

Abstract
Glutathione peroxidase 3 (GPx3) accounts for the major antioxidant activity in the plasma. Here, we demonstrate that down-regulation of GPx3 in the plasma of obese subjects is associated with adipose GPx3 dysregulation, resulting from the increase of inflammatory signals and oxidative stress. Although GPx3 was abundantly expressed in kidney, lung, and adipose tissue, we observed that GPx3 expression was reduced selectively in the adipose tissue of several obese animal models as decreasing plasma GPx3 level. Adipose GPx3 expression was greatly suppressed by prooxidative conditions such as high levels of TNFα and hypoxia. In contrast, the antioxidant N-acetyl cysteine and the antidiabetic drug rosiglitazone increased adipose GPx3 expression in obese and diabetic db/db mice. Moreover, GPx3 overexpression in adipocytes improved high glucose-induced insulin resistance and attenuated inflammatory gene expression whereas GPx3 neutralization in adipocytes promoted expression of proinflammatory genes. Taken together, these data suggest that suppression of GPx3 expression in the adipose tissue of obese subjects might constitute a vicious cycle to expand local reactive oxygen species accumulation in adipose tissue potentially into systemic oxidative stress and obesity-related metabolic complications.
ADIPOSE TISSUE contributes to maintaining the energy homeostasis of the whole body not only by buffering lipid metabolites but also by secreting several adipocytokines in response to the inputs of the central nervous system and periphery (1). In obesity, however, adipose tissue fails to accommodate fatty acids effectively according to the changing metabolic requirements, resulting in excessive accumulation of lipid metabolites in peripheral tissues, including the liver and muscle tissues, along with adipocytokine dysregulation (2). The abnormal regulation of adipocytokines occurring in obesity affects the functions of other tissues, including the liver, muscle, central nervous system, and vasculatures, thus increasing the risks for metabolic complications. Therefore, it appears that examining the molecular regulatory mechanisms for adipocytokines in obesity is crucial for understanding metabolic disorders and for developing effective therapeutic interventions for obesity and its related complications.
One of the main clinical manifestations of obesity is increased systemic oxidative stress (3, 4, 5). Oxidative stress has been implicated in several forms of tissue damage and leads to pathological conditions such as irradiation damage and ischemia reperfusion injury, as well as neurodegenerative diseases (6, 7). However, accumulating evidence indicates that increased oxidative stress is also strongly associated with metabolic disorders, including atherosclerosis, thrombosis, liver steatosis, and diabetes mellitus, which are often observed in morbid obesity (8, 9, 10, 11). Reactive oxygen species (ROS) can rapidly inactivate vascular nitrogen oxide (NO), a major vasorelaxant and inhibitor of platelet function, and can increase the risks for atherosclerosis and stroke. Moreover, posttranslational modifications of fibrinogen by ROS and NO-derived oxidants enhance fibrinogen activity, thus accelerating clot formation and thrombosis (12, 13). In addition, oxidative stress impairs insulin secretion by the pancreatic β-cells (14) as well as glucose transport into the muscle (15) and adipose tissue (16).
Recently, it has been shown that ROS generation is selectively increased in the fat tissues of obese mice, resulting in insulin resistance and dysregulation of adipocytokine gene expression (5). Interestingly, several insulin resistance-inducing factors such as TNFα and dexamethasone, as well as free fatty acids and high glucose levels, potently stimulate ROS production in adipocytes (5, 17, 18). On the other hand, antioxidant molecules such as N-acetyl cysteine (NAC), manganese (III) tetrakis (4-benzoic acid) porphyrin, and apocynin not only reverse TNFα-induced dysregulation of adipocytokine gene expression but also ameliorate insulin resistance, hyperlipidemia, and liver steatosis in obese animals, without altering the body weight (5, 17). Thus, it appears that increased systemic oxidative stress stemming from the expansion of adipose tissue during developing obesity may play a role in mediating obesity-related metabolic complications.
The cellular redox potential is maintained by a balanced regulation of prooxidative and antioxidative enzymes. The catalytic triad of superoxide dismutase, catalase (CAT), and glutathione peroxidase (GPx) is an antioxidant system for removing superoxide anions and is well conserved from prokaryotes to eukaryotes (12). Superoxide dismutase converts superoxide anions to H2O2, which is further catalyzed by GPx and CAT into a harmless product, H2O. CAT recognizes only H2O2 as its substrate and functions with very low affinity (19). Thus, it mainly functions only at H2O2 levels above the physiological level; these conditions may arise during oxidative burst in response to stress. On the other hand, GPx metabolizes peroxidized organic molecules as well as H2O2, recycles some of the molecules attacked by H2O2 with a relatively high affinity, and catalyzes these molecules even at the normal physiological concentrations (20). Therefore, GPx activity is considered to represent the initial protective response required for adjusting the H2O2 concentration under normal physiological conditions as well as after oxidative insult.
To date, seven isoforms of GPx proteins have been identified in mice (21). Of these, only GPx3 is found in the plasma and accounts for a major part of the plasma GPx activity (21). A large amount of GPx3 is synthesized in and secreted from the kidneys and lungs; it maintains the bioavailability of vascular NO and scavenges H2O2 and peroxidized organic molecules in the plasma to reduce systemic oxidative stress (22, 23).
In this study, we demonstrated that GPx3 was highly expressed in the adipose tissue, and its expression was reduced in both sera and fat tissues of obese subjects. Because GPx3 was reduced selectively in the fat tissues of obese mice, we propose that elevated systemic oxidative stress in obesity is associated with reduced circulating GPx3 expression, probably by diminished adipose GPx3 expression.
RESULTS
Systemic Oxidative Stress in Obesity Is Associated with Reduced Circulating GPx3 Expression
Recently, it has been reported that a systemic increase in oxidative stress is often observed in obese subjects and is regarded to be directly involved in increasing incidence of obesity-related metabolic complications including diabetes mellitus and cardiovascular diseases (3, 4, 5). Consistent with these reports, we observed that obese and diabetic db/db mice exhibited increased plasma ROS and oxidative damages, which were assessed in terms of thiobarbituric acid-reactive substances (TBARS) concentration (Fig. 1, A and B). While analyzing protein expression profile in the plasma of normal and obese mice, we found that the circulating GPx3 level was greatly decreased in the obese subjects (Fig. 1C). Concurrently, we observed that the total GPx activity in the plasma was reduced in the obese mice (Fig. 1D). Similar to the data obtained from the obese animal models, the plasma levels of the GPx3 protein and the total plasma GPx activity were substantially diminished even in obese human subjects (Fig. 1, E and F). These results suggest that increased oxidative stress in obesity appears to be linked with reduced circulating GPx3 expression.
Fig. 1.

Systemic Increase in Oxidative Stress in Obese Subjects Is Associated with Decreasing Circulating GPx3 Expression
A, ROS concentration in the serum samples obtained from lean and obese db/db mice. B, Systemic oxidative damage as reflected by the TBARS concentrations in the serum samples from lean and obese db/db mice. The values represent the means ± sem (n = 4 or 8). C, Western blot analyses of GPx3 protein expression in the serum samples of lean, ob/ob, and db/db mice. D, GPx activity assays on the serum samples from lean and obese mice. E and F, GPx3 protein expression (E) and GPx activity (F) are reduced in the serum of obese human subjects. Lean, body mass index <25; obese, body mass index >30. Error bars indicate the sem (n = 3 or 4). *, P < 0.05; **, P < 0.01. AdipoQ, Adiponectin.
GPx3 Is Abundantly Expressed in Adipose Tissue as Well as Kidney and Lung
To examine tissue distribution of the GPx3, we performed Northern blot analyses with mouse tissues. Previously, it has been shown that GPx3 is abundantly expressed in kidney, lung, and fat tissue in humans (24). Consistently, we observed that GPx3 mRNA was highly expressed in the kidney, lung, and white adipose tissue (WAT) of B6 mice (Fig. 2A). Moreover, GPx3 was highly expressed in brown adipose tissue (BAT) (Fig. 2A). In addition, GPx3 expression in the WAT was remarkably reduced in diet-induced obese mice than in the control mice (Fig. 2A).
Fig. 2.

GPx3 Is Abundantly Expressed in Adipose Tissue
A, Total RNA was isolated from several tissues of lean C57BL6 mice and was subjected to Northern blot analyses. DIO, Diet-induced obese. B, GPx3 mRNA expression in the two fractions—SVCs and adipocytes (Adipo)—obtained by collagenase digestion of WAT. Total RNA isolated from each fraction was analyzed by quantitative real-time RT-PCR. Adiponectin (AdipoQ) was used as a maker gene of adipocytes. C, GPx3 mRNA expression in the preadipocytes (Pre) and adipocytes (Adipo) of 3T3-L1 was analyzed by quantitative real-time RT-PCR. Error bars indicate the sem (n = 3).
Next, we determined relative expression of GPx3 in adipocytes and stromal vascular cells (SVCs) isolated from the adipose tissue. Recently, it was reported that GPx3 is induced during adipogenesis of human and bovine preadipocytes (25, 26). In accordance with this report, GPx3 was more abundantly expressed in adipocytes than in SVCs of B6 mouse adipose tissue (Fig. 2B). Moreover, its mRNA expression was elevated during adipocyte differentiation of 3T3-L1 (Fig. 2C). We also examined the expression of adiponection mRNA as a control for adipocyte marker gene (27). However, the GPx3 mRNA level was considerably lower (<250-fold less) in the 3T3-L1 adipocytes than in the mouse primary adipocytes, as frequently observed in other cell lines derived from the tissues abundantly expressing GPx3 (28).
GPx3 Expression Is Reduced in the Adipose Tissue, But Not Kidney, of Obese Mice
Because GPx3 mRNA was expressed most abundantly in kidney, we decided to determine whether the diminished circulating GPx3 level in obese subjects is associated with GPx3 expression in the kidneys. When we analyzed GPx3 expression, both mRNA and protein levels of GPx3 were not altered or even slightly increased in the kidneys of ob/ob and db/db obese mice (Fig. 3A) (see supplemental Figs. 1 and 2 published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals. org data). In contrast, GPx3 mRNA was significantly reduced in the WAT of obese animal models including ob/ob and db/db mice, as compared with that in lean mice (Fig. 3B); this result is consistent with the previous report that GPx3 is suppressed in the adipose tissue of obese fatty OLETF rats (29). Further, a similar reduction in the GPx3 expression was selectively observed in several adipose tissues of obese mice (supplemental Fig. 2). Furthermore, reduced GPx3 expression was mainly observed in the adipocytes but not in the SVCs isolated from the WAT of db/db mice (Fig. 3C) (supplemental Fig. 3). Notably, TNFα was induced in the WAT of ob/ob and db/db mice (Fig. 3B) as expected (30). Moreover, the GPx3 protein level and total GPx activity in the WAT of obese ob/ob and db/db mice were significantly diminished (Fig. 3, D and E). Concurrent with these findings, the levels of total ROS and TBARS found to be elevated in the WAT of db/db mice (Fig. 3, F and G). Together, it is likely that reduced plasma GPx3 levels observed in obese subjects is presumably due to reduced GPx3 expression in the fat tissues rather than in the kidneys (Figs. 1–3).
Fig. 3.

GPx3 Is Reduced in the Fat Tissue, but not in the Kidney of Obese Mice
A, Northern blot analysis of GPx3 mRNA expression in the kidneys of lean, ob/ob, and db/db mice. B, Northern blot analysis of GPx3 mRNA expression in the fat tissues of lean, ob/ob, and db/db mice. C, GPx3 mRNA expression was analyzed in the two fractions (SVCs and adipocytes) obtained by collagenase digestion of adipose tissue from lean and db/db mice. Total RNA isolated from each fraction was analyzed by quantitative real-time RT-PCR. D, Western blot analyses of GPx3 protein expression in the fat tissues obtained from lean, ob/ob, and db/db mice. E, GPx activity assays on the total lysates of epididymal fat tissues from lean and obese mice. F, ROS concentration in the total lysates of fat tissue from lean and obese db/db mice. G, Systemic oxidative damage as reflected by the TBARS concentrations in the total lysates of fat tissues obtained from lean and obese db/db mice. The values represent the means ± sem (n = 4 or 8). *, P < 0.05 for lean mice vs. ob/ob or db/db mice, indicating a significant difference between the groups. AdipoQ, Adiponectin.
Adipose GPx3 Expression Is Suppressed by TNFα, Lipopolysaccharide (LPS), and Hypoxia, Whereas It Is Stimulated by the Antioxidant NAC
It is well established that obesity is closely associated with ROS accumulation and with increased proinflammatory gene expression in adipose tissue (5, 30). Functioning as a key proinflammatory cytokine, TNFα expression is elevated in the fat tissues of obese mice, and it stimulates the expression of prooxidative genes such as inducible nitric oxide synthase (iNOS) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (30). To determine whether this augmented expression of TNFα is related to the reduced adipose GPx3 expression observed in obese mice, we treated 3T3-L1 adipocytes as well as mouse epididymal fat tissue with TNFα and analyzed their GPx3 mRNA expression. As previously reported (31, 32), TNFα induced inflammatory genes, including TNFα, SAA3, and iNOS, whereas it decreased the peroxisomal proliferator-activated receptor (PPAR)γ and adiponectin mRNA levels (Fig. 4, A and B). Interestingly, TNFα significantly decreased the GPx3 expression in both 3T3-L1 adipocytes and epididymal fat tissue (Fig. 4, A and B; and see supplemental Fig. 4).
Fig. 4.
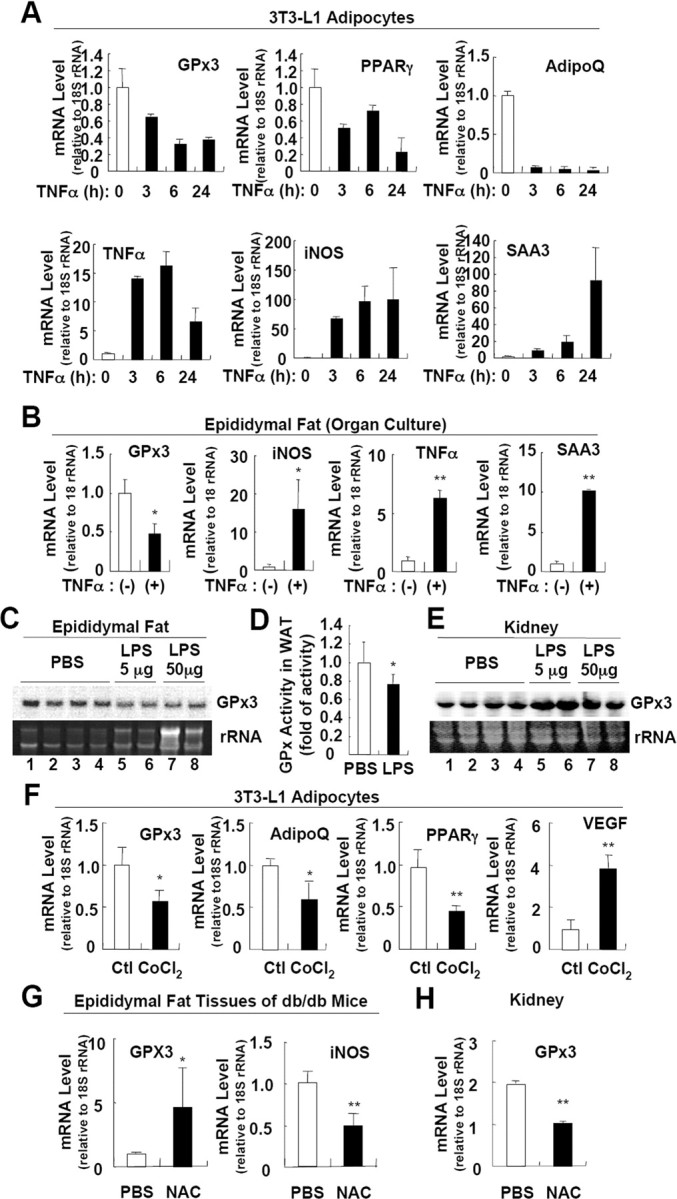
Adipose GPx3 Expression Is Suppressed by TNFα, LPS, and Hypoxia and Is Stimulated by the Antioxidant NAC
A and B, Adipose GPx3 is suppressed by TNFα. 3T3-L1 adipocytes (A) or epididymal fat tissue (B) was incubated with TNFα (10 ng/ml), and the expression of each gene was analyzed by quantitative real-time RT-PCR. +, TNFα treatment for 8 h. Error bars indicate the sem (n = 2). C, Northern blot analysis of GPx3 mRNA expression in fat tissues obtained from vehicle- and LPS-treated lean mice. LPS was administered to lean C57/BL6 mice as an ip injection for 24 h (5 μg) or 4 h (50 μg). D, GPx activity assays for total lysates of fat tissues obtained from control and LPS-treated (5 μg, 24 h) mice. Error bars indicate the sem (n = 4). E, Northern blot analysis of GPx3 mRNA expression in kidney from vehicle- and LPS-treated lean mice. LPS was administered to lean C57/BL6 mice as an ip injection for 24 h (5 μg) or 4 h (50 μg). F, GPx3 expression in adipocytes is repressed by hypoxia. 3T3-L1 adipocytes were incubated with CoCl2 (100 μm) for 24 h. The expression of each gene was analyzed by quantitative real-time RT-PCR. Error bars indicate the sem (n = 2). G and H, Differential regulation of GPx3 expression in the adipose tissue (G) and the kidneys (H) by the antioxidant NAC. db/db mice were ip injected with NAC for 1 wk. Expression of each mRNA was analyzed by quantitative real-time RT-PCR. Error bars indicate the sem (n = 3). *, P < 0.05; **, P < 0.01 for vehicle- vs. drug-treated group, indicating a significant difference between the groups. AdipoQ, Adiponectin; Ctl, control.
To directly ascertain whether inflammatory signals affect the adipose GPx3 expression in vivo, lean mice were treated with LPS, and the GPx3 mRNA expression levels and total GPx activities in fat tissues were measured. As shown in Fig. 4, C and D, both GPx3 expression and GPx activity were repressed in the adipose tissue by LPS-induced systemic inflammation, whereas the level of kidney GPx3 mRNA was enhanced by LPS (Fig. 4E). These results imply that increased inflammatory signals present in obesity could selectively down-regulate adipose GPx3 expression.
Hypoxic conditions stimulate ROS generation in several cell types and induce inflammatory gene expression (33). Recent reports have demonstrated the adipose tissue of obese mice to be hypoxic (34, 35), and hypoxia interferes with adipocyte differentiation as well as adipocytokine expression (36, 37, 38). To examine the effects of hypoxia on adipose GPx3 expression, 3T3-L1 adipocytes were treated with CoCl2, a well-known hypoxia mimetic (37, 39, 40, 41). As expected, treatment of CoCl2 reduced the expression of PPARγ and adiponectin, whereas it stimulated vascular endothelial growth factor expression (36) (Fig. 4F). Simultaneously, CoCl2 markedly suppressed GPx3 expression (Fig. 4F); this implies that hypoxia might also be a possible factor for the reduced adipose GPx3 expression observed in obese animals.
To clarify whether the decreased adipose GPx3 expression occurring in obesity may be due to oxidative stress with chronic inflammation and/or hypoxia, we administered a potent antioxidant chemical, NAC, to obese db/db mice, and we examined the GPx3 expression in the fat tissues. In accordance with the in vitro and in vivo data described above, NAC treatment enhanced GPx3 expression but reduced iNOS expression in the fat tissues of db/db mice (Fig. 4G). In contrast, GPx3 expression in the kidneys was reduced by NAC treatment in db/db mice (Fig. 4H), implying that GPx3 may be regulated by distinct mechanisms in adipose tissue and kidney in response to oxidative stress. Taken together, these in vitro, ex vivo, and in vivo data suggest that the reduced adipose GPx3 expression observed in obese mice would be tightly regulated by oxidative insults as well as inflammatory signals in an adipose tissue-specific manner.
Rosiglitazone and PPARγ Stimulate GPx3 Expression
Functioning as a master transcription factor for adipogenesis, PPARγ is involved in regulating the expression of several adipocytokine genes, including leptin, resistin, and adiponectin (42). PPARγ activation by its ligand thiazolidinediones (TZDs) improves insulin sensitivity and protects cells from oxidative stress-induced apoptosis (43, 44, 45, 46, 47). Because GPx3 was abundantly expressed in the WAT and BAT, we investigated the effects of PPARγ activation on adipose GPx3 expression. After treating 3T3-L1 adipocytes with rosiglitazone, the GPx3 mRNA level was dramatically elevated (Fig. 5A). Moreover, the administration of rosiglitazone to db/db mice restored the GPx3 expression in the fat tissues up to the level observed in lean mice (Fig. 5B) and abated oxidative stress, as reflected by the TBARS levels (Fig. 5C). However, GPx3 expression in kidney was not altered by rosiglitazone treatment (supplemental Fig. 5). To assess whether PPARγ is involved in the expression of adipose GPx3, we analyzed the mouse GPx3 promoter and found, at least, three putative PPAR response element (PPRE) motifs localized approximately at −1.4 kb (PPRE1), −2.4 kb (PPRE2), and −3.1 kb (PPRE3) away from the transcription start site (Fig. 5D). Next, we performed gel shift assays with ARE7 containing an endogenous PPRE from aP2 gene (48). As shown in Fig. 5E, all the three putative PPRE motifs from mouse GPx3 promoter competed with ARE7 probe, suggesting that the three PPREs might be potential binding sites for PPARγ/retinoid X receptor (RXR)α in vitro. For further analysis of the PPREs in mouse GPx3 promoter, PPARγ binding activity was determined with chromatin immunoprecipitation (ChIP) assays in adipocytes. As shown in Fig. 5F, substantial binding of PPARγ was detected only around PPRE3, but not PPRE1 and PPRE2, in adipocytes in the presence of rosiglitazone. These results suggest that PPARγ could stimulate GPx3 expression through binding to the mouse GPx3 promoter, and that the activation of PPARγ with TZD could reduce systemic oxidative stress, at least partially, by up-regulating the GPx3 expression in the fat tissues.
Fig. 5.

GPx3 Is Regulated by Rosiglitazone and PPARγ
A, GPx3 expression in adipocytes is induced by rosiglitazone. 3T3-L1 adipocytes were incubated with or without rosiglitazone for 18 h. The mRNA levels were analyzed by quantitative real-time RT-PCR. Error bars indicate the sem (n = 2). B, Adipose GPx3 expression is stimulated by rosiglitazone in db/db mice. db/db Mice were administered an oral gavage of rosiglitazone (5 mg/kg) for 10 d. Total RNA was prepared from the epididymal fat tissues and was subjected to real-time RT-PCR analysis. Error bars indicate the sem (n = 4). **, P < 0.01. C, Systemic oxidative damage as reflected by the TBARS concentrations in the serum samples obtained from normal and db/db mice. The values represent the means ± sem (n = 4). *, P < 0.05. D, Schematic presentation of putative PPRE motifs in the mouse GPx3 promoter region. E, Gel shift assays were performed with 32P-labeled ARE7 oligonucleotide containing an endogenous PPRE on the enhancer region of mouse aP2 gene as a probe and in vitro translated PPARγ and RXRα proteins. The specific probe-PPARγ/RXRα complex was abolished by addition of 100-fold molar excess of unlabeled ARE7 (lane 8) and the three putative PPRE motifs (lanes 3–5), but not E-box (lane 7) and PPRE3-mutant. In competitors, 3, PPRE3; 2, PPRE2; 1, PPRE1; 3m, PPRE3 mutant; NS, nonspecific competitor (E-box); SC, specific competitor (ARE7). F, ChIP analysis of GPx3 promoter. 3T3-L1 preadipocytes or fully differentiated adipocytes were incubated with or without rosiglitazone (10 μm) for 48 h. For each experiment, 0.5% of input was used. IgG, Purified nonspecifc IgG; PPARγ, anti-PPARγ antibody; AdipoQ, adiponectin; Rosi, rosiglitazone; Veh, vehicle.
GPx3 Overexpression in Adipocytes Ameliorates High-Glucose-Induced Inflammatory Gene Expression and ROS Accumulation, Whereas GPx3 Neutralization Enhances Inflammatory Gene Expression
To gain further insights into the roles of GPx3 in adipocytes, we adenovirally overexpressed GPx3 in 3T3-L1 adipocytes and incubated the cells with sodium selenite because GPx3 is a selenocystein-containing protein (10). Analysis of the inflammatory gene expression profiles in the presence or absence of a hyperglycemic challenge revealed that GPx3 overexpression in 3T3-L1 adipocytes significantly repressed the high-glucose-induced expression of proinflammatory genes such as SAA3, resistin, and CCR2 (18, 49) (Fig. 6). Additionally, it suppressed the expression of the p47 and p67 subunits of the NADPH-oxidase complex (Fig. 6), which are regulated by nuclear factor-κB and TNFα (50). More interestingly, ROS released from the adipocytes into the medium were reduced by GPx3 overexpression under high glucose conditions (Fig. 7A). Because ROS has been reported to trigger insulin resistance in adipocytes (17, 18), we examined the effects of GPx3 overexpression on high-glucose-induced insulin resistance. Incubation of 3T3-L1 adipocytes in high-glucose media substantially reduced the insulin-stimulated glucose uptake (by ∼33%) (18), whereas GPx3 overexpression in 3T3-L1 adipocytes restored the insulin-stimulated glucose uptake under the same conditions (Fig. 7, B and C).
Fig. 6.

Adenoviral Overexpression of GPx3 in Adipocytes Reduces Proinflammatory Gene Expression in Adipocytes
3T3-L1 adipocytes were infected with a green fluorescent protein (GFP)- or GPx3-expressing adenovirus (multiplicity of infection = 50). Two days after infection (>80% of cells were GFP positive), the cells were incubated with low (5.5 mm)- or high (25 mm)-glucose DMEM for additional 24 h. The medium was supplemented with sodium selenite (0.1 μm) throughout the experiments. The mRNA levels of proinflammatory genes were analyzed by quantitative real-time RT-PCR. sem are indicated by the error bars (n = 2). **, P < 0.01. Ad-GFP, Adenoviral GFP.
Fig. 7.
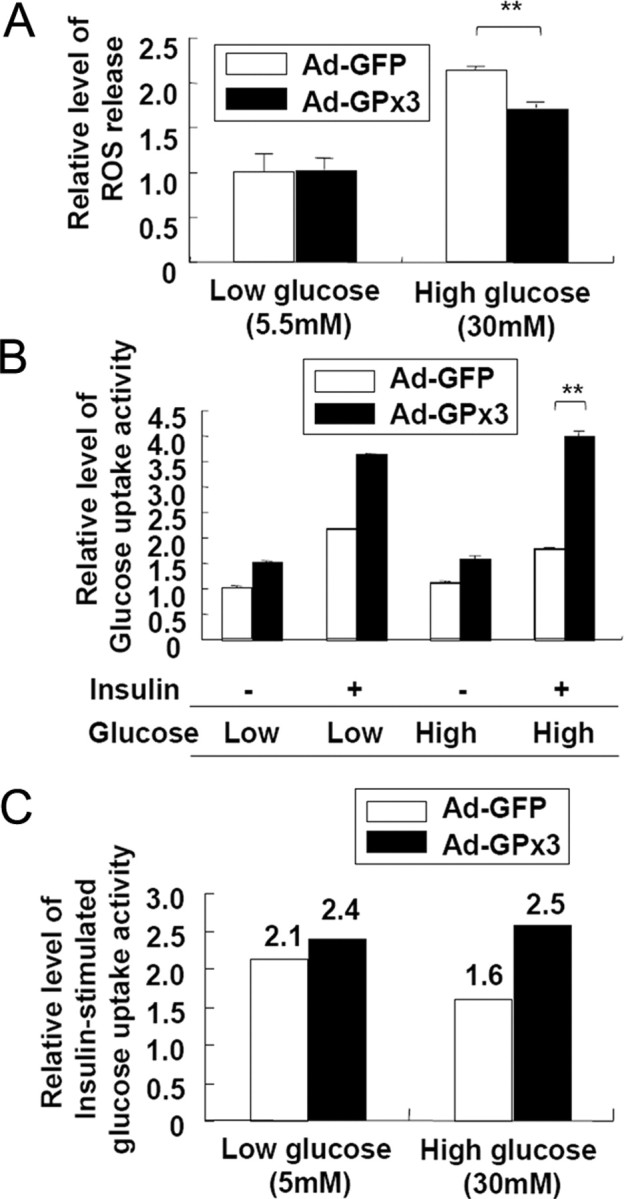
GPx3 Overexpression Reduces ROS Accumulation and Improves High-Glucose-Induced Insulin Resistance in 3T3-L1 Adipocytes
3T3-L1 adipocytes were infected with a green fluorescent protein (GFP)- or GPx3-expressing adenovirus (multiplicity of infection = 50). Two days after infection (>80% of cells were GFP positive), the cells were incubated with low- (5.5 mm) or high-glucose (25 mm) DMEM. The medium was supplemented with sodium selenite (0.1 μm) throughout the experiments. A, The level of ROS released from the adipocytes was measured using the Amplex Red hydrogen peroxide assay kit, as described in Materials and Methods. **, P < 0.01. B, Glucose uptake assays. Twenty-four hours after incubation in low- or high-glucose media, the cells were incubated in the presence or absence of insulin for 1 h; [14C]2-deoxy-glucose was added for 30 min and its uptake was measured. The values represent the means ± sem (n = 3). *, P < 0.05. C, Insulin-stimulated glucose uptake activity. The relative activities were determined by normalizing the values in panel B in the presence of insulin with the values obtained in the absence of insulin. Ad-GFP, adenoviral GFP; Ad-GPx3, adenoviral GPx3.
To confirm the effects of adipose GPx3 on inflammatory gene expression, we tried to perform loss-of-function experiments in adipocytes. Because 3T3-L1 adipocytes expressed extremely low levels of GPx3 compared with primary adipocytes, we failed to obtain substantial knockdown of GPx3 expression via siRNA. Alternatively, we adopted antibody-assisted neutralization with primary mouse adipocytes. As illustrated in Fig. 8A, treatment of GPx3-specific antibodies to neutralize secreted GPx3 from adipocytes promoted the expression of several inflammatory genes. Of interest, the level of GPx3 protein in the adipocyte-conditioned media was comparable to that observed in plasma of lean B6 mice, suggesting that GPx3 concentration might be enough to mediate antioxidative properties (Fig. 8B). Taken together, these results strongly indicate that GPx3 overexpression in adipocytes would alleviate the proinflammatory gene expression and oxidative burst induced by hyperglycemia and ameliorate high-glucose-induced insulin resistance.
Fig. 8.

Neutralization of GPx3 with Anti-GPx3 Antibodies Stimulates Proinflammatory Gene Expression in Mouse Primary Adipocytes
A, Mouse primary adipocytes were obtained by collagenase digestion of adipose tissue from lean C57/BL6 mice. Cells were incubated with a rabbit control IgG (mock) or purified anti-GPx3 antibodies (IgG fraction) for 24 h. Total RNA isolated from each group was analyzed by quantitative real-time RT-PCR. B, GPx3 protein concentration in each sample was assessed by using a GPx3 ELISA kit.
DISCUSSION
Accumulating evidence indicates that expanding stressful conditions in the adipose tissue of obesity, including hypoxia and macrophage infiltration, could induce local inflammation and ROS accumulation to affect dysregulation of adipocytokine genes and systemic oxidative stress, resulting in metabolic abnormalities. Recently, it has been also shown that oxidative stress increases selectively in the adipose tissue of obesity, which confers systemic oxidative stress to raise risks for obesity-related metabolic complications (5). As plausible candidates fetching oxidative stress in the adipose tissue of obesity, we and others reported that the activities of the NADPH oxidase and a NADPH-producing enzyme, glucose-6 phosphate dehydrogenase, are elevated in the fat tissues of obese mice, and they trigger local ROS accumulation and inflammation in adipose tissue (5, 51, 52). However, the molecular mechanism by which a local increase in oxidative stress in the fat tissues could bring about a systemic increase in the ROS accumulation in obesity remains unclear.
In this study, we demonstrated that the prooxidative conditions such as hypoxia and inflammation could reduce adipose GPx3 expression and, thereby, contribute to the decreased plasma GPx activity. As a major antioxidant enzyme in circulation, reduced plasma GPx3 level has been shown to be associated with enhanced systemic oxidative stress to increase susceptibility to childhood idiopathic stroke (53, 54), suggesting that extracellular GPx activity is critical for maintaining plasma oxidative tone and normal vascular function. Therefore, it seems that down-regulation of adipose GPx3 expression and subsequent decrease in circulating GPx activity might be associated with the obesity-related rise in systemic oxidative stress and incidence of metabolic complications.
Because excessive levels of ROS play causative roles in the development of insulin resistance and diabetes (55, 56), it has been speculated that increased GPx activity could have beneficial effects on glucose metabolism. However, GPx1-overexpressing transgenic mice develop insulin resistance along with hampered insulin function, probably due to overquenching of the intracellular ROS burst required for insulin sensitization (57). An acute intracellular ROS burst after insulin stimulation is required for sensitizing insulin signaling to suppress protein tyrosine phosphatase activity (58). Thus, it appears that reducing the ROS accumulation in circulation while maintaining proper intracellular ROS tone may be critical for managing glucose homeostasis in diabetic subjects. In this regard, we highlight the use of GPx3 as a potential target for intervention in insulin resistance. GPx3 functions as a major extracellular antioxidant enzyme, and its overexpression in adipocytes was observed to reduce ROS accumulation, diminish proinflammatory gene expression, and ameliorate hyperglycemia-induced insulin resistance (Figs. 6 and 7). Additionally, we observed that glucose tolerance was improved in db/db mice by administering the antioxidant NAC (data not shown), suggesting that increased systemic antioxidative activity may reverse obesity-related glucose intolerance. Therefore, it is likely that GPx3 would participate in controlling ROS-induced stress in circulation as well as in adipose tissue, thus modulating the energy homeostasis of the whole body, and that it would play a protective role in obesity-related metabolic disorders
Interestingly, we observed that circulating GPx3 levels closely correlated with adipose GPx3 expression rather than that in the kidneys of obese animals (Figs. 1 and 3) (supplemental Figs. 1 and 2). GPx3 is expressed most abundantly in the kidneys (Fig. 2). Further, anephric individuals show reduced plasma GPx activity and GPx3 protein expression, which is reversed by kidney transplantation (59, 60, 61). Thus, it had been suspected that circulating GPx3 appears to be derived mainly from the kidneys. However, to our surprise, kidney GPx3 expression was not altered or even slightly increased when circulating GPx3 expression was substantially reduced in obese ob/ob and db/db mice (Figs. 1 and 3). With these findings, it would be feasible to propose that reduced circulating GPx activity in obese animals would be primarily correlated with decreased adipose GPx3 expression. However, it remains to be elucidated whether kidney GPx3 expression might also affect reduced plasma GPx3 in obesity with decreased secretion of GPx3 proteins.
In obesity, adipose tissue gradually develops hypoxia due to rapid growth in the overall fat cell size and fat mass. Recently, it has been shown that hypoxia potently enhances GPx3 expression in Caki-2 renal cells, and this expression is blocked by treatment with the antioxidant molecule NAC (62). Because hypoxia stimulates ROS production, it has been proposed that increased GPx3 expression may induce an adaptive response mechanism to oxidative stress under hypoxic conditions in the kidneys. However, in the current study, we observed that GPx3 expression evidently decreased in the adipose tissue but increased slightly in the kidneys of obese animals such as ob/ob and db/db mice (Fig. 3) (supplemental Fig. 1). Additionally, GPx3 expression was specifically diminished in the adipocytes but remained unaltered in the SVCs of the fat tissues in obese mice (Fig. 3C). Moreover, the GPx3 expression in adipocytes was reduced in CoCl2-induced hypoxic conditions (Fig. 4F), indicating that cell type-specific regulation of GPx3 in adipocytes due to hypoxia could be at least partly responsible for the down-regulation of GPx3 mRNA in the adipose tissue of obese mice.
Another potential mechanism responsible for reduced GPx3 expression in the adipocytes of obese mice is related to chronically augmented local inflammation with increased macrophage infiltration into the adipose tissue. We observed that the adipose GPx3 mRNA level was diminished by inflammatory signals of TNFα and LPS in vitro and in vivo (Fig. 4). TNFα increases the ROS generation in adipose tissue by stimulating iNOS expression and the NADPH oxidase activity in adipocytes and macrophages (63, 64). Thus, it would be plausible that adipose GPx3 expression is reduced by prooxidative conditions such as inflammation and hypoxia, which may induce further ROS accumulation in the adipose tissue and serum of obese animals. Consistent with this hypothesis, administration of the antioxidant molecule NAC to db/db mice increased adipose GPx3 expression (Fig. 4G).
TZDs are prominent antidiabetic drugs that are widely used for decreasing the fasting glucose levels and improving insulin resistance (43, 44). Additionally, recent evidence indicates that TZDs also exert protective effects against cardiovascular diseases, including atherosclerosis, thrombosis, and stroke, due to their antiinflammatory and antioxidative properties (42). Of the TZDs, troglitazone reduces ROS accumulation by directly scavenging superoxide anions (65, 66). Moreover, pioglitazone and rosiglitazone reduce systemic oxidative stress in diet-induced obese mice by unknown mechanisms in the absence of direct ROS scavenging (65). Here, we demonstrated that rosiglitazone can reduce systemic ROS accumulation by inducing GPx3 expression via the stimulation of PPARγ in adipose tissue (Fig. 5). It is interesting to note that adipose GPx3 expression was suppressed under stressful conditions such as inflammation and hypoxia (Fig. 4), wherein PPARγ expression is substantially reduced in adipocytes (37, 67). Moreover, GPx3 down-regulation in obese subjects was shown to be restricted to the adipocytes where PPARγ is dominantly expressed, but not in SVCs. These results imply that PPARγ is one of the major transcriptional regulators of GPx3 expression, at least in adipocytes, and explain why GPx3 was decreased most severely and selectively in the adipose tissue of obese subjects.
In summary, we have provided the first evidence that defective GPx3 expression in adipose tissue is associated with reduced systemic GPx activity and increased oxidative stress in obesity. Furthermore, we demonstrated that hypoxia and TNFα regulate GPx3 in a tissue-specific manner that is possibly regulated by PPARγ; this may induce obesity-related down-regulation of adipose GPx3 expression, leading to augmented systemic oxidative stress and the onset of metabolic complications such as diabetes and cardiovascular diseases. In this respect, it is possible to propose that local ROS accumulation in the adipose tissue of obesity could be expanded into systemic oxidative stress by the vicious cycle wherein increasing local ROS accumulation suppresses adipose GPx3 expression. Thus, these results support further exploration of GPx3 expression in adipose tissue as a therapeutic target for obesity-related metabolic complications.
MATERIALS AND METHODS
Cell Culture
3T3-L1 preadipocytes were grown to confluence in DMEM supplemented with 10% bovine calf serum. At 2 d postconfluence, the 3T3-L1 cells were incubated for 48 h with DMEM containing 10% fetal bovine serum, methylisobutylxanthine (500 μm), dexamethasone (1 μm), and insulin (5 μg/ml). Every alternate day, the culture medium was replaced with DMEM containing 10% fetal bovine serum and insulin (1 μg/ml).
Adipose Tissue Culture
Mouse epididymal adipose tissue was evenly minced and incubated in DMEM with 0.1% BSA and antibiotics (penicillin and streptomycin), in the presence or absence of 10 ng/ml recombinant murine TNFα for 8 h.
Quantitative Real-Time RT-PCR
Quantitative real-time RT-PCR was performed as previously described (68). rRNA (18S) was used as the invariant control. The primers used were as follows: GPx3-forward (f), 5′-TAATTTCCAGCTCTTTGAGAAA-3′; GPx3-reverse (r), 5′-GG AACTTCTCAAAGTTCCAGCG-3′; iNOS-f, 5′-AATCTTGGGCGAGTTGTGG-3′; iNOS-r, 5′-CAGGAAGTAGGTGAGGGCTTG-3′; SAA3-f, 5′-AGTGATGCCAGAGAGGCTGT-3′; SAA3-r, 5′-ACCCAGTAGTTGCCCCTCTT-3′; resistin-f, 5′-CAGAAGGCACAGCAGTCTTG-3′; resistin-r, 5′-GACCGGAGGACATCAGACAT-3′; p47phox-f, 5′-AGTGTTCCCCATTGAGGCCCG-3′; p47phox-r, 5′-GTTTCAGGTCATCAGGCCGC-3′; p67phox-f, 5′-CTGGCTGAGGCCATCAGACT-3′; p67phox-r, 5′-AGGCCACTGCAGAGTGCTTG-3′; adiponectin-f, 5′-ATGCTACTGTTGCAAGCTCTC-3′; and adiponectin-r, 5′-GTTGGTATCATGGAAGAGAAG-3′.
GPx Activity Assay
GPx activity was measured according to the manufacturer’s protocol (Northwest Life Science Specialties, Vancouver, WA).
Lipid Peroxidation (LPO) and H2O2 Concentration
The tissue samples were homogenized and centrifuged, and the supernatant was used for the subsequent assays. The levels of LPO in the plasma and tissue homogenate were measured in terms of TBARS by performing the LPO test (Oxford Biomedical Research, Rochester Hills, MI). The H2O2 concentration was measured using an Amplex Red hydrogen peroxide assay kit (Invitrogen, Carlsbad, CA).
Gel Shift Assay
Gel shift assays were performed as described previously (69). One microgram of plasmid DNA expressing PPARγ or RXRα was used as template for in vitro translation. The DNA sequences of the double-stranded oligonucleotides used as probe or competitors were as follows (only one strand is shown): ARE7, 5′-GATCTGGAACTCTGATCCAGTAAG-3′; PPRE1, 5′-GCTGGAGGTTAGAAGTCAACTCT-3′; PPRE2, 5′-TAATGGGGTCACAGGTTATGCCA-3′; PPRE3, 5′-GAATTTGAACTTTAACCCGAGG-3′; PPRE3-mutant, 5′-GAATTAAAACTTAAACCCGAGG-3′. Probe was end labeled with [γ-32P]ATP. Purified probe (∼30,000 cpm) and proteins were used in 20 μl binding reactions containing reaction buffer [10 mm Tris (pH 7.5), 50 mm KCl, 2.5 mm MgCl2, 0.05 mm EDTA, 0.1% (vol/vol) Triton X-100, 8.5% (vol/vol) glycerol, 1 μg of polydeoxyinosinic deoxycytidylic acid, 1 mm dithiothreitol, and 0.1% (wt/vol) nonfat dry milk]. Samples were loaded onto a nondenaturing 4% polyacrylamide gel. The gels were dried and autoradiographed.
ChIP Analysis
ChIP assays were performed as described previously (68). Primers used were as follows: PPRE1-f, 5′-CAGCTAGTCACATGCCTCCA-3′; PPRE1-r, 5′-TCAGCAGGTAAAAGCCCTCA-3′; PPRE2-f, 5′-CCTGCCATTTATGTGGTGCT-3′; PPRE2-r, 5′-AACAAAACGGGGGAACAAAG-3′; PPRE3-f, 5′-TGCAGGTGAGGCTGAGCTAT-3′; PPRE3-r, 5′-GGAGGAGGCTGAAGCAGAAG-3′.
Animals and Treatments
All experiments were approved by the Seoul National University Animal Experiment Ethics Committee. Male C57BL/6J, ob/ob, and db/db mice were housed in colony cages under 12-h light, 12-h dark cycles. For rosiglitazone treatment, the mice received an oral gavage of the drug (5 mg/kg body weight) (Calbiochem, La Jolla, CA) daily for 10 d. After the final administration, the animals were fasted for 4 h, and the plasma glucose levels were tested to confirm the hypoglycemic effects (glucose level <200 mg/dl) of rosiglitazone. The animals were killed by cervical dislocation 1 d later. For LPS injection, the mice were ip injected with the vehicle (PBS) or with 5 μg (for 24 h) or 50 μg (for 4 h) LPS.
Human Serum Samples
Human serum samples provided by Samsung Medical Center were analyzed for GPx3 protein expression and GPx activity. The procedure for obtaining human serum samples was approved by the Samsung Medical Center Institutional Review Board (IRB file no. 2006-03-053), and written informed consent was obtained from the volunteers.
Measurement of Glucose Uptake
Insulin-stimulated glucose uptake in the 3T3-L1 adipocytes was determined by measuring the [14C]2-deoxyglucose uptake as described previously (51). In short, adenovirus-infected 3T3-L1 adipocytes were incubated in low- (5.5 mm) or high-glucose (25 mm) DMEM containing 0.1% BSA for 24 h at 37 C. The cells were stimulated with 100 nm insulin for 1 h at 37 C or were left untreated. Glucose uptake was initiated by treatment with [14C]2-deoxy-d-glucose at a final concentration of 3 μmol/liter in HEPES-buffered saline [140 mm NaCl, 5 mm KCl, 2.5 mm MgCl2, 1 mm CaCl2, and 20 mm HEPES (pH 7.4)] for 10 min. The reaction was terminated by separating the cells from the HEPES-buffered saline and [14C]2-deoxy-d-glucose. After three washings in ice-cold PBS, the cells were extracted with 0.1% sodium dodecyl sulfate and subjected to scintillation counting to determine their 14C radioactivity. The protein concentrations were determined using a bicinchoninic acid assay kit (Pierce Chemical Co., Rockford, IL), and the radioactivities were normalized by determining each protein concentration.
GPx3 Neutralization
Mouse primary adipocytes were prepared by collagen digestion. After washing three times with DMEM supplemented with 0.2% BSA, cells were treated with either the IgG fraction of a polyclonal rabbit antibody to mouse GPx3 (1 μg/ml) or with equivalent mounts of normal rabbit IgG as a control for 24 h.
Measurement of GPx3 Concentration
GPx3 protein concentration was measured using ELISA according to the manufacturer’s protocol (Adipogen, Seoul, Korea).
Statistical Analysis
All the results are presented as mean ± sem. Statistical significance was assessed by Student’s t test. Differences were considered statistically significant at P < 0.05.
Acknowledgments
We thank Joseph Loscalzo for providing the human GPx3 expression vector and the GPx3 promoter luciferase vector.
Footnotes
This work was supported by grants from the Korea Science and Engineering Foundation funded by the Korean government (Ministry of Education, Science and Technology, MEST) (nos. R0A-2004-000-10359-0, R11-2005-009-01002-0, M10748000258-07N4800-25810). A.Y.K., J.W.C., K.S.P., and J.B.K. were supported by a BK21 Research Fellowship from the Ministry of Education and Human Resources Development.
Disclosure Statement: the authors have nothing to disclose.
First Published Online June 18, 2008
Abbreviations: BAT, Brown adipose tissue; CAT, catalase; ChIP, chromatin immunoprecipitation; f, forward; GPx3, glutathione peroxidase 3; iNOS, inducible nitric oxide synthase; LPO, lipid peroxidation; LPS, lipopolysaccharide; NAC, N-acetyl cysteine; NADPH, reduced nicotinamide adenine dinucleotide phosphate; PPAR, peroxisomal proliferator-activated receptor; PPRE, PPAR response element; r, reverse; ROS, reactive oxygen species; RXR, retinoid X receptor; SVC, stromal vascular cell; TBARS, thiobarbituric acid-reactive substances; TZD, thiazolidinedione; WAT, white adipose tissue.
References
- 1.Scherer PE 2006. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes 55:1537–1545 [DOI] [PubMed] [Google Scholar]
- 2.Trujillo ME, Scherer PE 2006. Adipose tissue-derived factors: impact on health and disease. Endocr Rev 27:762–778 [DOI] [PubMed] [Google Scholar]
- 3.Keaney Jr JF, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ 2003. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol 23:434–439 [DOI] [PubMed] [Google Scholar]
- 4.Fujita K, Nishizawa H, Funahashi T, Shimomura I, Shimabukuro M 2006. Systemic oxidative stress is associated with visceral fat accumulation and the metabolic syndrome. Circ J 70:1437–1442 [DOI] [PubMed] [Google Scholar]
- 5.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I 2004. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114:1752–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mates JM, Sanchez-Jimenez F 1999. Antioxidant enzymes and their implications in pathophysiologic processes. Front Biosci 4:D339–D345 [DOI] [PubMed]
- 7.Simonian NA, Coyle JT 1996. Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol 36:83–106 [DOI] [PubMed] [Google Scholar]
- 8.Stocker R, Keaney Jr JF 2004. Role of oxidative modifications in atherosclerosis. Physiol Rev 84:1381–1478 [DOI] [PubMed] [Google Scholar]
- 9.Grattagliano I, Palmieri VO, Portincasa P, Moschetta A, Palasciano G 2008. Oxidative stress-induced risk factors associated with the metabolic syndrome: a unifying hypothesis. J Nutr Biochem 19:491–504 [DOI] [PubMed] [Google Scholar]
- 10.Leopold JA, Loscalzo J 2005. Oxidative enzymopathies and vascular disease. Arterioscler Thromb Vasc Biol 25:1332–1340 [DOI] [PubMed] [Google Scholar]
- 11.Roskams T, Yang SQ, Koteish A, Durnez A, DeVos R, Huang X, Achten R, Verslype C, Diehl AM 2003. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am J Pathol 163:1301–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Upchurch Jr GR, Ramdev N, Walsh MT, Loscalzo J 1998. Prothrombotic consequences of the oxidation of fibrinogen and their inhibition by aspirin. J Thromb Thrombolysis 5:9–14 [DOI] [PubMed] [Google Scholar]
- 13.Vadseth C, Souza JM, Thomson L, Seagraves A, Nagaswami C, Scheiner T, Torbet J, Vilaire G, Bennett JS, Murciano JC, Muzykantov V, Penn MS, Hazen SL, Weisel JW, Ischiropoulos H 2004. Pro-thrombotic state induced by post-translational modification of fibrinogen by reactive nitrogen species. J Biol Chem 279:8820–8826 [DOI] [PubMed] [Google Scholar]
- 14.Matsuoka T, Kajimoto Y, Watada H, Kaneto H, Kishimoto M, Umayahara Y, Fujitani Y, Kamada T, Kawamori R, Yamasaki Y 1997. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J Clin Invest 99:144–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maddux BA, See W, Lawrence Jr JC, Goldfine AL, Goldfine ID, Evans JL 2001. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of α-lipoic acid. Diabetes 50:404–410 [DOI] [PubMed] [Google Scholar]
- 16.Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, Bashan N 1998. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 47:1562–1569 [DOI] [PubMed] [Google Scholar]
- 17.Houstis N, Rosen ED, Lander ES 2006. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440:944–948 [DOI] [PubMed] [Google Scholar]
- 18.Lin Y, Berg AH, Iyengar P, Lam TK, Giacca A, Combs TP, Rajala MW, Du X, Rollman B, Li W, Hawkins M, Barzilai N, Rhodes CJ, Fantus IG, Brownlee M, Scherer PE 2005. The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species. J Biol Chem 280:4617–4626 [DOI] [PubMed] [Google Scholar]
- 19.Cohen G, Hochstein P 1963. Glutathione peroxidase: the primary agent for the elimination of hydrogen peroxide in erythrocytes. Biochemistry 2:1420–1428 [DOI] [PubMed] [Google Scholar]
- 20.Izawa S, Inoue Y, Kimura A 1996. Importance of catalase in the adaptive response to hydrogen peroxide: analysis of acatalasaemic Saccharomyces cerevisiae Biochem J 320:61–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drevet JR 2006. The antioxidant glutathione peroxidase family and spermatozoa: a complex story. Mol Cell Endocrinol 250:70–79 [DOI] [PubMed] [Google Scholar]
- 22.Maddipati KR, Marnett LJ 1987. Characterization of the major hydroperoxide-reducing activity of human plasma. Purification and properties of a selenium-dependent glutathione peroxidase. J Biol Chem 262:17398–17403 [PubMed] [Google Scholar]
- 23.Takahashi K, Avissar N, Whitin J, Cohen H 1987. Purification and characterization of human plasma glutathione peroxidase: a selenoglycoprotein distinct from the known cellular enzyme. Arch Biochem Biophys 256:677–686 [DOI] [PubMed] [Google Scholar]
- 24.Maeda K, Okubo K, Shimomura I, Mizuno K, Matsuzawa Y, Matsubara K 1997. Analysis of an expression profile of genes in the human adipose tissue. Gene 190:227–235 [DOI] [PubMed] [Google Scholar]
- 25.Urs S, Smith C, Campbell B, Saxton AM, Taylor J, Zhang B, Snoddy J, Jones Voy B, Moustaid-Moussa N 2004. Gene expression profiling in human preadipocytes and adipocytes by microarray analysis. J Nutr 134:762–770 [DOI] [PubMed] [Google Scholar]
- 26.Yamasaki T, Tahara K, Takano S, Inoue-Murayama M, Rose MT, Minashima T, Aso H, Ito S 2006. Mechanism of plasma glutathione peroxidase production in bovine adipocytes. Cell Tissue Res 326:139–147 [DOI] [PubMed] [Google Scholar]
- 27.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF 1995. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270:26746–26749 [DOI] [PubMed] [Google Scholar]
- 28.Chu FF, Esworthy RS, Doroshow JH, Doan K, Liu XF 1992. Expression of plasma glutathione peroxidase in human liver in addition to kidney, heart, lung, and breast in humans and rodents. Blood 79:3233–3238 [PubMed] [Google Scholar]
- 29.Asayama K, Nakane T, Dobashi K, Kodera K, Hayashibe H, Uchida N, Nakazawa S 2001. Effect of obesity and troglitazone on expression of two glutathione peroxidases: cellular and extracellular types in serum, kidney and adipose tissue. Free Radic Res 34:337–347 [DOI] [PubMed] [Google Scholar]
- 30.Hotamisligil GS 2006. Inflammation and metabolic disorders. Nature 444:860–867 [DOI] [PubMed] [Google Scholar]
- 31.Zhang B, Berger J, Hu E, Szalkowski D, White-Carrington S, Spiegelman BM, Moller DE 1996. Negative regulation of peroxisome proliferator-activated receptor-γ gene expression contributes to the antiadipogenic effects of tumor necrosis factor-α. Mol Endocrinol 10:1457–1466 [DOI] [PubMed] [Google Scholar]
- 32.Maeda N, Takahashi M, Funahashi T, Kihara S, Nishizawa H, Kishida K, Nagaretani H, Matsuda M, Komuro R, Ouchi N, Kuriyama H, Hotta K, Nakamura T, Shimomura I, Matsuzawa Y 2001. PPARγ ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 50:2094–2099 [DOI] [PubMed] [Google Scholar]
- 33.Murdoch C, Muthana M, Lewis CE 2005. Hypoxia regulates macrophage functions in inflammation. J Immunol 175:6257–6263 [DOI] [PubMed] [Google Scholar]
- 34.Fleischmann E, Kurz A, Niedermayr M, Schebesta K, Kimberger O, Sessler DI, Kabon B, Prager G 2005. Tissue oxygenation in obese and non-obese patients during laparoscopy. Obes Surg 15:813–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kabon B, Nagele A, Reddy D, Eagon C, Fleshman JW, Sessler DI, Kurz A 2004. Obesity decreases perioperative tissue oxygenation. Anesthesiology 100:274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, Shimomura I 2007. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56:901–911 [DOI] [PubMed] [Google Scholar]
- 37.Kim KH, Song MJ, Chung J, Park H, Kim JB 2005. Hypoxia inhibits adipocyte differentiation in a HDAC-independent manner. Biochem Biophys Res Commun 333:1178–1184 [DOI] [PubMed] [Google Scholar]
- 38.Wang B, Wood IS, Trayhurn P 2007. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflugers Arch 455:479–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT 1998. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 95:11715–11720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM 1998. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature 392:405–408 [DOI] [PubMed] [Google Scholar]
- 41.Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL 1997. Transactivation and inhibitory domains of hypoxia-inducible factor 1α. Modulation of transcriptional activity by oxygen tension. J Biol Chem 272:19253–19260 [DOI] [PubMed] [Google Scholar]
- 42.Kelly AS, Bank AJ 2007. The cardiovascular effects of the thiazolidinediones: a review of the clinical data. J Diabetes Complications 21:326–334 [DOI] [PubMed] [Google Scholar]
- 43.Berger J, Bailey P, Biswas C, Cullinan CA, Doebber TW, Hayes NS, Saperstein R, Smith RG, Leibowitz MD 1996. Thiazolidinediones produce a conformational change in peroxisomal proliferator-activated receptor-γ: binding and activation correlate with antidiabetic actions in db/db mice. Endocrinology 137:4189–4195 [DOI] [PubMed] [Google Scholar]
- 44.Nolan JJ, Ludvik B, Beerdsen P, Joyce M, Olefsky J 1994. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med 331:1188–1193 [DOI] [PubMed] [Google Scholar]
- 45.Saitoh Y, Chun-Ping C, Noma K, Ueno H, Mizuta M, Nakazato M 2007. Pioglitazone attenuates fatty acid-induced oxidative stress and apoptosis in pancreatic β-cells. Diabetes Obes Metab 10:564–573 [DOI] [PubMed] [Google Scholar]
- 46.Sommer M, Wolf G 2007. Rosiglitazone increases PPARγ in renal tubular epithelial cells and protects against damage by hydrogen peroxide. Am J Nephrol 27:425–434 [DOI] [PubMed] [Google Scholar]
- 47.Jung TW, Lee JY, Shim WS, Kang ES, Kim SK, Ahn CW, Lee HC, Cha BS 2007. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. J Neurol Sci 253:53–60 [DOI] [PubMed] [Google Scholar]
- 48.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM 1994. mPPAR γ2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 8:1224–1234 [DOI] [PubMed] [Google Scholar]
- 49.Lin Y, Rajala MW, Berger JP, Moller DE, Barzilai N, Scherer PE 2001. Hyperglycemia-induced production of acute phase reactants in adipose tissue. J Biol Chem 276:42077–42083 [DOI] [PubMed] [Google Scholar]
- 50.Gauss KA, Nelson-Overton LK, Siemsen DW, Gao Y, Deleo FR, Quinn MT 2007. Role of NF-κB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-α. J Leukoc Biol 82:729–741 [DOI] [PubMed] [Google Scholar]
- 51.Park J, Rho HK, Kim KH, Choe SS, Lee YS, Kim JB 2005. Overexpression of glucose-6-phosphate dehydrogenase is associated with lipid dysregulation and insulin resistance in obesity. Mol Cell Biol 25:5146–5157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park J, Choe SS, Choi AH, Kim KH, Yoon MJ, Suganami T, Ogawa Y, Kim JB 2006. Increase in glucose-6-phosphate dehydrogenase in adipocytes stimulates oxidative stress and inflammatory signals. Diabetes 55:2939–2949 [DOI] [PubMed] [Google Scholar]
- 53.Freedman JE, Loscalzo J, Benoit SE, Valeri CR, Barnard MR, Michelson AD 1996. Decreased platelet inhibition by nitric oxide in two brothers with a history of arterial thrombosis. J Clin Invest 97:979–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kenet G, Freedman J, Shenkman B, Regina E, Brok-Simoni F, Holzman F, Vavva F, Brand N, Michelson A, Trolliet M, Loscalzo J, Inbal A 1999. Plasma glutathione peroxidase deficiency and platelet insensitivity to nitric oxide in children with familial stroke. Arterioscler Thromb Vasc Biol 19:2017–2023 [DOI] [PubMed] [Google Scholar]
- 55.Rosen P, Nawroth PP, King G, Moller W, Tritschler HJ, Packer L 2001. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab Res Rev 17:189–212 [DOI] [PubMed] [Google Scholar]
- 56.Packer L, Kraemer K, Rimbach G 2001. Molecular aspects of lipoic acid in the prevention of diabetes complications. Nutrition 17:888–895 [DOI] [PubMed] [Google Scholar]
- 57.McClung JP, Roneker CA, Mu W, Lisk DJ, Langlais P, Liu F, Lei XG 2004. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc Natl Acad Sci USA 101:8852–8857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mahadev K, Zilbering A, Zhu L, Goldstein BJ 2001. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem 276:21938–21942 [DOI] [PubMed] [Google Scholar]
- 59.Avissar N, Ornt DB, Yagil Y, Horowitz S, Watkins RH, Kerl EA, Takahashi K, Palmer IS, Cohen HJ 1994. Human kidney proximal tubules are the main source of plasma glutathione peroxidase. Am J Physiol 266:C367–C375 [DOI] [PubMed]
- 60.Yoshimura S, Suemizu H, Nomoto Y, Sakai H, Katsuoka Y, Kawamura N, Moriuchi T 1996. Plasma glutathione peroxidase deficiency caused by renal dysfunction. Nephron 73:207–211 [DOI] [PubMed] [Google Scholar]
- 61.Whitin JC, Tham DM, Bhamre S, Ornt DB, Scandling JD, Tune BM, Salvatierra O, Avissar N, Cohen HJ 1998. Plasma glutathione peroxidase and its relationship to renal proximal tubule function. Mol Genet Metab 65:238–245 [DOI] [PubMed] [Google Scholar]
- 62.Bierl C, Voetsch B, Jin RC, Handy DE, Loscalzo J 2004. Determinants of human plasma glutathione peroxidase (GPx-3) expression. J Biol Chem 279:26839–26845 [DOI] [PubMed] [Google Scholar]
- 63.Kapur S, Marcotte B, Marette A 1999. Mechanism of adipose tissue iNOS induction in endotoxemia. Am J Physiol 276:E635–E641 [DOI] [PubMed]
- 64.Green SP, Hamilton JA, Uhlinger DJ, Phillips WA 1994. Expression of p47-phox and p67-phox proteins in murine bone marrow-derived macrophages: enhancement by lipopolysaccharide and tumor necrosis factor α but not colony stimulating factor 1. J Leukoc Biol 55:530–535 [DOI] [PubMed] [Google Scholar]
- 65.Da Ros R, Assaloni R, Ceriello A 2004. The preventive anti-oxidant action of thiazolidinediones: a new therapeutic prospect in diabetes and insulin resistance. Diabet Med 21:1249–1252 [DOI] [PubMed] [Google Scholar]
- 66.Noguchi N, Sakai H, Kato Y, Tsuchiya J, Yamamoto Y, Niki E, Horikoshi H, Kodama T 1996. Inhibition of oxidation of low density lipoprotein by troglitazone. Atherosclerosis 123:227–234 [DOI] [PubMed] [Google Scholar]
- 67.Petruschke T, Hauner H 1993. Tumor necrosis factor-α prevents the differentiation of human adipocyte precursor cells and causes delipidation of newly developed fat cells. J Clin Endocrinol Metab 76:742–747 [DOI] [PubMed] [Google Scholar]
- 68.Lee YS, Sohn DH, Han D, Lee HW, Seong RH, Kim JB 2007. Chromatin remodeling complex interacts with ADD1/SREBP1c to mediate insulin-dependent regulation of gene expression. Mol Cell Biol 27:438–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee YS, Lee HH, Park J, Yoo EJ, Glackin CA, Choi YI, Jeon SH, Seong RH, Park SD, Kim JB 2003. Twist2, a novel ADD1/SREBP1c interacting protein, represses the transcriptional activity of ADD1/SREBP1c. Nucleic Acids Res 31:7165–7174 [DOI] [PMC free article] [PubMed] [Google Scholar]