

Abstract
Several posttranslational modifications including phosphorylation have been detected on the glucocorticoid receptor (GR). However, the interdependence and combinatorial regulation of these modifications and their role in GR functions are poorly understood. We studied the effects of c-Jun N-terminal kinase (JNK)-dependent phosphorylation of GR on its sumoylation status and the impact that these modifications have on GR transcriptional activity. GR is targeted for phosphorylation at serine 246 (S246) by the JNK protein family in a rapid and transient manner. The levels of S246 phosphorylation of endogenous GR increased significantly in cells treated with UV radiation that activates JNK. S246 GR phosphorylation by JNK facilitated subsequent GR sumoylation at lysines 297 and 313. GR sumoylation increased with JNK activation and was inhibited in cells treated with JNK inhibitor. GR sumoylation in cells with activated JNK was mediated preferentially by small ubiquitin-like modifier (SUMO)2 rather than SUMO1. An increase in GR transcriptional activity was observed after inhibition of JNK or SUMO pathways and suppression of GR transcriptional activity after activation of both pathways in cells transfected with GR-responsive reporter genes. Endogenous GR transcriptional activity was inhibited on endogenous target genes IGF binding protein (IGFBP) and glucocorticoid-induced leucine zipper (GILZ) when JNK and SUMO pathways were induced individually or simultaneously. Activation of both of these signals inhibited GR-mediated regulation of human inhibitor of apoptosis gene (hIAP), whereas simultaneous activation had no effect. We conclude that phosphorylation aids GR sumoylation and that cross talk of JNK and SUMO pathways fine tune GR transcriptional activity in a target gene-specific manner, thereby modulating the hormonal response of cells exposed to stress.
THE GLUCOCORTICOID RECEPTOR (GR) is a ligand-regulated transcription factor and a member of the nuclear hormone receptor superfamily that plays an important role in physiology and development, and is implicated in the regulation of metabolism, inflammation, and stress responses (1, 2). GR is widely expressed in vertebrate cells and, in the absence of ligand, is complexed with chaperones such as heat shock protein 90. Upon binding to the ligand, GR dissociates from chaperones and translocates into the nucleus, where it binds specifically to DNA sequences known as glucocorticoid-responsive elements (GREs). The GR modulates the expression of numerous target genes depending on cell type, promoter context, and physiological settings. The magnitude and direction of GR-mediated transcriptional activity is determined by multiple factors including the GRE sequence, balance between coactivators and corepressors, and posttranslational modifications (1, 2, 3, 4).
Phosphorylation of the N-terminal region in the transactivation domain AF-1 (activation function 1) of the GR is an important modification potentially affecting GR protein stability, nuclear location, and transcriptional activity (4, 5, 6, 7, 8, 9, 10). However, the role of phosphorylation in regulating the transcriptional function of GR remains controversial with several kinases proposed to be involved. Cyclin-dependent protein kinases (CDKs) and MAPKs phosphorylate rat GR and modulate its function (4, 11, 12). MAPKs receive signals from a diverse range of extracellular stimuli such as stress and mitogens, thereby controlling the cellular response to environmental changes. c-Jun N-terminal kinase (JNK) represents a subgroup of the MAPK family that is activated primarily by cytokines and environmental stresses such as osmotic or redox stress and UV radiation. These kinases are essential for embryonic morphogenesis and contribute to the control of cellular proliferation and apoptosis. JNK plays a crucial role in regulating the activity of numerous transcription factors such as AP-1, activating transcription factor 2, Elk-1, and GR (13, 14, 15).
GR and JNK pathways regulate common processes including inflammation, proliferation, and apoptosis, often in opposing ways. Cross talk of JNK signaling pathways with GR was observed in U2OS cells, where introduction of activated JNK protein resulted in inhibition of consensus GRE-driven, GR-mediated transcription (8), whereas Itoh and colleagues (7) observed increased cytoplasmic GR subcellular localization in UV-treated cells after withdrawal of dexamethasone (Dex). In accord with these reports, treatment of the hippocampal HT22 cells with JNK inhibitors enhanced mouse mammary tumor virus GRE-driven, glucocorticoid-dependent transcription (16). In addition, glucocorticoids negatively regulate the JNK pathway through transcriptional and nontranscriptional mechanisms (17, 18). These findings suggest that the JNK family is an important regulator of GR function, but the molecular mechanisms and biological significance of this cross talk have not been defined in detail.
In addition to phosphorylation, other posttranslational modifications, such as ubiquitination and sumoylation, play a role in the regulation of GR function (19, 20, 21, 22). The small ubiquitin-like modifier (SUMO) pathway includes E1-activating enzymes, an E2-conjugating enzyme, and E3 ligases that control addition of SUMO to its targets, as well as proteases that remove SUMO protein from modified substrates (23, 24, 25). SUMO modification of numerous transcription factors (Elk-1, heat shock factor-1 and -2, p53 and steroid receptors) has been associated with regulation of protein stability, subcellular location, and transcriptional control (26, 27, 28, 29). SUMO modification sites have been mapped to the N-terminal region of GR, termed “synergy control motifs,” that act as inhibitory elements from multiple but not single GREs. However, both inhibitory and stimulatory effects of SUMO addition on GR function have been reported (19, 20, 21).
In this study we have analyzed the effects of cellular stress on GR activity and have identified a novel mechanism by which JNK regulates GR function through SUMO conjugation. Our results elucidate the importance of a combinatorial role of phosphorylation and sumoylation in the cellular response to glucocorticoid hormones. These observations provide further insight into the intricate network of tightly controlled signals that converge on GR to adjust its various activities to cellular needs.
RESULTS
The JNK Family of Proteins Target GR at Serine 246 (S246) for Phosphorylation
It has been reported that the GR transcriptional activation domain is a potential target for several families of kinases (4, 7, 8). Glycogen synthase kinase-3 phosphorylates T171, whereas CDKs phosphorylate S224 and S232 of the rat GR (4, 10). MAPKs also phosphorylate the GR, and JNK pathway has been shown to phosphorylate S246 residue most efficiently (8). Receptor-dependent transcriptional enhancement is reduced in yeast strains deficient in the CDKs whereas deletion of MAPK homologs in yeast results in increased GR-mediated transcription (4).
To study in more details the effects of JNK activation on GR we performed in vitro kinase assays using wild-type (WT) GR AF-1 domain or derivatives of AF-1 carrying mutated phosphorylation sites T171A, S224A, S232A, and S246A (Fig. 1A). We observed efficient phosphorylation of all the GR recombinant proteins except the S246A mutant using either recombinant JNK1 purified from bacteria (Fig. 1A, upper panel, lane 7) or precipitated JNK1 from transfected cells (Fig. 1B, lane 2). Equal amounts of target proteins were used in the in vitro kinase assay (Fig. 1A, lower panel).
Fig. 1.

GR Is Phosphorylated by the JNK Family of Proteins
A, GST-GR fusion protein carrying WT AF-1 domain of GR (lane 3) or T171A (lane 4), S224A (lane 5), S232A (lane 6), and S246A (lane 7) were expressed and purified from Escherichia coli, and resolved on SDS-PAGE and Coomassie blue stained (lower panel). Indicated proteins were used in in vitro kinase assays with radiolabeled ATP as substrates for JNK1α1 kinase (Upstate Biotechnology), together with GST-c-Jun fusion protein (lane 1) that served as positive control and BSA (lane 2) that served as negative control (top panel). B, COS-7 cells were transfected with control vector pcDNA3 (lane 1) or PEBG JNK1α1 (lane 2), together with pcDNA3HA-MLK3 plasmids. Top panel shows Western blot analysis of JNK1α1 kinase immunoprecipated with GST-specific antibody. GST GR AF-1 WT, T171A, and S246A (middle panels), or c-Jun (lower panel) purified proteins were phosphorylated in vitro with immunoprecipitated kinases in the presence of radiolabeled ATP. Proteins were resolved on SDS-PAGE and exposed to film. C, GST-GR AF-1 WT (lanes 1, 2, 5, and 6) and S246A fusion proteins (lanes 3 and 4) were used as substrates in in vitro kinase assays with nonradiolabeled ATP and JNK1α1 kinase (Upstate Biotechnology). GR was detected by Western blot analysis using preimmune serum (PI) or antibody raised against phosphorylated S246 (S246-P). D, COS-7 cells were transfected with control vector pcDNA3 (lane 1) or PEBG JNK1α1 (lane 2), JNK2α2 (lane 3), and JNK3α1 (lane 4), together with pcDNA3HA-MLK3 plasmids (lanes 2–4). Top panel shows Western blot analysis of JNK1α1 kinase immunoprecipated with GST-specific antibody. GST GR AF-1 S246A (second panel), GST GR AF-1 WT (third panel), or c-Jun (lower panel) purified proteins were phosphorylated in vitro with immunoprecipitated kinases, and Western blot was developed using S246-P antibody (middle panels) and c-Jun phosphospecific antibody (lower panel). IB, Immunoblot; IP, immunoprecipitation.
To test whether JNK targets GR in vivo and to investigate relevance of this phosphorylation to GR function and cellular response to signaling pathways, we generated an antibody against GR phosphorylated at serine S246 (S246-P). The specificity of this antibody was studied in in vitro kinase assays shown in Fig. 1C where only GR WT phosphorylated by JNK1 was detected by this antibody (Fig. 1C, lane 2), whereas neither the S246A derivative nor GR WT that had not been phosphorylated by JNK1 was detected (Fig. 1C, lanes 4 and 6, respectively). The preimmune serum did not react with any of the GR substrates (Fig. 1C, lanes 1, 3, and 5).
The JNK family is encoded by three genes and is composed of JNK 1, 2, and 3 proteins that have different expression patterns and potentially similar, but not identical, substrate specificity and functions (13). To test whether there is differential GR phosphorylation by individual JNK family members, we transfected PEBG-JNK1α1, JNK2α2, JNK3α1 together with the JNK upstream activator-mixed lineage kinase 3 (MLK3) expression vector pcDNA3-HAMLK3 into COS-7 cells and isolated these kinases using glutathione-S-transferase (GST) affinity resin. As shown in Fig. 1D (GR S246-P panel) and supplemental Fig. 1 (published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org), all three family members of the JNK family of kinases phosphorylate GR S246 residue with similar efficiency.
The in vivo regulation of S246 GR phosphorylation was analyzed by transient transfection of COS-7 cells with GR and PEBG-JNK1α1/pcDNA3-HAMLK3. As shown in Fig. 2A, JNK1 activation leads to a substantial increase in phosphorylation of GR protein in the absence (Fig. 2A, top panel; compare lanes 3 and 5) and some increase in the presence of synthetic glucocorticoid hormone Dex (compare lanes 4 and 6), whereas total GR protein slightly increased (Fig. 2A, middle panel). This GR phosphorylation is not observed in cells transfected with the phosphorylation-deficient mutant S246A (Fig. 2A, lanes 9 and 10). Treatment of cells with JNK inhibitor abolished this increase in cells not exposed to hormone to a greater extent than in cells treated with hormone (Fig. 2A, lanes 7 and 8). JNK1 expression was confirmed by GST antibody (data not shown), and the levels of actin served as loading control (Fig. 2A, bottom panel). Finally, no reactivity was detected in the extracts from cells transfected with the pcDNA3 control vector (Fig. 2A, lanes 1 and 2) or when Western blot membranes were probed with preimmune serum (data not shown). Quantitative analysis of the extent of GR phosphorylation when normalized to total GR and actin levels indicated that S246 phosphorylation shows an increase of 40% in the absence and 10% in the presence of hormone in cells overexpressing GR and activated JNK (Fig. 2B, compare lanes 5 to 3 and lanes 6 to 4). This increase was not seen if cells were treated with JNK inhibitor or when S246A mutant was transfected (Fig. 2B, lanes 7–10).
Fig. 2.

JNK Regulates Phosphorylation of the S246 Residue on GR in Vivo
A, COS-7 cells were transfected with pcDNA3 (lanes 1 and 2), pcDNA3-WTGR (lanes 3–8), or pcDNA3-S246AGR (lanes 9 and 10) in the presence of JNK1α1/MLK3 (lanes 5–10). Cells were treated with vehicle (lanes 1, 3, 5, 7, and 9), 100 nm Dex for 1 h (lanes 2, 4, 6, 8, and 10), or 10 μm JNK inhibitor SP600125 30 min before hormone treatment, cell extracts were resolved by SDS-PAGE, and Western blots were probed with antibody specific for phosphorylated S246, total GR, and actin for loading control. B, Quantitative analysis of the GR phosphorylation. Graph represents intensity of the bands corresponding to the GR phosphorylated on the S246 vs. total GR normalized to the actin values shown in panel A. C, H4 cells were treated with vehicle (lane 1), 100 nm Dex (lanes 2–5), 100 nm RU486 (lanes 6–8), and UV (60 J/m2) (lanes 2–8). D, Quantitative analysis of the GR phosphorylation. Graph represents intensity of the bands corresponding to the GR phosphorylated on the S246 vs. total GR normalized to the actin values shown in panel C. E, The same as in panel C except that UV treatment was omitted. Equal amounts of total cellular proteins were analyzed by Western blotting. Immunoblots were probed with antibody specific for phosphorylated S246 (S246-P), GR-specific antibody, and actin antibody. F, H4 cells were incubated in the absence of hormone and treated with UV, and cellular extracts were made as described in panel C. Western blot was stained with Ponceau dye, cut along dotted lines, and strips were probed with antibody specific for phosphorylated S246 in the absence (lanes 1 and 2) or presence of specific peptide used as antigen for this antibody (lane 3, pep). Strips were incubated with preimmune serum (lane 4, PI) or with nonspecific IgG (lane 5, NS) (top panel). Blot was stripped of primary antibodies and probed with GR-specific antibody (middle panel). Lower part of this blot was probed with actin antibody.
Endogenous GR phosphorylation was assessed in H4 rat hepatoma cells treated with UV radiation under conditions that activate JNK (14). Data presented in Fig. 2, C and D, demonstrate that GR expressed at physiological levels was phosphorylated at S246 because it could be detected by the S246-P antibody. A substantial increase in GR phosphorylation was observed in the cells treated with UV radiation in the absence of hormone with maximal levels detected after the first hour of UV exposure suggesting that this modification is rapid and transient (Fig. 2C, compare lanes 1 and 2, and our unpublished observations). A gradual reduction of both phosphorylated and total GR protein levels was observed in H4 cells treated with UV in the presence of Dex (Fig. 2C, lanes 3–5) and the GR agonist triamcinolone acetonide (data not shown). On the contrary, this down-regulation did not occur in UV-treated cells in the presence of the GR antagonist RU486 (Fig. 2C, lanes 6–8) or progesterone (data not shown). Quantitative analysis of the S246 phosphorylation shows that UV radiation caused 46% increase in GR phosphorylation in the absence of hormone (Fig. 2D, compare lanes 1 and 2). This phosphorylation increased further 22% after 1 h and 2 h of Dex treatment and 42% after 6 h of Dex treatment (Fig. 2D, compare lanes 3, 4, and 5 with lane 2). RU486 treatment did not cause further increase in this phosphorylation (Fig. 2D, lanes 6–8). Basal S246 phosphorylation levels were observed in untreated cells and were slightly higher in cells treated with antagonist RU486 (Fig. 2E). The specificity of S246-P antibody was confirmed by incubating the Western blot membrane strips with S246-P antibody in the absence and presence of specific peptide, preimmune serum, or unrelated antibody (Fig. 2F, top panel). Total GR and actin proteins were readily detectable (Fig. 2F, middle and bottom panels, respectively). These results demonstrated that S246-P antibody is specific for GR isoforms phosphorylated at S246 in vivo and that JNK family preferentially targets endogenous GR at S246 for phosphorylation. The fact that the GR phosphorylation levels increased in the UV-radiated cells in the absence of hormone or in the presence of GR antagonists suggests that S246 phosphorylation is associated with the repression of GR activity, implying its importance in the regulation of GR activity under cellular stress.
S246 Phosphorylation Affects GR Sumoylation
Both JNK activation and GR sumoylation have been linked to repression of GR function, and lysines 297 and 313 in the vicinity of S246 phosphorylation site of GR have been identified as target sites for sumoylation (19). To explore the molecular basis for the effects of JNK-dependent phosphorylation of GR and whether phosphorylation affects GR sumoylation, we performed in vitro sumoylation assays using SUMO2 enzyme and purified GST-GR-AF1 recombinant proteins (Fig. 1A and supplemental Fig. 2) as substrates. As shown in Fig. 3A, lane 3, two GR isoforms with molecular masses of approximately 110 kDa and 65 kDa were detected in this assay (indicated with a and b arrows, respectively) and were not present when mutant SUMO2 was used as source of the enzyme (Fig. 3A, middle panel). These isoforms represent sumoylated GR because they are not present when the K297R/K313R double mutant GR derivative was used as substrate in the sumoylation assays (Fig. 3A, compare lanes 7 and 9). Single K297R substitution did not change the GR sumoylation pattern substantially, except for a modest reduction in the intensity of the isoform a (Fig. 3A, compare lanes 7 and 8). The efficiency and specificity of this sumoylation assay were estimated using BSA as a negative and p53 as a positive control (Fig. 3A, lanes 1 and 2, respectively). p53 sumoylation resulted in two isoforms, the mono SUMO-p53 conjugate at approximately 68 kDa and the poly-SUMO-p53 conjugate at about 98 kDa, in line with the manufacturer’s data. There are other bands corresponding to E1 and E2 SUMO enzyme conjugates that are present in samples containing negative control BSA (Active Motif, Carlsbad, CA) and no substrate (Fig. 3A, lane 1, and data not shown). Next we performed in vitro sumoylation assay using GR WT and GR-S246A as substrates prephosphorylated in vitro by JNK1. JNK1-mediated phosphorylation resulted in the increase in intensity of both GR a and b SUMO isoforms (Fig. 3B, lane 2). This increase was not seen when ATP was omitted from the kinase assays or when the GR-S246A derivative was used as substrate. The phosphorylation of the WT GR derivative by JNK1 was confirmed by blotting with antibody specific for GR phosphorylated at S246 residue (Fig. 3B, lane 7). The same approach was used to confirm that the GR-S246A mutant was not phosphorylated (Fig. 3B, lane 10). Taken together, these results indicate that JNK-dependent phosphorylation of GR at S246 facilitates sumoylation at K279 and K313.
Fig. 3.

Prephosphorylated GR Shows Increased Sumoylation
A, In vitro sumoylation assays were performed as indicated in Material and Methods, and samples were resolved on SDS-PAGE and probed with SUMO2-specific antibody as indicated by the supplier (Active Motif). BSA and p53 purified proteins served as negative and positive control, respectively, in the reaction with the WT SUMO2 (left panel) or catalytically inactive mutant SUMO2 (middle panel). Indicated GST fusion to GR AF-1 region derivatives were purified from E. coli, and the same amount of proteins was used in each assay. GR-specific bands labeled by arrows are lacking in the K297R/K313R mutant derivative (right panel). B, GST GR AF-1 was prephosphorylated by JNK1 as indicated in Fig. 1 and then sumoylated as in Fig. 3A. Membranes were incubated with SUMO2-specific antibody (left panel) to detect sumoylated proteins and then stripped and reprobed with antibody against phosphorylated S246 (right panel). *, Nonspecific band.
To determine whether these events take place in vivo, his-tagged SUMO1/SUMO2 and GR expression plasmids were transfected in COS-7 cells in the presence or absence of Dex. Sumoylated proteins were isolated by nickel affinity chromatography under denaturing conditions. GR was detected by probing the blots with a GR-specific antibody (Fig. 4). A sumoylated GR isoform at about 130 kDa was observed only in cells transfected with the SUMO2 expression plasmid (Fig. 4A, lanes 7 and 8, arrow 3). Another isoform with molecular mass of about 140 kDa was detected with higher intensity in the SUMO2 than in SUMO1-expressing cells (Fig. 4A, compare lanes 5 and 6 with lanes 7 and 8, arrow 2). The presence of hormone did not have a significant effect on GR sumoylation under these experimental conditions. The effect of JNK-mediated phosphorylation of GR on its sumoylation status was followed in the next set of experiments where, in addition to GR, SUMO1 (Fig. 4B) or SUMO2 (Fig. 4C) and JNK1 and MLK3 expression plasmids were transfected in COS-7 cells. A new GR-sumoylated isoform at about 180 kDa was observed (Fig. 4B, arrow 1) in the JNK1- and SUMO1-expressing cells. SUMO2 conjugation resulted in a marked increase of the intensity and the number of GR isoforms (compare Fig. 4B with 4C). In hormone-treated cells activation of JNK1 and SUMO2 pathways resulted in isoforms 1 and 3 peaking at 1 h of hormone treatment (Fig. 4C, compare lanes 1–5 with lanes 6–10). Consistent with the notion that JNK1-mediated phosphorylation of GR induces its sumoylation, GR sumoylation level was significantly lower in cells treated with the JNK inhibitor SP600125, as compared with cells treated with the p38 inhibitor SB203580 (Fig. 4D, compare lanes 3 and 4 with lanes 5 and 6).
Fig. 4.
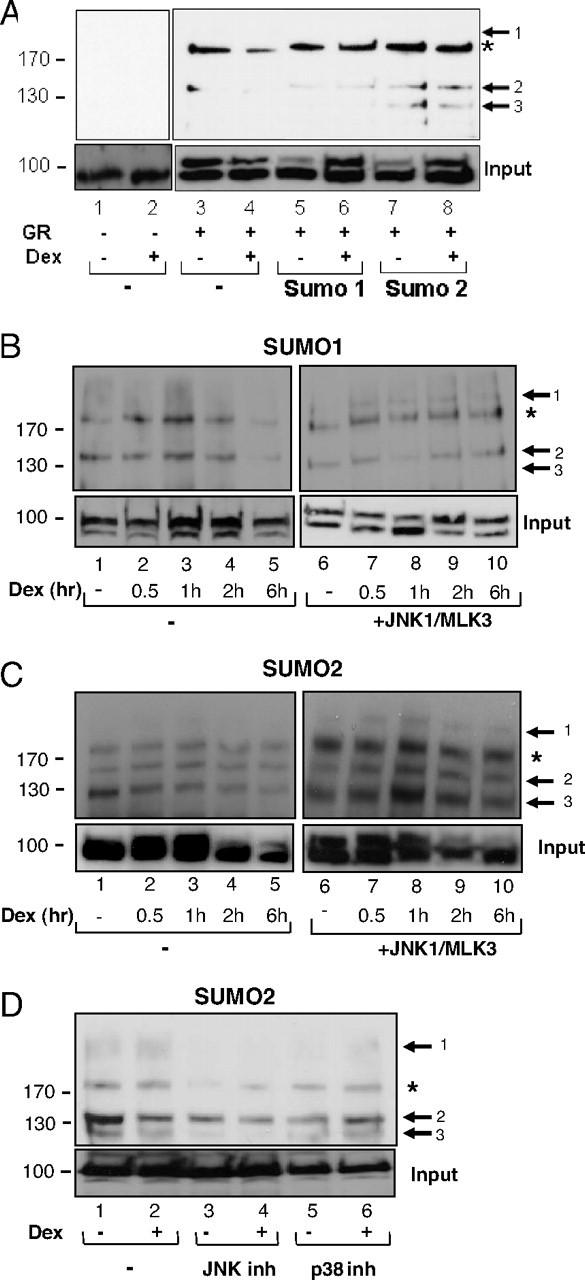
GR Sumoylation Pattern Depends on SUMO Family Members and JNK Activity
A, pcDNA3 (lanes 1 and 2), His-SUMO1 (lanes 5 and 6), and His-SUMO2 (lanes 7 and 8) were transfected in COS-7 cells, together with the pcDNA3-GR (lanes 3–8), in the presence (lanes 2, 4, 6, and 8) or absence (lanes 1, 3, 5, and 7) of 100 nm Dex for 1 h. Sumoylated proteins were isolated from cellular extracts under denaturing conditions in 6 m guanidinium HCl, with nickel affinity column and SUMO-modified GR detected by GR-specific antibody. Bottom panels show input protein levels. Lower band of the doublet is nonspecific because it was detected in untransfected cells and was not competed with specific peptide (49 ). B, His-SUMO1 plasmid was transfected in COS-7 cells together with the pcDNA3-GR in the presence (right panel) and absence of JNK1α1/MLK3 (left panel). Cells were treated with vehicle (lanes 1 and 6) or 100 nm Dex (lanes 2–5 and 7–10) for indicated times, and proteins were isolated as described above and detected by GR-specific antibody. Arrows indicate GR SUMO isoforms and asterisk indicates nonspecific band. C, The same as in panel B except that His-SUMO2 plasmid was transfected in COS-7 cells together with the pcDNA3-GR. D, COS-7 cells were transfected with His-SUMO-2 and JNK1α1 together with MLK3 and pcDNA3-GR expression plasmids. Cells were treated (lanes 2, 4, and 6) or not (lanes 1, 3, and 5) with 100 nm Dex for 1 h in the presence of 10 μm JNK inhibitor SP600125 (lanes 3 and 4) or p38 inhibitor SB203580 (lanes 5 and 6) that were added 30 min before the hormone addition. Inh, Inhibitor.
To substantiate these conclusions, we immunoprecipitated GR from COS-7 cells ectopically expressing GR and SUMO2 under high salt/detergent conditions, to minimize potential coimmunoprecipitation of other sumoylated proteins. Monoclonal antibody against SUMO2/3 was used to detect sumoylated polypeptides among the precipitated proteins (Fig. 5, top panel) or GR-specific antibody to detect GR in cell lysates (Fig. 5, bottom panel). A modest increase in the higher molecular weight forms was observed when SUMO2 was cotransfected with GR WT and a substantial decrease when GR-S246A mutant was used (Fig. 5, compare lanes 7 and 8 with lanes 3–6). When COS-7 cells were transfected with activated JNK in addition to GR and SUMO2 expression vectors, overall sumoylation of the GR WT (Fig. 5, lanes 9 and 10) increased to a greater extent than sumoylation of GR-S246A (Fig. 5, lanes 11 and 12). Despite lower resolution of individual isoforms observed by nondenaturing vs. denaturing protocol (shown in Figs. 5 and 4, respectively), these results show that JNK activation increases overall GR sumoylation and that GR-S246A mutant is sumoylated less that the WT GR. Lower exposure of the right upper blot (supplemental Fig. 3) indicated complex patterns of the higher molecular weight forms of sumoylated GR in the presence of activated JNK.
Fig. 5.

Cross Talk of JNK and SUMO Pathways
Whole-cell extracts prepared from COS-7 cells transfected with the pcDNA3 (lanes 1 and 2), WT GR (lanes 3–6 and 9–10), or S246A mutant GR (lanes 7–8 and 11–12) in the absence (lanes 1–8) or presence (lanes 9–12) of PEBG-JNK1α1 and pcDNA3HA-MLK3 expression plasmids. Cells were treated with 100 nm Dex for 1 h, and samples were immunoprecipitated under nondenaturing conditions in HSL buffer, using anti-GR-specific antibody and proteins were analyzed by SDS-PAGE. Sumoylated proteins were detected by developing Western blot membranes with SUMO2/3-specific monoclonal antibody (top panels), and input lysates were probed with GR-specific antibody (bottom panels). Ab, Antibody.
Taken together, results shown in Figs. 4 and 5 strengthened the notion that GR sumoylation and phosphorylation are two linked modifications and that the efficiency of GR sumoylation is modulated by JNK-dependent phosphorylation of this transcription factor.
Interplay of JNK and SUMO Pathways in the Regulation of GR-Mediated Transcription
The physiological significance of the interplay between phosphorylation and sumoylation of GR was investigated using luciferase reporter gene analysis (Fig. 6). For this purpose the activity of the Gal4 luciferase reporter gene was monitored in COS-7 cells transfected with Gal4 DNA-binding domain fused to the GR AF-1 domain (residues 106–318, labeled as Gal4-GR in Fig. 6A) together with (Fig. 6A, lanes 4 and 6) or without (Fig. 6A, lanes 1, 2, 3, and 5) JNK1 and MLK3 expression plasmids. JNK1 activation inhibited GR transactivation (Fig. 6A, compare lane 3 with lane 4). The inhibition of SUMO pathway by a dominant-negative (DN) form of SUMO-conjugating enzyme Ubc9 (DN-Ubc9) (30) resulted in a 4-fold increase of GR transactivation (Fig. 6A, lane 5) as well as loss of the repressive function of JNK on GR transcriptional activity (Fig. 6A, lane 6), suggesting that JNK-mediated repression of GR is dependent on the ability of GR to be sumoylated. Gam-1 and Ssp3 SUMO inhibitors (23, 30) also induced GR transcriptional activity, and Gam-1 elicited the most pronounced effect of all SUMO inhibitors (Fig. 6B, lanes 4 and 5). Inhibition of both signaling pathways, JNK1 phosphorylation by SP600125 and sumoylation by DN-Ubc9, stimulated Gal4-GR fusion protein activity, but to no higher extent than that achieved by the use of the DN-Ubc9 SUMO inhibitor alone, suggesting that their inhibitory mechanisms are linked (Fig. 6C, lanes 4 and 5). The nonphosphorylatable (GR-S246A), nonsumoylatable [GR-K297R/K313R; double mutant (DM)] and the mutant unable to be either phosphorylated or sumoylated [GR-S246A/K297R/K313R; triple mutant (TM)], all exhibited increased Gal4 luciferase reporter activity, compared with GR WT (Fig. 6D, compare lanes 2, 3, and 4 with lane 1). The protein levels for all GR derivatives were similar (Fig. 6E), suggesting that both modifications have an inhibitory effect on GR transcriptional activity. In the context of the full-length GR transcriptional activity the trend was similar as shown in tyrosine aminotransferase (TAT) luciferase reporter gene assays (Fig. 6F), with significantly smaller differences in activity of all derivatives. Although requiring further investigation, these results suggest that direct or indirect intramolecular communication (31), or other posttranslational modifications or other signals within the full-length protein, may contribute to the altered phenotype of these mutants through JNK- and SUMO-dependent modifications. Detailed analysis of the full-length GR mutant derivatives indicated that they display increased transcriptional activity compared with the WT receptor, across a range of concentrations and duration of hormone treatments, whereas their steady state protein levels were not significantly affected (Fig. 6, G and H, and data not shown). Reporter gene analysis therefore demonstrated that JNK-dependent phosphorylation of GR at S246 and GR sumoylation are linked and required for efficient repression of GR transcriptional activity.
Fig. 6.
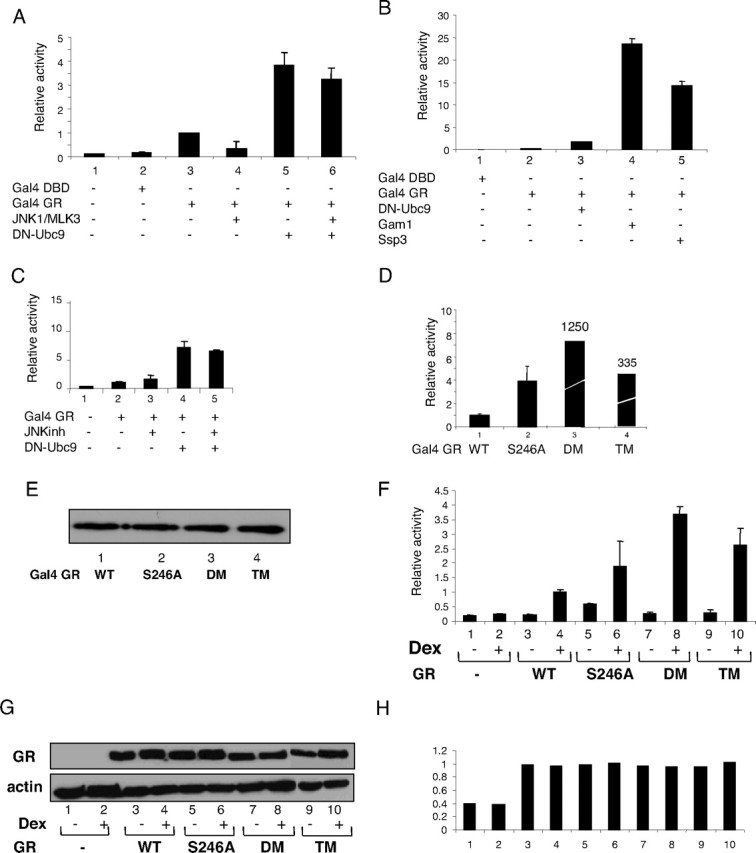
Effect of Phosphorylation/Sumoylation on GR-Mediated Transcription
A, COS-7 cells were transfected with pcDNA3 (lane 1), 100 ng of Gal4-DBD (lane 2), and Gal4-DBD-GR/AF-1 fusion constructs (Gal4 GR, lanes 3–6) together with 100 ng of Gal4-luciferase and β-galactosidase plasmids. Effects of JNK activation or SUMO inhibition on GR were followed by coexpression of 100 ng of PEBG-JNK1α1 and pcDNA3HA-MLK3 (lanes 4 and 6) or 50 ng of DN-Ubc9 (lanes 5 and 6). Luciferase assays were performed as indicated in Materials and Methods and represent the average of the relative values of three or more separate experiments. B, Fifty nanograms of DN-Ubc9 (lane 3), Gam-1 (lane 4), and Ssp3 (lane 5) were transfected as in panel A. C, Cells were treated with 10 μm JNK inhibitor SP600125 (lanes 3 and 5) for 16 h and transfected with 50 ng of DN-Ubc9 (lanes 4 and 5). D, Transcriptional activity of Gal4-WT (lane 1), Gal4-S246A (lane 2), Gal4-DM (K297R/K313R) (lane 3), and Gal4-TM (S246A/K297R/K313R) (lane 4) constructs was analyzed as described above. E, Protein levels of Gal4-WT (lane 1), Gal4-S246A (lane 2), Gal4-DM (lane 3), and Gal4-TM (lane 4) were determined by Western blotting of transfected cell extracts with GAL4-specific antibodies. F, Fifty nanograms of pcDNA3 (lanes 1 and 2), pcDNA3-GR-WT (lanes 3 and 4), pcDNA3-GR-S246A, (lanes 5 and 6), pcDNA3-GR-DM (lanes 7 and 8), and pcDNA3-GR-TM (lanes 9 and 10) were transfected together with 100 ng of TAT3-luciferase and β-galactosidase plasmids. Cells were treated with vehicle (lanes 1, 3, 5, 7, and 9) or 100 nm Dex (lanes 2, 4, 6, 8, and 10) for 6 h. G, pcDNA3HA (lanes 1 and 2) pcDNA3-HAGR-WT (lanes 3 and 4), pcDNA3-HAGR-S246A (lanes 5 and 6), pcDNA3-HAGR-DM (lanes 7 and 8), and pcDNA3-HAGR-TM (lanes 9 and 10) were transfected and cells were treated with vehicle (lanes 1, 3, 5, 7, and 9) or 100 nm Dex (lanes 2, 4, 6, 8, and 10) for 6 h. Cell extracts were analyzed using SDS-PAGE and Western blots developed with HA (top panel), or actin specific antibodies (bottom panel). H, Results obtained in panel G were quantified using Image J analysis, and ratio of total GR vs. actin levels is shown. DBD, DNA-binding domain.
To assess the effect of phosphorylation and sumoylation of endogenous GR on expression of endogenous GR target genes, we have transfected human lung A549 cells with JNK1/MLK3 expression vectors and analyzed by quantitative RT-PCR (qRT-PCR) the expression of three genes previously suggested to be the GR targets (Fig. 7) (32). Kinetic analysis revealed that JNK activation inhibits GR-mediated induction of IGFBP, GILZ, and human inhibitor of apoptosis (hIAP) genes in a transient manner and that this effect is most pronounced within first 2 h of hormone treatment whereas longer exposure to hormone did not change expression levels of these genes (Fig. 7A). Similar inhibition and kinetic patterns of above mentioned gene expression were detected when SUMO2 was overexpressed in these cells (Fig. 7B). In the next series of experiments cells were treated for 1 h with hormone and transfected with JNK1/MLK3 and SUMO2 expression plasmids (Fig. 7C). Activation of JNK or SUMO pathways individually inhibited GR-mediated transcription of all genes tested (Fig. 7C, compare lanes 3–6 with lanes 1 and 2). Activation of both pathways simultaneously inhibited GR-mediated expression of IGFBP and glucocorticoid-induced leucine zipper (GILZ) to a similar extent as individual treatments (Fig. 7C, compare lanes 7 and 8 with lanes 3–6). The repressive effects of JNK and SUMO activation on GR-directed transcription were not additive, suggesting that they function through a common mechanism. Individual activation of JNK or SUMO pathways resulted in the inhibition of GR-mediated induction of the hIAP gene. Surprisingly, activation of both pathways had no effect on transcription of hIAP. Further to this, the effect of GR phosphorylation and sumoylation on transcription of its targets seems to be gene specific.
Fig. 7.

Regulation of Endogenous GR Target Genes by Phosphorylation/Sumoylation
A549 cells were transfected with pcDNA3 (indicated by the solid line in panels A and B and lanes 1 and 2 in panel C), PEBG-JNK1α1 (1 μg), and pcDNA3HA-MLK3 (0.5 μg), (indicated by the dashed line in panel A and lanes 3, 4, 7, and 8 in panel C) and SUMO2 (1 μg, indicated by the dotted line in panel B, and lanes 5–8 in panel C). Cells were treated with 100 nm Dex for the times indicated (1 h for 7C), RNA was isolated, and reverse transcription was performed. Obtained cDNA was used for qRT-PCR reactions as described in Materials and Methods. Graphs represent the average of three or more separate experiments.
DISCUSSION
GR phosphorylation occurs in its N-terminal transcriptional-activating domain and has been hypothesized to both inhibit and stimulate its activity (4). To shed light on the molecular mechanisms involved in GR phosphorylation and dissect the pathways controlling the transcriptional activity of the posttranslationally modified GR, we studied the JNK1-dependent phosphorylation of GR at S246. We present evidence that JNK1 preferentially phosphorylates GR at S246 in vivo (Fig. 1), thus complementing and extending results reported previously (4, 31).
The JNK family consists of several proteins encoded by three genes. Jnk1 and Jnk2 genes are ubiquitously expressed, and the Jnk3 gene is selectively expressed mostly in the brain, testis, and heart (13). There are numerous splice variants of these kinases but whether there is differential target specificity and kinase activity of these isoforms and family members is a matter of debate. Here we observe that there is no differential kinase activity of these isoforms and family members in phosphorylating GR residue S246 (Fig. 1D and supplemental Fig. 1), suggesting that this amino acid is the target of all members of this family.
Activation of JNK by UV radiation substantially increases GR S246 phosphorylation of transfected and endogenous GR, providing strong evidence for the physiological role of this phosphorylation in the GR-mediated stress response (Fig. 2A). Phosphorylation of GR at S246 in the absence of hormone is maximal within the first hour of JNK activation (Fig. 2C, lanes 1 and 2 and data not shown). Increase in S246 phosphorylation is more evident in the absence than in the presence of hormone, possibly due to differential availability of activated GR to JNK due to subcellular compartmentalization, feedback loops between the two pathways, or negative influence of modifications occurring on other sites (7, 17, 18). When normalized to the total level of GR, this phosphorylation increases with UV and Dex treatment whereas it does not change in cells treated with antagonist, which could potentially be due to the difference in down-regulation of total GR levels under these conditions (Fig. 2C).
GR is also subject to ubiquitination and sumoylation (19, 20, 21, 22). SUMO conjugation has been mapped to synergy control motifs of GR that are localized in close proximity to S246 (19). Our results indicate that GR is sumoylated by SUMO1 and SUMO2 in a hormone-independent manner (Figs. 4 and 5) in accord with reports by Holmstrom et al. (19) and Le Drean et al. (20), and in disagreement with Tian et al. (21). It is possible that in different cell types or under different experimental conditions SUMO modification can display varying degrees of ligand dependency but, nevertheless, provides an important level of regulation of GR activity. SUMO-1- and SUMO-2-expressing cells exhibit different patterns of GR-sumoylated isoforms, suggesting that these two family members take part in different regulatory mechanisms that fine tune GR activity and possibly link GR to diverse extracellular signals. Activation of JNK increases the intensity of GR sumoylation in vitro and in vivo, providing evidence for the interplay between GR phosphorylation and sumoylation (Figs. 3 and 4). In addition, JNK is more efficient than p38 kinase in stimulating GR SUMO modification, suggesting preferential involvement of the JNK pathway in GR sumoylation (Fig. 4D). Sumoylation of the GR-S246A mutant was significantly lower than that of the WT protein (Figs. 3 and 5), indicating that phosphorylation and sumoylation pathways converge to regulate GR.
Identification of phosphorylation-dependent sumoylation in proteins carrying the ψKxExxSP motif, as well as studies reporting the existence of this interplay in a large number of proteins (33, 34), argues that this cross talk is an important part of control mechanisms that adjust the activity of proteins to extracellular signals. One characteristic example is the transcription factor nuclear factor-κB, which translocates into the nucleus after its inhibitor IκB is degraded by a cascade of phosphorylation and ubiquitination events. However, if the ubiquitinated lysine in IκB, which targets the protein for degradation, is instead sumoylated, IκB is stabilized and prevents nuclear factor-κB nuclear translocation (30). Other steroid receptors modified by SUMO addition include the androgen receptor, which is sumoylated on two sites in its N-terminal domain, and each one of these sites exhibits a differential effect on androgen receptor activity (27, 35). Sumoylation of the progesterone receptor in its N terminus and of the estrogen receptor in the hinge region results in both inhibitory and stimulatory effects on transcriptional activity of these proteins (26, 36, 37). Positive and negative effects of sumoylation on GR transcriptional activity perhaps depend on experimental settings, the cell type, or target gene used for analysis. Our results indicate that JNK activation reduces whereas inhibition of SUMO up-regulates GR AF-1 activity and prevents repression by JNK, providing additional evidence for the cross talk of these two pathways (Fig. 6A). S246A mutant phenotype showed a weak or nonsignificant increase in transcriptional activity, whereas GR DM and TM exhibited increased transcriptional activity compared with the WT protein in the tyrosine aminotransferase (TAT)3 reporter gene assay (Fig. 6, D and F).
Inhibitory effects of JNK and SUMO activation were observed in the study of endogenous GR transcriptional function. We have chosen endogenous GR primary target genes and observed similar effects of JNK and SUMO activation on expression of these genes (Fig. 7). Activation of both pathways at the same time did not result in additive effects on IGFBP gene expression, suggesting that phosphorylation and sumoylation act together (Fig. 7C). Effects of JNK and SUMO activation individually or simultaneously on GILZ gene expression were of a similar nature as described for IGFBP, but were more subtle. However, hIAP effects of JNK and SUMO activation suggested different mechanism, because individually these pathways had slight inhibitory effect, and simultaneous activation of both pathways restored WT level of gene expression, suggesting that these modifications counteract each other at this particular gene. This could be caused by different cofactors being recruited to different promoters, further posttranslational modifications, or differences in promoter architecture affecting GR binding, resulting in the ability of posttranslational modifications to affect transcription in a gene-specific manner. Future experiments will have to address and reconcile these different possibilities.
Although these data indicate that phosphorylation aids sumoylation, it is likely that other pathways can influence these GR modifications (Fig. 8). The fact that we have detected residual sumoylation in S246A mutant GR supports this possibility (Fig. 5). Potentially, JNK-dependent GR phosphorylation alters GR structure in a way that makes it more susceptible to SUMO modification, possibly through increased interaction with components of the SUMO pathway such as Ubc9 or protein inhibitor of activated signal (PIAS) transducer and activator of transcription proteins. This could lead to efficient repression of its transcriptional activity, through recruitment of histone deacetylase (HDAC), as has been shown for the transcription factor Elk (38). Alternatively, unknown GR modulatory factor(s) could selectively interact with GR modified by both signaling pathways, thereby altering its subcellular location, stability, or transcriptional activity. In fact, results by Itoh (7) and our own unpublished observations suggest that GR nuclear export is stimulated, whereas nuclear import is impaired by JNK activation. Although these results suggest that JNK exerts a negative influence on GR nuclear translocation, comprehensive assessment of the physiological significance of these events awaits further experimentation.
Fig. 8.

Interplay of Modifications on GR
Schematic representation of interdependency of phosphorylation and sumoylation. CDK-dependent phosphorylation of S224 stimulates phosphorylation of S232 and has negative influence on JNK-dependent phosphorylation of S246 and vice versa. JNK activation facilitates GR sumoylation and, at the same time, GR potentially responds to other signals from the environment that may involve intra- and intermolecular interactions with ligand binding domain. P, Phosphorylation; S, sumoylation.
Phosphorylation of GR has also been linked to the control of its protein stability (Ref. 6 and our unpublished observations). It is therefore possible that JNK activation affects nuclear localization, sumoylation, and degradation of GR, and the final level of GR activity will have different outcomes depending on type and strength of the signal and the GR target gene. This mechanism would provide the cell with a rapid, reversible response to stress signals without the need for synthesis of new GR molecules. This model also connects proteasome with kinases and SUMO pathways in regulation of GR and other transcription factor activities (39, 40).
Results from our and other laboratories suggest the existence of a posttranslational modification code of the receptor (41, 42) (Fig. 8), meaning that one modification influences another, resulting in increased plasticity and dynamic regulation of the receptor. Together, these results highlight the importance of studying individual isoforms of the receptor, because understanding their varying activities could unravel the complex regulation of this transcription factor, involved in such a diverse range of processes. Through further analysis of subsets of genes targeted by particular GR-modified isoforms, potential therapeutic targets could be discovered, undesirable side effects of glucocorticoid treatment could be reduced, and more effective drugs to combat glucocorticoid resistance could be developed.
MATERIALS AND METHODS
Cell Culture and Transfections
COS-7 monkey kidney fibroblast, A549 human lung adenocarcinoma, and H4 rat hepatoma cell lines were cultured in DMEM (Life Technologies, Inc., Gaithersburg, MD) supplemented with 10% fetal calf serum and 10 U/ml each of penicillin and streptomycin (Life Technologies, Inc.). Mammalian expression vectors were transfected using calcium phosphate and/or Polyfect (QIAGEN, Chatsworth, CA) (43). cells were transferred 16 h after transfection into DMEM supplemented with 10% charcoal-stripped fetal bovine serum (Hyclone Laboratories, Inc., Logan, UT) and treated with 100 nm Dex (Sigma Chemical Co.) for indicated time, and/or 60 J/m2 of UV radiation.
Plasmids
The rat GR AF-1 domain (residues 106–318) carrying individual phosphorylation site mutations was isolated from PG-N795 (4) and cloned into BamHI and EcoRI restriction enzyme sites of pGEX2T or pcDNA3-Gal4-DBD (1–93) vectors. K297R/K313R and S246A/K297R/K313R mutations were generated using QuikChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA). pcDNA3-GR WT and S246A mutant were subcloned as BamHI fragments from PGN795. pcDNA3-HAGR was generated by PCR amplification of the full-length WT and mutant GR cDNA and insertion of the PCR product into the BamH1 and Xho1 site of PHA1 plasmid (43). PEBG-JNK1α1, JNK2α2, JNK3α1 and pcDNA3-HAMLK3, and Gam1 expression vectors were previously described (44, 45). His-SUMO-1 and -2, DN-Ubc9, and Ssp3 mammalian expression vectors were kindly provided by Ron Hay (29, 30). TAT3-luc reporter plasmid was a gift from K.R. Yamamoto (19, 46).
Kinase Assays
GR fusion proteins were purified using GST affinity chromatography as previously described, and their purity was analyzed by Coomassie staining (43). In vitro kinase assays were performed as previously described (47) using GR and c-Jun as substrates and purified JNK1 kinase (Upstate Biotechnology, Inc., Lake Placid, NY) as a source of enzyme. Reactions were carried out in 25 mm HEPES (pH 7.4), 25 mm β-glycerophosphate, 25 mm MgCl2, 0.1 mm NaVO4, 0.5 mm dithiothreitol (DTT), 25 μm ATP, and 10 μCi of [γ-32P] ATP. Samples were incubated for 30 min at 30 C, and reaction was terminated by addition of SDS sample buffer. Labeled GR was visualized by autoradiography.
For analysis of GR phosphorylation by JNK family members, COS-7 cells were transfected with PEBG-JNK1α1, JNK2α2, JNK3α1, and pcDNA3-HAMLK3 expression vectors, and kinases were precipitated using GST affinity resin. Cells were lysed in TLB buffer (20 mm Tris, pH 7.4; 137 mm NaCl; 2 mm EDTA; 1% Triton X-100; 10% glycerol, 25 mm β-glycerophosphate, 2 mm sodium pyrophosphate; 1 mm sodium vanadate; 1 mm phenylmethylsulfonylfluoride; 1 μg/ml aprotinin, leupeptin, and pepstatin A; and 0.5 mm DTT). Lysates were incubated with GST beads for 2 h at 4 C, followed by three washes with TLB buffer and once with the kinase buffer. These GST beads were used as source of enzymes and purified GR and c-Jun as substrate in kinase reactions performed as described above.
Immunoprecipitation and Immunoblotting
Immunoprecipitations and immunoblotting procedures were as described previously with following modifications (48). Cells were lysed in HSL buffer (45 mm HEPES, pH 7.5; 400 mm NaCl; 1 mm EDTA; 10% glycerol; 0.5% Nonidet P-40; 1 mm DTT; 1 mm phenylmethylsulfonylfluoride; protease inhibitor cocktail as above; 20 mm β-glycerophosphate; 5 mm sodium pyrophosphate; 2 mm sodium orthovanadate; and 10 mm N-ethylmaleimide). Equal amounts of protein were loaded and resolved by SDS-PAGE, transferred to Immobilon-P membrane (Millipore Corp., Bedford, MA) and probed with indicated antibodies. Blots were developed with the enhanced chemiluminescence substrate according to manufacturer’s instructions (Pierce Chemical Co., Rockford, IL), and intensity of the bands was quantified using Image J software. For immunoprecipitation, antibody was added to cell extracts together with the protein-A sepharose (Sigma) and rotated at 4 C overnight. The following antisera were used: S246-P antibody was raised against phosphorylated peptide LLIDENLLpSPLAGEDDP (amino acid residues 238–254 of the rat GR), purified by epitope affinity chromatography by Sigma and its specificity confirmed by ELISA as specified by the manufacturer; M-20 anti-GR (49) and actin antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); anti-GST goat antibody was a gift from Dr. Whitmarsh (The University of Manchester).
Sumoylation Assays
In vitro sumoylation assays were performed using SUMO link according to manufacturer’s instructions (Active Motif). In vivo sumoylation assays under denaturing conditions were performed as described previously (29). Briefly, COS-7 cells were transfected with His-tagged SUMO expression vectors, pcDNA3 GR, PEBG-JNK1α1, and pcDNA3-HAMLK3 expression vectors as indicated in the legend of Fig. 4 and harvested in lysis buffer containing 6 m guanidine-HCl. His-tagged sumoylated proteins were bound to Ni-agarose beads, extensively washed with buffers containing 8 m urea, followed by washes in PBS, eluted, resolved by SDS-PAGE, and transferred onto nitrocellulose membranes. Western blots were developed with GR-specific antibody.
Luciferase Reporter Gene Assays
Luciferase assays were as described previously (43). Briefly, COS-7 cells were plated in 60-mm dishes and transfected with indicated plasmids using Polyfect reagent (QIAGEN). Cells were washed twice with PBS and harvested in the lysis solution. Luciferase and β-galactosidase activity was measured according to manufacturer’s instruction [Tropix Dual-Light reporter gene assay system purchased from Applied Biosystems (Foster City, CA)]. An orion microplate luminometer from Berthold detection systems [Berthold Technologies (U.K.) Ltd., Harpenden, UK] was used for measurements of luciferase and β-galactosidase activity.
qRT-PCR
A549 cells were plated into 35-mm well dishes. Cells were treated with 100 nm Dex for the times indicated. RNA was extracted using the RNeasy plus mini kit (QIAGEN). RNA concentrations were measured, and 1 μg of RNA was used for reverse transcription reaction according to the two-step protocol (ABgene Ltd., Epsom, UK) using an oligo-deoxythymidine primer (ABgene). cDNA was diluted 4-fold and used for qPCR analysis performed with SYBR Green JumpStart Taq ReadyMix (Sigma). The specific primers used for amplification were as follows. Rpl19 forward (F): atgtatcacagcctgtacctg; reverse (R), ttcttggtctcttcctccttg; hIAP (F): gacaggagttcatccgtcaag; (R), ttccacggcagcattaatc; IGFBP (F): ccaaggcacaggagacatcag; (R), agggtagacgcaccagcagagt; GILZ (F): ggacttcacgtttcagtggaca; (R), aatgcggccacggatg. Analysis was performed using Opticon monitor 3 software.
Acknowledgments
We thank K.R. Yamamoto for plasmids and reagents; D. Ray and A. White for GR plasmids and antibodies; A. Whitmarsh, C. Tournier, S-H Yang, and A. Sharrocks for c-Jun, JNK, and MLK3 constructs, antibodies, and helpful criticisms. We thank R. Hay for SUMO constructs and sumoylation protocols; S. Chiocca for the Gam-1 plasmid; and A. Dickson for H4 cells.
NURSA Molecule Pages:
Ligands: Dexamethasone | Progesterone | RU486;
Nuclear Receptors: GR.
Footnotes
This project was funded by grants from the Wellcome Trust and the Royal Society; M.K.D. and N.K. are supported by the Wellcome Trust Research Fellowship (069024); L.D. is supported by The Biotechnology and Biological Sciences Research Council; A.G. is supported by Leonardo Da Vinci program. J.T.L. is supported by Medical Research Council, and C.D. is supported by the School of Pharmacy.
Disclosure Statement: The authors have nothing to disclose.
First Published Online March 12, 2008
Abbreviations: AF-1, Activation function 1; CDK, cyclin-dependent protein kinase; DDT, dithiothreitol; Dex, dexamethasone; DM, double mutant; DN, dominant negative; GILZ, glucocorticoid-induced leucine zipper; GR, glucocorticoid receptor; GRE, glucocorticoid-responsive element; GST, glutathione-S-transferase; hIAP, human inhibitor of apoptosis; IGFBP, IGF-binding protein; JNK, c-Jun N-terminal kinase; MLK3, mixed lineage kinase 3; qRT-PCR, quantitative RT-PCR; S246, serine 246; S246-P, phosphorylated at S246; SUMO, small ubiquitin-like modifier; TAT, tyrosine aminotransferase; TM, triple mutant; WT, wild type.
References
- 1.Yamamoto KR 1985. Steroid receptor regulated transcription of specific genes and gene networks. Annu Rev Genet 19:209–252 [DOI] [PubMed] [Google Scholar]
- 2.Munck A 2005. Glucocorticoid receptors and physiology: a personal history. Steroids 70:335–344 [DOI] [PubMed] [Google Scholar]
- 3.McKenna NJ, Lanz RB, O'Malley BW 1999. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev 20:321–344 [DOI] [PubMed] [Google Scholar]
- 4.Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ 1997. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol Cell Biol 17:3947–3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bodwell JE, Orti E, Coull JM, Pappin DJC, Smith LI, Swift F 1991. Identification of phosphorylation sites in the mouse glucocorticoid receptor. J Biol Chem 266:7549–7555 [PubMed] [Google Scholar]
- 6.Webster JC, Jewell CM, Bodwell JE, Munck A, Sar M, Cidlowski JA 1997. Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J Biol Chem 272:9287–9293 [DOI] [PubMed] [Google Scholar]
- 7.Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K 2002. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol 16:2382–2392 [DOI] [PubMed] [Google Scholar]
- 8.Rogatsky I, Logan SK, Garabedian MJ 1998. Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc Natl Acad Sci USA 95:2050–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garabedian MJ, Rogatsky I, Hittelman A, Knoblauch R, Trowbridge JM, Krstic MD 1998. Regulation of glucocorticoid and estrogen receptor activity by phosphorylation. In: Freedman L, ed. Molecular biology of steroid and nuclear hormone receptors. Boston: Birkauser; 238–260
- 10.Ismaili N, Garabedian MJ 2004. Modulation of glucocorticoid receptor function via phosphorylation. Ann NY Acad Sci 1024:86–101 [DOI] [PubMed] [Google Scholar]
- 11.Miller AL, Webb MS, Copik AJ, Wang Y, Johnson BH, Kumar R, Thompson EB 2005. p38 mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol 19:1569–1583 [DOI] [PubMed] [Google Scholar]
- 12.Kino T, Ichijo T, Amin ND, Kesavapany S, Wang Y, Kim N, Rao S, Player A, Zheng Y-Li, Garabedian MJ, Kawasaki E, Pant HC, Chrousos GP 2007. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol 21:1552–1568 [DOI] [PubMed] [Google Scholar]
- 13.Davis RJ 2000. Signal transduction by the JNK group of MAP kinases. Cell 103:239–252 [DOI] [PubMed] [Google Scholar]
- 14.Karin M, Gallagher E 2005. From JNK to pay dirt: Jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 57:283–295 [DOI] [PubMed] [Google Scholar]
- 15.Yang S-H, Sharrocks AD, Whitmarsh AJ 2003. Transcriptional regulation by the MAP kinase signaling cascades. Gene 320:3–21 [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Wu H, Lakdawala VS, Hu F, Hanson ND, Miller AH 2005. Inhibition of Jun N-terminal kinase (JNK) enhances glucocorticoid receptor-mediated function in mouse hippocampal HT22 cells. Neuropsychopharmacology 30:242–249 [DOI] [PubMed] [Google Scholar]
- 17.Bruna A, Nicolas M, Munoz A, Kyriakis JM, Caelles C 2003. Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. EMBO J 22:6035–6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark AR, Lasa M 2003. Crosstalk between glucocorticoids and mitogen-activated protein kinase signalling pathways. Curr Opinion Pharm 3:404–411 [DOI] [PubMed] [Google Scholar]
- 19.Holmstrom S, Antwerp ME, Iniguez-Lluhi JA 2003. Direct and distinguishable inhibitory roles for SUMO isoforms in the control of transcriptional synergy. Proc Natl Acad Sci USA 100:15758–15763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Drean Y, Mincheneau N, Le Goff P, Michel D 2002. Potentiation of glucocorticoid receptor transcriptional activity by sumoylation. Endocrinology 143:3482–3489 [DOI] [PubMed] [Google Scholar]
- 21.Tian S, Poukka H, Palvimo JJ, Janne OA 2002. Small ubiquitin related modifier-1 (SUMO-1) modification of the glucocorticoid receptor. Biochem J 367:907–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallace AD, Cidlowski JA 2001. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem 276:42714–42721 [DOI] [PubMed] [Google Scholar]
- 23.Boggio R, Colombo R, Hay RT, Draetta GF, Chiocca S 2004. A mechanism for inhibiting the SUMO pathway. Mol Cell 16:549–561 [DOI] [PubMed] [Google Scholar]
- 24.Gill G 2004. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev 18:2046–2059 [DOI] [PubMed] [Google Scholar]
- 25.Hay R 2005. SUMO: a history of modification. Mol Cell 18:1–12 [DOI] [PubMed] [Google Scholar]
- 26.Chauchereau A, Amazit L, Quesne M, Guiochon-Mantel A, Milgrom E 2004. Sumoylation of the progesterone receptor and of the steroid receptor coactivator SRC-1. J Biol Chem 278:12335–12343 [DOI] [PubMed] [Google Scholar]
- 27.Poukka H, Karvonen U, Janne OA, Palvimo JJ 2000. Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (SUMO-1). Proc Natl Acad Sci USA 97:14145–14150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang S-H, Jaffray E, Hay RT, Sharrocks AD 2003. Dynamic interplay of the SUMO and ERK pathways in regulating ELK-1 transcriptional activity. Mol Cell 12:63–74 [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez MS, Desterro JMP, Lain S, Midgley CA, Lane DP, Hay RT 1999. SUMO-1 modification activates the transcriptional response of p53. EMBO J 18:6455–6461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Desterro JM, Rodriguez MS, Hay RT 1998. SUMO-1 modification of IκBα inhibits NF-κB activation. Mol Cell 2:233–239 [DOI] [PubMed] [Google Scholar]
- 31.Wang Z, Chen W, Kono E, Dang T, Garabedian MJ 2007. Modulation of glucocorticoid receptor phosphorylation and transcriptional activity by a C-terminal-associated protein phosphatase. Mol Endocrinol 21:625–634 [DOI] [PubMed] [Google Scholar]
- 32.Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Ha CM, Darimont BD, Garabedian MJ, Yamamoto KR 2003. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci USA 100:13845–13850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hietakangas V, Anckar J, Blomster HA, Fujimoto M, Palvimo JJ, Nakai A, Sistonen L 2006. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc Natl Acad Sci USA 103:45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang X-J, Gregoire S 2006. A recurrent phosphor-sumoyl switch in transcriptional repression and beyond. Mol Cell 23:779–786 [DOI] [PubMed] [Google Scholar]
- 35.Callewaert L, Verrijdt G, Haelens A, Claessens F 2004. Differential effect of small ubiquitin-like modifier (SUMO)-ylation of the androgen receptor in the control of cooperativity on selective versus canonical response elements. Mol Endocrinol 18:1438–1449 [DOI] [PubMed] [Google Scholar]
- 36.Sentis S, Le Romancer M, Bianchin C, Rostan MC, Corbo L 2005. Sumoylation of the estrogen receptor α hinge region regulates its transcriptional activity. Mol Endocrinol 19:2671–2684 [DOI] [PubMed] [Google Scholar]
- 37.Abdel-Hafiz H, Takimoto GS, Tung L, Horwitz KB 2002. The inhibitory function in human progesterone receptor N-termini binds SUMO-1 protein to regulate autoinhibition and transrepression. J Biol Chem 277:33950–33956 [DOI] [PubMed] [Google Scholar]
- 38.Yang S-H, Sharrocks AD 2004. SUMO promotes HDAC-mediated transcriptional repression. Mol Cell 13:611–617 [DOI] [PubMed] [Google Scholar]
- 39.Muratani M, Tansey WP 2003. How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol 4:192–201 [DOI] [PubMed] [Google Scholar]
- 40.Hay RT, Shen LN, Tatham MH, Dong C, Jaffray E, Golebiowski F, Geoffroy M-C, Hattersley N, Naismith JH, Role of SUMO modification in the control of gene expression. Biochemical Society Focused Meeting, Regulation of protein function by SUMO modification, Manchester Conference Centre, Manchester, UK, June 25–27, 2007
- 41.Wang Z, Frederick J, Garabedian MJ 2002. Deciphering the phosphorylation code of the glucocorticoid receptor in vivo. J Biol Chem 277:26573–26580 [DOI] [PubMed] [Google Scholar]
- 42.Rochette-Egly C 2005. Dynamic combinatorial networks in nuclear receptor-mediated transcription. J Biol Chem 280:32565–32568 [DOI] [PubMed] [Google Scholar]
- 43.Demonacos C, Krstic-Demonacos M, La Thangue NB 2001. A TPR motif cofactor contributes to p300 activity in the p53 response. Mol Cell 8:71–84 [DOI] [PubMed] [Google Scholar]
- 44.Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ 2000. Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J Neurosci 20:7602–7613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ 1998. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science 281:1671–1674 [DOI] [PubMed] [Google Scholar]
- 46.Rigaud G, Roux J, Picted R, Grange T 1991. In vivo footprinting of rat TAT gene: dynamic interplay between the glucocorticoid receptor and a liver-specific factor. Cell 67:977–986 [DOI] [PubMed] [Google Scholar]
- 47.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ 2000. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288:870–874 [DOI] [PubMed] [Google Scholar]
- 48.Demonacos C, Krstic-Demonacos M, Smith L, Xu D, O'Connor D, Jansson M, La Thangue NB 2004. A new effector pathway links ATM kinase with the DNA damage response. Nat Cell Biol 6:968–976 [DOI] [PubMed] [Google Scholar]
- 49.Waters CE, Stevens A, White A, Ray DW 2004. Analysis of co-factor function in a glucocorticoid-resistant small cell carcinoma cell line. J Endocrinol 183:375–383 [DOI] [PubMed] [Google Scholar]