

Abstract
Peroxisome proliferator-activated receptor-α (PPARα) is a central regulator of lipid metabolism. Fibrate drugs act on PPARα to modulate dyslipidemias. A natural variant (V227A) affecting the PPARα hinge region was associated with perturbations in blood lipid levels in Asian populations. In this study, we investigated the functional significance of the V227A substitution. The variant significantly attenuated PPARα-mediated transactivation of the cytochrome P450 4A6 and mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS2) genes in the presence of fibrate ligands. Screening of a panel of PPARα coregulators revealed that V227A enhanced recruitment of the nuclear corepressor NCoR. Transactivation activity of V227A could be restored by silencing NCoR or by inhibition of its histone deacetylase activity. Deletion studies indicated that PPARα interacted with NCoR receptor-interacting domain 1 (ID1) but not ID2 or ID3. These interactions were dependent on the intact consensus nonapeptide nuclear receptor interaction motif in NCoR ID1 and were enhanced by the adjacent 24 N-terminal residues. Novel corepressor interaction determinants involving PPARα helices 1 and 2 were identified. In hepatic cells, the V227A substitution stabilized PPARα/NCoR interactions and caused defective release of NCoR in the presence of agonists on the HMGCS2 promoter. These results provide the first indication that defective function of a natural PPARα variant was due, at least partially, to increased corepressor binding. Our data suggest that the PPARα/NCoR interaction is physiologically relevant and can produce a discernable phenotype when the magnitude of the interaction is altered by a naturally occurring variation.
PEROXISOME PROLIFERATOR-activated receptor-α (PPARα) is one of three pharmacologically and genetically distinct subtypes, namely PPARα, PPARβ/δ, and PPARγ, that have critical roles in lipid and carbohydrate metabolism (1). Although PPARγ regulates glucose levels and adipogenesis, PPARα is highly expressed in tissues involved in fatty acid metabolism where it regulates several key proteins in fatty acid oxidation and ketogenesis. Endogenous ligands for PPARα include polyunsaturated fatty acids (PUFA) such as α-linolenic acid. Synthetic fibric acid derivatives, such as WY14,643, activate PPARα strongly, and the fibrate class of drugs have lipid-lowering properties and are used to maintain cardiovascular health (2). When activated by ligand, the receptor binds to specific PPAR response elements (PPRE) in promoters of PPARα-responsive genes to regulate their transcription. Whereas function of the conserved DNA-binding domain (DBD) and the ligand-binding domain (LBD) in the transactivation process are well known, the role of other regions such as the poorly conserved PPARα hinge region (residues after the DBD and incorporating LBD helices 1 and 2) (3) is less well understood (4). Liganded PPARα binds chromatin as a heterodimer with the receptor for 9-cis retinoic acid [retinoid X receptor (RXR)]. The consensus PPRE is a direct repeat (DR) of the hexameric nucleotide core consensus sequence AGGTCA separated by 1 bp (DR1), where PPAR occupies the 5′ motif. Consensus PPAR/RXR heterodimeric sites have been identified in the promoters of several genes, such as cytochrome P450 4A6 (CYP4A6) (5) encoding an enzyme in the microsomal fatty acid ω-oxidation pathway, which is particularly active in the fasted and diabetic states (6, 7). Mice totally lacking PPARα are relatively healthy in the fed state but on starvation become hypoketonemic with profound hypoglycemia, indicating a defect in activation of the ketogenic pathway in response to glucopenia (8, 9, 10). A key enzyme in the highly regulated ketogenesis pathway is mitochondrial 3-hydroxy-3-methylglutaryl coenzyme A synthase (HMGCS2) gene, which mediates the first critical irreversible step in the formation of ketone bodies as the alternative body fuel (11, 12). The HMGCS2 gene is among the most PPAR responsive (13).
After ligand binding, the PPARα LBD forms a docking surface for the assembly of a host of coregulator molecules on chromatin. Coactivators such as cAMP response element-binding protein (CBP)/p300 (14), the steroid receptor coactivators such as steroid receptor coactivator-1 (SRC-1) (14), transcriptional intermediate factor 2 (TIF2), (15) and PPARγ coactivator-1 (PGC-1) (16) interact with PPARα to modify histones by acetylation resulting in active transcription. PPAR-related coactivators such as PPAR-interacting protein (PRIP) (17) act as linkers with other coactivators to the preinitiation complex (18). On the other hand, repressor complexes assembled around nuclear receptor corepressor (NCoR) (19) and silencing mediator for retinoid and thyroid hormone receptors (SMRT) (20) possess histone deacetylase (HDAC) activities resulting in gene silencing (21).
The locus encoding PPARα (PPARA) is polymorphic in humans, and over a dozen missense polymorphisms resulting in amino-acid changes have been described (22). Two polymorphisms occur with high allele frequency and have been associated with variations in lipid metabolism. The L162V polymorphism at the DBD of PPARα was found in the Caucasian populations (23, 24) and in several populations from the Indian subcontinent (25). V162 was associated with atherogenic/hyperapolipoprotein B dyslipidemia (24) and can influence progression of coronary atherosclerosis and risk of coronary artery disease (26). The functionality of this polymorphism has been examined in vitro (23, 27) and has been shown to affect PPARα-mediated gene-transcription. The L162V polymorphism is rare in Asian populations (22, 28, 29). On the other hand, a nonsynonymous variant at the PPARA locus encoding a substitution of valine for alanine at residue 227 (V227A) in the hinge region of the receptor has been observed in Singapore (28) and Japanese (22, 29) populations with relatively high allelic frequencies. This variant was associated with perturbations in plasma lipid levels and modulated the association between dietary PUFA and high-density lipoprotein cholesterol (28). The impact of this variant on the function of PPARα is not known (22, 28). The aims of this study were to examine the effects of the V227A variant on PPARα function and to elucidate the mechanisms for any observed effects.
RESULTS
The PPARα V227A Variant Induced Lower Transcriptional Activity
To investigate transcription function of the variant, HeLa cells were cotransfected with plasmids encoding either wild-type (WT) or V227A PPARα and a reporter gene CYP4A6-PPRE-Luc containing two copies of the consensus PPRE derived from the CYP4A6 promoter (5). In HeLa cells, V227A was 35–49% weaker than WT on exposure to the potent fibrate WY14,643 (Fig. 1A). To further evaluate the effects of V227A variant on lipid metabolism, transactivation activity was further examined on the PPARα-responsive HMGCS2 gene. Cells were cotransfected with WT and V227A PPARα and a luciferase-reporter driven by residues −1081 to +22 of HMGCS2 promoter (HMGCS2-Luc) (12). In this system, V227A was about 33% weaker than WT with WY14,643 (Fig. 1B). HeLa cells contain low levels of endogenous PPARα, and HepG2 is a functionally relevant cell line. To understand the physiological implications of the substitution, HepG2 cells were infected with adenovirus expressing WT PPARα, V227A, or LacZ before transfection with reporter plasmids. In HepG2 cells, V227A was 37–50% weaker with WY14,643 (Fig. 1C) with CYP4A6-PPRE and 47–56% weaker than WT at the HMGCS2-Luc reporter (Fig. 1D). Immunoblots for PPAR and actin protein indicate differences in WT and V227A bioactivity were not associated with any changes in relative receptor content (Fig. 1E).
Fig. 1.

Transactivation Activity of PPARα V227A Variant on a Consensus CYP4A6-PPRE and the Mitochondria HMGCS2 Promoter
HeLa cells were cotransfected with reporter vector CYP4A6-PPRE-Luc (100 ng) (A) or a reporter construct driven by residues −1081 to +22 of the HMGCS2 promoter (100 ng) (B) and full-length WT or V227A PPARα (50 ng) before treatment with indicated doses of WY14,643. HepG2 cells were infected with adenovirus expressing WT PPARα, V227A, or LacZ before transfection with CYP4A6-PPRE-Luc (100 ng) (C) or HMGCS2-Luc (100 ng) (D) and treatment with WY14,643 as above. PPARα and actin protein levels from total protein lysates (20–25 μg) of representative replicates were detected with specific antibodies. Values are mean ± sd of three replicates and expressed as percentage of maximal WT activity. *, P < 0.05; **, P < 0.01. E, Western blot for PPARα protein content. HepG2 cells were transfected with empty vector and 50 ng of WT or V227A PPARα, treated with increasing doses of WY14,643, and harvested 48 h after treatment. PPARα protein was detected with specific antibody and quantified using ScionImage analyzer. PPARα expression was normalized with actin and expressed as the ratio of PPARα and actin. Values are mean ± sd of eight replicates done on different occasions. TK, Thymidine kinase basal promoter.
Weaker Transactivation of V227A Resided in the LBD and Was Independent of Ligand Binding and PPAR/RXR Content
To dissect the molecular basis of reduced transactivation, we systematically studied the effects of the V227A polymorphism on PPARα-mediated signaling pathways. Chimeric receptors consisting of the PPARα LBD (residues 171–468) of the WT or V227A variants and the yeast Gal4DBD were constructed. We examined the effects of the V227A polymorphism in the LBD on transcriptional activation in the mammalian one-hybrid assay. In the presence of WY14,643, V227A LBD exhibited 62–66% lower transactivation activity compared with the WT (Fig. 2A). Similarly, Gal-V227A was 28% weaker than Gal-WT with high doses of α-linolenic acid, a PPARα natural ligand, suggesting that the mechanism for defective transactivation resides in reduced LBD activation function. To determine whether the V227A substitution affected ligand affinity, the ligand-binding properties of the mutant receptor was examined in a competition assay. In this assay, [3H]WY14,643 binding to WT PPARα was largely replaced by unlabeled WY14,643 at doses of at least 5 nm, whereas the non-PPAR ligand, dihydrotestosterone (DHT), did not affect [3H]Wy14,643 binding (Fig. 2B). No significant differences in the relative binding affinity of WY14,643 to WT or V227A PPARα were observed, indicating the differences in V227A transactivation function were unlikely to be due to defective ligand binding. To examine whether endogenous RXR was limiting, cells were cotransfected with a plasmid encoding this obligate heterodimer. Differences between WT and V227A were still observed, suggesting that weaker activity of V227A was not stoichiometrically limited by RXR (data not shown). Because a natural PPARα splice variant has been reported to affect nuclear location (30), we investigated whether the variant affected subcellular localization. Immunofluorescence studies showed that both WT and V227A PPARα reside in the nucleus, in the presence and absence of ligand, and no differences in subcellular localization patterns were observed (Fig. 2C).
Fig. 2.
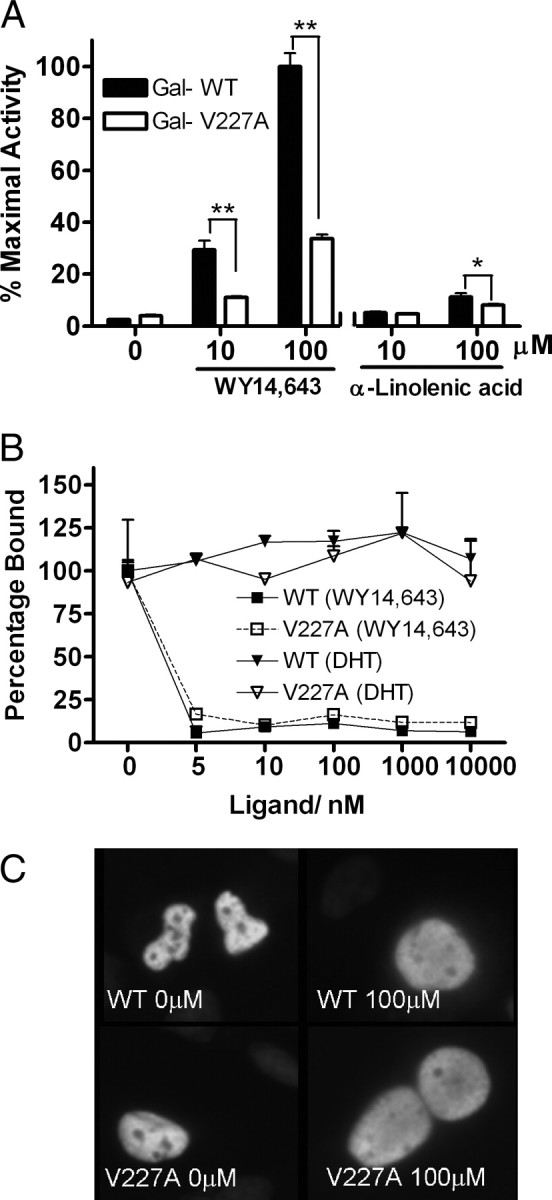
Effect of V227A Variant on LBD Function, Ligand Binding, and Nuclear Localization
A, HeLa cells were cotransfected with chimeric Gal-PPARαWT-LBD or Gal-PPARαV227A-LBD (50 ng) together with 100 ng (UASG)5-Luc reporter before treatment with ligands in the mammalian one-hybrid assay. Values are mean ± sd of three replicates and expressed as percentage of maximal WT activity. *, P < 0.05; **, P < 0.01. B, Ligand-binding affinity. WT or V227A PPARα in HeLa cells was exposed to 3 nm [3H]WY14,643 alone or with increasing concentrations of unlabeled WY14,643. The control was DHT. The amount of [3H]WY14,643 specifically bound after 24 h incubation was measured and expressed as a percentage relative to controls not exposed to cold hormone. C, Subcellular localization of PPARα proteins. Full-length WT or V227A PPARα (50 ng) was transfected into HepG2 cells before treatment with WY14,643. The resulting cells were stained with PPARα polyclonal antibody and goat antirabbit antibody conjugated with FITC. Nucleus was stained with 4′,6-diamidino-2-phenylindole.
The V227A Variant Enhanced Recruitment of the Corepressor NCoR
To further elucidate the mechanism(s) for the decrease in transcriptional activity of the variant, we compared interactions of WT and V227A with major coregulator proteins known to interact with PPARα (18). Interactions between PPARα LBD (residues 167–468) linked to VP16 domain, and Gal-nuclear receptor (NR) interacting domains of major coregulators (14, 15, 16, 17, 19, 20) known to interact with PPARα (Table 1), were measured in the mammalian two-hybrid assay. No significant differences between WT and V227A were observed in the recruitment of the coactivators SRC-1, TIF2, PGC-1, p300, and PRIP, in the absence or presence of ligand (Table 1). Strikingly, marked differences (about 2-fold higher for V227A) were observed in the relative binding of the corepressors NCoR and SMRT in the absence of ligand (Table 1). Lesser but still significantly increased interactions with V227A were still observed with a low dose of ligand, suggesting that increased corepressor recruitment contributed to reduced transactivation function of V227A.
Table 1.
Interaction of Coregulators with PPARα in the Mammalian Two-Hybrid Assay
| VP16-PPARα (167–468) | |||||||
|---|---|---|---|---|---|---|---|
| WT | V227A | ||||||
| − | + | − | + | ||||
| Ligand | |||||||
| Gal-coactivator1 | |||||||
| SRC-1 (213–1061) | 3.1 ± 0.3 | 5.5 ± 0.8 | 4.9 ± 0.5 | 6.0 ± 0.4 | |||
| TIF2 (622–869) | 1.6 ± 0.0 | 2.2 ± 0.1 | 1.8 ± 0.1 | 2.4 ± 0.1 | |||
| PGC1α (120–284) | 1.1 ± 0.1 | 1.5 ± 0.1 | 1.6 ± 0.1 | 2.3 ± 0.1 | |||
| p300 (1–117) | 26.5 ± 1.0 | 27.6 ± 3.0 | 22.7 ± 0.1 | 24.3 ± 0.1 | |||
| PRIP (819–1096) | 4.8 ± 0.5 | 7.4 ± 0.6 | 6.9 ± 1.1 | 8.2 ± 0.8 | |||
| Gal-corepressor1 | |||||||
| SMRT (2004–2517) | 1.9 ± 0.1 | 1.9 ± 0.2 | 4.0 ± 0.3 | 2.8 ± 0.3 | |||
| NCoR (1575–2453) | 5.3 ± 0.8 | 4.3 ± 0.7 | 10.4 ± 0.4 | 6.7 ± 0.5 | |||
HeLa cells were cotransfected with chimeric VP16-WT, VP16-V227A (100 ng), and Gal-coregulator (100 ng) together with (UASG)5-Luc reporter (500 ng). Cells were exposed to the absence or presence of WY14,643 (10 μm) for 48 h. Interaction was expressed as fold (mean ± se of three replicates) of VP16 alone.
The coregulator regions, cloned downstream of GAL4DBD, were based on residues reported to interact with PPARα: SRC-1 (residues 579–932) (14 ), TIF2 (residues 624–869) (15 ), PGC1α (residues 120–284) (16 ) and p300 (residues 39–117) (14 ), PRIP (residues 887–891) (17 ), SMRT (residues 2124–2149, 2329–2358) (20 ), and NCoR (residues 2110–2453) (19 ).
To understand the mechanistic basis of increased corepressor recruitment, we undertook detailed experiments to examine NCoR/PPARα interactions. In the absence of ligand, WT PPARα bound strongly to NCoR, whereas a LBD mutant (residues 245–468) lacking helices 1 and 2 exhibited minimal binding (Fig. 3A). The presence of ligand induced release of NCoR from WT PPAR-LBD in a dose-dependent manner; and at maximal ligand dose, no NCoR binding was observed. Strikingly, V227A bound with significantly higher avidity to NCoR compared with WT, both in the absence and the presence of low doses of WY14,643 (Fig. 3A) and α-linolenic acid (Fig. 3B).
Fig. 3.
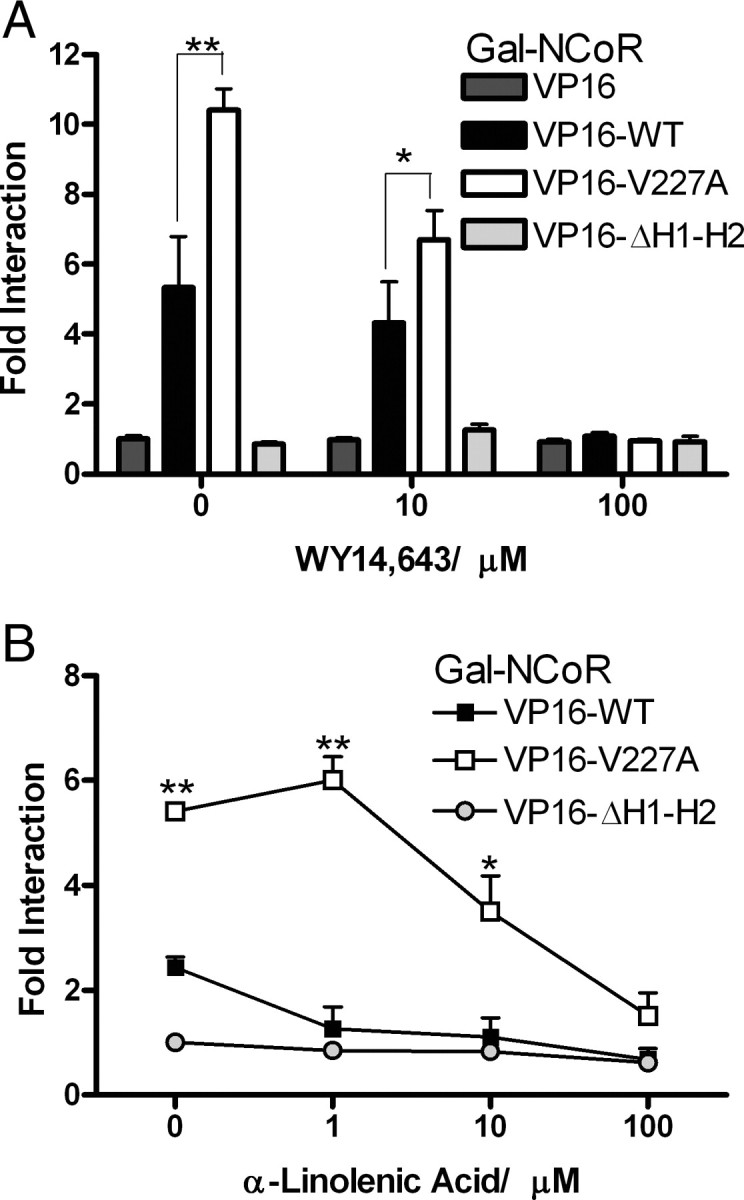
Interactions of NCoR in the Mammalian Two-Hybrid Assay
HeLa (A) and HepG2 (B) cells were cotransfected with chimeric VP16-WT or VP16-V227A (100 ng) and Gal-NCoR (100 ng) together with (UASG)5-Luc reporter (500 ng). VP16-ΔH1-H2 (100 ng) was used as a negative control. Cells were exposed to indicated doses of WY14,643 (A) or α-linolenic acid (B) for 48 h. Interaction was expressed as fold (mean ± sd of three replicates) of VP16 alone. *, P < 0.05; **, P < 0.01.
To further investigate the role of corepressor in modulation of V227A activity, we investigated the effects of silencing endogenous NCoR expression on transcriptional activity of V227A. HepG2 cells were transiently infected with lentiviral constructs encoding short hairpin (sh) sequences complementary to NCoR [small interfering NCoR (siNCoR)] or a randomized sequence (siScram). Cell extracts immunoblotted with anti-NCoR confirmed that expression of the corepressor protein was reduced in cells expressing siNCoR compared with siScram (Fig. 4A). Reduction of NCoR was associated with a restoration of V227A activity, such that differences between WT and variant PPARα were no longer apparent (Fig. 4A). Because repression of transactivation activity by corepressors is known to be dependent on their HDAC properties, we investigated the effects of the HDAC inhibitor trichostatin A. The presence of trichostatin A restored ligand-activated V227A activity (Fig. 4B). Immunoblots indicate that these changes were not due to variations in WT or V227A PPARα protein content. These data suggested that increased corepressor activity contributed to deficient function of the V227A mutant.
Fig. 4.
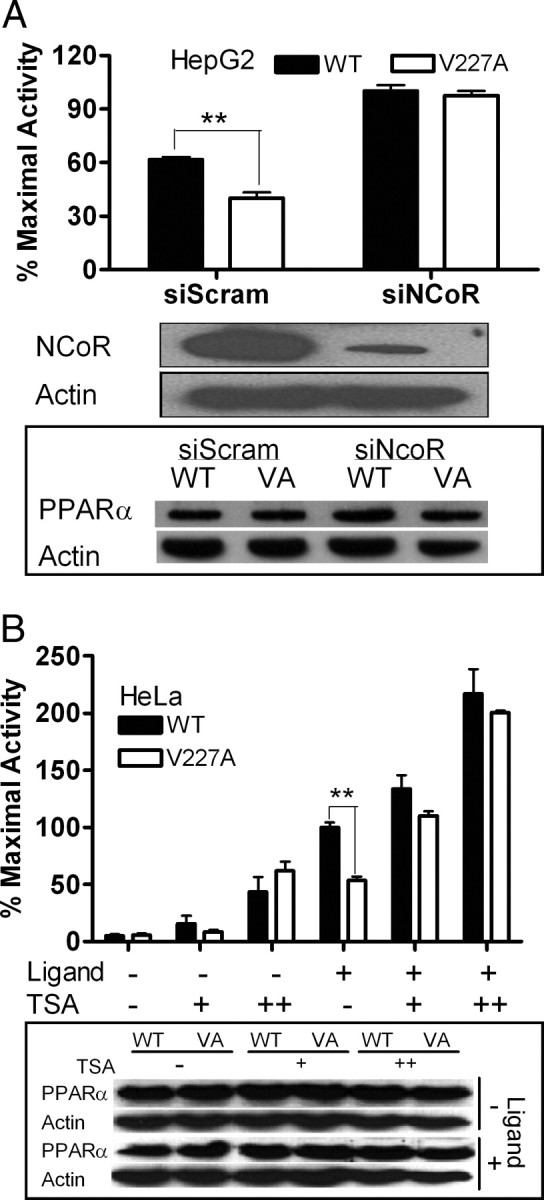
Effect of NCoR Silencing and HDAC Inhibition on Transcriptional Activity of V227A
A, NCoR silencing. HepG2 cells were transiently infected by pLenti6-GW/U6-scrambleshRNA (siScram) or pLenti6-GW/U6-NCoRshRNA (siNCoR) and cotransfected with either full-length WT or V227A (VA) PPARα (50 ng) and reporter vector CYP4A6-PPRE-Luc (100 ng) before exposure to WY14,643 (100 μm). Cell lysates were immunoblotted with antibodies to NCoR, PPARα, and actin to evaluate effects of NCoR knockdown. B, Effects of HDAC inhibitor. HeLa cells overexpressing WT or V227A PPARα (50 ng) together with CYP4A6-PPRE-Luc (100ng) were exposed to an increasing dose of TSA (100 and 200 nm) in the absence or presence of WY14,643 (100 μm). Cell lysates were immunoblotted with antibodies to PPARα and actin to evaluate effects of TSA on protein content. Values are mean ± sd of three replicates and expressed as percentage of maximal WT activity. *, P < 0.05; **, P < 0.01.
Receptor-Interacting Domain of NCoR Was Dependent on Residues 2251–2280 in ID1 but Was Independent of ID2 and ID3
Although the nuclear receptor interaction domains (ID) on NCoR have been described for thyroid hormone receptor (TR) and retinoic acid receptor (RAR), RXR, and PPARγ (31, 32, 33, 34, 35, 36, 37, 38), it is not clear whether any, or specific combinations, of the three identified domains interacts with PPARα. To characterize domains mediating NCoR/PPARα interactions, we constructed Gal-NCoR expression plasmids containing one, two, or all three receptor ID (Fig. 5A). The ID contained a core consensus nonapeptide motif (LXXI/HIXXXI/L) termed CoRNR box that mediates corepressor assembly on nuclear receptors (31, 32). Chimeric Gal-NCoR proteins were coexpressed with VP-PPARα-LBD in the mammalian two-hybrid interaction assay. The absence of interactions with NCoR fragments G5 and G6 (containing ID2 and ID3) and G7 (containing ID3) indicated that ID2 and ID3 did not interact with PPARα (Fig. 5B). On the other hand, PPARα exhibited the strongest interactions with the N-terminal NCoR truncation fragments G3 and G4 (containing ID1), both in the absence and presence of ligand, suggesting that ID1 is the most critical ID (Fig. 5B). Fragment G2 (containing the CoRNR box and C-terminal flanking sequences of ID1) exhibited more than 9-fold stronger binding compared with G1 (without an intact CoRNR box), indicating the critical importance of the CoRNR box to PPARα interactions. Fragment G3 (containing the CoRNR box and both C- and N-terminal flanking sequences of ID1) induced 2.7-fold higher interactions than fragment G2, indicating a role of the first 24 amino acids (2251–2274) immediately N-terminal of the ID1 CoRNR box for PPARα/NCoR interactions. Intriguingly fragment G4 (containing ID1 and ID2) exhibited stronger interactions compared with fragment G8 (containing all three ID and N-terminal sequences) suggesting a potential suppressive function in NCoR residues 1575–2039. Assuming these deletion fragments have functional ID, the aggregate data indicate that ID1, but not ID2 and ID3, was the PPARα ID. In particular, NCoR residues (residues 2251–2280) were necessary and sufficient for optimal interactions with PPARα.
Fig. 5.

Interactions of PPARα with Receptor ID of NCoR
A, Schematic diagram of Gal-NCoR truncation fragments and the mammalian two-hybrid system. Gray boxes represent unique CoRNR box sequences within each receptor ID, residues 2277–2285, 2073–2081, and 1949–1953 for ID1, ID2, and ID3, respectively. Black boxes represents the first 24 amino acids immediately N-terminal of the ID1 CoRNR box. B, HeLa cells were cotransfected with 100 ng PPARα LBD chimeras, VP16-WT or VP-V227A (100 ng), and indicated C-terminal Gal-NCoR truncated fragments (100 ng) with the (UASG)5-Luc reporter gene (500 ng) in the absence or presence of WY14,643 (10 μm). Fold interaction (mean ± sd of three replicates) was expressed as fold of G1 at 0 μm. *, P < 0.05; **, P < 0.01. C, GST pulldown assay. GST-NCoR truncated fragments were incubated with equal amounts of in vitro translated full-length PPARα WT or V227A and GST pulldown performed using Sepharose 4B beads. Bound proteins were identified using anti-PPARα rabbit polyclonal antibody. Input shows the immunoblot for in vitro translated PPARα and the Coomassie blue staining of GST proteins expressed.
Compared with WT, the V227A variant showed stronger interaction with NCoR fragments, both in the absence and presence of ligand (Fig. 5B), consistent with previous experiments and its lower transactivation activity. To further confirm residues 2251–2280 of ID1 is the PPARα ID, we constructed glutathione-S-transferase (GST)-NCoR fragments in the GST pulldown assay. Chimeric NCoR fragments were incubated with in vitro translated full-length PPARα WT or V227A, and bound proteins were evaluated by Western blot analysis using anti-PPARα rabbit polyclonal antibody. Consistent with previous results, interaction of the NCoR truncated mutants with WT and V227A was strongest with NCoR fragment G3, containing the entire ID1. Strikingly, interactions of PPARα V227A with G3 were stronger than WT in the GST pulldown assay, confirming the importance of this residue to PPAR/corepressor binding (Fig. 5C). Our results showed that PPARα/NCoR interactions are mediated predominantly by ID1 on the NCoR and that the V227A substitution significantly increased this interaction.
Residue 227A Created a Potent Novel Interaction Site for NCoR ID1
To further define the role of residue 227 for NCoR interactions, several VP16-PPARα truncation fragments, centered on the hinge region (residues 167–244) (3) that began at the termination of DBD and extending to helix 2 were constructed for the mammalian two-hybrid assay (Fig. 6A). The Gal4-NCoR truncated fragment G3 (NΔ2250) was used for the interaction study because this fragment exhibited the highest binding to PPARα (Fig. 5). Fragments containing PPARα-DBD and the pre-helix 1 region (residues 94–198) by themselves did not interact with NCoR (Fig. 6B). Interactions were strongest with the PPARα fragment containing the entire helices 1–12 (residues 196–468) but excluding the pre-helix 1 region (Fig. 6B). Unexpectedly, inclusion of the pre-helix 1 region 167–198 to the LBD reduced interaction with NCoR. Most strikingly, the V227ANΔ195 variant bound NCoR with more than 4-fold stronger avidity than the corresponding WTNΔ195 fragment (Fig. 6B), indicating that the V227A substitution introduces a residue that stabilized PPARα-NCoR ID1 interactions.
Fig. 6.

Interactions of NCoR with Deletion Fragments of PPARα Hinge
A, Schematic diagram of VP16-PPARα truncation fragments. Black boxes represent DBD (residues 94–166); gray boxes represent residues 167–195 before helix 1. White boxes represent residues 196–468 of helix 1 to helix 12. B, WT or V227A PPARα VP16 truncated fragments (100 ng) were coexpressed with G3 (NΔ2250) (Fig. 6) (100 ng) and the (UASG)5-Luc reporter gene (500 ng) in the mammalian two-hybrid assay. Fold interaction (mean ± sd of three replicates) was expressed as fold of VP16 empty vector. *, P < 0.05; **, P < 0.01.
Ternary Interactions between Coactivator, Corepressor, and PPARα
To understand the dynamics of corepressor/coactivator interactions with PPAR, we examined the effects of SRC-1 on PPARα-regulated transactivation on the HMGCS2 promoter. Exogenous SRC-1 can restore transactivation defect of V227A, indicating interactions between NCoR and the coactivator (Fig. 7A). To measure the effects of SRC-1 on NCoR/PPARα interactions, we performed the mammalian two-hybrid assay in the presence of increasing doses of full-length SRC-1. Low doses of exogenous coactivator increased transactivity of the Gal-NCoR/VP16-PPARα complex as expected (Fig. 7B). However, increasing doses of SRC-1 reduced WT PPARα/NCoR interactions to below baseline, consistent with a model in which NCoR and SRC-1 competed for the same PPARα binding site. However, the pattern of binding of NCoR to V227A was similar to WT except that it was higher at all doses of SRC-1, suggesting that the substitution did not directly affect SRC-1 recruitment.
Fig. 7.
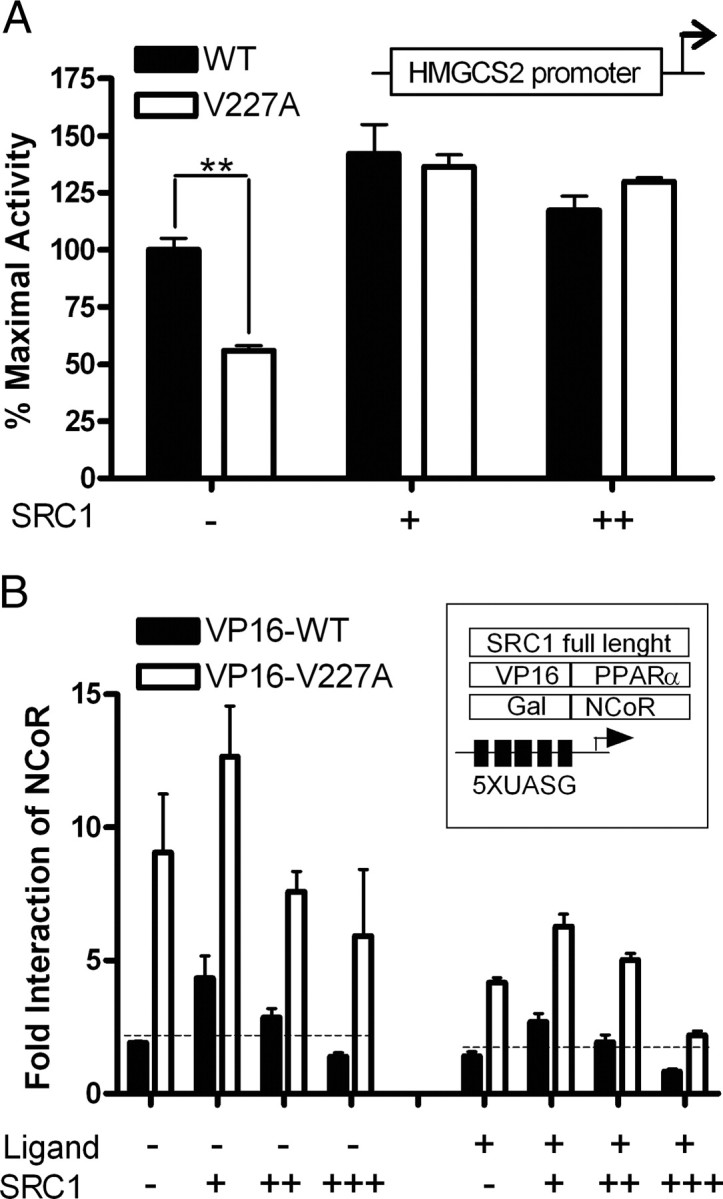
Ternary Interactions between SRC-1, NCoR, and PPAR
A, SRC-1 overexpression. HeLa cells were cotransfected with either full-length WT or V227A PPARα (50 ng), a reporter construct driven by residues −1081 to +22 of the HMGCS2 promoter (100 ng) and increasing amounts of full-length SRC-1 (50 and 100 ng) with 100 μm WY 14,643. Values (mean ± se of three replicates) are expressed as percentage of maximal WT activity. B, Effects of SRC-1 on NCoR/PPARα interactions. HeLa cells were cotransfected with chimeric VP16-WT, VP16-V227A (100 ng), and Gal-NCoR (100 ng) together with the (UASG)5-Luc reporter (500 ng). Cells were exposed to WY14,643 (10 μm) at increasing doses (50, 100, and 200 ng) of full-length SRC-1. Fold interaction (mean ± sd of three replicates) was expressed as fold of VP16 empty vector. *, P < 0.05; **, P < 0.01.
Association and Recruitment of NCoR to Chromatin Was Increased by V227A
To compare corepressor binding between WT and V227A, coimmunoprecipitation of PPARα with NCoR was performed in HepG2 cells. Cell lysates was precipitated with NCoR antibody, and bound corepressor was quantified by immunoblotting with anti-PPARα. In the absence of ligand, both WT and V227A PPARα bound NCoR (Fig. 8A, lowest panel). The addition of 100 μm WY14,643 resulted in almost complete dissociation of NCoR from WT, whereas V227A retained observable NCoR, suggesting that ligand-dependent release of NCoR from V227A was reduced. To evaluate PPARα V227A-NCoR complex formation on the HMGCS2 promoter, we performed chromatin immunoprecipitation (ChIP) assays. HepG2 cells overexpressing WT or V227A were cultured for 24 h in the presence or absence of ligand. Transcription factors interacting with genomic DNA were cross-linked with formaldehyde and chromatin immunoprecipitated with specific antibodies against PPARα, p300, SRC-1, and NCoR. Genomic DNA fragments recovered after immunoprecipitation served as template for PCR with primers for the PPRE (−198 to −34 of the HMGCS2 promoter), whereas primers for a fragment −5 kb upstream of the PPRE served as control (Fig. 8B, upper panels). With anti-PPAR, the amount of PPRE pulled down by WT or V227A PPARα was similar, indicating that receptor-DNA binding did not contribute to weaker transactivation activity of the V227A (Fig. 8B, lower panel). Binding of coactivator p300 to PPRE was not affected by the substitution. Strikingly, both SRC-1 and NCoR were bound to the WT PPARα/PPRE complex without added ligand, suggesting partial activation by endogenous ligand in HepG2 cells. In comparison, V227A preferentially bound NCoR and much less SRC-1 (Fig. 8B, lower panel). The presence of ligand induced complete release of NCoR from the WT PPARα/PPRE transcription complex. In contrast, defective release of NCoR resulted in both NCoR and SRC-1 being present simultaneously in the V227A PPARα/PPRE transcription complex in the presence of ligand (Fig. 8B, lowest two panels). To correlate PPARα-regulated gene expression with recruitment of NCoR to the PPRE of the HMGCS2 promoter, HepG2 cells were infected with adenovirus expressing PPARα WT, V227A, or LacZ, and HMGCS2 mRNA expression was measured with quantitative real-time RT-PCR. PPARα V227A-regulated HMGCS2 mRNA expression was 60% lower compared with WT in the presence of WY14,643 (Fig. 9A). Coimmunoprecipitation experiments performed on replicates indicated that decreased HMGCS2 mRNA expression in the presence of ligand was associated with defective release of NCoR from V227A (Fig. 9B). Simultaneous ChIP with anti-NCoR showed that decreased mRNA expression was associated with increased recruitment of NCoR to the PPRE in the HMGCS2 promoter (Fig. 9C). Increased binding of the V227A/NCoR complex to PPRE in the presence and absence of hormone was also evident in ChIP-re-ChIP experiments, wherein chromatin complexes were sequentially immunoprecipitated with antibodies to PPARα and then NCoR (Fig. 9D). In aggregate, our experiments indicate that the mechanism of reduced transactivation of the substitution was due to increased binding of NCoR to V227A/PPRE chromatin complex.
Fig. 8.

Effect of V227A on the Recruitment of Coregulators to the Mitochondria HMG-CoA Promoter
A, Coimmunoprecipitation. HepG2 cells transfected with full-length PPARα-WT or V227A (24 μg) were incubated with anti-PPARα rabbit monoclonal antibody in the absence or presence of 100 μm WY14,643. Precipitates were probed with anti-NCoR rabbit polyclonal antibody. Inputs were 5 and 10% cell lysate Western blotted for PPARα and NCoR expression, respectively. B, ChIP. HepG2 cells were transfected with 24 μg full-length PPARα-WT or V227A, with or without 100 μm WY14,643. ChIP analysis was carried out using antibodies against PPARα, p300, SRC-1, or NCoR. PCR for the HMGCS2 PPRE were performed up to 33 cycles. Amplification (36 cycles) of a fragment 5 kb upstream of the PPRE served as control (upper panels). The input was a representative of soluble chromatin (1%) that was reverse cross-linked and amplified under the same PCR conditions.
Fig. 9.
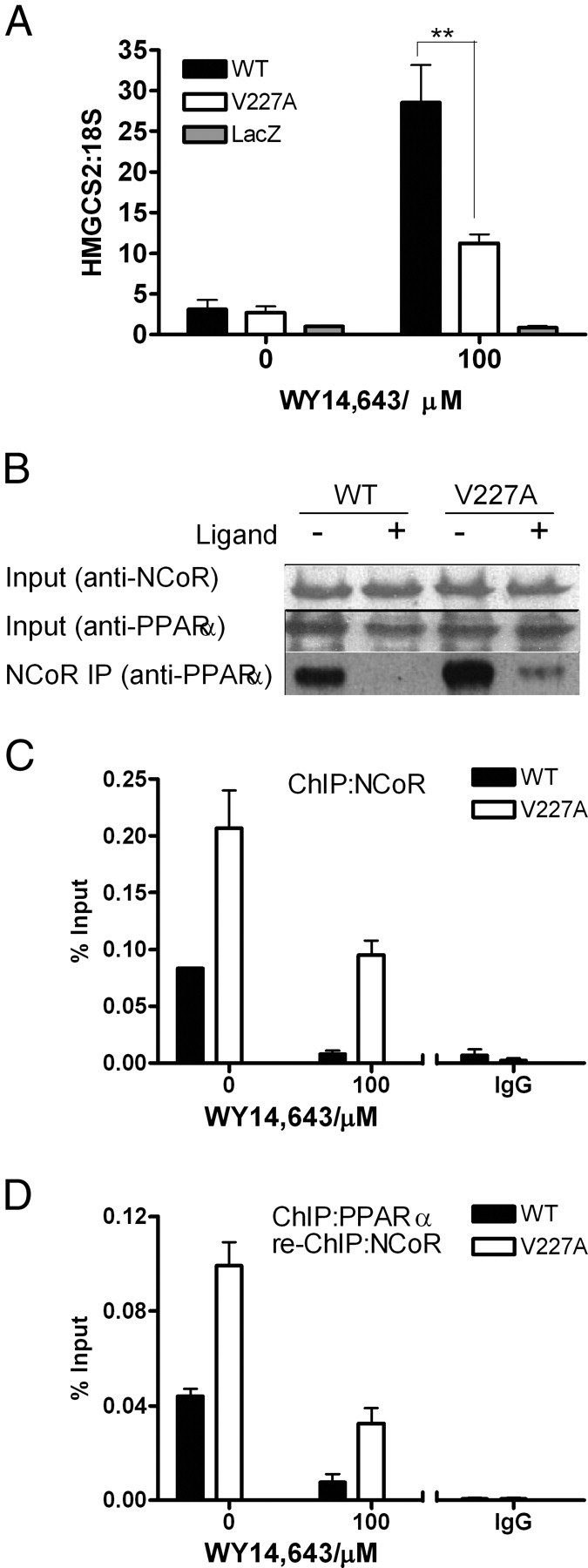
Correlation of PPARα-Regulated Gene Expression with Recruitment of NCoR to PPRE in the HMGCS2 Promoter
HepG2 cells were infected with adenovirus expressing WT PPARα, V227A, or empty LacZ vector and treated with, or without, WY14,643 for 24 h. A, HMGCS2 mRNA levels were measured by quantitative RT-PCR and normalized to 18S rRNA. Values are mean ± sd of three replicates and expressed in relative quantities to LacZ in the absence of ligand. B, Immunoprecipitation. Cells from representative replicates in A were incubated with anti-NCoR rabbit polyclonal antibody overnight, and precipitates were probed with anti-PPARα mouse monoclonal antibody. The input was a representative of soluble chromatin (1%) that was reverse cross-linked and amplified under the same PCR conditions. C, Recruitment of NCoR to HMGCS2 promoter. ChIP analyses were performed on parallel replicates depicted in A, using antibodies against NCoR or rabbit IgG. Real-time PCR was used to quantify amount of PPRE of the HMGCS2 promoter bound to NCoR. D, ChIP-re-ChIP experiments. ChIP experiments were carried out as in C, and PPARα ChIP complexes were eluted and subjected again to the ChIP procedure using NCoR antibody. Values are mean ± sd of three replicates.
DISCUSSION
Although several natural PPARα variants are known (22), mechanistic studies have not been performed on any of them. To our knowledge, this is the first demonstration that a natural PPARα variant attenuates transcription because of increased interaction with the corepressors. The V227A polymorphism induced significantly weaker transcriptional effects on a promoter containing the consensus CYP4A6-PPRE in the presence of a fibrate drug in physiologically relevant cells. The transactivation defect was about 35–56% weaker compared with the WT. The V227A mutant exhibited significantly weaker activity on the HMGCS2 proximal promoter in reporter gene assays and lower mRNA expression in hepatic cells. Attenuated mutant transactivation was not associated with defective ligand binding; neither were differences detected in protein expression or localization of mutant PPARα relative to WT. Rather, screening of major PPARα coregulators in the mammalian two-hybrid assay revealed that V227A LBD fragments bound corepressors with about 2-fold greater avidity than WT. The substitution also induced defective release of NCoR from PPAR in the presence of the ligands WY 14,643 and α-linolenic acid. The latter is consistent with our previous findings (28) that the polymorphism modulates the effect of dietary PUFA on plasma lipids. Silencing of NCoR, or inhibition of its HDAC activity, partially restored mutant function. Taken together, these data indicate that attenuated transactivation function of V227A was physiologically relevant and mediated through increased corepressor recruitment and activity.
Corepressors, like NCoR, suppress transactivation activity through their amino-terminal repressor domains and interact with nuclear receptors through carboxyl-terminal ID. NCoR has three ID (36), two of which contain core CoRNR box motifs with the consensus sequence LXXI/HIXXXI/L (31) and a third CoRNR box that lacks the extended helical motif (36). The crystal structure of antagonist-bound PPARα revealed that the CoRNR motif fits tightly into a groove formed by PPARα helices 3, 3′, 4, and 5 (20). Interestingly, this corepressor groove has also been identified as the critical docking site for coactivators. Although there is overlap, the corepressor site has a larger interaction interface occupied by the three α-helical turns generated by LXXI/HIXXXI/L of the CoRNR motif, compared with two turns of the SRC-1 coactivator motif, LXXLL. It is hypothesized that nuclear receptors distinguish corepressors from coactivators by the length of their interaction motifs. Consistent with this model, our data indicate that NCoR and SRC-1 can be detected at same PPARα binding site in solution and in chromatin and that the V227A substitution favored the binding of the corepressor.
There is preliminary evidence that PPARα interacts with ID1 (19), but the specific ID preference pattern for PPARα is still uncertain, because interaction studies comparing all three IDs have not been performed. Here we show that NCoR ID1, not ID2 or ID3, was necessary and sufficient for maximal PPARα-NCoR interactions in mammalian two-hybrid and GST pulldown assays. An intact CoRNR box was necessary, because deletion of the terminal four residues of the ID1 CoRNR box nonapeptide abolished PPARα-NCoR interactions. In addition, our deletion mutants indicate the importance of the N-terminal sequences (2251–2274) flanking the CoRNR box, because their deletion leads to weaker PPARα/NCoR interactions. Interaction with PPARα was lower with NCoR fragment G8 (residues 1575–2453) compared with G4 (2039–2453), suggesting a potential suppressive function in NCoR residues 1575–2039. Intriguingly, this fragment has been reported to harbor a sin3/HDAC3 recruiting site (39, 40). Nuclear receptors have distinct binding preferences for the three IDs of NCoR. The TR specifically requires the presence of all three, ID1, ID2, and ID3, all of which act cooperatively to induce maximal TR-NCoR interaction (36). On the other hand, RAR requires ID2 and interacts only minimally with ID1, whereas its heterodimeric partner RXR interacts with ID1 and not ID2, giving rise to the model that RXR/RAR heterodimer could be bound to the same corepressor molecule via the two adjacent ID1 and ID2 (32). However, this model is not applicable to PPARα, because both PPARα and its heterodimeric partner RXR interacts with ID1 and not ID2. Detailed knowledge of the ID preferences of PPARα may lead to the design of peptidomimetics that can block one of the other of the NCoR ID resulting in targeted effects on PPARα function (41).
Artificial mutations of PPARα in the C-terminal helix 12 are known to increase interactions with NCoR. Deletion of the terminal 13 residues encoding helix 12 results in a truncation mutant (PPARαΔ13) that was able to bind agonists but did not stimulate transcription (42). This PPARαΔ13 mutant exerted dominant-negative effects, in that it was able to repress the activity of coexpressed WT PPAR. Similarly substitutions in PPARα helix 12 (L459A and G462A), which were created based on PPARγ variants associated with severe insulin resistance, diabetes mellitus, and hypertension (43), resulted in an artificial dominant-negative PPARα mutant that recruited NCoR in a ligand-dissociable manner (44). These mutations may block helix 12 from assuming an active conformation, resulting in a larger pocket that can accommodate the three-turn corepressor motif, as was observed for antagonist-bound PPARα (20). Besides helix 12, our data indicate that the N-terminal portion of LBD, the short hinge region that begins at the termination of DBD and extending to helix 2 (45), plays an important repressor role in PPARα function. Evidence exists from other nuclear receptors that the hinge region plays a key role in receptor/NCoR interactions. Deletion of the androgen receptor hinge results in a mutant that is hyperactive, suggesting a repressor function in this domain (46). The estrogen (47) and progesterone receptor (48) hinge regions recruit NCoR in the presence of partial agonists, and hinge deletion mutants abolished interactions with NCoR. Intriguingly, sequence alignment (Fig. 10) indicates that PPAR residue 227 is located between two TRβ hinge residues 234 and 243 that were encountered in patients with resistance to thyroid hormone (49). High-resolution crystallography indicates that mutations affecting TRβ hinge residues 234 and 243 modulated the flexibility of the N-terminal region such that higher concentrations of ligand are required for optimal LBD assembly and stability (45). Our finding that V227A substitution in helix 2 enhances PPARα/NCoR interactions supports the concept that this region has a role in mediating corepressor function. The exact structural basis whereby V227A mediates increased corepressor recruitment to PPARα, whether by directly modifying contact points with NCoR or as a secondary structural determinant, requires crystallographic analysis.
Fig. 10.

Sequence Comparison of TRβ and PPARα
Sequence alignment is shown of helices 1 and 2 of human TRβ (hTR3β) and PPARα (hPPARα). The V227A PPARα variants TRβ (A234, R243) mutants associated with resistance to thyroid hormones are marked with asterisks.
The dynamic interactions between coactivators and corepressors in PPARα-regulated promoters are controversial. TRβ, RAR, RXR, and PPARγ receptors interact with SMRT and/or NCoR in the absence of hormone. Others, such as the androgen, estrogen, and progesterone receptors (48) do not bind NCoR with hormone but bind corepressors in the presence of hormone antagonists. In solution, PPARα can recruit corepressors in the absence of hormone but displays still higher corepressor affinity in the presence of antagonists. Whereas it is clear that PPARα can interact with corepressor in solution (19) or in the presence of antagonists (50), it has not been demonstrated conclusively that NCoR-PPARα binding occurs in PPRE, and their presence in chromatin is controversial (19, 44). It has been suggested that WT PPARα, like PPARγ (51), interacts with SMRT and NCoR in solution but not on DNA. Our ChIP assays show that WT PPARα interacts with NCoR on the HMGCS2 promoter in the unliganded state, and exogenous ligands induced complete release of NCoR and the simultaneous recruitment of SRC-1. The V227A substitution disrupted this process causing incomplete release of NCoR. Defective V227A transactivation function observed with high doses of ligand where NCoR no longer binds to V227A suggest that corepressors other than NCoR may have a role in defective mutant function. One such corepressor may be SMRT, which also binds avidly to V227A.
Although the specific genetic pathways that are differentially modulated by the mutant PPARα receptor are currently unclear, our data that V227A enhances PPARα/NCoR interaction in solution and on chromatin reveal a novel mechanism whereby a natural PPARα mutant may affect human health through its weaker transactivation activity. More studies are required to understand whether the response to fibrate drugs and other PPARα-regulated pathways are affected by the V227A polymorphism.
MATERIALS AND METHODS
Cell Culture, Transfection, and Reagents
HepG2 and HeLa cells were obtained from American Type Culture Collection (Rockville, MD). All cells were cultured in Eagle-modified MEM (Sigma Chemical Co., St. Louis, MO) with 2 mm [scap]l-glutamine, 1.5 g/liter sodium bicarbonate, 0.1 mm nonessential amino acids, 1 mm sodium pyruvate, 10% and charcoal-treated fetal bovine serum.
Cells were transfected using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) or GenePORTER 2 (Gelantis, San Diego, CA) according to manufacturer’s instructions. Briefly, cells were cultivated in charcoal-treated medium for 3 d before seeding at 40,000 cells per well in 24-well microtiter plates and incubated for 24 h before transfection. Transfected cells were then exposed to test samples in charcoal-treated medium. In reporter gene bioassays, cells were lysed after treatment with 100 μl of M-Per (Pierce, Rockford, IL) per well for 5 min. Luciferase activity was then measured using Glomax 96 Microplate Luminometer (Promega, Madison, WI). Experiment results obtained from reporter gene assays were reconfirmed with measurements of either renilla luciferase (Promega) or β-galactosidase (Promega) to ensure similar transfection efficiency.
WY14,643 was purchased from Tocris (Ellisville, MO). α-Linolenic acid and trichostatin A (TSA) were purchased from Sigma. [3H]WY14,643 was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO).
Plasmid Constructs
The full-length human PPARα cDNA was cloned by RT-PCR from HepG2 cell RNA using primers with BamHI and ApaI sites into pcDNA3.1/myc-His A (Invitrogen) to generate pcDNA3.1-PPARα WT. The valine at position 227 was mutated into alanine by PCR site-directed mutagenesis using QuikChange kit (Stratagene, La Jolla, CA) according to the manufacturer’s instructions to generate pcDNA3.1-PPARαV227A. The pCYP4A6-PPRE-Luc was a gift of Dr. W. Wahli, University of Lausanne, Switzerland (5). pCMX-hRXRα was generated after excision of VP16 and religation of the HindIII site of pCMX-VP16 hRXRα, a gift of Dr. R. M. Evans, The Salk Institute, San Diego, CA (52). pCMX-Gal-C-SMRT was also a gift of Dr. R. M. Evans (53). pSG424-C′ NCoR (amino acids 1575–2453) was a gift of Dr. Tetsuya Tamagi, National Hospital Organization, Japan (54). pSG5-NCoR (full-length) was a gift of Dr. R. N. Cohen, University of Chicago, Chicago, IL (55). The plasmid encoding PRIP was pSG5-Gal4-hRAP250-del4, a gift of Dr. J. A. Gustafsson, Karolinska Institute, Sweden (17). The promoter of mitochondria HMG-CoA synthase (−1081 to + 22) was cloned by RT-PCR of HepG2 cell genomic DNA using primers with NheI and HindIII sites into pGL3 basic vector (Promega) to generate pHMGCS2-Luc. pGal-PPARαWT-LBD (residues 171–468) was previously described (56), and pGal-PPARαV227A-LBD was generated through the QuikChange kit using pGal-WT-LBD as a template. The p[upstream activating sequence of Gal4 (UASG)]5-Luc reporter was previously described (57). The hinge and LBD (residues 167–468) of PPARα WT or V227A was cloned into the BamHI and HindIII site of pVP16 (Clontech, Palo Alto, CA) to give pVP16-WT and pVP16-V227A. The VP16 control plasmid, pVP16-ΔH1-H2, of helices 3–12 of PPARα LBD (residues 245–468) was also cloned using the above strategy. Residues 213-1061 of pCR3.1-hSRC-1A (58) (a gift from Dr. B. W. O’Malley, Baylor College of Medicine, Houston, TX) was cloned into the Sal1 and XbaI of pM vector. Residues 622–869 of TIF2 (59) and residues 120–284 of pSV-PGC1α (Addgene, Cambridge, MA) were cloned into the BamHI and MluI site of pM vector. Residues 1–117 of E1A binding protein p300 from pCMVb-p300 HA (Addgene) were cloned into the EcoRI of pSG424 to generate Gal-p300. Gal-NCoR truncated mutants were generated by cloning respective fragments into the EcoRI and SalI sites of the pM vector (Clontech). GST-NCoR mutants were generated by subcloning each NCoR truncated mutant into pGEX-4T-1 (Amersham Pharmacia Biotech, Piscataway, NJ). VP16-PPARα truncated mutants were generated by cloning respective fragments into the BamHI and HindIII sites of the pVP16 vector.
Recombinant Adenovirus Construction
Recombinant Adeno-X adenovirus containing LacZ, human full-length PPARα WT, or V227A was constructed according to manufacturer’s protocol (Clontech). Viruses were propagated in 293A packaging cells. Viral titer was determined by plaque assay. Cells were infected with adenovirus at a multiplicity of infection of 10.
Sequence Alignment
Helix 2 of PPARα was defined according to Xu et al. (20) and helix 2 of TRβ according to Huber et al. (45). PPARα was aligned against TRβ according to Wurtz et al. (60). PPARα domains were defined according to Desvergne and Wahli (3).
Quantitative Real-Time RT-PCR
HepG2 cells were infected with adenovirus expressing WT PPARα, V227A, or LacZ and treated with WY14,643 for 24 h. RNA was extracted from treated cells using RNeasy Mini Kit (QIAGEN, Valencia, CA) after treatment. Samples of total RNA were reverse transcribed using Superscript II (Invitrogen) according to manufacturers’ instructions. The levels of HMGCS2 were measured, using TaqMan Gene Expression Assays Hs00194145_m1 (Applied Biosystems, Foster, City, CA). Amplifications were performed and mRNA content measured with an ABI Prism 7000 Sequence Detection System. For each sample, the target mRNA level was normalized to the 18S rRNA level.
Competitive Binding Assay
Competitive PPARα ligand binding assay was performed as previously described (61). Briefly, HeLa cells in 24-well microtiter plates were transfected with pcDNA3.1-PPARαWT or PPARαV227A and incubated in 3 nm [3H]WY14,643 with increasing doses of unlabeled WY14,643 or DHT at 37 C. The amount of radiolabeled WY14,643 specifically bound was measured after 24 h incubation.
Immunofluorescence
HepG2 cells seeded on coverslips in 24-well microtiter plates were transfected with pcDNA3.1-PPARαWT or V227A and treated in the absence or presence of WY14,643. At 24 h after treatment, the cells were fixed with cold absolute methanol for 10 min, followed by the incubation with anti-PPARα rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) (1:400) in a humidity chamber for 1 h at 37 C. After washing, the cell monolayer on the coverslips was incubated with secondary antibodies conjugated with fluorescein isothiocyanate (FITC) (Amersham) (1:600). An optical immunofluorescence microscope (Olympus IX-81) was used to view the specimens at ×100 magnification, excitation wavelengths of 480 nm, for FITC using oil immersion objectives. The nucleus was stained with 4′,6-diamidino-2-phenylindole.
siRNA Knockdown
BLOCK-iT Lentiviral RNAi Expression System (Invitrogen) was used according to the manufacturer’s instructions to produce lentiviruses for gene silencing of NCoR in HepG2 cells. Double-stranded oligonucleotide encoding the shRNA of NCoR (siNCoR) (top strand, 5′-CACCGCTGAAGAGGGTTCTGTTTGTCGAAACAAACAGAACCCTCTTCAGC-3′; bottom strand, 5′-AAAAGCTGAAGAGGGTTCTGTTTGTTTCGACAAACAGAACCCTCTTCAGC-3′) were synthesized, and oligo duplexes were cloned into a lentivirus expression vector (pLenti6/BLOCK-iT-DEST). The expression plasmid and other virus-packaging plasmids were cotransfected into 293FT cells to generate replication-defective lentivirus. The resulting lentivirus was then concentrated and used to infect HepG2 for gene silencing of NCoR. Successful knockdown of NCoR was assessed using Western blotting. A randomized sequence (siScram) (forward, 5′-CACCAGGATGCAACACGTGGAATAGCGAACTATTCCACGTGTTGCATCCT-3′; reverse, 5′-AAAAAGGATGCAACACGTGGAATAGTTCGCTATTCCACGTGTTGCATCCT-3′) not targeting any known human transcript sequences was used as a control.
GST Pulldown
GST fusion proteins were expressed in BL21-DE3 Escherichia coli by induction with 0.4 mm isopropylthio-β-d-galactosidase at 37 C. Proteins were isolated after disruption through sonication and affinity purified on glutathione-Sepharose 4B beads (Amersham) according to the manufacturer’s protocol. WT and V227A PPARα were in vitro translated in reticulocyte lysate (Promega) using T7 polymerase, according to the manufacturer’s protocol, and 25 μl of the in vitro translated proteins was incubated with equal amounts of GST fusion proteins overnight at 4 C. After this, GST fusion protein complexes were affinity purified with 50 μl glutathione-Sepharose 4B beads for 2 h at 4 C. After extensive washing, bound proteins were eluted by boiling in 2× loading buffer and analyzed by Western blotting for PPARα.
ChIP
HepG2 cells grown on 100-mm plates were transfected with pcDNA3.1-PPARαWT or V227A or infected with adenovirus expressing WT PPARα or V227A and treated in the absence or presence of WY14,643. At 24 h after treatment, the cells were treated with formaldehyde to cross-link proteins to DNA, and ChIP was performed using the ChIP Assay Kit (Upstate Biotechnology, Lake Placid, NY) according to the manufacturer’s instructions. ChIP was performed using the following antibodies at 2 μg/ml: normal rabbit IgG, PPARα, SRC-1, NCoR, and p300 (Santa Cruz). The following primers were used to amplify the PPRE region (−198 to −34) of the HMGCS2 promoter: forward, 5′-CAGCCATTCCCACACATGCTCA-3′, and reverse, 5′-CAGACTTTATAAAGCCCCAAGACT-3′). The following primers were used to amplify a control 196-bp fragment −5 kb upstream region of the HMGCS2 promoter: forward, 5′-AGAGGAGGAACATTGTAAAAGCCCTA-3′, and reverse, 5′-CTGGAGACTCCTGTGACCAAGGTT-3′. In ChIP-re-ChIP experiments, PPARα ChIP complexes were eluted by incubation for 30 min at 37 C in 50 μl 10 mm dithiothreitol. After centrifugation, the supernatant was diluted 20 times with re-ChIP buffer (1% Triton X-100, 2 mm EDTA, 150 mm NaCl, 20 mm Tris-HCl, pH 8.1) and subjected again to the ChIP procedure using NCoR antibody. Real-time PCR was performed using appropriate primers and 2× SYBR green qPCR SuperMix for ABI Prism (Invitrogen) according to manufacturer’s directions.
Immunoprecipitation
HepG2 cells grown on 100-mm plates were transfected with pcDNA3.1-PPARαWT or V227A or infected with adenovirus expressing WT PPARα or V227A and treated in the absence or presence of WY14,643. At 24 h after treatment, the cells were washed with cold PBS and lysed using RIPA buffer. After centrifugation at 1500 × g for 15 min, the supernatants were precleared with protein A/G PLUS agarose beads (Santa Cruz) for 30 min and incubated with 2 μg/ml anti-NCoR rabbit polyclonal antibody (Santa Cruz) for HepG2 transfected cells or anti-PPARα rabbit polyclonal (Santa Cruz) for HepG2 adenovirus infected cells at 4 C overnight. After this, the antibody-protein complex was pulled down using A/G agarose beads and washed five times with RIPA buffer before elution of bound proteins by boiling in 2× loading buffer and analyzed by Western blotting for NCoR or PPARα. For Western blotting of immunoprecipitates of adenovirus-infected cells, anti-PPARα mouse monoclonal antibody (Affinity Bioreagents, Golden, CO) was used.
Western Analysis
Immunoblotting of total cell lysates were performed according to standard Western blotting protocol, using anti-PPARα rabbit polyclonal antibody (Santa Cruz), anti-NCoR rabbit polyclonal antibody (Santa Cruz), and anti-β-actin mouse monoclonal antibody (Sigma).
Statistical Analysis
All luciferase experiments were done in duplicates or triplicates at least three times on separate occasions. The difference between WT and V227A at each treatment was analyzed by two-sample t test. Adjustment for type 1 error due to multiple comparisons was done by Bonferroni procedure on groups with a significant difference.
Acknowledgments
We thank Edmund Chan for construction of plasmids, Dr. Y. H. Chan and Dr. Tai Bee Choo for statistical help, and Dr. Gong Yinhan and Wong Shih Peng for discussions of the manuscript.
NURSA Molecule Pages:
Coregulators: AIB1 | ASC-2 | GRIP1 | NCOR | SMRT | SRC-1 | p300;
Ligands: Pirinixic acid;
Nuclear Receptors: PPARα.
Footnotes
This work was supported by a grant from the Singapore Biomedical Research Council (BMRC 04/1/21/19/304). E.L.Y. and E.S.T are BMRC Clinician-Scientist investigators. L.M.H. is an A*STAR Graduate Scholar.
Disclosure Statement: The authors have nothing to disclose.
First Published Online February 21, 2008
Abbreviations: ChIP, Chromatin immunoprecipitation; CoRNR, core consensus nonapeptide motif (LXXI/HIXXXI/L) on the corepressor for receptor interaction; CYP4A6, cytochrome P450 4A6; DBD, DNA-binding domain; DHT, dihydrotestosterone; DR, direct repeat; FITC, fluorescein isothiocyanate; GST, glutathione-S-transferase; HDAC, histone deacetylase; HMGCS2, mitochondrial 3-hydroxy-3-methylglutaryl coenzyme A synthase; ID, interaction domain; LBD, ligand-binding domain; NCoR, nuclear receptor corepressor; NR, nuclear receptor; PGC-1, PPARγ coactivator-1; PPARα, peroxisome proliferator-activated receptor-α; PPARA, locus encoding for PPARα; PPRE, PPAR response elements; PRIP, PPAR interacting protein; PUFA, polyunsaturated fatty acids; RAR, retinoic acid receptor; RXR, retinoid X receptor; siNCoR, small interfering NCoR; sh, short hairpin; SMRT, silencing mediator for retinoid and thyroid hormone receptors; SRC-1, steroid receptor coactivator-1; TIF2, transcriptional intermediate factor 2; TR, thyroid hormone receptor; TSA, trichostatin A; UASG, upstream activating sequence of Gal4; WT, wild type.
References
- 1.Michalik L, Auwerx J, Berger JP, Chatterjee VK, Glass CK, Gonzalez FJ, Grimaldi PA, Kadowaki T, Lazar MA, O’Rahilly S, Palmer CN, Plutzky J, Reddy JK, Spiegelman BM, Staels B, Wahli W 2006. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol Rev 58:726–741 [DOI] [PubMed] [Google Scholar]
- 2.Lefebvre P, Chinetti G, Fruchart JC, Staels B 2006. Sorting out the roles of PPARα in energy metabolism and vascular homeostasis. J Clin Invest 116:571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desvergne B, Wahli W 1999. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev 20:649–688 [DOI] [PubMed] [Google Scholar]
- 4.Germain P, Staels B, Dacquet C, Spedding M, Laudet V 2006. Overview of nomenclature of nuclear receptors. Pharmacol Rev 58:685–704 [DOI] [PubMed] [Google Scholar]
- 5.Krey G, Keller H, Mahfoudi A, Medin J, Ozato K, Dreyer C, Wahli W 1993. Xenopus peroxisome proliferator activated receptors: genomic organization, response element recognition, heterodimer formation with retinoid X receptor and activation by fatty acids. J Steroid Biochem Mol Biol 47:65–73 [DOI] [PubMed] [Google Scholar]
- 6.Kroetz DL, Yook P, Costet P, Bianchi P, Pineau T 1998. Peroxisome proliferator-activated receptor α controls the hepatic CYP4A induction adaptive response to starvation and diabetes. J Biol Chem 273:31581–31589 [DOI] [PubMed] [Google Scholar]
- 7.Yu S, Rao S, Reddy JK 2003. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr Mol Med 3:561–572 [DOI] [PubMed] [Google Scholar]
- 8.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ 1995. Targeted disruption of the α-isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol 15:3012–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W 1999. Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J Clin Invest 103:1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Djouadi F, Weinheimer CJ, Saffitz JE, Pitchford C, Bastin J, Gonzalez FJ, Kelly DP 1998. A gender-related defect in lipid metabolism and glucose homeostasis in peroxisome proliferator-activated receptor α-deficient mice. J Clin Invest 102:1083–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu MH, Savas U, Griffin KJ, Johnson EF 2001. Identification of peroxisome proliferator-responsive human genes by elevated expression of the peroxisome proliferator-activated receptor α in HepG2 cells. J Biol Chem 276:27950–27958 [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez JC, Gil-Gomez G, Hegardt FG, Haro D 1994. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J Biol Chem 269:18767–18772 [PubMed] [Google Scholar]
- 13.Juge-Aubry C, Pernin A, Favez T, Burger AG, Wahli W, Meier CA, Desvergne B 1997. DNA binding properties of peroxisome proliferator-activated receptor subtypes on various natural peroxisome proliferator response elements. Importance of the 5′-flanking region. J Biol Chem 272:25252–25259 [DOI] [PubMed] [Google Scholar]
- 14.Dowell P, Ishmael JE, Avram D, Peterson VJ, Nevrivy DJ, Leid M 1997. p300 functions as a coactivator for the peroxisome proliferator-activated receptor α. J Biol Chem 272:33435–33443 [DOI] [PubMed] [Google Scholar]
- 15.Voegel JJ, Heine MJ, Tini M, Vivat V, Chambon P, Gronemeyer H 1998. The coactivator TIF2 contains three nuclear receptor-binding motifs and mediates transactivation through CBP binding-dependent and -independent pathways. EMBO J 17:507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vega RB, Huss JM, Kelly DP 2000. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol 20:1868–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caira F, Antonson P, Pelto-Huikko M, Treuter E, Gustafsson JA 2000. Cloning and characterization of RAP250, a novel nuclear receptor coactivator. J Biol Chem 275:5308–5317 [DOI] [PubMed] [Google Scholar]
- 18.Yu S, Reddy JK 2007. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim Biophys Acta 1771:936–951 [DOI] [PubMed] [Google Scholar]
- 19.Dowell P, Ishmael JE, Avram D, Peterson VJ, Nevrivy DJ, Leid M 1999. Identification of nuclear receptor corepressor as a peroxisome proliferator-activated receptor α interacting protein. J Biol Chem 274:15901–15907 [DOI] [PubMed] [Google Scholar]
- 20.Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, McKee DD, Galardi CM, Plunket KD, Nolte RT, Parks DJ, Moore JT, Kliewer SA, Willson TM, Stimmel JB 2002. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARα. Nature 415:813–817 [DOI] [PubMed] [Google Scholar]
- 21.Perissi V, Rosenfeld MG 2005. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol 6:542–554 [DOI] [PubMed] [Google Scholar]
- 22.Naito H, Yamanoshita O, Kamijima M, Katoh T, Matsunaga T, Lee CH, Kim H, Aoyama T, Gonzalez FJ, Nakajima T 2006. Association of V227A PPARα polymorphism with altered serum biochemistry and alcohol drinking in Japanese men. Pharmacogenet Genomics 16:569–577 [DOI] [PubMed] [Google Scholar]
- 23.Flavell DM, Pineda Torra I, Jamshidi Y, Evans D, Diamond JR, Elkeles RS, Bujac SR, Miller G, Talmud PJ, Staels B, Humphries SE 2000. Variation in the PPARα gene is associated with altered function in vitro and plasma lipid concentrations in type II diabetic subjects. Diabetologia 43:673–680 [DOI] [PubMed] [Google Scholar]
- 24.Vohl MC, Lepage P, Gaudet D, Brewer CG, Betard C, Perron P, Houde G, Cellier C, Faith JM, Despres JP, Morgan K, Hudson TJ 2000. Molecular scanning of the human PPARα gene: association of the L162v mutation with hyperapobetalipoproteinemia. J Lipid Res 41:945–952 [PubMed] [Google Scholar]
- 25.Lacquemant C, Lepretre F, Pineda Torra I, Manraj M, Charpentier G, Ruiz J, Staels B, Froguel P 2000. Mutation screening of the PPARα gene in type 2 diabetes associated with coronary heart disease. Diabetes Metab 26:393–401 [PubMed] [Google Scholar]
- 26.Flavell DM, Jamshidi Y, Hawe E, Pineda Torra I, Taskinen MR, Frick MH, Nieminen MS, Kesaniemi YA, Pasternack A, Staels B, Miller G, Humphries SE, Talmud PJ, Syvanne M 2002. Peroxisome proliferator-activated receptor α gene variants influence progression of coronary atherosclerosis and risk of coronary artery disease. Circulation 105:1440–1445 [DOI] [PubMed] [Google Scholar]
- 27.Sapone A, Peters JM, Sakai S, Tomita S, Papiha SS, Dai R, Friedman FK, Gonzalez FJ 2000. The human peroxisome proliferator-activated receptor α gene: identification and functional characterization of two natural allelic variants. Pharmacogenetics 10:321–333 [DOI] [PubMed] [Google Scholar]
- 28.Chan E, Tan CS, Deurenberg-Yap M, Chia KS, Chew SK, Tai ES 2006. The V227A polymorphism at the PPARA locus is associated with serum lipid concentrations and modulates the association between dietary polyunsaturated fatty acid intake and serum high density lipoprotein concentrations in Chinese women. Atherosclerosis 187:309–315 [DOI] [PubMed] [Google Scholar]
- 29.Yamakawa-Kobayashi K, Ishiguro H, Arinami T, Miyazaki R, Hamaguchi H 2002. A Val227Ala polymorphism in the peroxisome proliferator activated receptor α (PPARα) gene is associated with variations in serum lipid levels. J Med Genet 39:189–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gervois P, Torra IP, Chinetti G, Grotzinger T, Dubois G, Fruchart JC, Fruchart-Najib J, Leitersdorf E, Staels B 1999. A truncated human peroxisome proliferator-activated receptor α splice variant with dominant negative activity. Mol Endocrinol 13:1535–1549 [DOI] [PubMed] [Google Scholar]
- 31.Hu X, Lazar MA 1999. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 402:93–96 [DOI] [PubMed] [Google Scholar]
- 32.Hu X, Li Y, Lazar MA 2001. Determinants of CoRNR-dependent repression complex assembly on nuclear hormone receptors. Mol Cell Biol 21:1747–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, Rosenfeld MG 1995. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377:397–404 [DOI] [PubMed] [Google Scholar]
- 34.Seol W, Mahon MJ, Lee YK, Moore DD 1996. Two receptor interacting domains in the nuclear hormone receptor corepressor RIP13/N-CoR. Mol Endocrinol 10:1646–1655 [DOI] [PubMed] [Google Scholar]
- 35.Cohen RN, Putney A, Wondisford FE, Hollenberg AN 2000. The nuclear corepressors recognize distinct nuclear receptor complexes. Mol Endocrinol 14:900–914 [DOI] [PubMed] [Google Scholar]
- 36.Webb P, Anderson CM, Valentine C, Nguyen P, Marimuthu A, West BL, Baxter JD, Kushner PJ 2000. The nuclear receptor corepressor (N-CoR) contains three isoleucine motifs (I/LXXII) that serve as receptor interaction domains (IDs). Mol Endocrinol 14:1976–1985 [DOI] [PubMed] [Google Scholar]
- 37.Cohen RN, Brzostek S, Kim B, Chorev M, Wondisford FE, Hollenberg AN 2001. The specificity of interactions between nuclear hormone receptors and corepressors is mediated by distinct amino acid sequences within the interacting domains. Mol Endocrinol 15:1049–1061 [DOI] [PubMed] [Google Scholar]
- 38.Makowski A, Brzostek S, Cohen RN, Hollenberg AN 2003. Determination of nuclear receptor corepressor interactions with the thyroid hormone receptor. Mol Endocrinol 17:273–286 [DOI] [PubMed] [Google Scholar]
- 39.Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG 1997. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature 387:43–48 [DOI] [PubMed] [Google Scholar]
- 40.Li J, Wang J, Wang J, Nawaz Z, Liu JM, Qin J, Wong J 2000. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J 19:4342–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mettu NB, Stanley TB, Dwyer MA, Jansen MS, Allen JE, Hall JM, McDonnell DP 2007. The nuclear receptor-coactivator interaction surface as a target for peptide antagonists of the peroxisome proliferator activated receptors. Mol Endocrinol 21:2361–2377 [DOI] [PubMed] [Google Scholar]
- 42.Michalik L, Feige JN, Gelman L, Pedrazzini T, Keller H, Desvergne B, Wahli W 2005. Selective expression of a dominant-negative form of peroxisome proliferator-activated receptor in keratinocytes leads to impaired epidermal healing. Mol Endocrinol 19:2335–2348 [DOI] [PubMed] [Google Scholar]
- 43.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O’Rahilly S 1999. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402:880–883 [DOI] [PubMed] [Google Scholar]
- 44.Semple RK, Meirhaeghe A, Vidal-Puig AJ, Schwabe JW, Wiggins D, Gibbons GF, Gurnell M, Chatterjee VK, O’Rahilly S 2005. A dominant negative human peroxisome proliferator-activated receptor (PPAR)α is a constitutive transcriptional corepressor and inhibits signaling through all PPAR isoforms. Endocrinology 146:1871–1882 [DOI] [PubMed] [Google Scholar]
- 45.Huber BR, Desclozeaux M, West BL, Cunha-Lima ST, Nguyen HT, Baxter JD, Ingraham HA, Fletterick RJ 2003. Thyroid hormone receptor-β mutations conferring hormone resistance and reduced corepressor release exhibit decreased stability in the N-terminal ligand-binding domain. Mol Endocrinol 17:107–116 [DOI] [PubMed] [Google Scholar]
- 46.Wang Q, Lu J, Yong EL 2001. Ligand- and coactivator-mediated transactivation function (AF2) of the androgen receptor ligand-binding domain is inhibited by the cognate hinge region. J Biol Chem 276:7493–7499 [DOI] [PubMed] [Google Scholar]
- 47.Webb P, Nguyen P, Kushner PJ 2003. Differential SERM effects on corepressor binding dictate ERα activity in vivo. J Biol Chem 278:6912–6920 [DOI] [PubMed] [Google Scholar]
- 48.Jackson TA, Richer JK, Bain DL, Takimoto GS, Tung L, Horwitz KB 1997. The partial agonist activity of antagonist-occupied steroid receptors is controlled by a novel hinge domain-binding coactivator L7/SPA and the corepressors N-CoR or SMRT. Mol Endocrinol 11:693–705 [DOI] [PubMed] [Google Scholar]
- 49.Safer JD, Cohen RN, Hollenberg AN, Wondisford FE 1998. Defective release of corepressor by hinge mutants of the thyroid hormone receptor found in patients with resistance to thyroid hormone. J Biol Chem 273:30175–30182 [DOI] [PubMed] [Google Scholar]
- 50.Xu J, Xiao G, Trujillo C, Chang V, Blanco L, Joseph SB, Bassilian S, Saad MF, Tontonoz P, Lee WN, Kurland IJ 2002. Peroxisome proliferator-activated receptor α (PPARα) influences substrate utilization for hepatic glucose production. J Biol Chem 277:50237–50244 [DOI] [PubMed] [Google Scholar]
- 51.Zamir I, Zhang J, Lazar MA 1997. Stoichiometric and steric principles governing repression by nuclear hormone receptors. Genes Dev 11:835–846 [DOI] [PubMed] [Google Scholar]
- 52.Forman BM, Umesono K, Chen J, Evans RM 1995. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell 81:541–550 [DOI] [PubMed] [Google Scholar]
- 53.Chen JD, Evans RM 1995. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377:454–457 [DOI] [PubMed] [Google Scholar]
- 54.Tagami T, Gu WX, Peairs PT, West BL, Jameson JL 1998. A novel natural mutation in the thyroid hormone receptor defines a dual functional domain that exchanges nuclear receptor corepressors and coactivators. Mol Endocrinol 12:1888–1902 [DOI] [PubMed] [Google Scholar]
- 55.Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN 2005. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor γ transcriptional activity and repress 3T3-L1 adipogenesis. J Biol Chem 280:13600–13605 [DOI] [PubMed] [Google Scholar]
- 56.Shen P, Liu MH, Ng TY, Chan YH, Yong EL 2006. Differential effects of isoflavones, from Astragalus membranaceus and Pueraria thomsonii, on the activation of PPARα, PPARγ, and adipocyte differentiation in vitro. J Nutr 136:899–905 [DOI] [PubMed] [Google Scholar]
- 57.Ghadessy FJ, Lim J, Abdullah AA, Panet-Raymond V, Choo CK, Lumbroso R, Tut TG, Gottlieb B, Pinsky L, Trifiro MA, Yong EL 1999. Oligospermic infertility associated with an androgen receptor mutation that disrupts interdomain and coactivator (TIF2) interactions. J Clin Invest 103:1517–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jenster G, Spencer TE, Burcin MM, Tsai SY, Tsai MJ, O’Malley BW 1997. Steroid receptor induction of gene transcription: a two-step model. Proc Natl Acad Sci USA 94:7879–7884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loy CJ, Sim KS, Yong EL 2003. Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. Proc Natl Acad Sci USA 100:4562–4567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, Moras D, Gronemeyer H 1996. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol 3:87–94 [DOI] [PubMed] [Google Scholar]
- 61.Lim J, Ghadessy FJ, Abdullah AA, Pinsky L, Trifiro M, Yong EL 2000. Human androgen receptor mutation disrupts ternary interactions between ligand, receptor domains, and the coactivator TIF2 (transcription intermediary factor 2). Mol Endocrinol 14:1187–1197 [DOI] [PubMed] [Google Scholar]