is rapidly and strongly induced in response to hormonal stimulation, including LH in Leydig cells (29, 30, 33), and because it is known to bind elements similar to those recognized by SF-1, we hypothesized that NUR77 could constitute a de novo synthesized transcription factor involved in the LH/cAMP-dependent activation of the StAR promoter in steroidogenic cells.
In the present study, we have established the requirement of de novo protein synthesis in cAMP-induced StAR transcription. We also provide evidence that the immediate-early orphan nuclear receptor NUR77 regulates StAR expression and promoter activity in steroidogenic cells in response to cAMP and that this is mediated through a Ca2+/calmodulin-dependent protein kinase (CaMK) pathway.
RESULTS
Requirement of de Novo Protein Synthesis for StAR Expression in Leydig Cells
To determine whether StAR transcription in response to cAMP requires de novo protein synthesis, quantitative real-time PCR was used to quantify StAR mRNA levels in mouse MA-10 Leydig cells treated or not with cAMP in the presence or absence of an inhibitor of transcription, actinomycin-D (Act-D) or an inhibitor of translation, cycloheximide (CHX). In the presence of dibutyryl cAMP [(Bu)2-cAMP] (Fig. 1A) or forskolin (data not shown), a slight increase in StAR mRNA was first detected after 1 h. StAR mRNA levels then kept on rising to approximately 50-fold induction at 6 h (Fig. 1A, lanes 11–14). As expected, addition of the transcriptional inhibitor Act-D abrogated the (Bu)2-cAMP-mediated StAR transcription (Fig. 1A, lanes 22–28). Similar results were also obtained in the presence of 1 μm methyl α-amanitin oleate, a specific inhibitor of RNA polymerase II (data not shown). In the presence of CHX, we observed a significant decrease (∼50%) in the induction of StAR mRNA in response to (Bu)2-cAMP (Fig. 1A, compare lanes 40–42 with 12–14). Thus, these results indicate that de novo protein synthesis is required for maximal StAR transcription in response to cAMP signaling.
Fig. 1.

cAMP-Dependent Induction of Nur77 Precedes that of StAR
A, cAMP-induced StAR expression requires de novo protein synthesis. MA-10 Leydig cells were treated with (Bu)2-cAMP (0.5 mm), Act-D (8 μm), and CHX (25 μm) individually or in combination for the indicated times, and total RNA was isolated and used in quantitative real-time PCR using primers specific for StAR cDNA as described in Materials and Methods. B and C, MA-10 cells were treated with (Bu)2-cAMP (0.5 mm) as indicated, and expression of Nur77 (B) and SF-1 (C) was determined by real-time PCR. Results were corrected with the Rpl19 cDNA. Results are the mean of three individual experiments performed in duplicate (±sem). An asterisk indicates a statistically significant difference from the respective controls. D, Several experiments were performed where MA-10 and rat primary Leydig cells were treated with 0.5 mm (Bu)2-cAMP as indicated. For detection of NUR77 and SF-1, nuclear extracts (because of location in the nucleus) were prepared, whereas for StAR detection (because of location in mitochondria), whole-cell extracts were used. Western blots were done as described in Materials and Methods, and tubulin was used as a loading control. In the representative data shown here, the experiment to detect NUR77 was run separately from the other markers. However, all experiments were repeated at least three times and produced identical results.
Activation of StAR Transcription by NUR77
As for StAR, expression of the transcription factor NUR77 (NR4A1, NGFI-B) in steroidogenic cells is regulated by ACTH and LH (28, 29, 30, 31, 32, 33). To test the possibility that NUR77 could regulate StAR transcription, we first compared Nur77 and StAR expression patterns after (Bu)2-cAMP stimulation in the MA-10 Leydig cell line and in primary Leydig cell cultures. As we have previously reported (29), Nur77 expression is rapidly induced both at the mRNA and protein levels in a time-dependent manner in response to (Bu)2-cAMP (Fig. 1, B and D). Expression of the nuclear receptor SF-1 (NR5A1, Ad4BP), a well known regulator of StAR transcription (23), was weakly but significantly increased at the mRNA (∼2.5-fold, Fig. 1C) and protein (∼2-fold at 1 h once corrected for loading, Fig. 1D) levels in response to (Bu)2-cAMP. In agreement with a potential role for NUR77 in StAR transcription, we found that the induction of Nur77 in response to cAMP precedes that of StAR both at the mRNA and protein levels in MA-10 Leydig cells (Fig. 1, A, B, and D). This is also true when primary Leydig cell cultures were used (Fig. 1D). Thus, these results suggest that NUR77 might represent a de novo synthesized transcription factor involved in StAR transcription in response to cAMP signaling in Leydig cells. Furthermore, our data indicate that MA-10 Leydig cells constitute an appropriate alternative to primary Leydig cells to study the role of NUR77 in StAR transcriptional regulation.
The role of NUR77 in StAR transcription in response to cAMP stimulation was further assessed using small interfering RNA (siRNA). As shown in Fig. 2, siRNA that decreased Nur77 expression (Fig. 2, A and C) led to a 30% decrease in StAR mRNA (Fig. 2B) and protein (Fig. 2C) levels after 2 and 4 h of treatment with 10 μm forskolin (FSK), a potent activator of adenylate cyclase leading to production of physiological concentrations of cAMP. A scrambled siRNA, used as control, had no effect. It should be noted that FSK-mediated stimulation of Nur77 expression is transient compared with cAMP analogs (compare Fig. 1B with the white bars in Fig. 2A). This is due to the fact that cAMP analogs are more resistant to degradation by phosphodiesterases than is endogenously produced cAMP in response to FSK. In agreement with the previously described role for SF-1 in StAR expression (23), SF-1 siRNA also decreased StAR mRNA levels (supplemental Fig. S1, published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend. endojournals.org). These data indicate that NUR77 contributes to the induction of StAR expression in response to FSK/cAMP signaling in Leydig cells.
Fig. 2.

Knockdown of NUR77 Reduces the cAMP-Mediated Induction of StAR Expression
MA-10 Leydig cells were transfected with siRNA directed against Nur77 and then treated with FSK (10 μm) for the indicated time. Total RNA were isolated, and expression of Nur77 (A) and StAR (B) was determined by quantitative real-time PCR. Results were corrected with the Rpl19 cDNA. Results are the mean of three individual experiments performed in duplicate (±sem). An asterisk indicates a statistically significant difference. In C, Western blots were performed to assess the efficiency of Nur77 siRNA knockdown at the protein level (middle panel) and the impact on StAR protein level (top panel). Tubulin was used as control (lower panel).
NUR77 Activates the StAR Promoter
To test whether NUR77 could directly regulate StAR promoter activity in steroidogenic cells, we performed transient transfection assays in MA-10 Leydig and Y-1 adrenal cells. As shown in Fig. 3A, NUR77 as well as other NUR77 family members, NURR1 (NR4A2) and NOR1 (NR4A3), could activate a −902-bp mouse StAR reporter construct (3-fold in MA-10 and Y-1 cells). SF-1 and LRH-1 had no effect on StAR promoter activity in MA-10 cells (P > 0.05). This may be explained by the already high levels of SF-1 and LRH-1 in these cells (38, 40, 41) because in heterologous cells that do not express SF-1, SF-1 can activate the StAR promoter (42, 43, 44) (our unpublished data). SF-1 can activate the StAR promoter in MA-10 cells in the presence of cAMP analogs (45).
Fig. 3.

NUR77 Activates the StAR Promoter
A, Activation of the StAR promoter by NUR77. MA-10 Leydig cells (left panel) and Y-1 adrenal cells (right panel) were cotransfected with a −902- to +17-bp mouse StAR reporter construct along with either an empty expression vector (CTL; white bars) or expression vectors (250 ng) for NUR77 family members (NUR77, NURR1, and NOR-1; black bars), SF-1 (hatched bars), and LRH-1 (stippled bars) as indicated. The positions of the previously characterized binding sites for SF-1 within the −902- to +17-bp StAR promoter fragment (−890, −135, −95, and −42 bp) are represented by triangles. The number of experiments, each performed in duplicate, is indicated. Results are shown as fold activation over control (±sem). Activations statistically different from control are indicated by an asterisk. B, Localization of the Nur77-responsive element in the StARpromoter. To locate the NUR77-responsive element, MA-10 Leydig (left panel) and Y-1 adrenal (right panel) cells were cotransfected with various 5′ deletion constructs of the mouse StAR promoter (the 5′-end point of each construct is indicated on the left of the graph) with 250 ng of either an empty expression vector (white bars) or an expression vector for NUR77 (black bars). The number of experiments, each performed in duplicate, is indicated. Results are shown as fold activation over control (±sem).
Next, to locate the NUR77-responsive element (NBRE), 5′ progressive deletions of the mouse StAR promoter were generated and tested for NUR77 responsiveness in MA-10 and Y-1 cells (Fig. 3B). A deletion construct to −104 bp that no longer contains two previously characterized SF-1 elements (at −890 bp and −135 bp) was still significantly activated by NUR77 in both cell types. Additional deletion to −71 bp that removes the SF-1 element at −95 bp while retaining the proximal (−42 bp) SF-1 element, could no longer be activated by NUR77. These results indicate that the NBRE is located between −104 and −71 bp, a region containing a previously characterized SF-1 element that could also mediate NUR77 responsiveness (Fig. 4A). The importance of this element in NUR77-mediated StAR promoter activation was further analyzed by introducing a two-nucleotide (in italics) mutation (ATCCTTGA to ATAATTGA), known to prevent binding by nuclear receptors (29), in the context of the −902 bp StAR reporter. As shown in Fig. 4B, mutation of the SF-1 element at −95 bp resulted in a complete loss of NUR77-mediated StAR promoter activation in both MA-10 and Y-1 cells. Thus, the element at −95 bp is necessary to confer NUR77 responsiveness to the mouse StAR promoter. We have therefore renamed this element the NBRE/SF-1 element.
Fig. 4.

An Element at −95 bp Is Important for NUR77 Responsiveness of the StAR Promoter
A, Sequence of the NUR77-responsive element denoted as NBRE/SF-1 located at position −95 bp. The sequence is compared with the NBRE element from the human HSD3B2 and INSL3 promoters (29 36 37 92 ). B, The NBRE/SF-1 element at −95 bp is necessary for NUR77-dependent activation of the StAR promoter. MA-10 Leydig (left panel) and Y-1 adrenal (right panel) cells were cotransfected with 250 ng of either an empty expression vector (−, white bars) or an expression vector for NUR77 (+, black bars) along with either a wild-type −902- to +17-bp StAR reporter or a reporter harboring a two-nucleotide mutation in the NBRE/SF-1 element at −95 bp (CCATCCTTGA to CCATAATTGA). The mutated element is represented by a large X. The number of experiments, each performed in duplicate, is indicated. Results are shown as fold activation over control (±sem). Activations statistically different from control are indicated by an asterisk. C, MA-10 Leydig cells were transfected with the two −902- to +17-bp StAR reporter constructs described above (shown on the left) and treated with either vehicle (−, white bars) or 0.5 mm (Bu)2-cAMP (+, black bars) 4 h before harvesting. Results are shown as percent activity (±sem) relative to the activity of the −902-bp wild-type reporter in the absence of cAMP treatment (which was arbitrarily set at 100%). *, Statistically significant difference from the wild-type promoter in the absence of cAMP; **, statistically significant differences between cAMP-treated and untreated (for each reporter); ***, statistically significant difference between the fold induction of each reporter by cAMP, which is indicated on top of the graph.
The NBRE/SF-1 Element Is Required for Full Basal Activity and for Maximal cAMP Induction of the StAR Promoter
Because Nur77 participates in the cAMP-induced StAR expression in Leydig cells (Fig. 2) and because NUR77 can activate the StAR promoter via the NBRE/SF-1 element at −95 bp (Fig. 4B), we postulated that this element should contribute to the cAMP-dependent regulation of the StAR promoter. A StAR reporter construct harboring a two-nucleotide mutation in the −95-bp NBRE/SF-1 element was therefore transfected in MA-10 cells and tested for cAMP responsiveness. As shown in Fig. 4C, (Bu)2-cAMP treatment led to a 2.9-fold stimulation of the wild-type StAR promoter. Mutation of the NBRE/SF-1 element caused a 44% decrease in basal StAR promoter activity and a 24% decrease in cAMP responsiveness (2.2-fold). These results indicate that although an intact NBRE/SF-1 element is necessary for maximal stimulation, additional regulatory elements/transcription factors contribute to the cAMP regulation of the mouse StAR promoter. The strong decrease in basal activity could be attributed to a failure in binding of SF-1, an important regulator of StAR promoter activity (44, 45).
Association of NUR77 with the StAR Promoter
To test whether NUR77 is recruited to the StAR promoter in vivo in MA-10 and primary Leydig cells in response to (Bu)2-cAMP, chromatin immunoprecipitation (ChIP) assays were performed. As shown in Fig. 5A, association of NUR77 was considerably increased in both MA-10 and primary Leydig cells treated with (Bu)2-cAMP compared with unstimulated cells as quantified by real-time PCR. Through in vitro approaches (DNA precipitation and EMSA), we found that NUR77 only weakly binds to the −95-bp element (supplemental Fig. S2). Altogether, these data indicate that NUR77 is recruited to the proximal StAR promoter in response to cAMP stimulation in Leydig cells but poorly binds to the −95-bp element in vitro despite the necessity of this element for NUR77 responsiveness of the StAR promoter (Figs. 3B and 4B). This suggests that recruitment of NUR77 to the StAR promoter is likely mediated by interactions with other DNA-bound transcription factors.
Fig. 5.

NUR77 Is Recruited to the Proximal StAR Promoter
A, In vivo recruitment of NUR77 to the proximal mouse StAR promoter was determined by ChIP assays using MA-10 and rat primary Leydig cells. An aliquot (10%) of chromatin preparation before immunoprecipitation (input) was used as positive control. Chromatin was precipitated with a NUR77 antiserum (αNUR77, black bars) or a preimmune serum (IgG, white bars), which serves as negative control. A 258-bp fragment of the StAR promoter, encompassing the NBRE/SF-1 element, was amplified by real-time PCR. Results are presented as a ratio of StAR promoter-immunoprecipitated DNA to input DNA levels and are representative of three independent experiments. *, Statistically significant difference from the control. B, NUR77 phosphorylation is decreased in response to cAMP stimulation. MA-10 Leydig cells were treated with 0.5 mm (Bu)2-cAMP as indicated. Nuclear extracts were prepared, separated by SDS-PAGE, transferred to PVDF membrane, and immunodetected using antisera specific for phosphorylated forms of NUR77 (Phospho S340 and Phospho S350) or total NUR77. All experiments were repeated three times and produced identical results.
In addition to increased NUR77 expression in response to cAMP stimulation in MA-10 Leydig cells (Fig. 1) (29), the enhancement in NUR77 association with the StAR promoter (Fig. 5A) might also be the result of changes in the phosphorylation status of two serine residues (Ser340 and Ser350; amino acid numbers as per the rat NUR77 protein). Increased phosphorylation of NUR77 Ser350 has been associated with a decrease in NUR77 DNA binding activity (46). To assess the phosphorylation status of Ser340 and Ser350, Western blots were performed using NUR77 phosphospecific antibodies. As shown in Fig. 5B, (Bu)2-cAMP stimulation led to a decrease in Ser340 phosphorylation levels (upper panel), whereas phosphorylation of Ser350 was not altered (middle panel) despite the significant increase in total NUR77 protein levels (bottom panel). These results indicate that NUR77 phosphorylation status is altered in response to cAMP signaling in Leydig cells.
Requirement of the PKA- and CaMK-Dependent Signaling Pathways for StAR Expression in Leydig Cells
We next sought to identify the signaling pathway(s) that are involved in the transcriptional regulation of StAR by NUR77 in response to cAMP signaling in Leydig cells. This was achieved by using various signaling pathway inhibitors in MA-10 Leydig cells that have been stimulated with FSK. As shown in Fig. 6A, we found that the FSK-mediated induction in StAR mRNA levels could be prevented by 10 μm H-89, an inhibitor of PKA, and 20 μm KN-93, an inhibitor of CaMKs. PD98059, a blocker of MAPK action, led to a decrease in StAR mRNA levels, although this decrease was not statistically significant. Altogether, these data confirm the involvement of PKA in StAR transcription in Leydig cells and identify a previously undescribed role for the CaMK pathway in this process.
Fig. 6.

Involvement of PKA and CaMK in StAR Induction
MA-10 cells were treated with different signaling pathway inhibitors in the presence or absence of 10 μm FSK. Total RNA was harvested, and StAR expression was analyzed by quantitative real-time PCR. Rpl19 was used to standardize the results. Results are the mean of three individual experiments performed in duplicate (± sem). Differences statistically significant from control are indicated by an asterisk. B, Increase in NUR77 and StAR protein levels in response to FSK requires CaMK activity. MA-10 Leydig cells were preincubated with various inhibitors as indicated for 30 min before addition of vehicle (CTL), 10 μm FSK, or 0.5 mm (Bu)2-cAMP followed by a 2-h incubation. The same incubation time was used for both NUR77 and StAR. Where indicated, ddFSK, a FSK analog that cannot activate adenylate cyclase, was used instead of FSK. Data are representative of three individual experiments.
Because we found that NUR77 contributes to the FSK/cAMP-mediated increase in StAR transcription (Figs. 2–5) and because KN-93 blunts the FSK-induced StAR mRNA levels (Fig. 6A), we tested whether KN-93 had any effect on NUR77 induction in response to cAMP. As shown in Fig. 6B, of the inhibitors tested, KN-93 resulted in a significant decrease (>85%) in FSK-mediated increase in NUR77 protein levels (Fig. 6B, upper panel, lane 12). A concomitant decrease in StAR protein levels was observed in the presence of KN-93 (Fig. 6B, middle panel, lane 12) which is in agreement with the RNA data (Fig. 6A). The PKA inhibitor H-89, which partially inhibited FSK-induced StAR mRNA (Fig. 6A), also partly blocked FSK- and (Bu)2-cAMP-mediated induction of StAR protein levels while having no effect on NUR77 induction (Fig. 6B, lanes 9, 11, 23, and 24). Furthermore, MDL-12,330A, an irreversible inhibitor of adenylate cyclase, prevented FSK-mediated NUR77 and StAR induction (compare lanes 20 and 21). No increase in NUR77 and StAR protein levels was observed when 1,9-dideoxyforskolin (ddFSK), a biologically inactive FSK analog that does not stimulate adenylyl cyclase, was used (Fig. 6B, lane 18), confirming that FSK acts through activation of adenylate cyclase and production of cAMP. These results suggest that NUR77 and StAR expression are both regulated, at least in part, by a CaMK-dependent pathway in Leydig cells in response to FSK/adenylate cyclase/cAMP.
CaMKI Is Expressed in Testicular Leydig Cells
Although KN-93 is widely promoted and used as an inhibitor specific for CaMKII, data from the literature indicate that KN-93 is an efficient inhibitor not only of CaMKII but also CaMKI and CaMKIV (47, 48). Additional approaches were therefore used to determine which CaMK family member is involved in FSK/cAMP-induced NUR77 and StAR expression. We first performed RT-PCR for the different CaMKs using cDNAs from mouse testis, MA-10 Leydig cells, and mouse brain as positive control. As shown in Fig. 7A, CaMKI mRNA was easily detected in MA-10 Leydig cells, whereas a weaker signal was observed in whole testis. Although some members of the CaMKII subfamily (CaMKIIβ, CaMKIIδ, and CaMKIIγ) were also detected in MA-10 Leydig cells by RT-PCR (Fig. 7A), none could be immunodetected at the protein level by Western blot (data not shown). Detection of CaMKIV in whole testis but not in Leydig cells (Fig. 7A) is consistent with the fact that this CaMK is found specifically in germ cells (49).
Fig. 7.
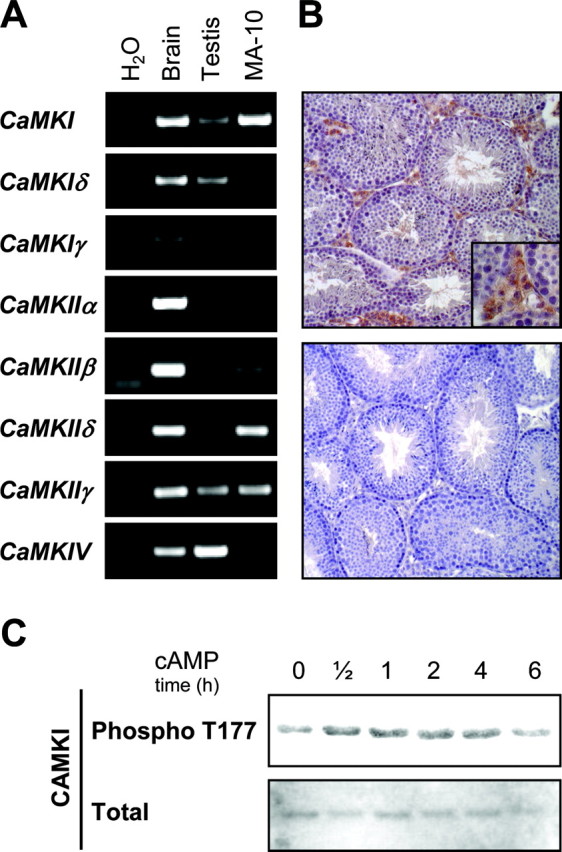
CaMKI Is Expressed in Leydig Cells
A, Semiquantitative RT-PCR using primers specific for different CaMK family members (listed in Table 1) was performed using cDNAs from mouse brain (used as positive control), mouse testis, and MA-10 Leydig cells. Results are representative of three individual experiments. B, Within the testis, CaMKI is specifically expressed in Leydig cells. Immunohistochemistry was performed on adult mouse testis sections using an anti-CaMKI antiserum (brownish staining in top panel). Omission of the primary antibody served as negative control (bottom panel). Magnifications, ×200 and ×400 (inset). C, MA-10 Leydig cells were treated with 0.5 mm (Bu)2-cAMP for the indicated times. Proteins were then isolated, separated by SDS-PAGE, transferred to PVDF membrane, and immunodetected using antisera specific for phospho-T177 CaMKI or total CaMKI.
To further confirm the expression of CaMKI in Leydig cells in vivo, we performed immunohistochemistry on adult mouse testis sections. In agreement with the RT-PCR results, CaMKI protein was specifically detected in interstitial Leydig cells (brownish staining in top panel of Fig. 7B). This labeling is specific because no signal was observed when the primary antibody was omitted (Fig. 7B, lower panel). Within Leydig cells, CaMKI appears to be located in the cytoplasmic compartment (Fig. 7B, inset in top panel), which is consistent with a previous report that established the intracellular localization of CaMKI (50). CaMKI is also expressed in MA-10 Leydig cells (Fig. 7C). In these cells, (Bu)2-cAMP treatment led to a slight but consistent increase in CaMKI threonine 177 phosphorylation (Phospho T177), which is known to stimulate its activity (51). Thus, within the testis, CaMKI appears to be the physiologically relevant CaMK in Leydig cells.
Cooperation between CaMKI and NUR77 Family Members in StAR Transcription
To directly assess the role of CaMK in the regulation of StAR transcription, MA-10 cells were cotransfected with a −902-bp StAR reporter construct along with expression vectors for wild-type CaMKI, CaMKII, and CaMKIV. Of the three CaMKs, only CaMKI significantly increased StAR promoter activity, up to 7-fold, which is similar to the activation observed with the cAMP analog (10-fold) (Fig. 8A). The combination of CaMKI and cAMP further enhanced StAR promoter activity to nearly 18-fold, whereas there were no such additive effects with CaMKII and CaMKIV (Fig. 8A). To better define the implication of CaMKI in StAR promoter activity, we used a calcium-independent constitutively active form of CaMKI (CaMKI CA) which is generated by removing an autoinhibitory domain (52). As expected, CaMKI CA was more efficient at activating the StAR promoter (14-fold compared with 4- to 7-fold for CaMKI WT) (Fig. 8B, white bars). Because NUR77 is likely a downstream effector of the FSK/cAMP pathway in StAR transcription in Leydig cells (Figs. 1–5) and because the cAMP pathway enhanced the CaMKI-dependent activation of the StAR promoter (Fig. 8A), we tested the possibility that CaMKI might directly cooperate with NUR77. As shown in Fig. 8B, the combination of NUR77 and CaMKI, either wild type or constitutively active, resulted in a synergistic activation of the StAR promoter (20- to 25-fold; black bars in Fig. 8B). A similar cooperation was also observed between CaMKI and the other NUR77 family members NURR1 and NOR-1 (Fig. 8B). No cooperation, however, was observed between CaMKI and SF-1 (Fig. 8B). Thus, transcriptional cooperation with CaMKI on the StAR promoter is specific to NUR77 family members.
Fig. 8.
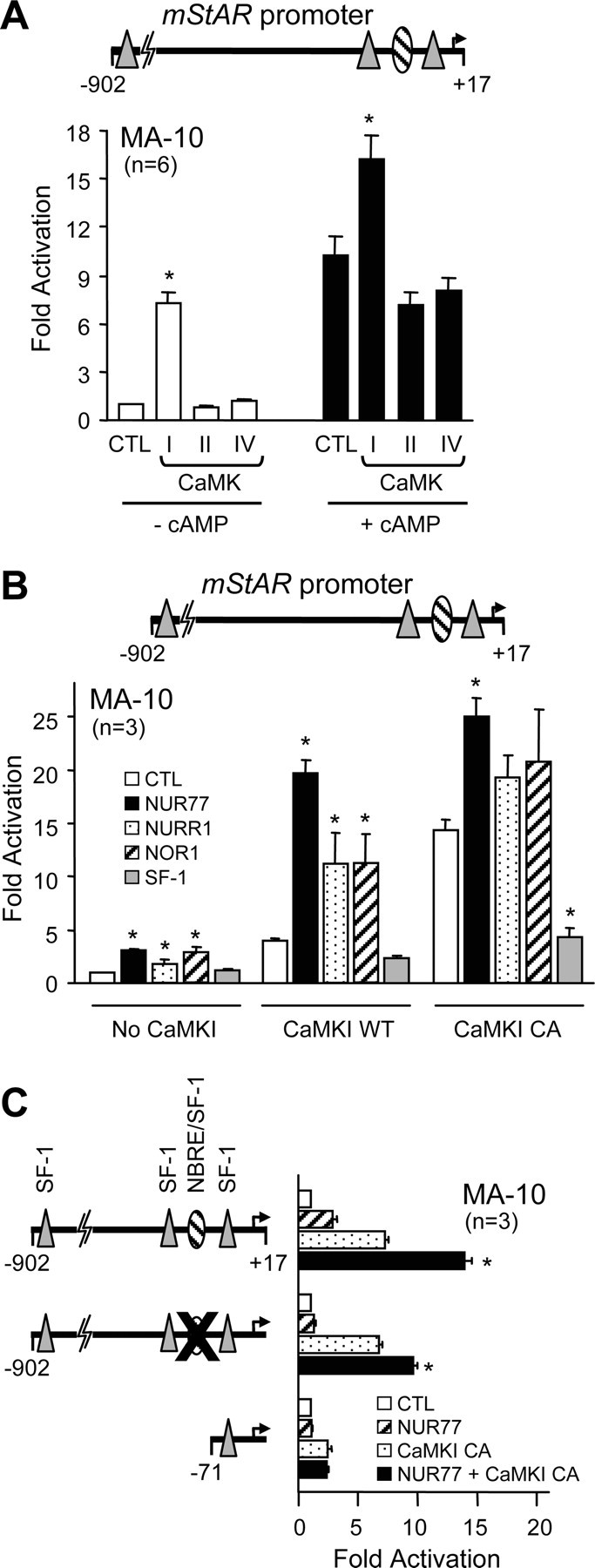
CaMKI Synergizes with NUR77 on the Mouse StAR Promoter
A, CaMKI, but not CaMKII or CaMKIV, activates the StAR promoter. MA-10 Leydig cells were cotransfected with a −902- to +17-bp mouse StAR promoter construct along with an empty expression vector (CTL) or expression vectors encoding wild-type CaMKI, CaMKII, or CaMKIV in the absence (white bars) or presence (black bars) of 0.5 mm (Bu)2-cAMP. B, NUR77 family members transcriptionally cooperate with CaMKI. MA-10 Leydig cells were cotransfected with a −902- to +17-bp mouse StAR promoter construct along with an empty expression vector (CTL) or expression vectors for NUR77 family members (NUR77, NURR1, and NOR1) and SF-1 in the absence or presence of expression vectors encoding either wild-type (WT) or a constitutively active (CA) form of CaMKI. C, The NBRE/SF-1 element at −95 bp contributes to the NUR77/CaMKI cooperation. MA-10 Leydig cells were cotransfected with an empty expression vector (white bars) or expression vectors for NUR77 (hatched bars), CaMKI CA (stippled bars), and NUR77 + CaMKI CA (black bars) along with a wild-type −902- to +17-bp StAR reporter, a reporter harboring a two-nucleotide mutation in the NBRE/SF-1 element at −95 bp (CCATCCTTGA to CCATAATTGA), or a −71- to +17-bp deletion construct. The mutated element is represented by a large X. The number of experiments, each performed in duplicate, is indicated. Results are shown as fold activation over control (±sem). *, Statistically significant difference from the respective controls (A and B); *, difference that is statistically significant from NUR77 or CaMKI alone (C).
The regulatory elements required for the CaMKI-dependent activation of the StAR promoter and for the transcriptional cooperation between NUR77 and CaMKI were next determined using modified StAR reporter constructs. A StAR reporter harboring a mutation in the NBRE/SF-1 element was still activated by CaMKI as efficiently as the wild-type StAR promoter (Fig. 8C), suggesting that CaMKI also regulates other transcription factors involved in StAR promoter activity. Consistent with the fact that NUR77 can no longer activate this reporter, the CaMKI/NUR77 cooperation was decreased on the StAR promoter containing a mutation in the NBRE/SF-1 element (Fig. 8C). A minimal StAR reporter construct (−71 bp) was only weakly activated by constitutively active CaMKI, and the cooperation with NUR77 was abolished (Fig. 8C). These results indicate that the NBRE/SF-1 element at −95 bp is essential for maximal CaMKI/NUR77 transcriptional cooperation.
DISCUSSION
StAR Induction in Response to Hormonal Stimulation Requires de Novo Protein Synthesis
There are conflicting data in the literature regarding the requirement of de novo protein synthesis in FSK/cAMP-mediated StAR induction in Leydig cells (21, 22, 23, 24). The use of nonquantitative methodologies, such as traditional RT-PCR, may be the cause of these discrepancies. To provide a more definitive answer, we have used a quantitative real-time PCR approach to show that maximal StAR transcription in response to FSK/cAMP stimulation in Leydig cells does require de novo protein synthesis. Therefore, in addition to transcription factors already present in the cell, FSK/cAMP-dependent stimulation of StAR expression in Leydig cells requires the production of newly synthesized transcription factors, a concept that is in agreement with previous data from the ovary (24).
The Mouse StAR Promoter Is a Target for NUR77 in Steroidogenic Cells
Several transcription factors constitutively present in Leydig cells have been shown to regulate StAR promoter activity (reviewed in Refs. 6 , 18 , and 19). On the other hand, the transcription factors whose expression is induced in response to hormonal stimulation and that contribute to StAR transcription have remained elusive. In the present study, we have identified the immediate-early response orphan nuclear receptor NUR77 as a novel regulator of cAMP-regulated StAR transcription. At first glance, the absence of any obvious steroidogenic and reproductive phenotypes in Nur77−/− mice (53) would argue against a role for NUR77 in StAR transcription. However, in these animals no analyses of testicular gene expression were performed. Therefore, it remains possible that in Nur77−/− mice, StAR expression could be decreased, as we have observed here using siRNA against Nur77 in Leydig cells, but not to a level causing severe steroid deficiency. Another possible explanation is that other members of the NUR77 family of nuclear receptors can compensate for the absence of NUR77. Indeed, Nurr1 was shown to be up-regulated in Nur77−/− mice (53). Furthermore, NUR77 and NOR-1 are functionally redundant in vivo in thymocyte gene expression (54). This compensatory mechanism likely occurs in steroidogenic cells as well where NURR1 is coexpressed with NUR77, albeit at lower levels, and where both factors respond to cAMP stimulation (32, 33). In support of this, we showed that all NUR77 family members can activate the StAR promoter and cooperate with CaMKI. Null mice for both Nur77 and Nurr1 genes are required to validate the role of these factors in steroidogenic cell function.
Consistent with a role for NUR77 in StAR transcription, both are coexpressed in several tissues. For instance, like StAR, Nur77 expression is zone specific in the adrenal; both are predominantly expressed in the zona glomerulosa and fasciculata and weakly in the zona reticularis (28, 55, 56, 57). Within the gonads, Nur77 is coexpressed with StAR in testicular Leydig cells (33, 57, 58) and in ovarian theca and luteinized granulosa cells (57, 59, 60). Finally, StAR and Nur77 are also found in the same areas of the brain (61, 62). In addition to having similar expression profiles in steroidogenic tissues (classic, e.g. gonads and adrenal, and nonclassic, such as the brain), Nur77 and StAR are also commonly regulated by hormones known to influence steroidogenesis. For instance, as for StAR (7), Nur77 expression is induced by LH/human chorionic gonadotropin in granulosa cells (31, 32), by LH/FSK/cAMP in testicular Leydig cells (Fig. 1 and Refs 29 and 33), and by ACTH/cAMP, angiotensin II, and K+ in adrenal cells (28, 55, 63, 64, 65). Finally, we found that in Leydig cells, Nur77 induction in response to cAMP precedes that of StAR, and both require a previously undescribed CaMK-dependent pathway. In addition to these classical regulators of steroidogenesis, StAR and Nur77 transcription is also up-regulated by other stimuli including phorbol esters/protein kinase C, arachidonic metabolites, growth factors, and calcium (reviewed in Refs. 7 , 25 , and 27).
Mechanisms of NUR77-Dependent Activation of StAR Transcription
NUR77 generally binds as a monomer to a DNA element similar to the binding site for the nuclear receptor SF-1 (35). Although the StAR promoter is known to be regulated by SF-1 through numerous binding sites (23, 45, 66), we found that the previously documented SF-1 element located at −95 bp is necessary to confer NUR77-responsiveness to the StAR promoter in steroidogenic cells. Mutation of this element led to a decrease in both basal activity and cAMP responsiveness of the StAR promoter in Leydig cells. The fact that about 75% of the cAMP-dependent activation of the StAR promoter was retained when the −95-bp NBRE/SF-1 was mutated is consistent with previous reports describing the involvement of other transcription factors (e.g. GATA4, CCAAT/enhancer binding protein-β, SF-1, AP-1, and CREB/CRE modulator) in hormonal regulation of StAR transcription (5, 18).
Even though the NUR77 responsiveness of the StAR promoter is exclusively conferred by the −95-bp element in transfection assays, our data also indicate that NUR77 only weakly binds to this element in in vitro assays. This is consistent with the fact that the −95-bp element is quite divergent from the consensus NBRE element and more closely conforms to the SF-1 binding motif (35, 67). In vivo ChIP assays, however, confirmed that NUR77 is actively and strongly recruited to the proximal StAR promoter region in response to cAMP stimulation in the MA-10 Leydig cell line and in primary Leydig cell cultures. This suggests that NUR77 recruitment to the StAR promoter likely requires association/stabilization with other transcription factors binding to, or in proximity of, the NBRE/SF-1 element to activate StAR transcription. In agreement with this, we found that NUR77 interacts with members of the AP-1 family known to bind to the proximal StAR promoter (Martin, L. J., and J. J. Tremblay, unpublished data). Posttranslational modifications such as phosphorylation may also differentially modulate protein-protein interactions as well as the DNA binding affinities of the factors. Consistent with this, phosphorylation of Ser340 and Ser350 in NUR77 DNA binding domain, which has been associated with a reduced affinity for DNA (46, 68, 69), is decreased in response to hormonal stimulation. This marked increase in the ratio of total NUR77 to phosphorylated NUR77 may enhance NUR77 recruitment to DNA in addition to facilitating protein-protein interactions with cofactors ultimately resulting in an increase in NUR77 transcriptional activity. A similar enhancement of NUR77 activation has been observed in ACTH-stimulated Y-1 adrenal cells and has been attributed to dephosphorylation of Ser350 (70). The phosphatase involved in this process, however, remains to be identified.
The CaMK Pathway in Testicular Leydig Cells
StAR expression involves various signaling pathways and protein kinases that seem to differ from one steroidogenic tissue to another (reviewed in Ref. 7). The best characterized is the cAMP/PKA pathway. Inactivation of PKA, either chemically (10 μm H-89) or biologically (protein kinase inhibitor) blunts the hormonal stimulation of StAR transcription in every steroidogenic cell type, including testicular Leydig cells. Although our data support a role for NUR77 as a downstream effector of FSK/cAMP in StAR transcription, the PKA inhibitor H-89 did not prevent FSK- and (Bu)2-cAMP-mediated NUR77 induction. The adenylate cyclase inhibitor MDL-12,330A, however, abolished NUR77 and StAR protein induction in response to FSK. Furthermore, 1,9-ddFSK, a forskolin analog that is devoid of adenylate cyclase-stimulating activity but retains cAMP-independent FSK effects, was inefficient at inducing NUR77 and StAR protein levels. Altogether, these data indicate that FSK- or (Bu)2-cAMP-mediated increase in NUR77 protein levels occurs independently of PKA activity but requires activation of adenylate cyclase and cAMP production. The CaMK inhibitor KN-93 (47) completely abrogated NUR77 induction in response to FSK, thus confirming the implication of CaMK in this process. The exact mechanism responsible for the cross talk between the cAMP and CaMK signaling pathways remains to be elucidated. An intriguing possibility that has recently been proposed (71) is that perhaps a cAMP mediator/adaptor molecule, such as exchange protein directly activated by cAMP, responsible for linking cAMP to the MAPK pathway (72, 73), could also be responsible for the cross talk between cAMP and CaMK. Such an adaptor molecule, however, has yet to be identified.
Consistent with a role for CaMK and NUR77 in Leydig cell steroidogenesis, we found that KN-93 blocked the FSK-mediated increase in StAR mRNA and protein levels. The role of the CaMK signaling pathway has mainly been studied in adrenal steroidogenic cells where it was found to be involved in the stimulation of aldosterone production in response to angiotensin II and K+ (71, 74, 75, 76, 77, 78). Much less is known regarding the CaMK pathway in Leydig cell steroidogenesis, although Ca2+ has been shown to be important for LH-stimulated testosterone production (13, 79, 80, 81).
CaMK family members have been described in numerous tissues, but no data were available regarding their expression in testicular Leydig cells. Here we found that the main CaMK present in mouse Leydig cells is CaMKI. Furthermore, cAMP stimulation of Leydig cells results in increased T177 phosphorylation of CaMKI known to enhance its kinase activity (51). The exact targets of CaMKI in Leydig cells and in StAR expression, however, remain to be fully elucidated. It is possible that CaMKI directly phosphorylates, and thus regulates, the activity of transcription factors involved in StAR transcription. In agreement with this, the transcription factor CREB, known to contribute to StAR expression in Leydig cells (82), can be phosphorylated by CaMKI (83). NUR77 itself might also be a target of CaMKI because it contains three consensus CaMKI phosphorylation sites. CaMKI might also regulate expression and/or nuclear localization of transcription factors involved in StAR transcription. Supporting this, we found that NUR77 protein levels in the nucleus of Leydig cells after FSK stimulation were severely decreased by the CaMK inhibitor. Because NUR77 participates in StAR transcription in Leydig cells and because CaMKI enhances NUR77 expression and activity, it is therefore likely that part of the effects of CaMKI on StAR transcription might be mediated by NUR77, although other yet to be identified transcription factors are also implicated.
MATERIALS AND METHODS
Chemicals
Protein kinase inhibitors bisindolylmaleimide I, H-89, KN-93, ML-7, protein kinase G inhibitor, staurosporine, and PD98059, the adenylate cyclase inhibitor MDL-12,330A, and the inactive FSK analog 1,9-ddFSK were purchased from Calbiochem (San Diego, CA). Act-D, CHX, (Bu)2-cAMP, and FSK were purchased from Sigma-Aldrich Canada (Oakville, Ontario, Canada).
Plasmids
The −902-bp murine StAR-luciferase promoter construct has been described previously (84). Deletions of the StAR promoter to −193, −144, −121, −104, and −71 bp were obtained by PCR using the −902-bp StAR promoter as template, along with a common reverse primer containing a KpnI (italicized) cloning site (5′-GAG GTA CCT GAG TCC TGC AGC TGT GGC-3′) and the following forward primers containing a BamHI cloning site: −193 bp, 5′-CGG GAT CCC TGC TTT CCC CTA CCT GCA GAG TC-3′; −144 bp, 5′-CGG GAT CCC CCT CCC ACC TTG GCC AGC-3′; −121 bp, 5′-CGG GAT CCA GGA TGA GGC AAT CAT TCC ATC CT-3′; −104 bp, 5′-CGG GAT CCT CCA TCC TTG ACC CTC TGC-3′; and −71 bp, 5′-ACG GAT CCT TTT TTA TCT CAA GTG ATG A-3′. The −902-bp StAR reporter construct harboring a mutation (underlined) inactivating the NBRE/SF-1 element at −95 bp (CATCCTTGA to CATAATTGA) was generated using the QuikChange XL mutagenesis kit (Stratagene, La Jolla, CA) with the following oligonucleotides (mutated nucleotides are underlined): sense, 5′-GGA TGA GGC AAT CAT TCC ATA ATT GAC CCT CTG CAC AAT GAC-3′; antisense, 5′-GTC ATT GTG CAG AGG GTC AAT TAT GGA ATG ATT GCC TCA TCC-3′. All promoter fragments were cloned into a modified pXP1 luciferase reporter plasmid (85) and subsequently verified by sequencing (Centre de génomique de Québec, CHUL Research Centre, Quebec City, Canada). The mouse SF-1 expression vector has been described previously (84). Rat NUR77, NOR1, and NURR1 expression vectors (86) were provided by Dr. Jacques Drouin (Laboratoire de Génétique Moléculaire, Institut de Recherches Cliniques de Montréal, Montréal, Canada). The human LRH-1 expression vector (87) was provided by Dr. Luc Bélanger (Centre de Recherche en Cancérologie, Centre de Recherche du CHUQ, Université Laval, Québec, Canada). Expression vectors encoding wild-type and constitutively active forms of CaMKI, CaMKII, and CaMKIV (52) were obtained from Dr. Thomas Soderling (Oregon Health Sciences University, Portland, OR).
RNA Isolation, Reverse Transcription, Quantitative Real-Time PCR, and Traditional PCR
Total RNA was isolated from MA-10 Leydig cells using RNeasy Plus extraction kit (QIAGEN Inc., Mississauga, Ontario, Canada). First-strand cDNAs were synthesized from a 5-μg aliquot of the various RNAs using the Superscript III Reverse Transcriptase System (Invitrogen Canada, Burlington, Ontario, Canada). MA-10 Leydig cells were cultured in serum-free media containing vehicle, 100 nm bisindolylmaleimide I, 10 μm H-89, 20 μm KN-93, 1 μm ML-7, 100 nm staurosporine, or 10 μm PD98059 for 30 min before addition of 10 μm FSK for 2 h before RNA isolation. Quantitative real-time PCR was performed using a LightCycler 1.5 instrument and the LightCycler FastStart DNA Master SYBR Green I kit (Roche Diagnostics, Laval, Canada) according to the manufacturer’s protocol. PCRs were performed using the following specific primers: StAR forward, 5′-TTG GGC ATA CTC AAC AAC CA-3′, and reverse, 5′-CCT TGA CAT TTG GGT TCC AC-3′; Nur77 forward, 5′-GGC TTC TTC AAG CGC ACA GT-3′, and reverse, 5′-GCT GCT TGG GTT TTG AAG GTA G-3′; and SF-1 forward, 5′-TCC AGT ACG GCA AGG AAG AC-3′, and reverse, 5′-GGC TGT GGT TGT TCA GGA AT-3′. As an internal control, PCRs were performed using previously described Rpl19-specific primers (88). The PCRs were done using the following conditions: 10 min at 95 C followed by 35 cycles of denaturation (5 sec at 95 C), annealing (5 sec at 60 C for Nur77 and 62 C for SF-1, StAR, and Rpl19), and extension (20 sec at 72 C) with single acquisition of fluorescence at the end of each extension step. The specificity of PCR products was confirmed by analysis of the melting curve and agarose gel electrophoresis. Quantification of gene expression was performed using the Relative Quantification Software (Roche Diagnostics) and is expressed as a ratio of StAR to Rpl19 mRNA levels. Each amplification was performed in duplicate using three different preparations of first-strand cDNAs for each of the three different RNA extractions. For the various CaMK family members, PCRs were done on a Tgradient thermocycler (Biometra) using Vent polymerase (New England Biolabs, Beverly, MA) and the following conditions: 3 min at 95 C followed by 30 cycles of denaturation (1 min at 95 C), annealing (1 min at 60 C), and extension (30 sec at 72 C) with a final extension step of 5 min at 72 C. PCR products were then analyzed by agarose gel electrophoresis and ethidium bromide staining. Primers used for amplification of each CaMK family member are presented in Table 1. Each amplification was performed three times using three different preparations of first-strand cDNAs resulting from three different RNA extractions.
Table 1.
Primers Used for Semiquantitative RT-PCR Analysis of CaMK Family Members
| Target Gene | Forward Primer | Reverse Primer |
|---|---|---|
| CaMKI | AAGCACCCCAACATTGTAGC | CTTGGAGAGGCCAAAGTCAG |
| CaMKIδ | TCAGTGACTTTGGCTTGTCG | AAGTCTTTGGCAGAGTCGGA |
| CaMKIγ | ATCTTCATGGAAGTGCTGGG | GCTCACCTCCAGAAACAAGC |
| CaMKIIα | TCTGAGAGCACCAACACCAC | CAGGTACTGAGTGATGCGGA |
| CaMKIIβ | TGAAGACATCGTGGCAAGAG | AGGCTTGAGGTCTCTGTGGA |
| CaMKIIδ | TGGACAAGAGTATGCTGCCA | CACCAAGTAATGGAAGCCCT |
| CaMKIIγ | TCAAAGCTGGAGCCTACGAT | ACTCTACCGTCTCTTGGCGA |
| CaMKIV | AGCTGGTCACAGGAGGAGAA | ATCAGCAATTTTGAGGGGTG |
Sequences are from 5′ to 3′ ends.
Protein Purification and Western Blots
Mouse MA-10 Leydig cells and rat primary Leydig cells were incubated in serum-free medium containing 0.5 mm (Bu)2-cAMP for times ranging from 0–6 h. In some experiments, vehicle, 100 nm bisindolylmaleimide I, 10 μm H-89, 20 μm KN-93, 1 μm ML-7, 100 nm staurosporine, 10 μm PD98059, or 10 μm MDL-12,330A was added to MA-10 cells for 30 min before addition of 10 μm FSK or 0.5 mm (Bu)2cAMP for 2 h before protein extraction. In some experiments, FSK was replaced by 10 μm ddFSK, an analog of FSK that does not activate adenylate cyclase. MA-10 and primary Leydig cells were then rinsed twice with ice-cold PBS and harvested for total and nuclear protein extractions. Total proteins were isolated by lysing the cells directly into RIPA buffer [50 mm Tris-HCl (pH 7.5), 1% Igepal, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 1 mm sodium fluoride, 1 mm sodium orthovanadate, and 1 μg/ml each for aprotinin, leupeptin, and pepstatin] for 1 h at 4 C, followed by centrifugation to remove cell debris. Nuclear proteins were prepared by the procedure outlined by Schreiber et al. (89). Protein concentrations were estimated using standard Bradford assay. Total proteins (40 μg for MA-10 cells and 20 μg for primary Leydig cells) or nuclear proteins (20 μg for MA-10 and 5 μg for primary Leydig cells) were boiled 10 min in a denaturing loading buffer, fractionated by SDS-PAGE, and transferred onto polyvinylidene difluoride (PVDF) membrane (Millipore, Bedford, MA). Immunodetection was performed using an avidin-biotin approach according to the manufacturer’s instructions (Vector Laboratories, Inc., Ontario, Canada). Detection of NUR77, SF-1, StAR, and α-TUBULIN was performed using a monoclonal anti-NUR77 antibody (which does not cross-react with NURR1 or NOR1; 1:500 dilution; BD Biosciences Pharmingen, San Diego, CA), an anti-SF-1 polyclonal antiserum (1:5000 dilution; kindly provided by Ken-Ichirou Morohashi, National Institute for Basic Biology, Japan), an anti-StAR antiserum (FL-285, 1:200 dilution; Santa Cruz Biotechnologies, Santa Cruz, CA), and a monoclonal anti-α-TUBULIN antibody (1:50,000 dilution; Sigma-Aldrich Canada), respectively. Phosphorylated forms of NUR77 (amino acid numbers correspond to the rat NUR77 protein) were immunodetected using polyclonal antisera against phosphoserine 340 and phosphoserine 350 NUR77 (1:200 dilution; Santa Cruz Biotechnologies). Detection of CaMKI was performed using an anti-CaMKI polyclonal antiserum (M-20, 1:200 dilution; Santa Cruz). Phospho-T177 CaMKI was detected using a monoclonal anti-phospho-T177-CaMKI antibody (1:1000 dilution; kindly provided by Thomas Soderling, Oregon Health Sciences University).
Cell Culture and Transfections
Mouse MA-10 Leydig cells (90), provided by Dr. Mario Ascoli (University of Iowa, Iowa City, IA), were grown in Waymouth’s MB752/1 medium supplemented with 1.2 g/liter NaHCO3, 15% horse serum, and 50 mg/liter gentamicin and streptomycin sulfate at 37 C in 5% CO2. MA-10 cells were transfected in 24-well plates using the calcium-phosphate precipitation method (91). Briefly, MA-10 cells were transfected 24 h after plating at a density of 120,000 cells per well, by using 0.5 μg StAR promoter construct fused to the Firefly luciferase reporter gene, 0.5 μg cytomegalovirus-driven expression vector, 10 ng phRL-TK Renilla luciferase expression vector used as an internal control for transfection efficiency, and pSP64 as carrier DNA up to 1.5 μg/well. Two days later, MA-10 cells were harvested and luciferase activities measured using the Dual Luciferase Assay System (Promega Corp., Madison, WI) and the EG&G Berthold LB 9507 luminometer (Berthold Technologies, Oak Ridge, TN). Mouse Y-1 adrenal cells were grown and transfected as previously described (84). In experiments with cAMP stimulation, cells were treated with 0.5 mm (Bu)2-cAMP for 4 h before harvesting. Data reported represent the average of at least three experiments, each performed in duplicate.
siRNA Transfection
A mixture of four RNA oligonucleotides (the sequences are shown in Table 2), each directed against Nur77 or SF-1, were purchased from Dharmacon, Inc. (Lafayette, CO) and transfected in MA-10 Leydig cells using BIO-Fectin Transfection Reagent (Bioshop, Burlington, Ontario, Canada). MA-10 cells were then incubated with vehicle or 10 μm FSK. As a negative control, scrambled siRNAs were used (Dharmacon). Less than 10% difference was observed between the scrambled siRNAs and no siRNA (data not shown).
Table 2.
Sequences of the siRNA Oligonucleotides Used
| Target Gene | Sense Oligonucleotide | Antisense Oligonucleotide |
|---|---|---|
| Nur77 | CCCUGGACGUUAUCCGAAAUU | PUUUCGGAUAACGUCCAGGGUU |
| CCGUGACACUUCCGGCAUUUU | PAAUGCCGGAAGUGUCACGGUU | |
| GUAAAUAAGCUGACGCUACUU | PGUAGCGUCAGCUUAUUUACUU | |
| GCACAUGGCUACCGUGGCAUU | PUGCCACGGUAGCCAUGUGCUU | |
| SF-1 | CAUUACACGUGCACCGAGAUU | PUCUCGGUGCACGUGUAAUGUU |
| GCAUUUGGGCAACGAGAUGUU | PCAUCUCGUUGCCCAAAUGCUU | |
| ACGCUGCCCUGUUGGAUUAUU | PUAAUCCAACAGGGCAGCGUUU | |
| CGUCAGAUUUACAGCUUAUUU | PAUAAGCUGUAAAUCUGACGUU |
Sequences are from 5′ to 3′ ends.
ChIP Assay
ChIP assays were performed as previously described (92). NUR77-immunoprecipitated DNA fragments were analyzed by quantitative real-time PCR using primers specific for the proximal region (−299 to −41 bp) of the mouse StAR promoter (forward, 5′-TGA TGC ACC TCA GTT ACT GG-3′; reverse, 5′-GCT GTG CAT CAT CAC TTG AG-3′). The PCR were done using the following conditions: 10 min at 95 C followed by 35 cycles of denaturation (5 sec at 95 C), annealing (5 sec at 62 C), and extension (20 sec at 72 C) with single acquisition of fluorescence at the end of each extension step. The specificity of PCR products was confirmed by analysis of the melting curve and agarose gel electrophoresis. Absolute quantification of StAR promoter DNA fragments was performed using a standard curve done from various concentrations of the −902-bp StAR promoter construct described previously and is expressed as a ratio of StAR promoter-immunoprecipitated DNA to input DNA levels. Input DNA represents 10% of total DNA used for a ChIP experiment. ChIP results were confirmed by three separate experiments.
Preparation of Primary Leydig Cells
Primary Leydig cells were isolated as described previously (93) from 35-d-old Sprague Dawley rats obtained on site. Serum-free medium 199 with Earle’s salts (Sigma-Aldrich Canada), l-glutamine, 1.5 mm HEPES, and 2.5 g/liter NaHCO3 containing antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin) was used during preparation and culturing. After 2 d in culture, the purity of the Leydig cell preparation was evaluated to be about 95% enriched as assessed by histochemical staining for 3β-hydroxysteroid dehydrogenase activity (94). All experiments were conducted according to the Canadian Council for Animal Care and have been approved by the Animal Care and Ethics Committee of Laval University (protocol 06-059).
Immunohistochemistry
Adult CD-1 mice (∼90 d old) were obtained on site and killed by CO2 inhalation. The testes were harvested and fixed with ice-cold 4% paraformaldehyde (wt/vol) for 24 h. Tissues were then dehydrated with ethanol, substituted with xylene, embedded in paraffin, and cut into 5-μm sections. After paraffin removal, tissues were blocked with 0.5% BSA in PBS for 1 h at 25 C. Immunodetection was performed using an avidin-biotin approach according to the manufacturer’s instructions for the Vectastain Elite ABC reagent (Vector). CaMKI protein localization was assessed using an anti-CaMKI polyclonal antiserum (M-20, 1:500 dilution; Santa Cruz). Negative control corresponds to the same procedure with the omission of anti-CaMKI antibody. Final revelation was done using 3-amino-9-acetylcarbazole as substrate (Sigma-Aldrich Canada), and the sections were counterstained with hematoxylin Gill 1 (VWR International, Mount-Royal, Quebec, Canada). All experiments were conducted according to the Canadian Council for Animal Care and have been approved by the Animal Care and Ethics Committee of Laval University (protocol 2003-068).
Statistical Analyses
To identify significant differences between multiple groups, statistical analyses were done using either a one-way ANOVA followed by Holm-Sidak test or a nonparametric Kruskal-Wallis one-way ANOVA followed by Mann-Whitney U tests when conditions of normality and/or equal variance between groups was not met. Single comparisons between two experimental groups were done using either a paired Student’s t test or again a Mann-Whitney U test when conditions of normality/variance failed. For all statistical analyses, P < 0.05 was considered significant. All statistical analyses were done using the SigmaStat software package (Systat Software Inc., San Jose, CA).
Acknowledgments
We thank Drs. Jacques Drouin, Luc Bélanger, Thomas Soderling, Ken-Ichirou Morohashi, and Mario Ascoli for generously providing expression plasmids, antiserum, and cell lines used in this study. We are also thankful to Nicholas Robert for his assistance in the preparation of primary Leydig cells.
NURSA Molecule Pages:
Nuclear Receptors: LRH-1 | NGFIB | NOR1 | NURR1 | SF-1.
Footnotes
L.J.M. holds a doctoral studentship from the Natural Sciences and Engineering Research Council of Canada (NSERC). N.B. held a studentship from the Chaire Jeanne et Jean-Louis Lévesque. J.J.T. holds a New Investigator scholarship from the Institute of Gender and Health of the Canadian Institutes of Health Research (CIHR). This work was supported by grants from NSERC and CIHR (MOP-81387) to J.J.T.
Author Disclosure: L.J.M., N.B., C.B., and J.J.T. have nothing to declare.
First Published Online July 3, 2008
Abbreviations: Act-D, Actinomycin D; AP-1, activation protein 1; (Bu)2-cAMP, dibutyryl cAMP; CaMK, Ca2+/calmodulin-dependent protein kinase; ChIP, chromatin immunoprecipitation; CHX, cycloheximide; CRE, cAMP response element; CREB, CRE-binding protein; ddFSK, 1,9-dideoxyforskolin; FSK, forskolin; NBRE, NUR77-responsive element; NGFI-B, nerve growth factor induced-B; NOR1, nuclear orphan receptor 1; NR4A1, nuclear receptor 4A1; PKA, protein kinase A; PVDF, polyvinylidene difluoride; SF-1, steroidogenic factor 1; siRNA, small interfering RNA; StAR, steroidogenic acute regulatory.
References
- 1.Lin D, Sugawara T, Strauss III JF, Clark BJ, Stocco DM, Saenger P, Rogol A, Miller WL 1995. Role of steroidogenic acute regulatory protein in adrenal and gonadal steroidogenesis. Science 267:1828–1831 [DOI] [PubMed] [Google Scholar]
- 2.Miller WL 1997. Congenital lipoid adrenal hyperplasia: the human gene knockout for the steroidogenic acute regulatory protein. J Mol Endocrinol 19:227–240 [DOI] [PubMed] [Google Scholar]
- 3.Hasegawa T, Zhao L, Caron KM, Majdic G, Suzuki T, Shizawa S, Sasano H, Parker KL 2000. Developmental roles of the steroidogenic acute regulatory protein (StAR) as revealed by StAR knockout mice. Mol Endocrinol 14:1462–1471 [DOI] [PubMed] [Google Scholar]
- 4.Stocco DM 2002. Clinical disorders associated with abnormal cholesterol transport: mutations in the steroidogenic acute regulatory protein. Mol Cell Endocrinol 191:19–25 [DOI] [PubMed] [Google Scholar]
- 5.Stocco DM 2001. StAR protein and the regulation of steroid hormone biosynthesis. Annu Rev Physiol 63:193–213 [DOI] [PubMed] [Google Scholar]
- 6.Manna PR, Stocco DM 2005. Regulation of the steroidogenic acute regulatory protein expression: functional and physiological consequences. Curr Drug Targets Immune Endocr Metabol Disord 5:93–108 [DOI] [PubMed] [Google Scholar]
- 7.Stocco DM, Wang X, Jo Y, Manna PR 2005. Multiple signaling pathways regulating steroidogenesis and steroidogenic acute regulatory protein expression: more complicated than we thought. Mol Endocrinol 19:2647–2659 [DOI] [PubMed] [Google Scholar]
- 8.Ascoli M, Fanelli F, Segaloff DL 2002 The lutropin/choriogonadotropin receptor, a 2002. perspective. Endocr Rev 23:141–174 [DOI] [PubMed] [Google Scholar]
- 9.Wang X, Stocco DM 1999. Cyclic AMP and arachidonic acid: a tale of two pathways. Mol Cell Endocrinol 158:7–12 [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Walsh LP, Stocco DM 1999. The role of arachidonic acid on LH-stimulated steroidogenesis and steroidogenic acute regulatory protein accumulation in MA-10 mouse Leydig tumor cells. Endocrine 10:7–12 [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Walsh LP, Reinhart AJ, Stocco DM 2000. The role of arachidonic acid in steroidogenesis and steroidogenic acute regulatory (StAR) gene and protein expression. J Biol Chem 275:20204–20209 [DOI] [PubMed] [Google Scholar]
- 12.Li J, Feltzer RE, Dawson KL, Hudson EA, Clark BJ 2003. Janus kinase 2 and calcium are required for angiotensin II-dependent activation of steroidogenic acute regulatory protein transcription in H295R human adrenocortical cells. J Biol Chem 278:52355–52362 [DOI] [PubMed] [Google Scholar]
- 13.Ramnath HI, Peterson S, Michael AE, Stocco DM, Cooke BA 1997. Modulation of steroidogenesis by chloride ions in MA-10 mouse tumor Leydig cells: roles of calcium, protein synthesis, and the steroidogenic acute regulatory protein. Endocrinology 138:2308–2314 [DOI] [PubMed] [Google Scholar]
- 14.Cooke BA, Ashford L, Abayasekara DR, Choi M 1999. The role of chloride ions in the regulation of steroidogenesis in rat Leydig cells and adrenal cells. J Steroid Biochem Mol Biol 69:359–365 [DOI] [PubMed] [Google Scholar]
- 15.Gyles SL, Burns CJ, Whitehouse BJ, Sugden D, Marsh PJ, Persaud SJ, Jones PM 2001. ERKs regulate cyclic AMP-induced steroid synthesis through transcription of the steroidogenic acute regulatory (StAR) gene. J Biol Chem 276:34888–34895 [DOI] [PubMed] [Google Scholar]
- 16.Seger R, Hanoch T, Rosenberg R, Dantes A, Merz WE, Strauss III JF, Amsterdam A 2001. The ERK signaling cascade inhibits gonadotropin-stimulated steroidogenesis. J Biol Chem 276:13957–13964 [DOI] [PubMed] [Google Scholar]
- 17.Jo Y, King SR, Khan SA, Stocco DM 2005. Involvement of protein kinase C and cyclic adenosine 3′,5′-monophosphate-dependent kinase in steroidogenic acute regulatory protein expression and steroid biosynthesis in Leydig cells. Biol Reprod 73:244–255 [DOI] [PubMed] [Google Scholar]
- 18.Manna PR, Wang XJ, Stocco DM 2003. Involvement of multiple transcription factors in the regulation of steroidogenic acute regulatory protein gene expression. Steroids 68:1125–1134 [DOI] [PubMed] [Google Scholar]
- 19.Stocco DM, Clark BJ, Reinhart AJ, Williams SC, Dyson M, Dassi B, Walsh LP, Manna PR, Wang X, Zeleznik AJ, Orly J 2001. Elements involved in the regulation of the StAR gene. Mol Cell Endocrinol 177:55–59 [DOI] [PubMed] [Google Scholar]
- 20.Nishida H, Miyagawa S, Vieux-Rochas M, Morini M, Ogino Y, Suzuki K, Nakagata N, Choi HS, Levi G, Yamada G 2008. Positive regulation of steroidogenic acute regulatory protein gene expression through the interaction between Dlx and GATA-4 for testicular steroidogenesis. Endocrinology 149:2090–2097 [DOI] [PubMed] [Google Scholar]
- 21.Clark BJ, Soo SC, Caron KM, Ikeda Y, Parker KL, Stocco DM 1995. Hormonal and developmental regulation of the steroidogenic acute regulatory protein. Mol Endocrinol 9:1346–1355 [DOI] [PubMed] [Google Scholar]
- 22.Clark BJ, Combs R, Hales KH, Hales DB, Stocco DM 1997. Inhibition of transcription affects synthesis of steroidogenic acute regulatory protein and steroidogenesis in MA-10 mouse Leydig tumor cells. Endocrinology 138:4893–4901 [DOI] [PubMed] [Google Scholar]
- 23.Caron KM, Ikeda Y, Soo SC, Stocco DM, Parker KL, Clark BJ 1997. Characterization of the promoter region of the mouse gene encoding the steroidogenic acute regulatory protein. Mol Endocrinol 11:138–147 [DOI] [PubMed] [Google Scholar]
- 24.Kiriakidou M, McAllister JM, Sugawara T, Strauss III JF 1996. Expression of steroidogenic acute regulatory protein (StAR) in the human ovary. J Clin Endocrinol Metab 81:4122–4128 [DOI] [PubMed] [Google Scholar]
- 25.Eells JB, Witta J, Otridge JB, Zuffova E, Nikodem VM 2000. Structure and function of the Nur77 receptor subfamily, a unique class of hormone nuclear receptor. Curr Genomics 1:135–152 [Google Scholar]
- 26.Giguere V 1999. Orphan nuclear receptors: from gene to function. Endocr Rev 20:689–725 [DOI] [PubMed] [Google Scholar]
- 27.Maxwell MA, Muscat GE 2005. The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nucl Recept Signal 4:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis IJ, Lau LF 1994. Endocrine and neurogenic regulation of the orphan nuclear receptors Nur77 and Nurr-1 in the adrenal glands. Mol Cell Biol 14:3469–3483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin LJ, Tremblay JJ 2005. The human 3β-hydroxysteroid dehydrogenase/Δ5-Δ4 isomerase type 2 promoter is a novel target for the immediate early orphan nuclear receptor NUR77 in steroidogenic cells. Endocrinology 146:861–869 [DOI] [PubMed] [Google Scholar]
- 30.Li W, Amri H, Huang H, Wu C, Papadopoulos V 2004. Gene and protein profiling of the response of MA-10 Leydig tumor cells to human chorionic gonadotropin. J Androl 25:900–913 [DOI] [PubMed] [Google Scholar]
- 31.Park JI, Park HJ, Choi HS, Lee K, Lee WK, Chun SY 2001. Gonadotropin regulation of NGFI-B messenger ribonucleic acid expression during ovarian follicle development in the rat. Endocrinology 142:3051–3059 [DOI] [PubMed] [Google Scholar]
- 32.Park JI, Park HJ, Lee YI, Seo YM, Chun SY 2003. Regulation of NGFI-B expression during the ovulatory process. Mol Cell Endocrinol 202:25–29 [DOI] [PubMed] [Google Scholar]
- 33.Song KH, Park JI, Lee MO, Soh J, Lee K, Choi HS 2001. LH induces orphan nuclear receptor Nur77 gene expression in testicular Leydig cells. Endocrinology 142:5116–5123 [DOI] [PubMed] [Google Scholar]
- 34.Klopotowska D, Matuszyk J, Rapak A, Gidzinska B, Cebrat M, Ziolo E, Strzadala L 2005. Transactivation activity of Nur77 discriminates between Ca2+ and cAMP signals. Neurochem Int 46:305–312 [DOI] [PubMed] [Google Scholar]
- 35.Wilson TE, Fahrner TJ, Milbrandt J 1993. The orphan receptors NGFI-B and steroidogenic factor 1 establish monomer binding as a third paradigm of nuclear receptor-DNA interaction. Mol Cell Biol 13:5794–5804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bassett MH, Suzuki T, Sasano H, De Vries CJ, Jimenez PT, Carr BR, Rainey WE 2004. The orphan nuclear receptor NGFIB regulates transcription of 3β-hydroxysteroid dehydrogenase. Implications for the control of adrenal functional zonation. J Biol Chem 279:37622–37630 [DOI] [PubMed] [Google Scholar]
- 37.Havelock JC, Smith AL, Seely JB, Dooley CA, Rodgers RJ, Rainey WE, Carr BR 2005. The NGFI-B family of transcription factors regulates expression of 3β-hydroxysteroid dehydrogenase type 2 in the human ovary. Mol Hum Reprod 11:79–85 [DOI] [PubMed] [Google Scholar]
- 38.Zhang P, Mellon SH 1997. Multiple orphan nuclear receptors converge to regulate rat P450c17 gene transcription: novel mechanisms for orphan nuclear receptor action. Mol Endocrinol 11:891–904 [DOI] [PubMed] [Google Scholar]
- 39.Hong CY, Park JH, Ahn RS, Im SY, Choi HS, Soh J, Mellon SH, Lee K 2004. Molecular mechanism of suppression of testicular steroidogenesis by proinflammatory cytokine tumor necrosis factor α. Mol Cell Biol 24:2593–2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aesoy R, Mellgren G, Morohashi K, Lund J 2002. Activation of cAMP-dependent protein kinase increases the protein level of steroidogenic factor-1. Endocrinology 143:295–303 [DOI] [PubMed] [Google Scholar]
- 41.Daggett MA, Rice DA, Heckert LL 2000. Expression of steroidogenic factor 1 in the testis requires an E box and CCAAT box in its promoter proximal region. Biol Reprod 62:670–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rust W, Stedronsky K, Tillmann G, Morley S, Walther N, Ivell R 1998. The role of SF-1/Ad4BP in the control of the bovine gene for the steroidogenic acute regulatory (StAR) protein. J Mol Endocrinol 21:189–200 [DOI] [PubMed] [Google Scholar]
- 43.Sugawara T, Holt JA, Kiriakidou M, Strauss JF, III 1996. Steroidogenic factor 1-dependent promoter activity of the human steroidogenic acute regulatory protein (StAR) gene. Biochemistry 35:9052–9059 [DOI] [PubMed] [Google Scholar]
- 44.Reinhart AJ, Williams SC, Clark BJ, Stocco DM 1999. SF-1 (steroidogenic factor-1) and C/EBPβ (CCAAT/enhancer binding protein-β) cooperate to regulate the murine StAR (steroidogenic acute regulatory) promoter. Mol Endocrinol 13:729–741 [DOI] [PubMed] [Google Scholar]
- 45.Manna PR, Eubank DW, Lalli E, Sassone-Corsi P, Stocco DM 2003. Transcriptional regulation of the mouse steroidogenic acute regulatory protein gene by the cAMP response-element binding protein and steroidogenic factor 1. J Mol Endocrinol 30:381–397 [DOI] [PubMed] [Google Scholar]
- 46.Hirata Y, Kiuchi K, Chen HC, Milbrandt J, Guroff G 1993. The phosphorylation and DNA binding of the DNA-binding domain of the orphan nuclear receptor NGFI-B. J Biol Chem 268:24808–24812 [PubMed] [Google Scholar]
- 47.Hidaka H, Yokokura H 1996. Molecular and cellular pharmacology of a calcium/calmodulin-dependent protein kinase II (CaM kinase II) inhibitor, KN-62, and proposal of CaM kinase phosphorylation cascades. Adv Pharmacol 36:193–219 [DOI] [PubMed] [Google Scholar]
- 48.Ledoux J, Chartier D, Leblanc N 1999. Inhibitors of calmodulin-dependent protein kinase are nonspecific blockers of voltage-dependent K+ channels in vascular myocytes. J Pharmacol Exp Ther 290:1165–1174 [PubMed] [Google Scholar]
- 49.Wu JY, Means AR 2000. Ca2+/calmodulin-dependent protein kinase IV is expressed in spermatids and targeted to chromatin and the nuclear matrix. J Biol Chem 275:7994–7999 [DOI] [PubMed] [Google Scholar]
- 50.Picciotto MR, Zoli M, Bertuzzi G, Nairn AC 1995. Immunochemical localization of calcium/calmodulin-dependent protein kinase I. Synapse 20:75–84 [DOI] [PubMed] [Google Scholar]
- 51.Soderling TR 1999. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci 24:232–236 [DOI] [PubMed] [Google Scholar]
- 52.Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR 2004. Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci 24:3786–3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crawford PA, Sadovsky Y, Woodson K, Lee SL, Milbrandt J 1995. Adrenocortical function and regulation of the steroid 21-hydroxylase gene in NGFI-B-deficient mice. Mol Cell Biol 15:4331–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng LE, Chan FK, Cado D, Winoto A 1997. Functional redundancy of the Nur77 and Nor-1 orphan steroid receptors in T-cell apoptosis. EMBO J 16:1865–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bassett MH, Suzuki T, Sasano H, White PC, Rainey WE 2004. The orphan nuclear receptors NURR1 and NGFIB regulate adrenal aldosterone production. Mol Endocrinol 18:279–290 [DOI] [PubMed] [Google Scholar]
- 56.Peters B, Clausmeyer S, Obermuller N, Woyth A, Kranzlin B, Gretz N, Peters J 1998. Specific regulation of StAR expression in the rat adrenal zona glomerulosa. An in situ hybridization study. J Histochem Cytochem 46:1215–1221 [DOI] [PubMed] [Google Scholar]
- 57.Pollack SE, Furth EE, Kallen CB, Arakane F, Kiriakidou M, Kozarsky KF, Strauss III JF 1997. Localization of the steroidogenic acute regulatory protein in human tissues. J Clin Endocrinol Metab 82:4243–4251 [DOI] [PubMed] [Google Scholar]
- 58.Clark BJ, Wells J, King SR, Stocco DM 1994. The purification, cloning, and expression of a novel luteinizing hormone-induced mitochondrial protein in MA-10 mouse Leydig tumor cells. Characterization of the steroidogenic acute regulatory protein (StAR). J Biol Chem 269:28314–28322 [PubMed] [Google Scholar]
- 59.Ronen-Fuhrmann T, Timberg R, King SR, Hales KH, Hales DB, Stocco DM, Orly J 1998. Spatio-temporal expression patterns of steroidogenic acute regulatory protein (StAR) during follicular development in the rat ovary. Endocrinology 139:303–315 [DOI] [PubMed] [Google Scholar]
- 60.Stocco CO, Zhong L, Sugimoto Y, Ichikawa A, Lau LF, Gibori G 2000. Prostaglandin F2α-induced expression of 20α-hydroxysteroid dehydrogenase involves the transcription factor NUR77. J Biol Chem 275:37202–37211 [DOI] [PubMed] [Google Scholar]
- 61.Lavaque E, Sierra A, Azcoitia I, Garcia-Segura LM 2006. Steroidogenic acute regulatory protein in the brain. Neuroscience 138:741–747 [DOI] [PubMed] [Google Scholar]
- 62.Xiao Q, Castillo SO, Nikodem VM 1996. Distribution of messenger RNAs for the orphan nuclear receptors Nurr1 and Nur77 (NGFI-B) in adult rat brain using in situ hybridization. Neuroscience 75:221–230 [DOI] [PubMed] [Google Scholar]
- 63.Enyeart JJ, Boyd RT, Enyeart JA 1996. ACTH and AII differentially stimulate steroid hormone orphan receptor mRNAs in adrenal cortical cells. Mol Cell Endocrinol 124:97–110 [DOI] [PubMed] [Google Scholar]
- 64.Kelly SN, McKenna TJ, Young LS 2004. Modulation of steroidogenic enzymes by orphan nuclear transcriptional regulation may control diverse production of cortisol and androgens in the human adrenal. J Endocrinol 181:355–365 [DOI] [PubMed] [Google Scholar]
- 65.Wilson TE, Mouw AR, Weaver CA, Milbrandt J, Parker KL 1993. The orphan nuclear receptor NGFI-B regulates expression of the gene encoding steroid 21-hydroxylase. Mol Cell Biol 13:861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wooton-Kee CR, Clark BJ 2000. Steroidogenic factor-1 influences protein-deoxyribonucleic acid interactions within the cyclic adenosine 3,5-monophosphate-responsive regions of the murine steroidogenic acute regulatory protein gene. Endocrinology 141:1345–1355 [DOI] [PubMed] [Google Scholar]
- 67.Wilson TE, Fahrner TJ, Johnston M, Milbrandt J 1991. Identification of the DNA binding site for NGFI-B by genetic selection in yeast. Science 252:1296–1300 [DOI] [PubMed] [Google Scholar]
- 68.Davis IJ, Hazel TG, Chen RH, Blenis J, Lau LF 1993. Functional domains and phosphorylation of the orphan receptor Nur77. Mol Endocrinol 7:953–964 [DOI] [PubMed] [Google Scholar]
- 69.Katagiri Y, Hirata Y, Milbrandt J, Guroff G 1997. Differential regulation of the transcriptional activity of the orphan nuclear receptor NGFI-B by membrane depolarization and nerve growth factor. J Biol Chem 272:31278–31284 [DOI] [PubMed] [Google Scholar]
- 70.Li Y, Lau LF 1997. Adrenocorticotropic hormone regulates the activities of the orphan nuclear receptor Nur77 through modulation of phosphorylation. Endocrinology 138:4138–4146 [DOI] [PubMed] [Google Scholar]
- 71.Gambaryan S, Butt E, Tas P, Smolenski A, Allolio B, Walter U 2006. Regulation of aldosterone production from zona glomerulosa cells by ANG II and cAMP: evidence for PKA-independent activation of CaMK by cAMP. Am J Physiol Endocrinol Metab 290:E423–E433 [DOI] [PubMed]
- 72.Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM 1998. A family of cAMP-binding proteins that directly activate Rap1. Science 282:2275–2279 [DOI] [PubMed] [Google Scholar]
- 73.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL 1998. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396:474–477 [DOI] [PubMed] [Google Scholar]
- 74.Clark BJ, Pezzi V, Stocco DM, Rainey WE 1995. The steroidogenic acute regulatory protein is induced by angiotensin II and K+ in H295R adrenocortical cells. Mol Cell Endocrinol 115:215–219 [DOI] [PubMed] [Google Scholar]
- 75.Morley SD, Hobkirk JL, Hall RJ, Nicol M, Mason JI, Williams BC 2000. Effects of cellular mediator agonists on cortisol and steroid acute regulatory (StAR) protein in bovine zona fasciculata (ZF) cells. Endocr Res 26:603–608 [DOI] [PubMed] [Google Scholar]
- 76.Nishikawa T, Omura M, Suematsu S 1997. Possible involvement of calcium/calmodulin-dependent protein kinase in ACTH-induced expression of the steroidogenic acute regulatory (StAR) protein in bovine adrenal fasciculata cells. Endocr J 44:895–898 [DOI] [PubMed] [Google Scholar]
- 77.Pezzi V, Clark BJ, Ando S, Stocco DM, Rainey WE 1996. Role of calmodulin-dependent protein kinase II in the acute stimulation of aldosterone production. J Steroid Biochem Mol Biol 58:417–424 [DOI] [PubMed] [Google Scholar]
- 78.Condon JC, Pezzi V, Drummond BM, Yin S, Rainey WE 2002. Calmodulin-dependent kinase I regulates adrenal cell expression of aldosterone synthase. Endocrinology 143:3651–3657 [DOI] [PubMed] [Google Scholar]
- 79.Kumar S, Blumberg DL, Canas JA, Maddaiah VT 1994. Human chorionic gonadotropin (hCG) increases cytosolic free calcium in adult rat Leydig cells. Cell Calcium 15:349–355 [DOI] [PubMed] [Google Scholar]
- 80.Sullivan MH, Cooke BA 1986. The role of Ca2+ in steroidogenesis in Leydig cells. Stimulation of intracellular free Ca2+ by lutropin (LH), luliberin (LHRH) agonist and cyclic AMP. Biochem J 236:45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Manna PR, Pakarinen P, El-Hefnawy T, Huhtaniemi IT 1999. Functional assessment of the calcium messenger system in cultured mouse Leydig tumor cells: regulation of human chorionic gonadotropin-induced expression of the steroidogenic acute regulatory protein. Endocrinology 140:1739–1751 [DOI] [PubMed] [Google Scholar]
- 82.Manna PR, Dyson MT, Eubank DW, Clark BJ, Lalli E, Sassone-Corsi P, Zeleznik AJ, Stocco DM 2002. Regulation of steroidogenesis and the steroidogenic acute regulatory protein by a member of the cAMP response-element binding protein family. Mol Endocrinol 16:184–199 [DOI] [PubMed] [Google Scholar]
- 83.Sheng M, Thompson MA, Greenberg ME 1991. CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 252:1427–1430 [DOI] [PubMed] [Google Scholar]
- 84.Tremblay JJ, Viger RS 2001. GATA factors differentially activate multiple gonadal promoters through conserved GATA regulatory elements. Endocrinology 142:977–986 [DOI] [PubMed] [Google Scholar]
- 85.Tremblay JJ, Viger RS 1999. Transcription factor GATA-4 enhances Müllerian inhibiting substance gene transcription through a direct interaction with the nuclear receptor SF-1. Mol Endocrinol 13:1388–1401 [DOI] [PubMed] [Google Scholar]
- 86.Philips A, Lesage S, Gingras R, Maira MH, Gauthier Y, Hugo P, Drouin J 1997. Novel dimeric Nur77 signaling mechanism in endocrine and lymphoid cells. Mol Cell Biol 17:5946–5951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Galarneau L, Pare JF, Allard D, Hamel D, Levesque L, Tugwood JD, Green S, Belanger L 1996. The α1-fetoprotein locus is activated by a nuclear receptor of the Drosophila FTZ-F1 family. Mol Cell Biol 16:3853–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guigon CJ, Coudouel N, Mazaud-Guittot S, Forest MG, Magre S 2005. Follicular cells acquire Sertoli cell characteristics after oocyte loss. Endocrinology 146:2992–3004 [DOI] [PubMed] [Google Scholar]
- 89.Schreiber E, Matthias P, Muller MM, Schaffner W 1989. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res 17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ascoli M 1981. Characterization of several clonal lines of cultured Leydig tumor cells: gonadotropin receptors and steroidogenic responses. Endocrinology 108:88–95 [DOI] [PubMed] [Google Scholar]
- 91.Jordan M, Schallhorn A, Wurm FM 1996. Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res 24:596–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Robert NM, Martin LJ, Tremblay JJ 2006. The orphan nuclear receptor NR4A1 regulates insulin-like 3 gene transcription in Leydig cells. Biol Reprod 74:322–330 [DOI] [PubMed] [Google Scholar]
- 93.Khan S, Teerds K, Dorrington J 1992. Growth factor requirements for DNA synthesis by Leydig cells from the immature rat. Biol Reprod 46:335–341 [DOI] [PubMed] [Google Scholar]
- 94.Klinefelter GR, Hall PF, Ewing LL 1987. Effect of luteinizing hormone deprivation in situ on steroidogenesis of rat Leydig cells purified by a multistep procedure. Biol Reprod 36:769–783 [DOI] [PubMed] [Google Scholar]