

Abstract
AMP-activated protein kinase (AMPK) is a key regulator of glucose and fatty acid homeostasis. In muscle cells, AMPK stimulates mitochondrial fatty acid oxidation and ATP production. The thyroid hormone T3 increases cellular oxygen consumption and is considered to be a major regulator of mitochondrial activities. In this study, we examined the possible involvement of AMPK in the stimulatory action of T3 on mitochondria. Treatment of C2C12 myoblasts with T3 rapidly led to phosphorylation of AMPK. Acetyl-coenzyme A carboxylase, a direct target of AMPK, was also phosphorylated after T3 treatment. Similar results were obtained with 3T3-L1, FRTL-5, and HeLa cells. Stable expression of T3 receptor (TR)-α or TRβ in Neuro2a cells enhanced this effect of T3, indicating the involvement of TRs. Because HeLa cells express only Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKKβ), one of two known AMPK kinases, it was suggested that the effect of T3 is mediated by CaMKKβ. Indeed, experiments using a CaMKK inhibitor, STO-609, and an isoform-specific small interfering RNA demonstrated the CaMKKβ-dependent phosphorylation of AMPK. Furthermore, T3 was found to rapidly induce intracellular Ca2+ mobilization in HeLa cells, and a Ca2+ chelator, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), suppressed T3- as well as ionomycin-dependent phosphorylation of AMPK. In addition, T3-dependent oxidation of palmitic acids was attenuated by BAPTA, STO-609, and the small interfering RNA for CaMKKβ, indicating that T3-induced activation of AMPK leads to increased fatty acid oxidation. These results demonstrate that T3 nontranscriptionally activates AMPK via intracellular Ca2+ mobilization and CaMKKβ activation, thereby stimulating mitochondrial fatty acid oxidation.
AMP-ACTIVATED protein kinase (AMPK) is a serine/threonine kinase that acts as a sensor of cellular energy status; that is, AMPK is activated by a rise in intracellular AMP/ATP ratio (1, 2). Once activated, AMPK increases energy supply by switching on ATP-generating pathways and decreases energy demand by switching off ATP-consuming pathways. Not only cellular ATP depletion but also various cellular and metabolic stresses such as glucose deprivation, heat shock, hypoxia, ischemia, and muscle contraction induce AMPK activation.
AMPK is a heterotrimeric complex composed of a catalytic α-subunit and regulatory β- and γ-subunits and is allosterically activated by the phosphorylation of Thr-172 on the α-subunit, which is catalyzed by upstream kinases such as LKB1. Binding of AMP to the γ-subunit makes AMPK a better substrate for LKB1 (3). Recently, Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKKβ) was identified as another upstream kinase (4, 5, 6). Upon an elevation of intracellular Ca2+, CaMKKβ is activated and directly phosphorylates the α-subunit of AMPK at Thr-172. This action does not require intracellular AMP elevation.
The role played by AMPK in the regulation of fatty acid oxidation has been intensively studied in muscle cells (2). Activated AMPK directly phosphorylates Ser-79 in acetyl-coenzyme A (CoA) carboxylase (ACC) and down-regulates its enzymatic activity, resulting in a decrease in intracellular malonyl-CoA levels. Because malonyl-CoA inhibits fatty acid uptake into mitochondria by preventing carnitine:palmitoyl-CoA acyl transferase present on the mitochondrial membrane, its decrease allows fatty acid uptake, leading to increased fatty acid oxidation and ATP production.
The major thyroid hormone (TH) produced by the thyroid gland is l-T4 (T4), which is deiodinated to T3 in peripheral tissues. TH impacts on a number of physiological events, including growth, development, differentiation, thermogenesis, and metabolism. T3 receptor (TR) belongs to the steroid/thyroid hormone receptor superfamily and functions as a ligand-dependent transcription factor. Two separate genes, THRA and THRB, encode TRα and TRβ, respectively. Multiple isoforms are generated from each gene by alternative splicing and promoter usage. Some distinct isoforms of TRα are localized to mitochondria (7).
TH stimulates cellular oxygen consumption and is considered to be a major regulator of mitochondrial activities (8, 9). Although some controversy remains, TH increases mitochondrial oxidative phosphorylation, membrane potential (Δψ), and uncoupling proton transport that generates heat. TH concomitantly enhances ATP production capacity as a result of increased oxidative phosphorylation (10). Although the molecular bases underlying these TH actions are still not fully understood, TH stimulates the expression of components of oxidative phosphorylation (11, 12, 13), proteins involved in uncoupling proton transport (14), and factors regulating mitochondrial activities (15). These proteins are encoded by nuclear and mitochondrial genes, and T3 modulates their expression via TRs present in the respective organelles. Furthermore, T3 stimulates mitochondrial DNA replication (8). In addition to such long-term, probably mainly transcriptional effects, TH exerts nontranscriptional, short-term effects on mitochondria. T3 rapidly increases oxygen consumption and oxidative phosphorylation in mitochondria, in a process that does not require protein synthesis (16). Several mitochondrial proteins have been shown to bind to TH with high affinity. However, their roles in the short-term effects of TH appear to be controversial (17). In the present study, we examined the possible involvement of AMPK in the stimulatory actions of T3 on mitochondria. We provide evidence that T3 rapidly activates AMPK via intracellular Ca2+ mobilization and CaMKKβ activation.
RESULTS
T3 Rapidly Induces Phosphorylation of Thr-172 in the α-Subunit of AMPK
Because the involvement of AMPK in fatty acid oxidation has been well documented in muscle and fat cells (1), we first chose myoblast C2C12 cells and preadipocyte 3T3-L1 cells to test the effect of T3 on AMPK. Differentiated cells were kept in serum-deprived media for 12 h and treated with 10 nm T3 for 30 min. Western blot analysis (Fig. 1A) revealed that serum deprivation induced a slight but significant increase in phosphorylation of Thr-172 in the α-subunit of AMPK. T3 treatment further increased the phosphorylation. Similar results were obtained with thyroid follicular FRTL-5 cells, indicating that T3-induced phosphorylation of AMPK is not a phenomenon that is restricted to a particular cell type. A time-course experiment (Fig. 1B) showed that T3 induced the phosphorylation of AMPK within 5 min and that its effect lasted for more than 120 min. The phosphorylation of AMPK by T3 was dose dependent, as shown in Fig. 1C.
Fig. 1.
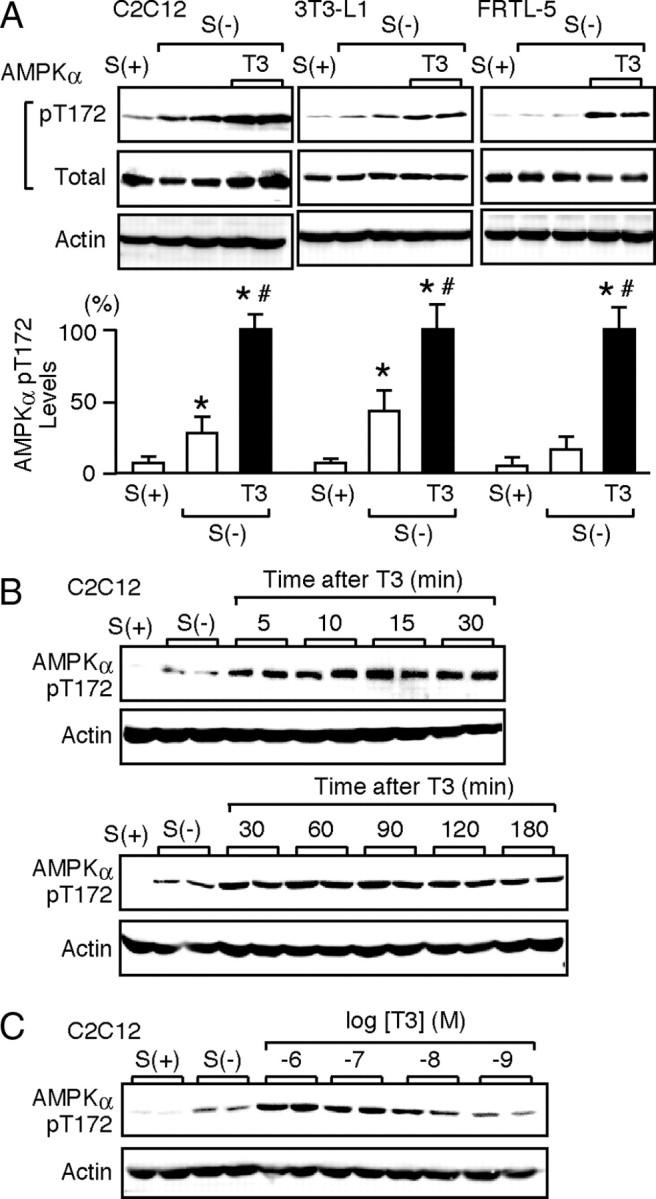
T3 Induces the Phosphorylation of Thr-172 in the α-Subunit of AMPK
Differentiated C2C12, 3T3-L1, and FRTL-5 cells were cultured with serum [S(+)] in serum-deprived media for 12 h [S(−)]. A, The cells were treated with 10 nm T3 for 30 min. Whole-cell lysates were subjected to Western blot analysis using antibodies directed against phospho-AMPK α-subunit (pT172), AMPK α-subunit (total), and actin. Most experiments were performed in duplicate cultures. Similar results were obtained from separate experiments. In densitometric analysis, the phospho-AMPK levels were normalized to the actin levels and expressed as percentages of the levels in T3-treated cells. Results are shown as means ± sd (n = 4). *, P < 0.05 vs. the levels in S(+) cells; #, P < 0.05 vs. the levels in S(−) cells. B, C2C12 cells were treated with 10 nm T3 for the indicated times, and Western blot analysis was performed. C, C2C12 cells were treated with 1 nm to 1 μm T3 for 30 min, and Western blot analysis was performed.
TRα and TRβ Mediate the T3-Induced Phosphorylation of AMPK
Several nontranscriptional effects of T3 have been demonstrated to be mediated by TRs (18, 19, 20, 21, 22). Thus, we studied the possible involvement of TR in the T3-induced phosphorylation of AMPK by using N2aTRα and N2aTRβ cells, which stably express TRα1 and TRβ1, respectively. As shown in Fig. 2A, serum deprivation significantly increased the phosphorylation of Thr-172 in the α-subunit of AMPK in wild-type N2a cells, but subsequent treatment with T3 had only a marginal effect. A similar tendency was observed for Ser-79 phosphorylation of ACC. By contrast, N2aTRα and N2aTRβ cells markedly responded to T3. Serum deprivation increased the phosphorylation of AMPK and ACC, and T3 further increased this phosphorylation.
Fig. 2.
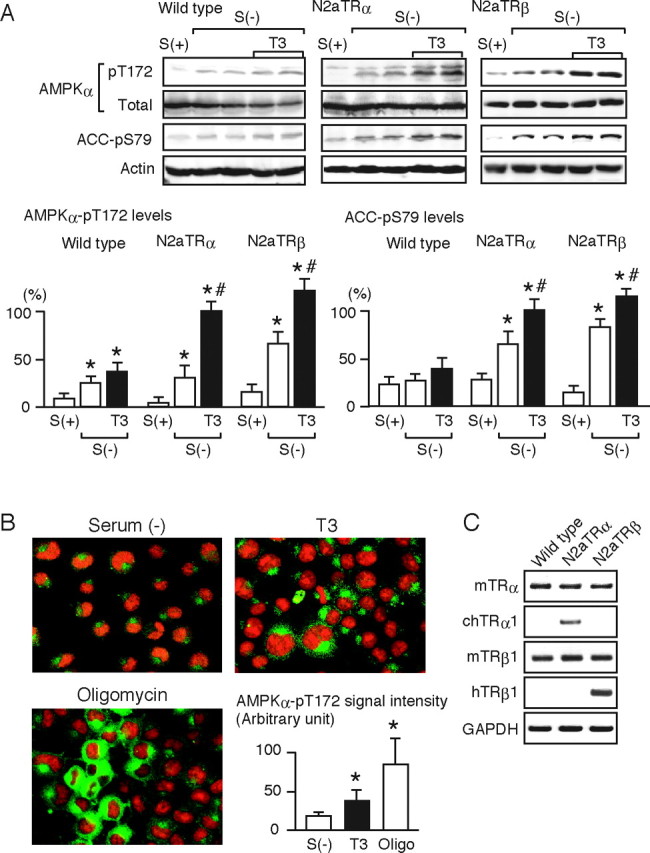
TR Mediates the T3-Induced Phosphorylation of AMPK
A, Wild-type N2a cells and N2aTRα and N2aTRβ cells were cultured with [S(+)] or without [S(−)] serum for 12 h and were then treated with 10 nm T3 for 30 min. Whole-cell lysates were subjected to Western blot analysis using antibodies directed against phospho-AMPK α-subunit (pT172), AMPK α-subunit (total), phospho-ACC (pS79), and actin. Most experiments were performed in duplicate cultures. Similar results were obtained from separate experiments. In densitometric analysis, the phospho-AMPK and phospho-ACC levels were normalized to the actin levels and expressed as percentages of the levels in T3-treated N2aTRα cells. Results are means ± sd (n = 4). *, P < 0.05 vs. the levels in S(+) cells; #, P < 0.05 vs. the levels in S(−) cells. B, N2aTRα cells cultured without serum for 12 h [S(−)] were treated with 10 nm T3 or 10 μm oligomycin for 30 min. Immunocytochemical analysis was performed using rabbit anti-phospho-AMPK α-subunit (pT172) antibody and antirabbit IgG antibody conjugated to Alexa Fluor 488 (green fluorescence). Propidium iodide staining was performed to identify nuclei (red fluorescence). Representative images are shown. Quantitative data on the amounts of phospho-AMPK in the cytoplasm are also presented. Several images were randomly captured, and the cytoplasmic signal densities were determined by Multi Gauge software on the LAS-1000 system. The values are expressed in arbitrary units, means ± sd (n = 20). *, P < 0.05 vs. S(−). Similar results were obtained from separate experiments. C, The mRNA levels of TRα and TRβ1 in wild-type N2a, N2aTRα, and N2aTRβ cells were determined by RT-PCR. N2aTRα and N2aTRβ cells are stable transformants expressing chicken TRα1 and human TRβ1, respectively. The PCR products were subjected to agarose gel electrophoresis and visualized by ethidium bromide.
Immunocytochemical analysis using anti-phospho-Thr-172 AMPK α-subunit showed that T3 significantly increased the fluorescence intensity in the cytoplasm, indicating that T3-induced phosphorylation of AMPK mainly occurred in the cytoplasm (Fig. 2B). A similar increase was observed in the cells treated with oligomycin, an ATP synthase inhibitor. The expression profiles of TR isoforms in wild-type N2a, N2aTRα, and N2aTRβ cells are shown in Fig. 2C. Together, these results indicate that both TRα and -β isoforms can mediate the T3-induced phosphorylation of AMPK.
The TR mRNA levels in N2a cells were compared with those in C2C12 and 3T3-L1 cells by RT-PCR. As shown in supplemental Fig. A (published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org), similar levels of TR mRNA were detected in all three cell types. Thus, it is unlikely that the lower response of N2a cells to T3 is due to a difference in TR expression. Some components of the downstream signaling pathway might be less functional in N2a cells.
CaMKKβ Mediates the T3-Induced Phosphorylation of AMPK
To explore the signaling cascade underlying the T3 action, we first examined the expression of LKB1 and CaMKKβ in the cells used in this study. As shown in Fig. 3A, RT-PCR analysis revealed that whereas CaMKKβ was expressed in all of the cell types tested, LKB1 was not expressed in HeLa cells, consistent with a previous report (6). Thus, we used HeLa cells to study whether LKB1 is required for the T3-induced phosphorylation of AMPK. TRα and TRβ1 expression was detected by RT-PCR in HeLa cells (data not shown). As shown in Fig. 3B, T3 induced a significant increase in the phosphorylation of Thr-172 in the α-subunit of AMPK. Ser-79 phosphorylation of ACC was also increased by T3 in HeLa cells, indicating that LKB1 was dispensable for the T3 action. We then tested a CaMKK inhibitor, STO-609. Hawley et al. (4) reported that 10 μm STO-609 inhibits a broad range of kinases, but 1 μm STO-609 is relatively selective for CaMKKα and -β. As shown in Fig. 3B, 1 μm STO-609 was sufficient to prevent the effect of T3 on AMPK and ACC, suggesting the involvement of CaMKKβ. This notion was validated by the following experiment using small interfering RNA (siRNA).
Fig. 3.

CaMKKβ Mediates the T3-Induced Phosphorylation of AMPK
A, The mRNA levels of LKB1 and CaMKKβ in C2C12, 3T3-L1, FRTL-5, N2a, and HeLa cells were determined by RT-PCR. The products were subjected to agarose gel electrophoresis and visualized by ethidium bromide. No band was detected when the reverse transcriptase was omitted (data not shown). B, HeLa cells were cultured without serum for 12 h. They were pretreated for 15 min with 1 μm STO-609 and treated for 30 min with 10 nm T3 in the presence of STO-609. Whole-cell lysates were subjected to Western blot analysis using antibodies against phospho-AMPK α-subunit (pT172), AMPK α-subunit (Total), phospho-ACC (pS79), and actin. Experiments were performed in duplicate cultures. Similar results were obtained from separate experiments. In densitometric analysis, the phospho-AMPK and phospho-ACC levels were normalized to the actin levels and expressed as percentages of the levels in cells treated with T3 alone. Results are means ± sd (n = 4). *, P < 0.05 vs. the control levels; #, P < 0.05 vs. the levels in cells treated with T3 alone. C, CaMKKβ siRNA and nontargeting, negative control siRNA were transfected into HeLa cells. The expression of CaMKKβ, CaMKKα, and TRβ1 was determined by RT-PCR 48 h after transfection. D, HeLa cells transfected with CaMKKβ or control siRNA for 48 h were cultured without serum for 12 h and then treated with 1 μm ionomycin or 10 nm T3 for 30 min. Whole-cell lysates were subjected to Western blot analysis using antibodies against phospho-AMPK α-subunit (pT172), phospho-ACC (pS79), and actin. The experiments were performed in duplicate cultures. Similar results were obtained from separate experiments.
The effects of CaMKKβ and control siRNAs on the expression of CaMKKβ, CaMKKα, and TRβ1 in HeLa cells were examined by RT-PCR (Fig. 3C). CaMKKβ siRNA markedly silenced CaMKKβ expression, whereas it did not affect the expression of CaMKKα and TRβ1. A nontargeting, control siRNA had no effect on the expression of these three proteins. To confirm the silencing effect of CaMKKβ siRNA, HeLa cells were treated with a Ca2+ ionophore, ionomycin, and the levels of AMPK and ACC phosphorylation were determined. As shown in Fig. 3D, the introduction of CaMKKβ siRNA markedly suppressed the ionomycin-dependent phosphorylation of AMPK and ACC, whereas control siRNA had no effect, demonstrating that the CaMKKβ siRNA successfully functioned in HeLa cells. When HeLa cells transfected with CaMKKβ siRNA were treated with T3, the phosphorylation of AMPK and ACC was markedly inhibited. Together, these results demonstrate that CaMKKβ mediates the T3-induced phosphorylation of AMPK.
T3-Dependent Intracellular Ca2+ Mobilization Is Associated with AMPK Phosphorylation
We next studied T3-dependent intracellular Ca2+ mobilization in HeLa cells by using a fluorescent indicator, Fluo-4. As shown in Fig. 4A, T3 induced a rapid and significant increase in intracellular Ca2+ levels, which gradually decreased after T3 withdrawal. When the cells were preincubated with 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), a Ca2+ chelator, the fluorescence signals from Ca2+/Fluo-4 complexes were markedly suppressed, possibly due to the competition of BAPTA with Fluo-4 to bind Ca2+. We repeated the experiment by using a ratiometric Ca2+ indicator, Indo-1 (Fig. 4B). The profiles of Ca2+ increase were similar to those obtained with Fluo-4. The lower degree of Ca2+ increase may be due to the lower sensitivity of Indo-1. These results demonstrate the T3 induction of intracellular Ca2+ mobilization in HeLa cells.
Fig. 4.
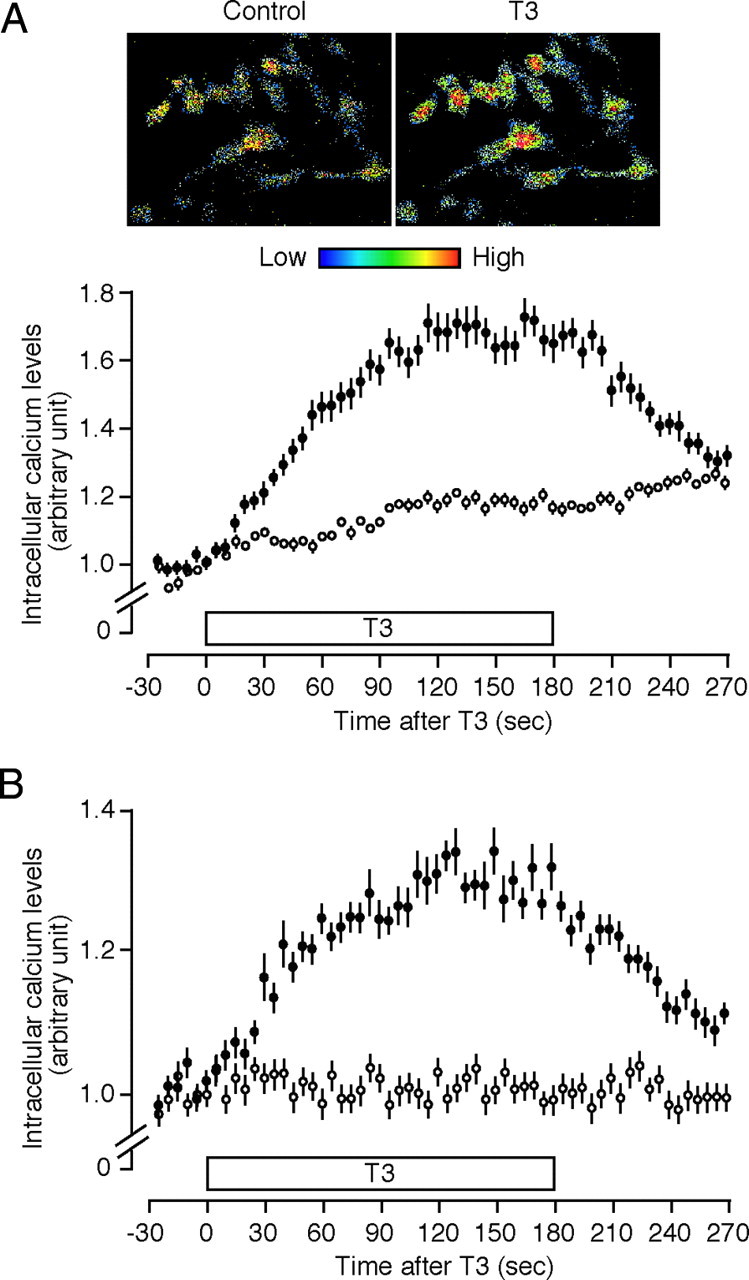
T3 Induces Intracellular Ca2+ Mobilization
A, HeLa cells labeled with Fluo-4-AM (•) or cells treated with both BAPTA-AM and Fluo-4-AM (○) were superfused with HEPES-buffered Ringer solution (see Materials and Methods). Five consecutive images with 6-sec intervals were obtained before T3 treatment. Cells were then treated with 10 nm T3 for 180 sec, and 45 consecutive images were obtained during and after T3 treatment for 270 sec. Representative images before (control) and 150 sec after T3 treatment are presented. Changes in fluorescence intensities are indicated by pseudo-colors. The fluorescence intensities of individual cells were measured and are expressed as a ratio of the averaged intensities obtained from five images before T3 stimulation. Results are means ± se (n = 38–45). B, HeLa cells labeled with the ratiometric Ca2+ indicator Indo-1-AM (•) or cells treated with both BAPTA-AM and Indo-1-AM (○) were treated with T3 as described above. Indo-1 was excited at 351 nm. Emissions at 400–445 nm (FS) and longer than 445 nm (FL) were captured simultaneously. Indo-1 ratios (a ratio of FS to FL) in individual cells were calculated and expressed relative to the averaged ratio obtained from five images before T3 stimulation. Results are means ± se (n = 30–35).
Then, the effects of BAPTA on the phosphorylation of AMPK and ACC were examined, as shown in Fig. 5A. BAPTA suppressed both ionomycin- and T3-dependent phosphorylation of AMPK and ACC, strongly suggesting that T3-dependent intracellular Ca2+ mobilization leads to CaMKKβ activation, followed by AMPK phosphorylation. Similarly, T3-dependent AMPK phosphorylation in N2aTRβ cells was suppressed by BAPTA and STO-609 (supplemental Fig. B).
Fig. 5.
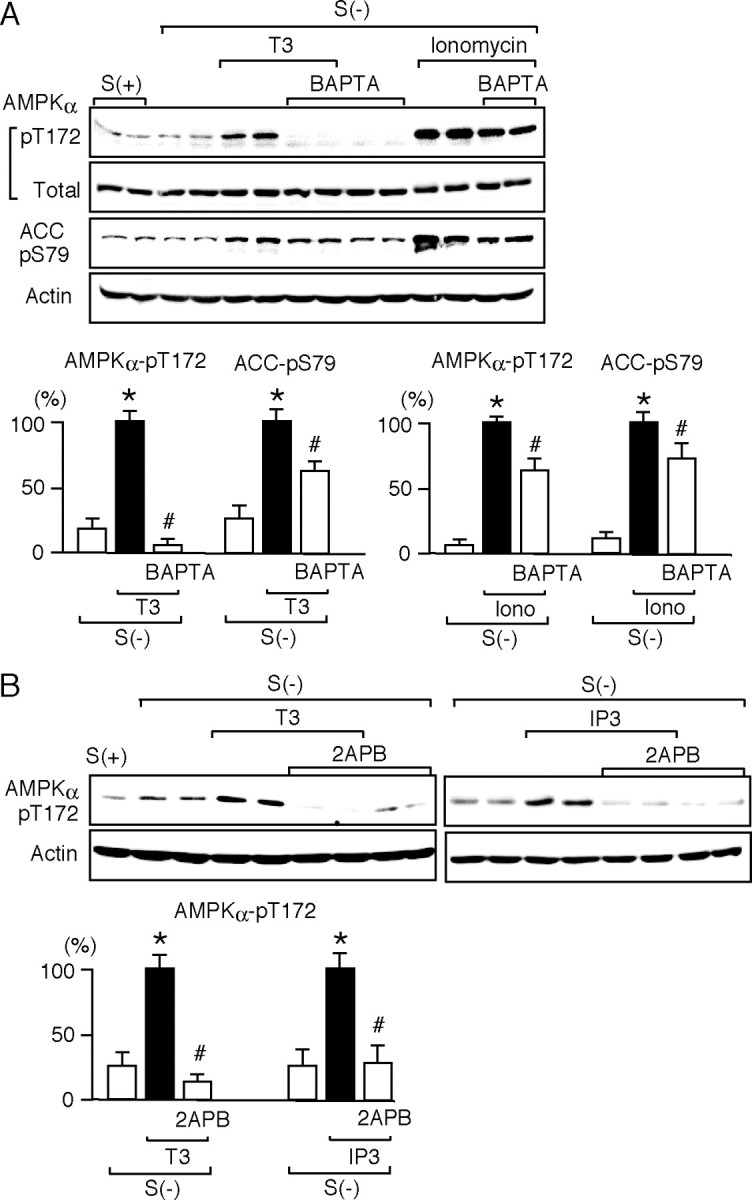
T3-Dependent Intracellular Ca2+ Mobilization Is Associated with the Phosphorylation of AMPK and ACC
A, HeLa cells were cultured with [S(+)] or without [S(−)] serum for 12 h. They were pretreated for 15 min with 10 μm BAPTA and treated for 30 min with 10 nm T3 or 1 μm ionomycin (Iono) in the presence of BAPTA. Whole-cell lysates were subjected to Western blot analysis using antibodies against phospho-AMPK α-subunit (pT172), AMPK α-subunit (total), phospho-ACC (pS79), and actin. The experiments were performed in duplicate cultures. Similar results were obtained from separate experiments. In densitometric analysis, the phospho-AMPK and phospho-ACC levels were normalized to the actin levels and expressed as percentages of the levels in cells treated with T3 or ionomycin alone. Results are means ± sd (n = 4). *, P < 0.05 vs. the levels in S(−) cells; #, P < 0.05 vs. the levels in cells treated with T3 or ionomycin alone. B, HeLa cells were cultured with [S(+)] or without [S(−)] serum for 12 h. They were pretreated for 15 min with 100 μm 2APB and treated with 10 nm T3 for 30 min or 1 μm IP3 for 5 min in the presence of 2APB. Whole-cell lysates were subjected to Western blot analysis using antibodies against phospho-AMPK α-subunit (pT172) and actin. The experiments were performed in duplicate cultures. Similar results were obtained from separate experiments. In densitometric analysis, the phospho-AMPK levels were normalized to the actin levels and expressed as percentages of the levels in cells treated with T3 or IP3 alone. Results are means ± sd (n = 4). *, P < 0.05 vs. the levels of S(−); #, P < 0.05 vs. the levels of T3 alone or the levels of IP3 alone.
We then studied the effects of an antagonist to inositol 1,4,5-triphosphate (IP3) receptor, 2-aminoethoxydiphenyl borate (2APB) (23), on T3- and IP3-dependent AMPK phosphorylation (Fig. 5B). AMPK phosphorylation was markedly suppressed by 2APB, suggesting that Ca2+ release from IP3 receptor is involved in the T3-induced phosphorylation of AMPK.
T3-Dependent Fatty Acid Oxidation Is Mediated by CaMKKβ
The above studies showed that T3 induced Ser-79 phosphorylation of ACC via AMPK activation, which would result in ACC inactivation and increase fatty acid oxidation (2). To confirm this notion, the effects of T3 on the oxidation of labeled palmitic acids were assessed in HeLa cells. As shown in Fig. 6A, the basal oxidation levels were significantly decreased by BAPTA, but not by STO-609, suggesting that intracellular Ca2+, but not CaMKK, might contribute to the basal fatty acid oxidation. T3 significantly increased the fatty acid oxidation. This increase was attenuated not only by BAPTA but also by STO-609. When HeLa cells transfected with CaMKKβ siRNA were subjected to the fatty acid oxidation assay, the T3-dependent increase was significantly reduced (Fig. 6B). These results indicate that T3-dependent fatty acid oxidation is mediated by CaMKKβ in HeLa cells.
Fig. 6.
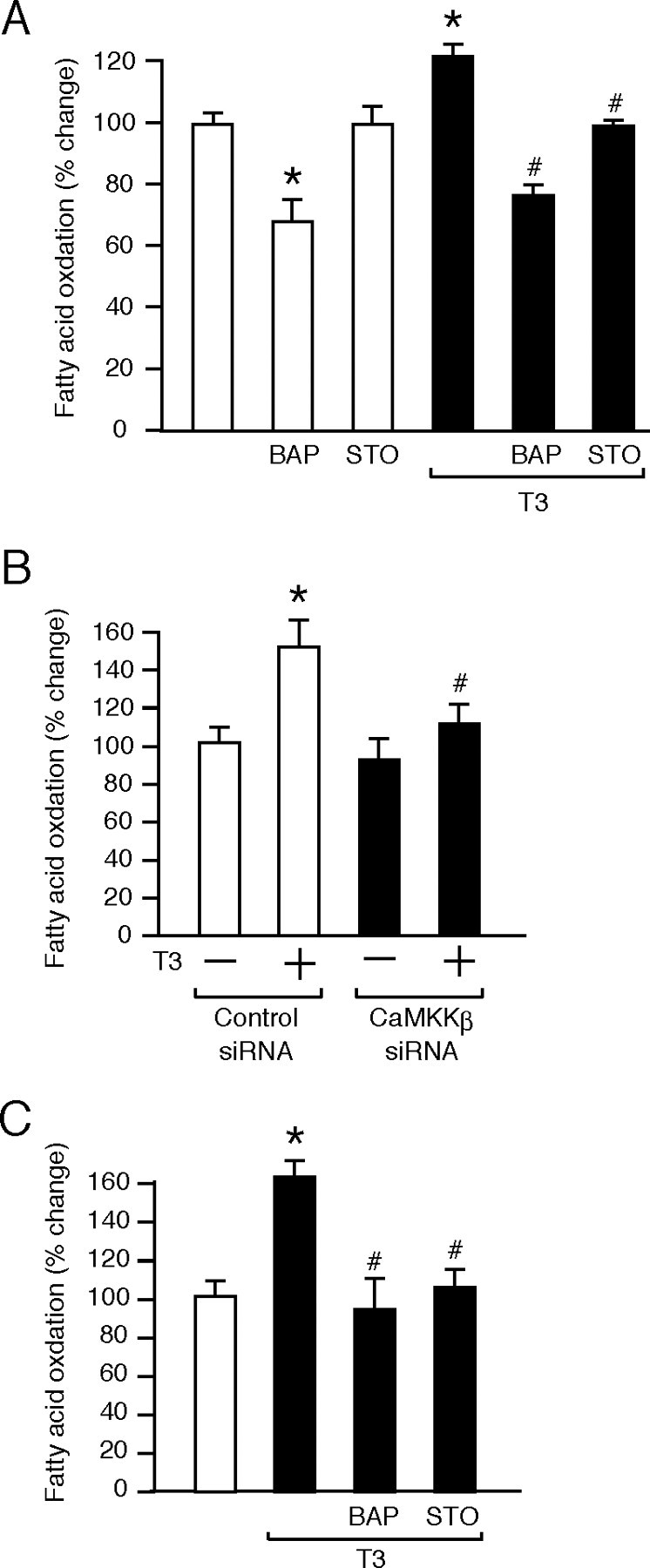
T3-Dependent Fatty Acid Oxidation Is Mediated by CaMKKβ
A, HeLa cells were preincubated for 2 h in DMEM containing 0.5% fatty acid-free BSA and were incubated for 3 h in DMEM containing 2% fatty acid-free BSA, 0.3 mm palmitic acid, and 1 μCi/ml [3H]palmitic acid in the presence or absence of 10 nm T3, 10 μm BAPTA (BAP), and 1 μm STO-609 (STO). After incubation, the media were collected, and the labeled palmitic acids remaining in the media were removed by precipitation with perchloric acid. The radioactivity of the supernatants was counted using a liquid scintillation counter. The levels of radioactive water production are expressed as percentages of the nontreated, control levels. Results are means ± sd (n = 3). *, P < 0.05 vs. the levels of the control; #, P < 0.05 vs. the levels in cells treated with T3 alone. Similar results were obtained from separate experiments. B, HeLa cells transfected with CaMKKβ or control siRNA for 48 h were subjected to the fatty acid oxidation assay as described above. Results are means ± sd (n = 3). *, P < 0.05 vs. the levels in cells treated with the control siRNA and T3 (−); #, P < 0.05 vs. the levels in cells treated with the control siRNA and T3 (+). Similar results were obtained from separate experiments. C, Differentiated C2C12 cells were subjected to the fatty acid oxidation assay as described above. Results are means ± sd (n = 3). *, P < 0.05 vs. the levels of the control; #, P < 0.05 vs. the levels in cells treated with T3 alone. Similar results were obtained from separate experiments.
As shown in Fig. 6C, T3-dependent fatty acid oxidation was also decreased by BAPTA and STO-609 in C2C12 cells that endogenously express both CaMKKβ and LKB1. Accordingly, T3-induced phosphorylation of AMPK was suppressed by BAPTA and STO-609 (supplemental Fig. C), suggesting that the effect of T3 on AMPK in C2C12 cells is mainly mediated by CaMKK. Similarly, the phosphorylation of AMPK by 2-deoxyglucose was also suppressed by BAPTA and STO-609 (supplemental Fig. C). Together, these results demonstrate that a signaling pathway involving CaMKK is important for the activation of AMPK by multiple stimuli such as T3 and 2-deoxyglucose in C2C12 cells.
DISCUSSION
In the present study, we show for the first time that T3 stimulates the phosphorylation of AMPK that is critical for kinase activation. This action occurred rapidly, within several minutes of the start of T3 treatment and was observed in cells in which the basal phosphorylation levels were already elevated by 12-h serum starvation. AMPK activation during long-term serum starvation is thought to be mainly due to cellular ATP depletion. It was thus suggested that the additive effect of T3 would be mediated by the enhancement of the signaling pathway elicited by ATP depletion or the activation of an alternative pathway leading to AMPK phosphorylation. The fact that the effect of T3 was extremely rapid does not support the involvement of alterations in intracellular adenine nucleotide levels. Using cells lacking LKB1, a CaMKK inhibitor, and an isoform-specific siRNA, we provide evidence that the T3-induced phosphorylation of AMPK is mediated by a signaling pathway involving CaMKKβ.
CaMKKβ was first identified as a kinase upstream of Ca2+/calmodulin-dependent protein kinase I and IV (24). Recent extensive studies involving yeast genetics have revealed that CaMKKβ can also phosphorylate AMPK in mammalian cells (4, 5, 6). These studies showed that ionomycin, hydrogen peroxide, sorbitol, and 2-deoxyglucose stimulated AMPK via CaMKKβ activation. These reagents mimic the situations of intracellular Ca2+ mobilization, oxidant stress, osmotic stress, and energy deprivation, respectively, thus suggesting that CaMKKβ-dependent AMPK activation would play an important role in cells exposed to various physiological and pathological conditions. However, the physiological stimuli activating the CaMKKβ-AMPK cascade remain poorly identified. It was recently reported that thrombin can activate this cascade in vein endothelial cells (25). Moreover, antigen receptor activation in T lymphocytes was reported to stimulate this cascade (26). T3 is another physiological stimulus activating AMPK via CaMKKβ.
T3 has been shown to nontranscriptionally modulate a variety of proteins, including ERKs, the sodium/proton exchanger, a voltage-dependent potassium channel (ether-a-go-go related gene channel), the sodium/potassium ATPase, and phosphoinositide 3 kinase (PI3K) (20, 22, 27, 28, 29). These proteins do not directly bind T3, so their functions are regulated by T3 indirectly, most likely through T3/TR complexes. The involvement of TR in the nontranscriptional effect of T3 was demonstrated in mice possessing a mutant TRβ that selectively lacks DNA-binding activity (30). Recent in vitro studies including ours have shown that TR directly interacts with a regulatory subunit of PI3K, suggesting an essential role of TR in the regulation of PI3K by T3 (19, 20, 21, 22). In addition, it was reported that T3-induced activation of Akt, a kinase downstream of PI3K, was greatly impaired in TRα and -β double-knockout mice (21). In the current study, the involvement of TRα and TRβ in the T3-induced activation of AMPK was suggested by using cells stably expressing TRα or TRβ. However, unlike the case of PI3K, T3/TR complexes would not directly contribute to AMPK activation; rather, they triggered intracellular Ca2+ mobilization.
This study demonstrates that T3 rapidly, within a minute, increases intracellular Ca2+ levels and that this is associated with the phosphorylation of AMPK and ACC. Several recent reports describe acute effects of T3 on intracellular Ca2+. D’Arezzo et al. (28) showed in the L-6 rat myoblast cell line that T3 induces intracellular Ca2+ mobilization within a few minutes, through activation of IP3 receptors and without contributions of extracellular Ca2+ and ryanodine receptors. Interestingly, T3-bound agarose, which cannot enter cells, reproduced this effect of T3, suggesting that its action is initiated at the plasma membrane, but the nature of T3-binding protein on the membrane was not identified. Saelim et al. (18) also reported that T3 increases the IP3-mediated Ca2+ wave period and amplitude in Xenopus oocytes expressing mitochondrial forms of TR and that this increase is mediated by mitochondrial Ca2+ uptake. They further showed that increased mitochondrial membrane potential (Δψ) contributes to the uptake. These reports indicate that T3 induces intracellular Ca2+ mobilization by cooperatively stimulating the endoplasmic reticulum and mitochondria.
The current study further shows that the T3-induced phosphorylation of AMPK leads to increased fatty acid oxidation, which would subsequently increase mitochondrial acetyl-CoA levels and stimulate oxidative phosphorylation. T3 exerts profound effects on cell growth, differentiation, and thermogenesis, processes that may consume a considerable amount of ATP or dissipate mitochondrial membrane potential. Thus, cells with limited energy turnover could not properly respond to T3. The activation of AMPK would allow cells to switch on ATP-generating pathways and render them acceptable for T3.
MATERIALS AND METHODS
Cell Culture
The mouse myoblast cell line C2C12 [American Type Culture Collection (ATCC), Manassas, VA; CRL-1772], the mouse preadipocyte cell line 3T3-L1 (ATCC CL-173), and the human cervical carcinoma cell line HeLa (ATCC CCL-2) were cultured in DMEM supplemented with 10% fetal bovine serum. The mouse neuroblastoma cell line Neuro 2a (ATCC CCL-131) was cultured in DMEM/F12 medium supplemented with 10% fetal bovine serum. The rat thyroid cell line FRTL-5 (ATCC CRL-1468) was cultured in Coon’s modified Ham’s F-12 medium supplemented with 5% heat-inactivated calf serum and six hormones as described previously (31). Neuro 2a cells stably transfected with chicken TRα1 (N2aTRα) were a generous gift from Professor J. Bernal (Instituto de Investigaciones Biomedicas, Madrid, Spain) (32). Neuro 2a cells stably transfected with human TRβ1 (N2aTRβ) were a generous gift from Professor J. Puymirat (Human Genetics Research Unit, Laval University, Quebec, Canada) (33). Differentiation of C2C12 cells into myotubes was induced by mitogen withdrawal (DMEM with 2% horse serum) and was confirmed by troponin T expression. The differentiation of 3T3-L1 cells into adipocytes was induced by combined treatment of the cells with 3-isobutyl-1-methylxanthine, dexamethasone, and insulin (34). Near-confluent cells were cultured in serum-deprived media for 12 h and treated with various hormones and reagents such as T3, oligomycin, ionomycin, STO-609, BAPTA-acetoxymethyl ester (BAPTA-AM), IP3, and 2APB, alone or in combination. These reagents were purchased from Sigma-Aldrich (St. Louis, MO), except for BAPTA-AM and IP3 (Dojindo Laboratories, Kumamoto, Japan).
Western Blot Analysis
The procedures used for preparation of whole-cell lysates and Western blot analysis were described in our previous report (22). In brief, whole-cell lysates (40 μg/lane) were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Amersham Pharmacia, Piscataway, NJ). The blots were probed with the first antibodies described below, followed by incubation with horseradish peroxidase-conjugated antirabbit IgG antibody. Rabbit anti-phospho-AMPK α-subunit (Thr-172), anti-AMPK α-subunit, and anti-phospho-ACC (Ser-79) antibodies were purchased from Cell Signaling (Beverly, MA). Rabbit anti-actin antibody was purchased from Sigma-Aldrich. Proteins were visualized using enhanced chemiluminescence (ECL) reagents (Pierce, Rockford, IL). Images of the membranes were obtained using an LAS-1000 lumino-image analyzer (Fuji Film, Tokyo, Japan), and densitometric analysis was performed using software on the LAS-1000 system.
Immunocytochemical Analysis
The procedures used for immunocytochemical analysis were described in our previous report (35). After fixation and blocking, cells were incubated with anti-phospho-AMPK (Thr-172) antibody followed by incubation with antirabbit IgG antibody conjugated to Alexa Fluor 488 (Molecular Probes, Eugene, OR). Counterstaining was performed using propidium iodide. Images were obtained using a confocal laser microscope (LSM510; Carl Zeiss, Jena, Germany) with the same set of optical parameters as described previously (36). Several images were captured, and densitometric analysis of the amounts of phosphorylated AMPK was performed using Multi Gauge software on the LAS-1000 system. Merged images were obtained using Adobe Photoshop software (version 7.0.1; Adobe Systems, San Jose, CA).
RT-PCR
The expression of LKB1, CaMKKβ, TRα, and TRβ1 was examined by RT-PCR using total RNA. The primer sequences are as follows: sense and antisense primers for LKB1, 5′-CTCATCGGCAAGTACCTGATG-3′ and 5′- TGGGAAACGCTTCTCCGGCAC-3′; primers for mouse CaMKK-β, 5′- TAGAGTACTTGCATTACCAGAAGA-3′ and 5′- TGGGTTTTTGTCCAACATCCGGGT-3′; primers for human CaMKK-β, 5′- TCGAGTACTTGCACTGCCAGAAGATC-3′ and 5′-GGGGTTCTTGTCCAGCATACGGGT-3′; primers for human CaMKK-α, 5′-TCGAGTACTTGCACTGCCAGAAGATC-3′ and 5′-GGGATTCTTGTCTAACATCTTCAG-3′; primers for TRα, 5′-AAGCCAAGCAAGGTGGAGTGT-3′ and 5′-ATGCACTTCTTGAAGCGGCACA-3′; primers for chicken TRα, 5′-GGTCGTGTCTGATGCCATCT-3′ and 5′- CTCCTGGTCCTCGAAGACCT-3′; primers for human TRβ1, 5′-GCTATGACCCAGAAAGTGAG-3′ and 5′-TGTCACCTTCATCAGGAGTT-3′; primers for mouse and rat TRβ1, 5′-ACAGAAAATGGCCTTCCAGC-3′ and 5′-TCTTGCTGTCATCCAGCACCA-3′; and primers for GAPDH (glyceraldehyde-3-phosphate dehydrogenase), 5′-GCACCGTCAAGGCTGAGAAC-3′ and 5′-GCCTTCTCCATGGTGGTGAA-3′.
siRNA
A predesigned siRNA directed against CaMKKβ (ID 110916, β3) and Silencer Negative Control no. 1 (nontargeting siRNA) were purchased from Ambion (Austin, TX). HeLa cells were seeded in six-well plates 24 h before transfection. siRNA (final concentration 80 nm) was introduced into cells using siPORT Lipid (Ambion).
Measurement of Intracellular Ca2+ Levels
Intracellular Ca2+ changes were measured using a fluorescent dye technique. The detailed procedures were described in our previous report (37). In brief, HeLa cells cultured on glass coverslips were treated for 45 min with 5 μm Fluo-4-AM (Molecular Probes), 0.02% pluronic acid F127 (Molecular Probes), and 1.25 mm probenecid in HEPES-buffered Ringer solution. In some experiments, cells were preincubated with 10 μm BAPTA-AM for 15 min; then, 5 μm Fluo-4 was added and incubated for 45 min. The coverslips were placed in a chamber mounted on the stage of an inverted microscope (Axiovert, Zeiss) equipped with a confocal laser scanning system (Oz; Noran, Middleton, WT) and then superfused with HEPES-buffered Ringer solution at a flow rate of 3 ml/min. Five consecutive images with 6-sec intervals were obtained before T3 stimulation. The cells were then treated with 10 nm T3 for 180 sec, and 45 consecutive images were obtained during and after T3 treatment for 270 sec. The averages of all the fluorescence intensities of individual cells were analyzed using the Intervision 2D program (Noran).
A ratiometric calcium indicator, Indo-1-AM (Dojindo), was also used instead of Fluo-4-AM. When Indo-1 binds Ca2+, the fluorescence emission at 405 nm increases, and the emission at 495 nm decreases. Measurement of the ratio of these emission intensities allows accurate estimation of intracellular Ca2+ concentrations, independent of intracellular dye concentrations. Indo-1-AM was loaded using the same protocol as Fluo-4-AM. To prevent photodegradation of Indo-1, 10 mm Trolox (EMD Biosciences, La Jolla, CA) was added to HEPES-Ringer. Indo-1 was excited at 351 nm. The emissions at 400–445 nm (FS) and longer than 445 nm (FL) were captured simultaneously using a dichroic mirror (445 nm) and a 400-nm longpass barrier filter, and the Indo-1 ratios (the ratio of FS to FL) in individual cells were calculated.
Measurement of Fatty Acid Oxidation
Fatty acid oxidation was determined using [9,10(n)-3H]palmitic acid (GE Healthcare Amersham, Buckinghamshire, UK). Palmitic acids were conjugated to fatty acid-free BSA (Sigma-Aldrich) by incubating DMEM containing 2% fatty acid-free BSA, 0.3 mm palmitic acid (Sigma-Aldrich), and 1 μCi/ml [3H]palmitic acid at 45 C. HeLa cells seeded in six-well plates on the day before the experiment were preincubated for 2 h in DMEM containing 0.5% fatty acid-free BSA. Then they were incubated for 3 h in DMEM containing [3H]palmitic acids conjugated to BSA in the presence or absence of 10 nm T3, 10 μm BAPTA, and 1 μm STO-609. After incubation, the media were collected, and the labeled palmitic acids remaining in the media were removed by precipitation with perchloric acid. Precipitation was performed twice. The radioactivity of the supernatants was determined using a liquid scintillation counter to quantify the radioactive water production.
Statistical Analysis
Statistical analysis was performed using ANOVA followed by Bonferroni’s multiple t test; a P value < 0.05 was considered to be statistically significant.
NURSA Molecule Pages:
Ligands: Thyroid hormone;
Nuclear Receptors: TRα | TRβ.
Footnotes
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture, Japan.
Disclosure Statement: The authors have nothing to disclose.
First Published Online January 10, 2008
Abbreviations: ACC, Acetyl-coenzyme A carboxylase; AM, acetoxymethyl ester; AMPK, AMP-activated protein kinase; 2APB, 2-aminoethoxydiphenyl borate; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; CaMKKβ, Ca2+/calmodulin-dependent protein kinase kinase-β; CoA, coenzyme A; IP3, inositol 1,4,5-triphosphate; TH, thyroid hormone; TR, T3 receptor.
References
- 1.Hardie DG, Sakamoto K 2006. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology (Bethesda) 21:48–60 [DOI] [PubMed] [Google Scholar]
- 2.Kahn BB, Alquier T, Carling D, Hardie DG 2005. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1:15–25 [DOI] [PubMed] [Google Scholar]
- 3.Hardie DG 2005. New roles for the LKB1→AMPK pathway. Curr Opin Cell Biol 17:167–173 [DOI] [PubMed] [Google Scholar]
- 4.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG 2005. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2:9–19 [DOI] [PubMed] [Google Scholar]
- 5.Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D 2005. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2:21–33 [DOI] [PubMed] [Google Scholar]
- 6.Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA 2005. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 280:29060–29066 [DOI] [PubMed] [Google Scholar]
- 7.Bassett JH, Harvey CB, Williams GR 2003. Mechanisms of thyroid hormone receptor-specific nuclear and extra nuclear actions. Mol Cell Endocrinol 213:1–11 [DOI] [PubMed] [Google Scholar]
- 8.Wrutniak-Cabello C, Casas F, Cabello G 2001. Thyroid hormone action in mitochondria. J Mol Endocrinol 26:67–77 [DOI] [PubMed] [Google Scholar]
- 9.Goglia F, Silvestri E, Lanni A 2002. Thyroid hormones and mitochondria. Biosci Rep 22:17–32 [DOI] [PubMed] [Google Scholar]
- 10.Short KR, Nygren J, Barazzoni R, Levine J, Nair KS 2001. T3 increases mitochondrial ATP production in oxidative muscle despite increased expression of UCP2 and -3. Am J Physiol Endocrinol Metab 280:E761–E769 [DOI] [PubMed]
- 11.Izquierdo JM, Cuezva JM 1993. Thyroid hormones promote transcriptional activation of the nuclear gene coding for mitochondrial β-F1-ATPase in rat liver. FEBS Lett 323:109–112 [DOI] [PubMed] [Google Scholar]
- 12.Dummler K, Muller S, Seitz HJ 1996. Regulation of adenine nucleotide translocase and glycerol 3-phosphate dehydrogenase expression by thyroid hormones in different rat tissues. Biochem J 317(Pt 3):913–918 [DOI] [PMC free article] [PubMed]
- 13.Li R, Luciakova K, Zaid A, Betina S, Fridell E, Nelson BD 1997. Thyroid hormone activates transcription from the promoter regions of some human nuclear-encoded genes of the oxidative phosphorylation system. Mol Cell Endocrinol 128:69–75 [DOI] [PubMed] [Google Scholar]
- 14.Lanni A, Moreno M, Lombardi A, Goglia F 2003. Thyroid hormone and uncoupling proteins. FEBS Lett 543:5–10 [DOI] [PubMed] [Google Scholar]
- 15.Weitzel JM, Iwen KA, Seitz HJ 2003. Regulation of mitochondrial biogenesis by thyroid hormone. Exp Physiol 88:121–128 [DOI] [PubMed] [Google Scholar]
- 16.Sterling K, Brenner MA, Sakurada T 1980. Rapid effect of triiodothyronine on the mitochondrial pathway in rat liver in vivo. Science 210:340–342 [DOI] [PubMed] [Google Scholar]
- 17.Goglia F, Moreno M, Lanni A 1999. Action of thyroid hormones at the cellular level: the mitochondrial target. FEBS Lett 452:115–120 [DOI] [PubMed] [Google Scholar]
- 18.Saelim N, John LM, Wu J, Park JS, Bai Y, Camacho P, Lechleiter JD 2004. Nontranscriptional modulation of intracellular Ca2+ signaling by ligand stimulated thyroid hormone receptor. J Cell Biol 167:915–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furuya F, Hanover JA, Cheng SY 2006. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proc Natl Acad Sci USA 103:1780–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Storey NM, Gentile S, Ullah H, Russo A, Muessel M, Erxleben C, Armstrong DL 2006. Rapid signaling at the plasma membrane by a nuclear receptor for thyroid hormone. Proc Natl Acad Sci USA 103:5197–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiroi Y, Kim HH, Ying H, Furuya F, Huang Z, Simoncini T, Noma K, Ueki K, Nguyen NH, Scanlan TS, Moskowitz MA, Cheng SY, Liao JK 2006. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci USA 103:14104–14109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao X, Kambe F, Moeller LC, Refetoff S, Seo H 2005. Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol Endocrinol 19:102–112 [DOI] [PubMed] [Google Scholar]
- 23.Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K 1997. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem (Tokyo) 122:498–505 [DOI] [PubMed] [Google Scholar]
- 24.Anderson KA, Means RL, Huang QH, Kemp BE, Goldstein EG, Selbert MA, Edelman AM, Fremeau RT, Means AR 1998. Components of a calmodulin-dependent protein kinase cascade. Molecular cloning, functional characterization and cellular localization of Ca2+/calmodulin-dependent protein kinase kinase β. J Biol Chem 273:31880–31889 [DOI] [PubMed] [Google Scholar]
- 25.Stahmann N, Woods A, Carling D, Heller R 2006. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase β. Mol Cell Biol 26:5933–5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamas P, Hawley SA, Clarke RG, Mustard KJ, Green K, Hardie DG, Cantrell DA 2006. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med 203:1665–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin HY, Davis FB, Gordinier JK, Martino LJ, Davis PJ 1999. Thyroid hormone induces activation of mitogen-activated protein kinase in cultured cells. Am J Physiol 276:C1014–C1024 [DOI] [PubMed]
- 28.D’Arezzo S, Incerpi S, Davis FB, Acconcia F, Marino M, Farias RN, Davis PJ 2004. Rapid nongenomic effects of 3,5,3′-triiodo-l-thyronine on the intracellular pH of L-6 myoblasts are mediated by intracellular calcium mobilization and kinase pathways. Endocrinology 145:5694–5703 [DOI] [PubMed] [Google Scholar]
- 29.Lei J, Mariash CN, Ingbar DH 2004. 3,3′,5-Triiodo-l-thyronine up-regulation of Na,K-ATPase activity and cell surface expression in alveolar epithelial cells is Src kinase- and phosphoinositide 3-kinase-dependent. J Biol Chem 279:47589–47600 [DOI] [PubMed] [Google Scholar]
- 30.Shibusawa N, Hashimoto K, Nikrodhanond AA, Liberman MC, Applebury ML, Liao XH, Robbins JT, Refetoff S, Cohen RN, Wondisford FE 2003. Thyroid hormone action in the absence of thyroid hormone receptor DNA-binding in vivo. J Clin Invest 112:588–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kambe F, Nomura Y, Okamoto T, Seo H 1996. Redox regulation of thyroid-transcription factors, Pax-8 and TTF-1, is involved in their increased DNA-binding activities by thyrotropin in rat thyroid FRTL-5 cells. Mol Endocrinol 10:801–812 [DOI] [PubMed] [Google Scholar]
- 32.Pastor R, Bernal J, Rodriguez-Pena A 1994. Unliganded c-erbA/thyroid hormone receptor induces trkB expression in neuroblastoma cells. Oncogene 9:1081–1089 [PubMed] [Google Scholar]
- 33.Lebel JM, Dussault JH, Puymirat J 1994. Overexpression of the β1 thyroid receptor induces differentiation in neuro-2a cells. Proc Natl Acad Sci USA 91:2644–2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green H, Kehinde O 1975. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell 5:19–27 [DOI] [PubMed] [Google Scholar]
- 35.Cao X, Kambe F, Lu X, Kobayashi N, Ohmori S, Seo H 2005. Glutathionylation of two cysteine residues in paired domain regulates DNA binding activity of Pax-8. J Biol Chem 280:25901–25906 [DOI] [PubMed] [Google Scholar]
- 36.Lu X, Kambe F, Cao X, Yoshida T, Ohmori S, Murakami K, Kaji T, Ishii T, Zadworny D, Seo H 2006. DHCR24-knockout embryonic fibroblasts are susceptible to serum withdrawal-induced apoptosis because of dysfunction of caveolae and insulin-Akt-Bad signaling. Endocrinology 147:3123–3132 [DOI] [PubMed] [Google Scholar]
- 37.Kozaki Y, Kambe F, Hayashi Y, Ohmori S, Seo H, Kumazawa T, Mizumura K 2007. Molecular cloning of prostaglandin EP3 receptors from canine sensory ganglia and their facilitatory action on bradykinin-induced mobilization of intracellular calcium. J Neurochem 100:1636–1647 [DOI] [PubMed] [Google Scholar]