

Abstract
Fibroblast growth factor (FGF) 21, a structural relative of FGF23 that regulates phosphate homeostasis, is a regulator of insulin-independent glucose transport in adipocytes and plays a role in the regulation of body weight. It also regulates ketogenesis and adaptive responses to starvation. We report that in a reconstituted receptor activation assay system using BaF3 cells, which do not endogenously express any type of FGF receptor (FGFR) or heparan sulfate proteoglycan, FGF21 alone does not activate FGFRs and that βKlotho is required for FGF21 to activate two specific FGFR subtypes: FGFR1c and FGFR3c. Coexpression of βKlotho and FGFR1c on BaF3 cells enabled FGF21, but not FGF23, to activate receptor signaling. Conversely, coexpression of FGFR1c and Klotho, a protein related to βKlotho, enabled FGF23 but not FGF21 to activate receptor signaling, indicating that expression of βKlotho/Klotho confers target cell specificity on FGF21/FGF23. In all of these cases, heparin enhanced the activation but was not essential. In 3T3-L1 adipocytes, up-regulation of glucose transporter (GLUT) expression by FGF21 was associated with expression of βKlotho, which was absent in undifferentiated 3T3-L1 fibroblasts. It is thus suggested that βKlotho expression is a crucial determinant of the FGF21 specificity of the target cells upon which it acts in an endocrine fashion.
THE FIBROBLAST GROWTH factor (FGF) family is comprised of 22 structurally related proteins in mammals that have a wide variety of functions in organogenesis, tissue remodeling, nervous system control, and angiogenesis (1, 2). In addition, recent analyses of the FGF19 subfamily, which consists of FGF19 (the human ortholog for mouse FGF15), FGF21, and FGF23, has revealed these mediators to function as metabolic regulators. For instance, FGF23 is a critical regulator of phosphate and vitamin D homeostasis (3), whereas FGF15/FGF19 secreted from the distal small intestine regulates bile acid homeostasis (4).
Fgf21 was originally identified as a new FGF family gene in mouse embryo using homology-based PCR (5). Thereafter, Kharitonenkov et al. (6) showed that FGF21 treatment increases expression of GLUT1 mRNA and stimulates glucose incorporation into differentiated mouse 3T3-L1 adipocytes, indicating the new FGF to be a glucose regulator. They further demonstrated that systematic administration of FGF21 reduces blood glucose and triglycerides to near normal levels in diabetic rodents and that transgenic mice overexpressing FGF21 were resistant to diet-induced obesity. It also has been reported that FGF21 regulates ketogenesis and lipolysis (7, 8). The clear therapeutic potential of FGF21 for the treatment of diabetes or other metabolic diseases has made elucidation of the FGF21 signaling pathway a pressing issue. However, the signaling pathway via which FGF21 acts has remained unclear. For instance, although it stimulates tyrosine phosphorylation of FGF receptor (FGFR) 1 and FGFR2 as well as phosphorylation of FGFR substrate 2α (FRS2α) and MAPK (Erk1/Erk2) in 3T3-L1 adipocytes, it has no effect in preadipocytes (6). In addition, the observation that FGF21 has little or no ability to stimulate proliferation of BaF3 cells expressing any of the FGFR subtypes (9) suggests that an unknown factor/mechanism may underlie FGF21-dependent activation of FGFRs. Interestingly, two research groups have recently demonstrated that Klotho, the product of a gene shown to be mutated in a mouse strain characterized by premature aging, functions as a coreceptor for FGF23 (10, 11). They observed that Klotho−/− and Fgf23−/− mice exhibit very similar phenotypes, which included elevated levels of calcium, phosphate, and 1,25-dihydroxyvitamin D in serum, ectopic calcification, generalized organ atrophy, infertility, and short lifespan (12, 13, 14). Klotho was shown to bind one or several FGFRs in the presence of heparin, and the putative FGF23-Klotho-FGFR-heparin signaling complex was postulated to transduce FGF signaling (10, 11). In the present work, we demonstrate that βKlotho, a single-span membrane protein belonging to the Klotho family, but not Klotho, serves as the coreceptor necessary for FGF21 signaling.
RESULTS AND DISCUSSION
We observed that FGF21 treatment leads to an increase in the expression of glucose transporter 1 (GLUT1) mRNA in 3T3-L1 adipocytes but not in undifferentiated fibroblasts (Fig. 1A) as previously reported (6). FGF signaling is thought to be transduced, at least in part, through activation of one or more FGFR tyrosine kinases (15). Kharitonenkov and colleagues (6, 16) showed that FGF21 activates both FGFR1 and FGFR2 tyrosine kinase and suggested they may function as FGF21 receptors. To test the possibility that a difference in FGFR levels underlies the difference in the sensitivity of 3T3-L1 adipocytes and fibroblasts to FGF21, we used quantitative RT-PCR to measure FGFR mRNA expression during 3T3-L1 differentiation. As shown in Fig. 1B, 3T3-L1 fibroblasts expressed both FGFR1 and FGFR2 mRNA, and the levels of both gradually declined as the cells differentiated into adipocytes. These data suggest that the level of FGFR1 or -R2 expression cannot, by itself, account for the unresponsiveness of 3T3-L1 fibroblasts to FGF21. 3T3-L1 fibroblasts express little or no FGFR3 and FGFR4 mRNA.
Fig. 1.
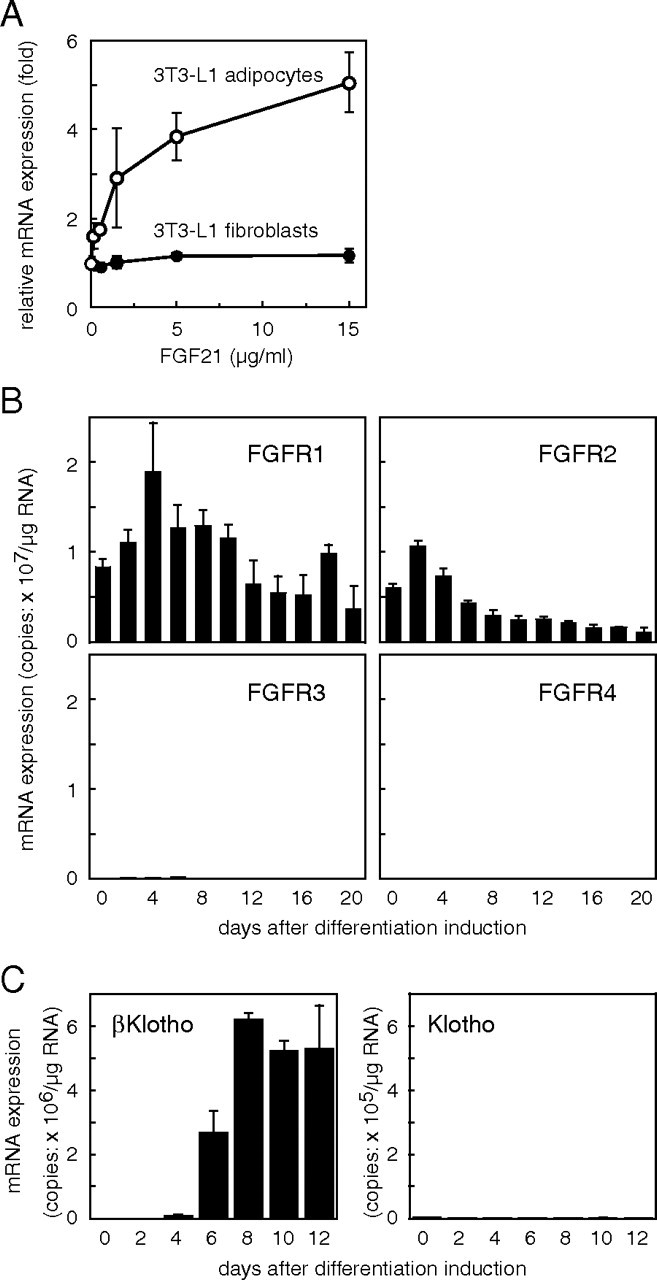
Gene Expression Differs in 3T3-L1 Fibroblasts and Adipocytes
A, FGF21 increases GLUT1 mRNA expression in 3T3-L1 adipocytes but not in fibroblasts. Fourteen days after induction of differentiation, 3T3-L1 fibroblasts or 3T3-L1 adipocytes were treated with the indicated concentrations of FGF21 for 6 h, after which the copy number of the expressed GLUT1 mRNA was measured. Values represent means ± sd and are normalized by the control value (no treatment). Essentially the same results were obtained in three separate experiments. B, Changes in FGFR mRNA expression during adipocyte differentiation of 3T3-L1 cells. 3T3-L1 fibroblasts were seeded into 35-mm dishes and cultured in differentiation-inducing medium. Every other day, the cells in three dishes were separately harvested, total cellular RNAs were prepared, and the copy number of the expressed mRNA encoding each FGFR was measured. Values represent mean ± sd of triplicate samples and are expressed as copy numbers per microgram of total RNA. Essentially the same results were obtained in two separate experiments. C, Expression of βKlotho and Klotho mRNAs during adipocyte differentiation of 3T3-L1 cells. The copy numbers of the expressed mRNA for βKlotho and Klotho were measured as described in the legend to Fig. 2. Values represent mean ± sd of triplicate samples and are expressed as copy numbers per microgram of total RNA. Essentially the same results were obtained in two separate experiments.
Among the 22 members of the FGF family, FGF21, FGF15/19 and FGF23 comprise the FGF19 subfamily (2). Recently the senescence-related molecule Klotho was reported to function as a coreceptor for FGF23 (10, 11). In vitro pull-down assays revealed that FGF23 binds to Klotho, and Klotho binds to FGFR1c (10, 11) and other FGFRs (10) independently of heparin (11). To test the possibility that Klotho functions as a coreceptor for FGF21, we assessed expression of Klotho mRNA during 3T3-L1 differentiation. Using real-time quantitative PCR, we found Klotho mRNA to be undetectable at all times before and during the differentiation, although it was abundantly expressed in the kidney (more than 3 × 107 copies/μg total RNA; Fig. 1C). The absence of Klotho from the differentiated 3T3-L1 adipocytes indicated that another molecule likely functions as a coreceptor for FGF21 in 3T3-L1 cells. Klotho is a type I membrane protein and is part of a family of proteins that all contain family 1 glycosidase-like domains. We assessed the mRNA expression of one of these, βKlotho, which exhibits 41.2% amino acid identity to Klotho (17). As shown in Fig. 1C, βKlotho mRNA was undetectable in undifferentiated 3T3-L1 fibroblasts, but its expression dramatically increased 6–8 d after the induction of differentiation, reaching a level higher than that in liver (2 × 106 copies/μg mRNA).
We investigated whether βKlotho is able to serve as a coreceptor for FGF21 using a BaF3 cell proliferation assay system that has been used extensively to investigate the activity of a variety of receptor tyrosine kinases. Wild-type BaF3 cells do not endogenously express FGFRs, although BaF3 transfectants expressing exogenous FGFRs can be propagated in the presence of appropriate FGFs. We transfected FGFR1c-expressing BaF3 cells with βKlotho and obtained several stable clones. Consistent with an earlier report (9), FGF21 had no mitogenic effect on BaF3 transfectants expressing FGFR1c alone (Fig. 2A, FGFR1c only and CL no. 10). On the other hand, FGF21 stimulated proliferation of FGFR1c/BaF3 transfectants expressing βKlotho (Fig. 2A, CL no. 1 and CL no. 7). Notably, FGF21 dose-dependently exerted its mitogenic effect even in the absence of heparin, a crucial cofactor necessary for most FGFs to activate their cognate receptors (Fig. 2B). Still, heparin did enhance FGF21’s ability to induce proliferation of FGFR1c/βKlotho-expressing BaF3 transfectants 1.5- to 3-fold (Fig. 2B). This finding is in contrast to the earlier finding that an FGF23-Klotho-FGFR1c complex is formed only in the presence of heparin (11). BaF3 transfectants overexpressing only βKlotho did not respond to FGF21, eliminating the possibility that βKlotho is able to transduce the FGF21 signal (Fig. 2C). These results suggest that βKlotho functions as a coreceptor for FGF21 in FGFR1c-expressing BaF3 cells.
Fig. 2.
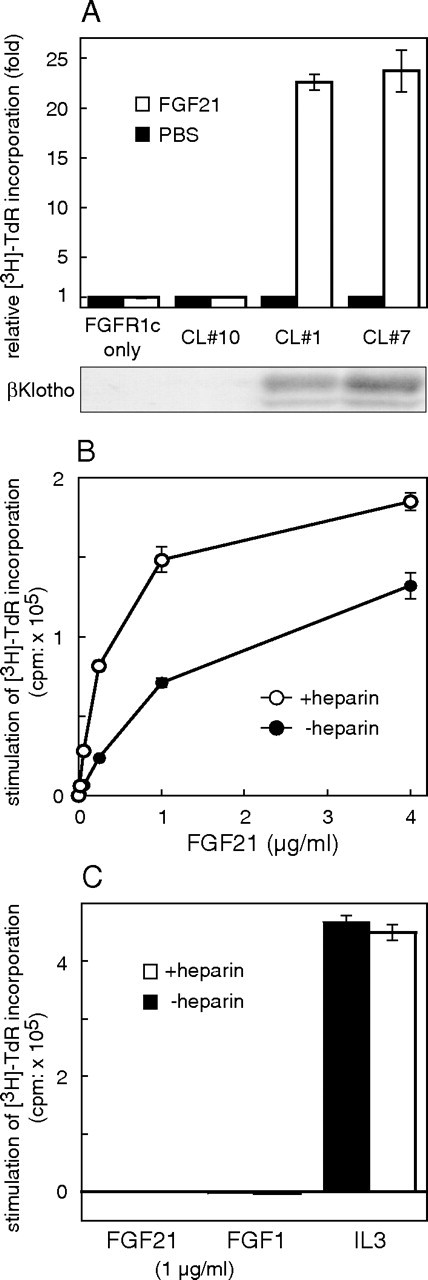
Coexpression of βKlotho Is Required for FGF21 to Stimulate DNA Synthesis in BaF3 Transfectants Expressing FGFR1c
A, BaF3 transfectants expressing FGFR1c were further transfected with a βKlotho expression vector, and stable clones showing different levels of βKlotho expression (shown by immunoblotting in the lower panel) were isolated and examined. Each clone and the parental cells were treated with 1 μg/ml FGF21 or PBS (control) in the presence of 10 μg/ml heparin, and DNA synthesis was analyzed based on [3H]thymidine incorporation. The FGF21-induced increases in DNA synthesis were expressed as fold increases from control (PBS). Each columnrepresents the mean ± sd of triplicate samples. Essentially the same results were obtained in two separate experiments. CL, Clone. B, Clone no. 1, which expressed both FGFR1c and βKlotho, was stimulated with the indicated concentration of FGF21 in the presence or absence of 10 μg/ml heparin, and DNA synthesis was analyzed based on [3H]thymidine incorporation, which was calculated by subtracting the value of the untreated (PBS) sample from those of the FGF-treated ones. Each symbol represents the mean ± sd of triplicate samples. Essentially the same results were obtained in three separate experiments. C, BaF3 cells expressing only βKlotho were treated with FGF21 (1 μg/ml), FGF1 (1 μg/ml), or IL-3-containing WEHI-3-conditioned medium (5%) in the presence or absence of 10 μg/ml heparin, and the DNA synthesis was analyzed based on [3H]thymidine incorporation, which was calculated by subtracting the value of untreated (PBS) sample from those of the FGF- and IL-3-treated ones. Each column represents the mean ± sd of triplicate samples.
In mammals, FGFRs with tyrosine kinase domains are encoded by four genes (2) whose translation products are classified into seven major protein forms, including the IIIb and IIIc splice forms of FGFRs 1–3. We next determined which FGFRs, in addition to FGFR1c, transduce the FGF21 signal in cooperation with βKlotho. BaF3 cells expressing βKlotho were further transfected with an expression plasmid encoding one of the seven FGFR forms, and clones stably expressing both βKlotho and each of the FGFRs were isolated. All of the BaF3 transfectants could be induced to proliferate with FGF1 plus heparin (Fig. 3), indicating that FGFR signaling was operating in these cells. Among the BaF3 transfectants, only those expressing FGFR3c or FGFR1c plus βKlotho proliferated in response to stimulation with FGF21 (Fig. 3). Moreover, this mitogenic activity was increased approximately 2-fold by the presence of heparin.
Fig. 3.
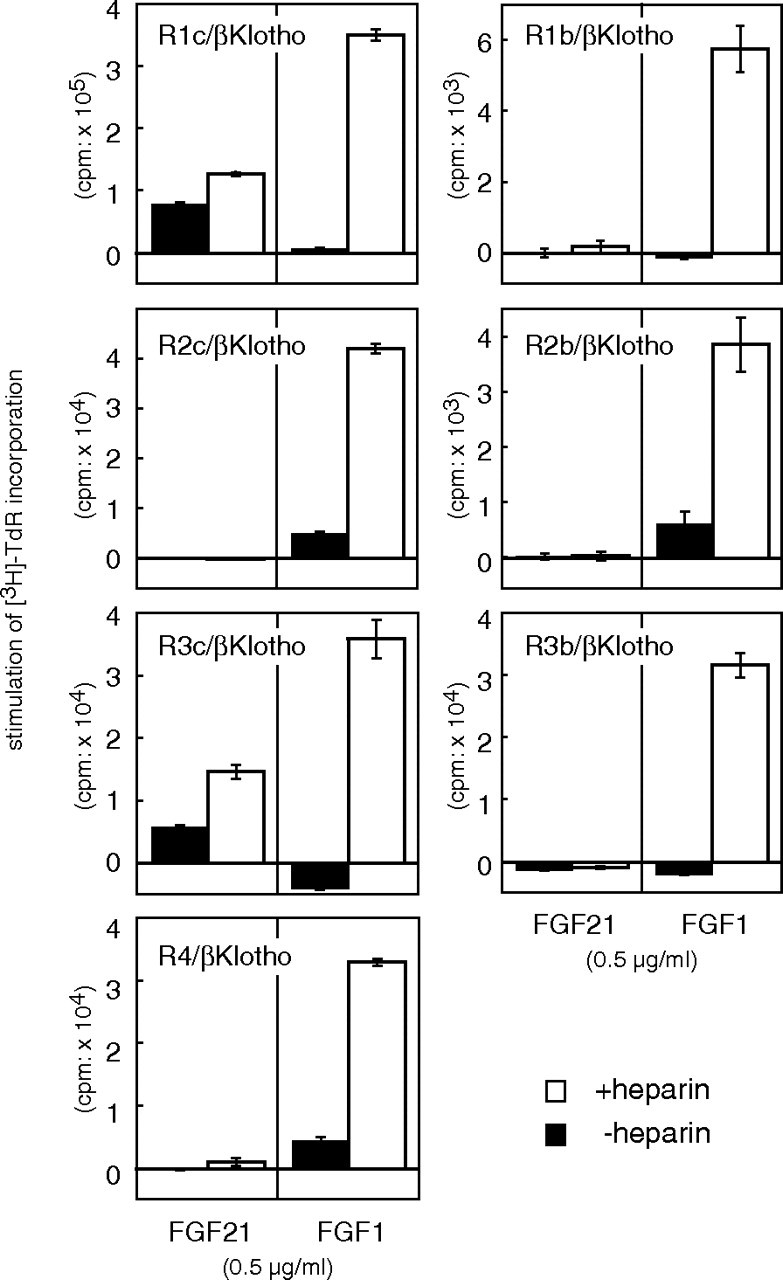
FGFR Specificity of FGF21-Induced DNA Synthesis in βKlotho-Expressing BaF3 Cells
BaF3 cells coexpressing βKlotho and one of the FGFR subtypes were isolated as described in the text and stimulated with 0.5 μg/ml FGF21 in the presence or absence of 10 μg/ml heparin, after which DNA synthesis was analyzed based on [3H]thymidine incorporation, which was calculated by subtracting the value of the untreated (PBS) sample from those of the FGF-treated ones. As a positive control, some cells were stimulated with 0.5 μg/ml FGF1. Each column represents the mean ± sd of triplicate samples. Essentially the same results were obtained using at least two independent clones for each FGFR.
We then analyzed the dose dependence of the responses to FGF21 in BaF3 transfectants expressing βKlotho in combination with the IIIc subtypes of FGFR1–3 or FGFR4 (Fig. 4). FGF1 was included as a positive control to stimulate all the FGFR transfectants, and FGF19 was also evaluated and compared with FGF21 in this experiment. Both FGF19 and FGF21 belong to FGF19 subfamily. We found that FGF21 dose-dependently induced DNA synthesis in FGFR1c/βKlotho/BaF3 and FGFR3c/βKlotho/BaF3 cells but did not induce DNA synthesis in FGFR2c/βKlotho/BaF3 or FGFR4/βKlotho/BaF3 cells (Fig. 4). By contrast, these cells were stimulated fully by FGF19, which induced maximal DNA synthesis at levels comparable to FGF1 in all four transfectants examined. We also examined FGF21-induced phosphorylation of FRS2α and MAPK in the BaF3 transfectants and found that FGF21 induced phosphorylation of both FRS2α and MAPK (Erk1/2) in FGFR1c/βKlotho/BaF3 and FGFR3c/βKlotho/BaF3 cells but not in FGFR2c/βKlotho/BaF3 or FGFR4/βKlotho/BaF3 cells (Fig. 5). We noted, however, that the phosphorylation of FRS2α and MAPK was weaker than that induced by FGF19 or FGF1. This observation was consistent with the results of the DNA synthesis assay summarized in Fig. 4, which confirms that signaling via FGFR1c and FGFR3c, but not FGFR2c or FGFR4, was activated by FGF21 in the presence of βKlotho in BaF3 cells.
Fig. 4.

Dose-Dependent Stimulation of DNA Synthesis by FGF21 in FGFR1c/βKlotho/BaF3 and FGFR3c/βKlotho/BaF3 Cells
BaF3 cells coexpressing βKlotho and one of the FGFR subtypes were treated with the indicated concentrations of FGF21, FGF19, or FGF1 in the presence of 10 μg/ml heparin. DNA synthesis was assessed based on [3H]thymidine incorporation. The incorporation by untreated cells was subtracted from that by FGF-treated cells to calculate stimulated [3H]thymidine incorporation. These values are presented as percentage of control, which was the stimulated [3H]thymidine incorporation in cells treated with 56 ng/ml FGF1. Each symbol represents the mean ± sd of quadruplicate samples. Essentially the same results were obtained with at least two independent clones for each transfectant.
Fig. 5.
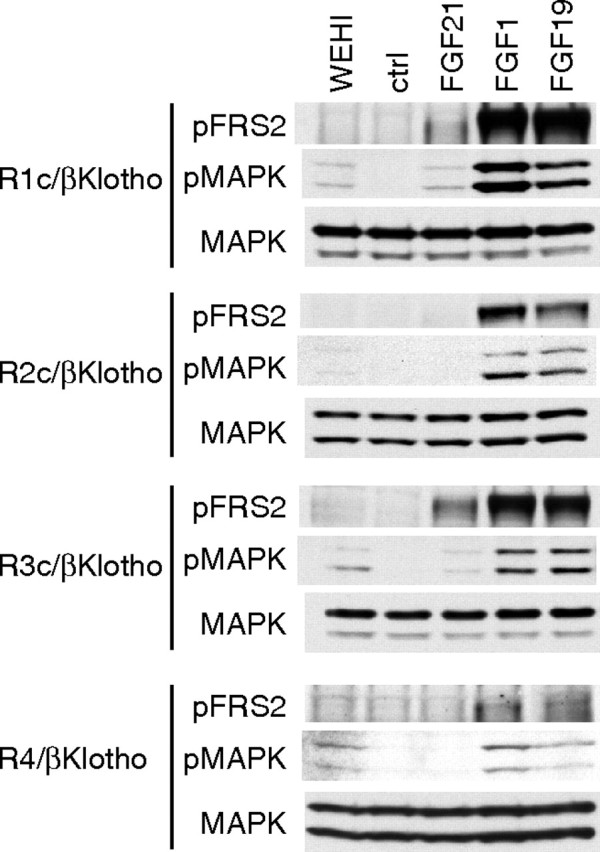
FGF21 Activates FGF Signaling Molecules FRS2α and MAPK in FGFR1c/βKlotho/BaF3 and FGFR3c/βKlotho/BaF3 Cells
BaF3 cells coexpressing βKlotho and one of the FGFR subtypes were stimulated with FGF21 (0.56 μg/ml), FGF19 (0.56 μg/ml), or FGF1 (56 ng/ml) in the presence of 10 μg/ml heparin for 10 min. The negative control cells were untreated (ctrl), and the positive control cells were stimulated with 5% WEHI-3-conditioned medium (WEHI). The cells were then harvested, and activation of the signaling molecules was analyzed by Western blotting using phosphospecific antibodies against FRS2α and MAPK. Essentially the same results were obtained in two separate experiments.
Earlier studies with FGF23 revealed that the Klotho-FGFR1c complex constitutes the functional FGF23 receptor (10, 11). Similarly, we found that the combination of βKlotho and FGFR1c constitutes a functional FGF21 receptor. With that in mind, we tested the functionality of different combinations of these receptor-coreceptor complexes by examining the capacity of FGF21 and FGF23 to stimulate proliferation of BaF3 transfectants expressing βKlotho/FGFR1c or Klotho/FGFR1c. As shown in Fig. 6, βKlotho/FGFR1c-expressing cells but not Klotho/FGFR1c-expressing cells proliferated in response to FGF21 stimulation. Conversely, FGF23 induced proliferation only of Klotho/FGFR1c-expressing cells. Thus, βKlotho and Klotho appear to selectively endow FGFR1c with sensitivity to FGF21 and FGF23, respectively.
Fig. 6.

FGF21 Stimulates DNA Synthesis in BaF3 Cells Coexpressing βKlotho and FGFR1c But Not Klotho and FGFR1c
BaF3 transfectants coexpressing FGFR1c and βKlotho (R1c/βKlotho) or Klotho (R1c/Klotho) were stimulated with 1 μg/ml FGF21 or FGF23 in the presence or absence of 10 μg/ml heparin, after which DNA synthesis was analyzed based on [3H]thymidine incorporation, which was calculated by subtracting the value of the untreated (PBS) sample from those of the FGF-treated ones. Each column represents the mean ± sd of triplicate samples. Essentially the same results were obtained in three separate experiments.
To determine whether βKlotho would also act as an FGFR coreceptor in undifferentiated 3T3-L1 fibroblasts, which expressed little or no endogenous βKlotho mRNA, 3T3-L1 fibroblasts stably expressing exogenous βKlotho were isolated and examined for their response to FGF21. The intracellular FGFR signal is mainly transduced via a specific docking protein, FRS2α and MAPK. We evaluated the activation of FRS2α and MAPK in βKlotho- and mock-transfected 3T3-L1 fibroblasts after confirming expression of βKlotho protein in an immunoblotting analysis (Fig. 7). Tyrosine phosphorylation and the molecular shift caused by threonine phosphorylation of FRS2α (18) were detected in the βKlotho transfectants 15 min after FGF21 treatment, as was phosphorylation of MAPK. Furthermore, the phosphorylation of these proteins was inhibited by SU5402, a specific FGFR tyrosine kinase inhibitor, indicating that FGF21 activates FRS2α and MAPK via FGFR activation. In the mock transfectants, neither FRS2α nor MAPK was activated by FGF21, although they were clearly activated by FGF1. Essentially the same result was obtained using 3T3-L1 fibroblasts transiently expressing βKlotho (data not shown). We also tested whether 3T3-L1 fibroblasts expressing βKlotho alone are able to express GLUT1 mRNA in response to FGF21, as was observed with 3T3-L1 adipocytes (Fig. 1A), which endogenously express βKlotho. As shown in Fig. 8, FGF21 dose-dependently increased expression of GLUT1 mRNA in βKlotho-transfected 3T3-L1 fibroblasts but not in mock-transfected fibroblasts. Thus, βKlotho endowed 3T3-L1 fibroblasts with the ability to respond to FGF21.
Fig. 7.

FGF21 Activates the FGF Signaling Molecules FRS2α and MAPK in 3T3-L1 Fibroblasts Expressing βKlotho
3T3-L1 fibroblasts stably expressing βKlotho (βKlotho/3T3-L1 fibroblasts) were serum starved in OptiMEM-I medium for 18 h, incubated with or without 10 μm SU5402 for 3 h, and then stimulated with 1.1 or 4.5 μg/ml FGF21 for 15 min. The cells were then harvested, and activation of the signaling molecules was analyzed using phosphospecific antibodies against FRS2α and MAPK. FGF1 (18 ng/ml) and PBS served as positive and negative controls, respectively. Essentially the same results were obtained in two separate experiments.
Fig. 8.

FGF21 Increases Expression of GLUT1 mRNA in 3T3-L1 Fibroblasts Expressing βKlotho
3T3-L1 fibroblasts stably overexpressing βKlotho (βKlotho/3T3-L1 fibroblasts) were treated with the indicated concentrations of FGF21 for 6 h, after which the copy number of the expressed GLUT1 mRNA was measured by quantitative real-time PCR. Values represent the mean ± sd of quadruplicate samples and are normalized by the control value (no treatment). *, P < 0.05; **, P < 0.01 (Student’s t test). Essentially the same results were obtained in two separate experiments.
While this manuscript was in preparation, Ogawa et al. (19) also reported that βKlotho is required for FGF21 activity. They demonstrated that knockdown of βKlotho expression using small interfering RNA in adipocytes diminished FGF21-induced glucose uptake. In addition, they showed that activation of MAPK induced by administration of FGF21 was restricted to tissues expressing βKlotho. These data clearly demonstrate the physiological importance of βKlotho for FGF21 activity. But although those investigators examined interactions among FGFRs, βKlotho, and FGF21 using immunoprecipitation and immunoblotting techniques, they did so in HEK293 cells, which, unlike BaF3 cells, endogenously express FGFRs (10) and heparan sulfate (20). They showed that FGFR1c and FGFR4, but not FGFR3c, bind to βKlotho and that FGF21 binds to the resultant complexes. In their experiments, however, the expressed level of βKlotho in the FGFR3c/βKlotho HEK293 cell transfectants was much lower than in the other transfectants, raising the possibility that the amount of βKlotho was undetectable after coprecipitation with FGFR3c.
After this manuscript was reviewed, a new report on FGF21 and its signaling in L6 cells was published (21). Using L6 transfectants overexpressing βKlotho, those investigators showed that FGFR3c signaling is activated in response to FGF21, as indicated by the phosphorylation of FRS2α. This confirms our observation of FGF21-induced FGFR3c signaling.
With regard to FGFR2, it has been reported FGF21 induces phosphorylation of FRS2α in both FGFR2c/βKlotho-expressing 3T3-L1 adipocytes and FGFR2c/βKlotho-transfected L6 cells (21, 22). We extensively examined several independent FGFR2c/βKlotho/BaF3 cell clones and found that FGF21 induced little or no DNA synthesis in these cells, even at the highest concentration tested (in Fig. 4D, <9% of the DNA synthesis induced by FGF19; several other clones did not respond at all, data not shown). We also tested whether inclusion of heparan sulfate (from bovine kidney) would influence the DNA synthesis in FGFR2c/βKlotho/BaF3 cells and found that FGF21 did not effectively induce DNA synthesis in these cells, even in the presence of up to 100 μg/ml heparan sulfate. Because 3T3-L1 and L6 cells endogenously express several subtypes of FGFR and heparan sulfate proteoglycan (described below), activation of FGFR2 on these cells could involve cross-talk between other FGFR subtypes or heparan sulfate sugar chains with cell-specific structures.
It also has been shown that although FGFR4 signaling is not activated in L6 transfectants overexpressing βKlotho, FGFR4 coprecipitates with FGF21 and βKlotho (21), which is consistent with our findings. Similarly, in liver, which abundantly expresses both FGFR4 and βKlotho, FGF21 does not induce phosphorylation of MAPK, although FGFR4 constitutively binds to βKlotho in liver lysates (21). Thus, the previously reported specificity of receptor binding to βKlotho (19) does not directly correlate with the specificity of receptor activation by FGF21 in the presence of βKlotho. The reason FGF21 does not activate FGFR4 remains unclear, however.
Although L6 cells are generally thought to endogenously express FGFRs at very low levels, we previously showed that the level of their expression is sufficient to mediate transduction of signals leading to suppression of myogenic differentiation of the cells and that these signals were quenchable with a dominant-negative FGFR1c mutant (23). Notably, L6 cells also express heparan sulfate (24). In the present study, we used BaF3 cells, which are free of endogenous FGFRs and heparan sulfate, which enabled us to express each FGFR separately, with or without coexpression of βKlotho, and with or without heparin or heparan sulfate. We found that, even in the presence of βKlotho, heparin potentiated the responses of FGFR1c and FGFR3c to FGF21, but how heparin contributes to the formation of FGF21/βKlotho/FGFR complexes remains unclear. We suggest that structural microheterogeneity of heparan sulfate could play a role in determining the ligand-receptor specificity for FGF21, as has been shown for other FGF family members (25). We found that FGF21 does not bind to heparin in physiological saline (data not shown). This feature may allow high levels of FGF21 to be sustained in the circulation under pathophysiological conditions. Similarly, FGF23 shows only weak affinity for heparin, so that once released from their expressing cells, both FGF21 and FGF23 may have access to various organs, where they could act in an endocrine fashion. In that context, we would expect that the crucial requirement for βKlotho as a coreceptor for FGF21, or Klotho for FGF23, is a key determinant of their respective target tissues when the tissues express FGFR subtypes that respond to both (e.g. FGFR1c) (Fig. 6).
The pancreas may be another organ in which βKlotho serves as a coreceptor for FGF21 because it expresses βKlotho at a high level (17) and FGF21 improves pancreatic β-cell function and survival (26). While in the liver, βKlotho likely serves as a coreceptor for FGF15/19 and FGFR4. βKlotho expression and activity in the liver have been reported (17, 27). When fed an atherogenic diet, βklotho−/− mice exhibited increased hepatic expression of Cyp7a1, a key bile acid synthetic enzyme; increased levels of bile acid; and increased resistance to gallstone formation. Similarly, FGFR4 is abundant in liver, and Fgfr4−/− exhibit increased bile acid synthesis and small gallbladders, suggesting that a βKlotho-FGFR4 system suppresses bile acid synthesis. Although FGF21 is expressed in the livers of normal mice (5), it is an unlikely ligand in this system because it does not activate FGFR4, even in the presence of βKlotho (Figs. 3 and 4). Instead, FGF15/19 appears to be the active ligand, because it is released from intestine (4), activates FGFR4, and suppresses Cyp7a1 expression (4, 28). A recent report by Kurosu et al. (21) demonstrating that administration of FGF19, but not FGF21, to mice activates MAPK in the liver strongly supports this idea.
Finally, given that FGF21 stimulates glucose uptake by adipocytes and lowers serum glucose and triglyceride levels, our findings, together with the latest publications, imply that βKlotho may be a novel target for development of therapeutic agents for the treatment of diabetes.
MATERIALS AND METHODS
Culture of 3T3-L1 Cells and Differentiation
3T3-L1 cells were grown and maintained in DME high glucose medium (Sigma-Aldrich, St. Louis, MO) containing 10% fetal bovine serum (FBS; Sigma-Aldrich) and 50 μg/ml gentamicin (Sigma-Aldrich) in a 5% CO2 environment. The cells were allowed to grow until postconfluent d 2 and were then induced to differentiate by incubation for 48 h in differentiation medium [growth medium plus 0.5 mm 3-isobutyl-1-methylxanthine (Sigma-Aldrich), 0.25 μm dexamethasone (Sigma-Aldrich), and 5 μg/ml insulin (Sigma-Aldrich)]. Thereafter, the medium was changed to maturation medium (growth medium supplemented only with 2.5 μg/ml insulin), which was subsequently replaced with fresh maturation medium every 2 d.
Plasmid Preparation
cDNAs encoding mouse FGF21 and human FGF19 and FGF23 were, respectively, amplified from mouse liver and SW480 and K562 cDNAs (Marathon-Ready cDNA; BD Bioscience/Clontech, Palo Alto, CA) by PCR using specific reverse primers containing an additional DNA sequence encoding a C-terminal His tag (TGHHHHHH) and then cloned into pcDNA3.1 vector (Invitrogen, Carlsbad, CA). An expression plasmid encoding the native form of FGF21, without the His tag, was also constructed using a specific reverse primer without the extra DNA sequence. FGF23R179Q, a proteolysis-resistant FGF23 mutant (29), was generated by PCR mutagenesis. cDNAs encoding the full-length mouse βKlotho and Klotho were, respectively, amplified from liver and kidney cDNAs by PCR and cloned into the pcDNA3.1/Pur(+) constructed based on pcDNA3.1 vector (30). To add the Flag and His tags tandemly at the C terminal of each protein, a DNA fragment encoding the amino acid sequence ARDYKDHDGDYKDHDIDYKDDDDKGHHHHHH was introduced into the XhoI-ApaI site of the βKlotho and Klotho expression vectors. MIRB expression vectors for FGFR1b (human), -R1c (mouse), -R2b (human), -R2c (mouse), -R3b (mouse), and -R3c (mouse) were kind gifts from Dr. David M. Ornitz (31). To facilitate mitogenic signaling in BaF3 cells, the transmembrane and intracellular tyrosine kinase domains of FGFR3b and -R3c were substituted with those of FGFR1c, as previously described (31). The amino acid sequence used to join FGFR3b (in bold) to FGFR1c (plain) was GPQAAEEELMETDEAGSLYLEIIIYCTGA; that used to join FGFR3c (bold) to FGFR1 (plain) was LVVLPAEEELMETDEAGSLYLEIIIYCTGA (the transmembrane domains are underlined). cDNA encoding the extracellular domain of mouse FGFR4 was amplified from a liver cDNA library and fused to the cDNA containing the transmembrane and cytoplasmic domains of the FGFR1c by PCR and cloned into the MIRB expression vector to produce the chimeric receptor FGFR4R1. The amino acid sequence used to join FGFR4 (bold) to FGFR1 (plain) was PEARYTDIIIYCTGA (the transmembrane domain is underlined).
Recombinant FGF Production
COS1 cells were cultured in DMEM (Sigma-Aldrich) supplemented with 10% FBS and 60 μg/ml kanamycin (GIBCO/Invitrogen). Cells were transiently transfected with each expression vector using Lipofectamine 2000 (Invitrogen) and cultured in a serum-free medium (OptiMEM-I; GIBCO/Invitrogen). Conditioned media were then collected every other day for 6 d, after which His-tagged FGFs secreted into the media were partially purified using Ni-beads (Ni-Sepharose 6 Fast Flow; GE Healthcare Bio-Sciences, Piscataway, NJ). FGF21 was further purified by reversed-phase column chromatography using a C18 column (GraceVydac, Hesperia, CA). Purified FGF21 and partially purified FGF23 were dialyzed against PBS and stored at −80 C until use.
Real-Time PCR Analysis
Total cellular RNA was isolated using Isogen (Nippon Gene, Toyama, Japan) according to the manufacturer’s protocol. First-strand cDNA was synthesized from 2 μg total RNA using SuperScript II with oligo(dT)12–14 (Invitrogen). Quantitative real-time PCR analysis of 3T3-L1 cell cDNAs was performed using the LightCycler (Roche Diagnostics, Mannheim, Germany) with Quantitect SYBR Green PCR mix (QIAGEN, Valencia, CA) as described previously (32). To verify the specificity of the PCR, melting-curve analysis was performed at the end of individual runs, and aliquots of the PCR products were analyzed by agarose gel electrophoresis. The specific primers were designed using a method developed by Prof. Akira Suyama (33). The forward and reverse primer sequences for each gene product were as follows: GLUT1, 5′-GGC TGC CTT GGA TGT CCT ATC TGA GCA TCG-3′ and 5′-AGA TGA TGA AGA CGT AGG GGC CGC ACA GTT-3′ (PCR fragment, 231 bp); FGFR1, 5′-CCA GTG CAT CCA TGA ACT CTG GGG TTC TCC-3′ and 5′-GGT CAC ACG GTT GGG TTT GTC CTT ATC CAG-3′ (PCR fragment, 230 bp); FGFR2, 5′-TGC CAC AGA GAA GGA CCT GTC TGA TCT GGT-3′ and 5′-GCT CCT CGG GGA CAC GGT TAA TGT CAT AGG-3′ (PCR fragment, 224 bp); FGFR3, 5′-GCG ACA GGT GTC CTT GGA ATC TAA CTC CTC-3′ and 5′-CCA ATA GCT TCT GCC ATG ACC ACC TGT CCA-3′ (PCR fragment, 219 bp); FGFR4, 5′-GAC CAA ACC AGC ACC GTG GCT GTG AAG ATG-3′ and 5′-GTT TCC CTT GGC GGC ACA TTC CAC AAT CAC-3′ (PCR fragment, 189 bp); Klotho, 5′-ATA CGA CCC GAC GAG GGC TGT TTT ATG TGG-3′ and 5′-ATA CAG GTG AGG ATC GGT AAA CTG CGG GGA-3′ (PCR fragment, 239 bp); and βKlotho, 5′-CTG GCT AAG GTT CAA GTA CGG AGA CCT CCC-3′ and 5′-GGA GCT GAG CGA TCA CTA AGT GAA TAC GCA-3′ (PCR fragment, 198 bp). To generate the reference standards for the quantitative real-time PCR experiments, PCR of the mouse tissue cDNAs was performed using each primer pair with KOD-plus-polymerase (Toyobo, Osaka, Japan), after which the amplified fragments were purified and cloned using the TOPO cloning system (pCR-Blunt II-TOPO; Invitrogen), and the cloned DNA sequences were verified.
Transfection of BaF3 Cells and DNA Synthesis Assay
BaF3 cells were maintained in RPMI 1640 (Sigma-Aldrich) medium supplemented with IL-3 (as 10% WEHI-3-conditioned medium) and 10% FBS. Cells were transfected with an FGFR, βKlotho, or Klotho expression vector, or a combination of these vectors, by electroporation [Bio-Rad (Hercules, CA) Gene Pulser II]. Transfectants expressing FGFR alone were selected by incubation in the presence of 1 mg/ml G418 and maintained in 0.6 mg/ml G418. Those expressing βKlotho or Klotho were selected and maintained in 1.5 μg/ml puromycin. Those expressing βKlotho or Klotho in combination with FGFR were selected and maintained in G418 and puromycin. DNA synthesis was assayed essentially as described previously (31). Briefly, the BaF3 transfectants were washed and plated to a density of 10,000 per well in 96-well plates. The cells were then stimulated with FGFs, with or without 10 μg/ml heparin, in a total volume of 200 ml/well and incubated at 37 C for 42 h. To verify the functionality of the FGFR signaling pathway in each transfectant, FGF1 (purified from bovine brains; R&D Systems, Minneapolis, MN) was used as a positive control. The cells were labeled by adding 37 kBq/well [3H]thymidine and were harvested 4 h later by filtration through glass fiber paper. The radioactivity incorporated into the DNA was counted on a scintillation counter (Wallac β-plate; PerkinElmer Life Sciences, Norwalk, CT).
Immunoblotting
The cells were harvested in a lysis buffer [50 mm Tris (pH 7.4), 150 mm NaCl, 1% Triton X-100, 5 mm EDTA] supplemented with a protease inhibitor cocktail (Roche Diagnostics) and a phosphatase inhibitor cocktail (Pierce, Rockford, IL), after which immunoblotting was performed using equal amounts of protein (50–100 μg) and the following primary antibodies: anti-phospho-FRS2α (Tyr196, no. 3864; Cell Signaling Technology, Beverly, MA), anti-phospho-Erk1/2 (Thr202/Tyr204, no. 9106; Cell Signaling), anti-Erk1/2 (no. 610030; BD Bioscience/Clontech), and anti-βKlotho (R&D Systems) and the following horseradish peroxidase-conjugated secondary antibodies: goat antimouse IgG (no. AP181P; Chemicon, Temecula, CA), goat antirabbit IgG [no. 074–1506; Kirkegaard & Perry Laboratories (KPL), Gaithersburg, MD], and rabbit antigoat IgG (no. 81–1620; Zymed, San Francisco, CA). The signals were detected using ECL Western blotting detection reagents (GE Healthcare Bio-Sciences) following the manufacturer’s instructions.
Acknowledgments
We thank E. Honda, S. Togayachi, and N. Watanabe for their technical assistance.
Footnotes
This work was supported in part by a National Institute of Advanced Industrial Science and Technology grant and by the Budget for Nuclear Research of the Ministry of Education, Culture, Sports, Science, and Technology.
Disclosure Statement: All authors have nothing to disclose.
First Published Online January 10, 2008
Abbreviations: FBS, Fetal bovine serum; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; FRS2, fibroblast growth factor receptor substrate 2; GLUT, glucose transporter.
References
- 1.Ornitz DM, Itoh N 2001. Fibroblast growth factors. Genome Biol 2:REVIEWS3005 [DOI] [PMC free article] [PubMed]
- 2.Itoh N, Ornitz DM 2004. Evolution of the Fgf and Fgfr gene families. Trends Genet 20:563–569 [DOI] [PubMed] [Google Scholar]
- 3.Yu X, White KE 2005. FGF23 and disorders of phosphate homeostasis. Cytokine Growth Factor Rev 16:221–232 [DOI] [PubMed] [Google Scholar]
- 4.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA 2005. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2:217–225 [DOI] [PubMed] [Google Scholar]
- 5.Nishimura T, Nakatake Y, Konishi M, Itoh N 2000. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta 1492:203–206 [DOI] [PubMed] [Google Scholar]
- 6.Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, Gromada J, Brozinick JT, Hawkins ED, Wroblewski VJ, Li DS, Mehrbod F, Jaskunas SR, Shanafelt AB 2005. FGF-21 as a novel metabolic regulator. J Clin Invest 115:1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, Elmquist JK, Gerard RD, Burgess SC, Hammer RE, Mangelsdorf DJ, Kliewer SA 2007. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab 5:415–425 [DOI] [PubMed] [Google Scholar]
- 8.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E 2007. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 5:426–437 [DOI] [PubMed] [Google Scholar]
- 9.Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM 2006. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem 281:15694–15700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M 2006. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281:6120–6123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T 2006. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444:770–77417086194 [Google Scholar]
- 12.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI 1997. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390:45–51 [DOI] [PubMed] [Google Scholar]
- 13.Yoshida T, Fujimori T, Nabeshima Y 2002. Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1α-hydroxylase gene. Endocrinology 143:683–689 [DOI] [PubMed] [Google Scholar]
- 14.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, Fukumoto S, Tomizuka K, Yamashita T 2004. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest 113:561–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eswarakumar VP, Lax I, Schlessinger J 2005. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 16:139–149 [DOI] [PubMed] [Google Scholar]
- 16.Moyers JS, Shiyanova TL, Mehrbod F, Dunbar JD, Noblitt TW, Otto KA, Reifel-Miller A, Kharitonenkov A 2007. Molecular determinants of FGF-21 activity-synergy and cross-talk with PPARγ signaling. J Cell Physiol 210:1–6 [DOI] [PubMed] [Google Scholar]
- 17.Ito S, Kinoshita S, Shiraishi N, Nakagawa S, Sekine S, Fujimori T, Nabeshima YI 2000. Molecular cloning and expression analyses of mouse βklotho, which encodes a novel Klotho family protein. Mech Dev 98:115–119 [DOI] [PubMed] [Google Scholar]
- 18.Lax I, Wong A, Lamothe B, Lee A, Frost A, Hawes J, Schlessinger J 2002. The docking protein FRS2α controls a MAP kinase-mediated negative feedback mechanism for signaling by FGF receptors. Mol Cell 10:709–719 [DOI] [PubMed] [Google Scholar]
- 19.Ogawa Y, Kurosu H, Yamamoto M, Nandi A, Rosenblatt KP, Goetz R, Eliseenkova AV, Mohammadi M, Kuro-o M 2007. βKlotho is required for metabolic activity of fibroblast growth factor 21. Proc Natl Acad Sci USA 104:7432–7437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung WF, Eriksson I, Kusche-Gullberg M, Lindhal U, Kjellen L 1996. Expression of the mouse mastocytoma glucosaminyl N-deacetylase/N-sulfotransferase in human kidney 293 cells results in increased N-sulfation of heparan sulfate. Biochemistry 35:5250–5256 [DOI] [PubMed] [Google Scholar]
- 21.Kurosu H, Choi M, Ogawa Y, Dickson AS, Goetz R, Eliseenkova AV, Mohammadi M, Rosenblatt KP, Kliewer SA, Kuro-o M 2007. Tissue-specific expression of βKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem 282:26687–26695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kharitonenkov A, Dunbar JD, Bina HA, Bright S, Moyers JS, Zhang C, Ding L, Micanovic R, Mehrbod SF, Knierman MD, Hale JE, Coskun T, Shanafelt AB 2008. FGF-21/FGF-21 receptor interaction and activation is determined by βKlotho. J Cell Physiol 215:1–7 [DOI] [PubMed] [Google Scholar]
- 23.Uruno T, Oki J, Ozawa K, Miyakawa K, Ueno H, Imamura T 1999. Distinct regulation of myoblast differentiation by intracellular and extracellular fibroblast growth factor-1. Growth Factors 17:93–113 [DOI] [PubMed] [Google Scholar]
- 24.Schubert D, LaCorbiere M 1980. Altered collagen and glycosaminoglycan secretion by a skeletal muscle myoblast variant. J Biol Chem 255:11557–11563 [PubMed] [Google Scholar]
- 25.Allen BL, Filla MS, Rapraeger AC 2001. Role of heparan sulfate as a tissue-specific regulator of FGF-4 and FGF receptor recognition. J Cell Biol 155:845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wente W, Efanov AM, Brenner M, Kharitonenkov A, Koster A, Sandusky GE, Sewing S, Treinies I, Zitzer H, Gromada J 2006. Fibroblast growth factor-21 improves pancreatic β-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways. Diabetes 55:2470–2478 [DOI] [PubMed] [Google Scholar]
- 27.Ito S, Fujimori T, Furuya A, Satoh J, Nabeshima Y, Nabeshima Y 2005. Impaired negative feedback suppression of bile acid synthesis in mice lacking βKlotho. J Clin Invest 115:2202–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, Goodwin B, Jones SA 2003. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev 17:1581–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T 2002. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo Endocrinology 143:3179–3182 [DOI] [PubMed] [Google Scholar]
- 30.Asada M, Honda E, Imamura T 2006. Construction of pcDNA3.1-based vectors with blasticidin and puromycin resistance markers. Anal Biochem 352:305–307 [DOI] [PubMed] [Google Scholar]
- 31.Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M 1996. Receptor specificity of the fibroblast growth factor family. J Biol Chem 271:15292–15297 [DOI] [PubMed] [Google Scholar]
- 32.Komi-Kuramochi A, Kawano M, Oda Y, Asada M, Suzuki M, Oki J, Imamura T 2005. Expression of fibroblast growth factors and their receptors during full-thickness skin wound healing in young and aged mice. J Endocrinol 186:273–289 [DOI] [PubMed] [Google Scholar]
- 33.Kurata K, Suyama A 1999. Probe design for DNA chips. Genome Informatics 10:225–226 [Google Scholar]