

Abstract
Glucocorticoids (GCs) play a key role in skin homeostasis and stress responses acting through the GC receptor (GR), which modulates gene expression by DNA binding-dependent (transactivation) and -independent (transrepression) mechanisms. To delineate which mechanisms underlie the beneficial and adverse effects mediated by GR in epidermis and other epithelia, we have generated transgenic mice that express a mutant GR (P493R, A494S), which is defective for transactivation but retains transrepression activity, under control of the keratin 5 promoter (K5-GR-TR mice). K5-GR-TR embryos exhibited eyelid opening at birth and corneal defects that resulted in corneal opacity in the adulthood. Transgenic embryos developed normal skin, although epidermal atrophy and focal alopecia was detected in adult mice. GR-mediated transrepression was sufficient to inhibit keratinocyte proliferation induced by acute and chronic phorbol 12-myristate 13-acetate exposure, as demonstrated by morphometric analyses, bromodeoxyuridine incorporation, and repression of keratin 6, a marker of hyperproliferative epidermis. These antiproliferative effects were mediated through negative interference of GR with MAPK/activator protein-1 and nuclear factor-κB activities, although these interactions occurred with different kinetics. However, phorbol 12-myristate 13-acetate-induced inflammation was only partially inhibited by GR-TR, which efficiently repressed IL-1β and MMP-3 genes while weakly repressing IL-6 and TNF-α. Our data highlight the relevance of deciphering the mechanisms underlying GR actions on epithelial morphogenesis as well as for its therapeutic use to identify more restricted targets of GC administration.
GLUCOCORTICOID (GC) HORMONES, acting through the GC receptor (GR), mediate profound and diverse physiological effects in vertebrate development, metabolism, neurobiology, and programmed cell death (1). GR is a ligand-activated transcription factor, which can modulate gene expression through mechanisms that are DNA-binding dependent (transactivation) and independent (transrepression). These two mechanisms can be genetically separated, and it was demonstrated that, contrary to GR−/− mice that die perinatally, mouse mutants with a point mutation in the DNA-binding domain of GR (GRdim/dim), unable to homodimerize and thus transactivate, were viable (2, 3). These mouse models revealed that the DNA-binding-dependent actions of GR are not essential for survival and allowed assignment of specific roles to the transactivation and transrepression functions of GR in different physiopathological systems (4).
GR signaling is key in early life because prenatal GCs alter the rate of maturation of organs such as the lung, heart, kidney, gut, and epidermis (5); accordingly, GR−/− mice died due to a respiratory failure. These early GC effects are routinely exploited in the clinic by antenatal administration of corticosteroid to premature infants to accelerate lung epithelium differentiation as well as skin barrier formation (reviewed in Ref. 6). Despite these beneficial effects, it is well known that maternal exposure to GCs causes birth defects by mechanisms that are still not well understood (5). We have previously reported that overexpression of wild-type (WT) GR in keratinocytes of the epidermis, hair follicles (HFs), and other stratified epithelia by means of the keratin k5 promoter (K5-GR mice) elicited major phenotypic alterations including epidermal hypoplasia, dysplastic HFs, incomplete closure of the eyelids, and maldevelopment of tooth, palate, and secretory glands (7, 8, 9). Altogether, it seems that a precise spatiotemporal expression pattern of GR and its ligand is required to modulate many morphogenetic processes, particularly epithelial morphogenesis.
On the other hand, GC analogs have been prescribed for more than 50 yr as a treatment for numerous disorders involving hyperproliferation and inflammation due to their therapeutic efficacy. However, the adverse side effects associated with the long-term treatments and/or elevated doses of GCs limit their clinical applications (1, 4). It has been long assumed that the therapeutic potential of GC analogs is exerted mostly through the transrepression function of GR, whereas adverse side effects are predominantly mediated by the transactivation function of GR (1, 10).
In an attempt to delineate the mechanisms responsible for the beneficial vs. adverse effects of GR in epidermis and other epithelia, we have generated a novel transgenic mouse model by expressing a mutant GR (P493R, A494S), which is unable to dimerize and transactivate but retains transrepression activity, under control of the k5 promoter (K5-GR-TR mice). Our data show that the transrepression function of GR was sufficient to cause eyelid opening at birth and corneal defects that resulted in corneal opacity in the adulthood. K5-GR-TR embryos developed normal skin, but adult mice exhibited epidermal atrophy, patchy alopecia, and underdeveloped vibrissae.
In adult transgenic mice, GR-TR was sufficient to inhibit keratinocyte proliferation induced by acute and chronic phorbol 12-myristate 13-acetate (PMA) exposure, as shown by morphometric analyses, bromodeoxyuridine (BrdU) in vivo incorporation, and drastic inhibition of the marker of hyperproliferative epidermis, keratin 6 (K6). GR-TR inhibited keratinocyte growth by interfering with MAPK/activator protein-1 (AP-1) and nuclear factor-κB (NF-κB) activities. In contrast, PMA-induced inflammation was not efficiently inhibited by GR-TR, which down-regulated IL-1β and MMP-3 but repressed IL-6 and TNF-α only weakly. This transgenic mouse model opens new perspectives in deciphering the mechanisms responsible for GR impact on epithelial morphogenesis and also may help to identify more restricted targets of GC administration in the treatment of cutaneous pathologies.
RESULTS
Generation of Skin-Targeted Mice Expressing the Transrepression Function of GR
We aimed to investigate the consequences of expressing a mutant GR in skin, which is defective in transactivation but effective in transrepression. To do this, we placed a mutant GR containing two adjacent amino acid substitutions (P493R, A494S) in the second half of the second zinc finger (11) under control of the keratinocyte-specific k5 regulatory sequences (Fig. 1A). To test the functionality of this construct in the keratinocyte cell line PB, we transiently transfected either K5-GR-TR, K5-GR (containing the WT form of GR under the k5 promoter) (7) or an empty vector along with the reporter plasmids MMTV-CAT or 8x-κB-CAT and evaluated CAT activity. Contrary to K5-GR, K5-GR-TR was unable to transactivate the MMTV promoter upon dexamethasone (Dex) treatment, whereas both constructs efficiently transrepressed a κB-containing promoter (Fig. 1B, upper panels). These results were not due to the different levels of transfected proteins because equivalent amounts of the WT and mutant GR were detected by immunoblotting using an anti-GR antibody (Fig. 1B, lower panel). We then microinjected the K5-GR-TR construct and obtained three founder transgenic mice (298, 306, and 599), of which two independent lines (298 and 306) were established. Transgene expression was analyzed by RT-PCR, immunoblotting, and immunolocalization (Fig. 1, C–E). RT-PCR analyses using specific primers distinguished the endogenous (mouse GR) and transgene (rat GR) transcripts in dorsal skin of WT and K5-GR-TR adult littermates (Fig. 1C, line 298). Transgene mRNA was exclusively present in K5-GR-TR mouse skin, whereas similar amounts of endogenous GR mRNA were detected in both WT and transgenic mice. At the protein level, line 298 showed the highest relative transgene expression in tail skin from founder mice as well as in back skin from adult transgenic mice of the F1 generation, as shown by immunoblotting (Fig. 1D). We thus performed most of the experiments in line 298, although results were also confirmed in line 306. Immunolocalization of GR in dorsal skin paraffin sections from newborn WT and transgenic mice demonstrated that endogenous GR was localized in the cytoplasm of suprabasal epidermal cells (Fig. 1E), whereas positive GR staining was additionally observed in interfollicular basal epidermis and outer root sheath of HFs in transgenic mice (Fig. 1E, arrows). Transgene GR-TR was predominantly localized in the nucleus of keratinocytes, as observed for K5-GR transgenic mice (7).
Fig. 1.

Generation of K5-GR-TR Transgenic Mice Expressing a Mutant GR in Keratinocytes that Is Defective in Transactivation but Effective in Transrepression
A, Scheme of the construct K5-GR-TR. The cDNA of GR containing a double mutation (P493R, A494S) was placed under control of bovine K5 (bk5) regulatory sequences. B, PB keratinocytes were transiently transfected with either MMTV-CAT or 8x-κB-CAT reporter plasmids along with expression vectors encoding for empty vector (pcDNA3), K5-GR, or K5-GR-TR. After depletion of endogenous GCs, cells were treated with either vehicle or 1 μm Dex for 6 h to determine MMTV-CAT activity. In transfections using 8x-κB-CAT reporter, vehicle or TNF-α (10 ng/ml) were added for 4 h before measuring CAT activity (a.u, arbitrary units). Data presented are average of three independent experiments, each performed in triplicate. Error bars indicate sd. Asterisks denote statistically significant differences relative to pCDNA3-transfected cells, as analyzed by Student’s t test (*, P < 0.0005). Expression levels for the WT and mutant GR upon transfection were checked by immunoblotting (lower panel). C, RT-PCR analysis showing the endogenous [mouse GR (mGR)] and transgene [rat GR (rGR)] transcript levels in the skin of WT and K5-GR-TR adult littermates. D, Immunoblotting using whole-cell extracts from tail and dorsal skin of three transgenic founders (298, 306, and 599) to check the relative expression levels of GR. E, Immunolocalization of GR in dorsal skin paraffin sections from newborn WT and transgenic mice. Arrows indicate the nuclear localization of GR in the interfollicular basal epithelium and the outer root sheath of HFs in transgenic skin. Scale bar, 25 μm.
GR Regulates Embryonic Eyelid Closure through Its Transrepression Function
K5-GR-TR transgenic mice were viable and macroscopically normal, except that approximately 5% of newborn heterozygous (HT) mice showed eyelid opening at birth (EOB). When homozygous (HM) mice were generated, a fully penetrant EOB phenotype was observed, and late embryos and newborn mice could be readily identified by this phenotype (Fig. 2 and data not shown). Histological analysis revealed that eyelid fusion had failed in transgenic mice, whereas the developing eyelids of control embryos had already fused at this stage (Fig. 2B). Figure 2B shows immunostaining using K5 to demonstrate that transgene expression was targeted to cornea (a and d), conjunctiva (b and e), and eyelid epithelia (c and f). Remarkably, corneal epithelium was almost absent in the transgenics (Fig. 2B, compare a and d), and the leading edge of the eyelid epithelium had a more restricted expression of K5 (Fig. 2B, compare c and f); however, K5 staining in the conjunctival epithelium was similar between controls and transgenics (Fig. 2B, b and e). In many cases, we observed microphthalmia and dysplastic retinal tissue, most likely owing to a secondary effect of eye exposure in utero (12). K5-GR-TR late embryos also had additional ocular malformations with severe cornea defects and attachment of eyelid epithelium tissue to the posterior side of the cornea (Fig. 2B, arrow).
Fig. 2.
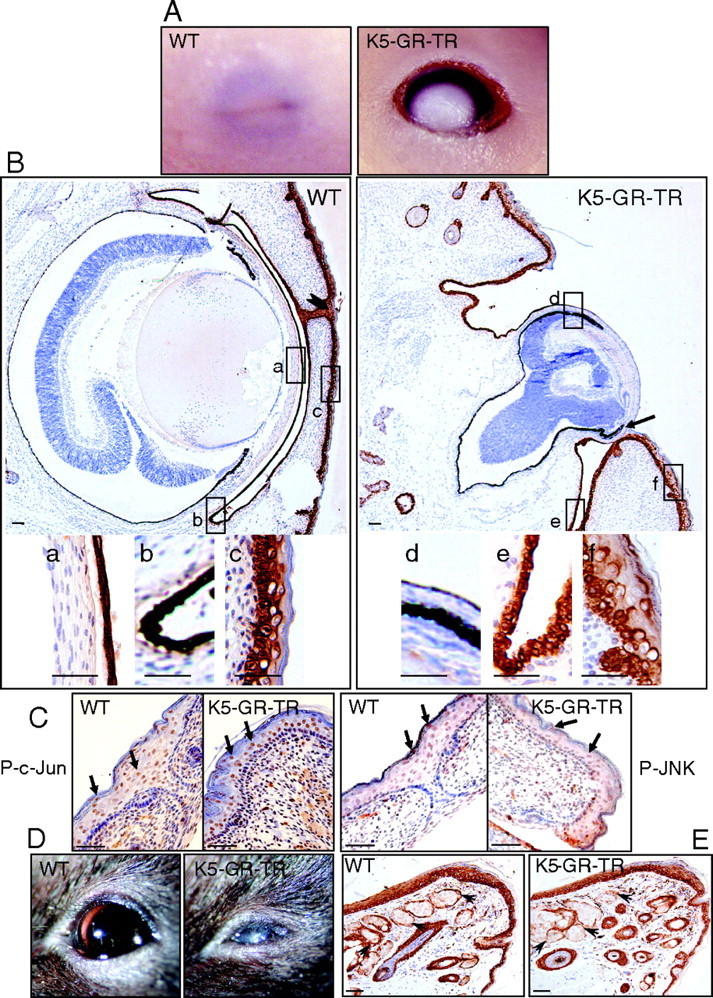
Lack of Eyelid Closure in K5-GR-TR Embryos
A, Eyelid fusion failed in 18.5-dpc K5-GR-TR HM embryos as compared with WT mice. B, Immunostaining using K5 in WT and K5-GR-TR embryos demonstrating the epithelia to which transgene expression is targeted: cornea (a and d), conjunctiva (b and e), and eyelid epithelium (c and f). Fused eyelids in WT embryos are pointed to by an arrowhead. a–f are also shown at higher magnification in the lower panel. Note that K5 staining was lacking in the cornea epithelium, whereas it appeared as more restricted in K5-GR-TR eye embryos. Additional ocular defects, including microphthalmia, dysplastic retinal tissue, and attachment of eyelid epithelium to the posterior side of the cornea were observed in K5-GR-TR transgenic embryos (arrow). C, Immunostaining using P-c-Jun and P-JNK antibodies revealed no differences in the expression of these phosphorylated proteins in the eyelid epithelium of K5-GR-TR vs. WT embryos. Arrows indicate nuclear localization of P-c-Jun and P-JNK. D, Adult transgenic mice exhibited corneal opacity. E, The secretory Meibomian glands of the eyelids were unaffected in the transgenics (arrowheads). Scale bar, 50 μm.
Because alterations in the developmental process of eyelid closure have been ascribed to defective c-Jun N-terminal kinase (JNK)/c-jun signaling in other mouse models (13, 14, 15), we examined these pathways in transgenic late embryos and newborn mice. Total and phosphorylated forms of JNK and c-Jun were detected at the leading edge of the eyelid epithelium regardless of whether the eyelid was fused or open (Fig. 2C, arrows, and data not shown). Therefore, our data suggest that the transrepression function of GR is sufficient to regulate ocular epithelial development although the precise mechanisms are still not known (see Discussion).
In adulthood, the observed ocular defects correlated with corneal opacity in 100% of HM mice and around 30% of HT mice (Fig. 2D). The Meibomian secretory glands of the eyelids, which also express K5 and are thus targeted for transgene expression, were not altered in transgenic adult mice (Fig. 2E, arrowheads).
Different Impact of the GR-Mediated Transrepression in Embryonic and Adult Skin
We analyzed the skin histopathology of late embryos and newborn HT and HM K5-GR-TR mice and found no significant changes in its architecture. Immunostaining showed no differences in the expression pattern of proliferation (K5) or differentiation (K10, filaggrin, loricrin, and involucrin) markers of the developing epidermis or HFs (Fig. 3A and data not shown). BrdU incorporation of interfollicular keratinocytes in transgenic late embryos was unchanged as compared with WT mice (14.5 and 16.2%, respectively). However, adult HM transgenic mice developed patchy alopecia along the whole coat and throughout the face, with scarce and underdeveloped vibrissae (Fig. 3B). Immunostaining using an anti-K5 antibody showed flattened keratinocytes with discontinuous K5 staining in the adult HM epidermis (Fig. 3C), and the number of HFs per millimeter was reduced approximately by 25% as compared with WT littermates (Fig. 4H). From these results, we conclude that overexpression of GR-TR in basal keratinocytes does not impair skin development during the embryogenesis but produces epidermal atrophy and reduced HF number in adult mice.
Fig. 3.
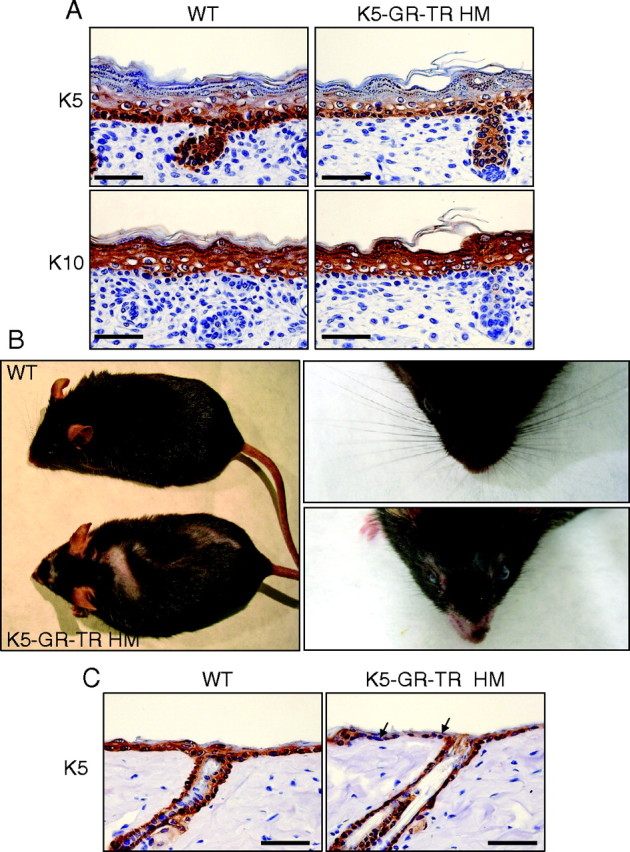
Normal Embryo Skin Development and Adult Skin Alterations in K5-GR-TR Mice
A, Normal development of the epidermis and HFs in 18.5-dpc K5-GR-TR HM mice. Immunostaining with K5 and K10 showed no alterations in transgenic embryo skin as compared with controls. B, Adult HM transgenic mice developed patchy alopecia along the whole coat (left) and throughout the face, with scarce and underdeveloped vibrissae (right). C, Immunostaining using an anti-K5 antibody showed a discontinuous pattern in the epidermis of adult HM mice (arrows). Scale bar, 50 μm.
Fig. 4.

GR-TR Inhibits PMA-Induced Proliferation upon Acute and Chronic Treatment.
A and B, Responses of adult K5-GR-TR and K5-GR to acute PMA treatment. Age-matched adult WT, K5-GR-TR HT mice of lines 298 and 306, and K5-GR mice (line 285) were used for these experiments. The proliferation rate of interfollicular epidermal keratinocytes was evaluated in dorsal skin paraffin sections from the indicated genotypes, as indicated in Materials and Methods. Arrows indicate BrdU-positive interfollicular keratinocytes. Results of the quantification were graphed as the percentage of positive BrdU keratinocytes vs. total nuclei. Mean values ± sd are shown (n = 5). Asterisks denote statistically significant differences relative to PMA-treated WT mice, as determined by Student’s t test (*, P < 0.05; **, P < 0.005). C, Responses of adult K5-GR-TR and K5-GR mice to chronic PMA (1 wk). Age-matched adult WT, K5-GR-TR HT and HM mice of line 298, and K5-GR mice (line 285) were used for these experiments. Hematoxylin and eosin staining shows dramatic epidermal hyperplasia in WT mice that was greatly reduced in HT and HM skin as well as in K5-GR mice. K6 staining was evaluated as a marker of hyperproliferative skin. Bar, 50 μm except for K6 immunostaining, where the bar represents 25 μm. D, Determination of the mRNA levels of k6β by RT-PCR in WT and K5-GR-TR HM skin that was treated with either vehicle (−) or PMA for 1 wk. Experiments were performed in at least three individuals of each genotype. Quantitation of RT-PCR is shown at right. Mean values ± sd are shown (n = 3), and statistically significant differences (P < 0.05) are indicated by asterisks. E–H, Responses of adult K5-GR-TR and K5-GR to PMA (1 wk) and PMA plus Dex (1 wk) (P+D). Age-matched adult WT, K5-GR-TR HT and HM mice of line 298, K5-GR-TR mice of line 306, and K5-GR mice (line 285) were used for these experiments. Morphometric analyses were performed to determine epidermal width (E), dermal cellularity (G), and number of HFs per millimeter (H) in mouse skin of the indicated genotypes. Percentage of BrdU incorporation was also examined in the same skin samples (F). Note that the inhibition of epidermal keratinocyte growth correlated with transgene dosage (compare HT vs. HM) and was consistently observed in the two independent transgenic lines (298 and 306). Morphometric and BrdU measurements were performed in at least five individuals of each genotype. Mean values ± sd are shown. Asterisks indicate statistical significance of the response of each genotype as compared with PMA-treated WT mice, as determined by Student’s t test (*, P < 0.05; **, P < 0.005; ***, P < 0.0005).
GR Transrepression Is Sufficient to Inhibit Keratinocyte Proliferation in Vivo
We next investigated the hyperproliferative and inflammatory responses of adult K5-GR-TR mice to acute and chronic PMA treatment (Fig. 4). The basal proliferation rate of interfollicular epidermal keratinocytes was equivalent in transgenic and control mice, as quantitated in paraffin sections incubated with an anti-BrdU antibody (Fig. 4, A and B; 4%). After a single dose of PMA for 48 h, the number of suprabasal layers was augmented in control skin, correlating with an increase of up to 45% in the labeling index of basal cells. However, PMA-induced proliferation was notably reduced (30%) in two independent K5-GR-TR transgenic lines (Fig. 4, A and B), thus demonstrating that the transrepression function of GR is sufficient to inhibit induced keratinocyte growth. Moreover, when K5-GR mice were subjected to this PMA treatment, we observed a similar response, indicating that the inhibitory effects of the WT form of GR and mutant GR-TR on keratinocyte proliferation were comparable (Fig. 4B). We also analyzed whether GR-TR effectively inhibited epidermal hyperplasia elicited by a chronic PMA treatment in HT and HM mice from lines 298 and 306 (Fig. 4, C and E). In addition, we studied the response of K5-GR mice to PMA in analogous experiments. Mice were treated with 2 μg PMA every other day for 1–3 wk, and morphometric analyses were performed at various time points (Fig. 4, C and E–H, and data not shown). PMA caused maximal epidermal hyperplasia and increased number of HFs after 1 wk (Fig. 4, C and E–H, and data not shown). We also analyzed the expression of K6 as a marker of hyperproliferative skin and found positive staining throughout all epidermal layers of WT mice treated with PMA. In sharp contrast, K6 expression was drastically reduced in HT mice and K5-GR mice and was almost undetectable in the skin of HM animals (Fig. 4C, lower panel). We also found that the mRNA levels of k6β, the isoform of k6 being induced by PMA, among other hyperproliferative stimuli, were strongly reduced in PMA-treated skin of K5-GR-TR and K5-GR mice (Fig. 4D).
At 1 wk, epidermal width increased up to 53.1 ± 11.7 μm in WT mice as compared with 36.2 ± 7.9 μm in HT mice and 20.7 ± 4.0 μm in HM animals. The reduction of epidermal width was also statistically significant in transgenic line 306 and in K5-GR mice (Fig. 4E). Consistent with this, BrdU incorporation was reduced by 50% in K5-GR-TR HT skin of line 298 and line 306 and by 80% in K5-GR-TR HM line 298 and K5-GR skin (Fig. 4F). We also investigated whether the presence of a GR ligand could modulate the effect of the transgenes by treating the mice with PMA plus Dex for 1 wk. In all genotypes and transgenic lines, Dex significantly inhibited both epidermal hyperplasia and keratinocyte proliferation to a similar extent (Fig. 4, E and F).
We also evaluated the effect of GR-TR in the PMA-induced anagenic growth of HFs. After 1 wk of PMA treatment, approximately 40% of HFs had entered into anagen, as quantitated by morphometric analysis, in agreement with previous reports (16). However, only 20% of HFs in K5-GR-TR HM mice and K5-GR mice was in the growing phase. Overall, these results support the notion that GR-TR exerts a potent antiproliferative effect in keratinocytes of the epidermis and HFs. In contrast, dermal cellularity induced by PMA was reduced by only 15–20% in K5-GR-TR mice, as compared with WT littermates, whereas this reduction reached approximately 30% in skin of K5-GR animals (Fig. 4G). The addition of Dex inhibited the effects of both transgenes on dermal cellularity to a similar extent.
GR-Mediated Transrepression Discriminates among Proinflammatory Cytokines in Skin
We next asked whether GR and GR-TR differentially regulated the transcript levels of specific genes that are markers of proliferation and inflammation after PMA treatment, as compared with WT skin (Fig. 5). Given that effects on transcription may occur earlier than the time points used in our previous experiments (Fig. 4), we analyzed the effect of PMA 4–48 h after the treatments by semiquantitative RT-PCR (Fig. 5). Additionally, as a control for ligand-induced activation of endogenous GR, groups of transgenic and WT mice were pretreated with Dex for 24 h before PMA application (Fig. 5). In WT mice, the induction of the proinflammatory cytokines IL-6, TNF-α, and IL-1β followed the expected kinetics, reaching a peak at 4 h upon PMA treatment (Fig. 5). Pretreatment with Dex diminished cytokine induction in WT skin, although to a different extent, with IL-1β being the cytokine most effectively repressed by addition of the ligand (Fig. 5). In K5-GR-TR transgenic mice, PMA-induction of IL-6 and TNF-α transcripts was marginally reduced, although the differences relative to PMA-treated WT mice were not statistically significant, either in the absence or presence of Dex. However, mRNA levels of IL-6 and TNF-α were significantly reduced (1.7-fold) after 4 h of PMA exposure in K5-GR skin as compared with WT mice. Interestingly, the ability of PMA to induce IL-1β (4 and 48 h) was significantly inhibited by GR-TR and GR when compared with WT treated skin (Fig. 5, IL-1β). Transcriptional repression was equivalent for the K5-GR-TR line 298 and K5-GR (5-fold) but less potent for K5-GR-TR line 306 (3.5-fold). Dex further inhibited IL-1β gene induction by PMA at 4 h in both K5-GR-TR and K5-GR skin (4-fold repression for K5-GR-TR line 298 and K5-GR and 2-fold repression for K5-GR-TR line 306).
Fig. 5.

GR-TR Differentially Regulates Proinflammatory Cytokines in Vivo
Determination of the mRNA levels of IL-6, TNF-α, cycD1, IL-1β, and MMP-3 in mouse skin of the indicated genotypes. Mice were treated with either vehicle or PMA for 4 or 48 h or pretreated with Dex for 24 h before PMA application. For quantitation of RT-PCR, experiments were performed in at least three individuals of each genotype. Mean values ± sd are shown. Values representing gene expression levels of untreated mice from each genotype were set to 1; fold induction is thus represented relative to this basal level. Asterisks denote statistically significant differences relative to WT mice with the same treatment, as determined by Student’s t test (*, P < 0.05; **, P < 0.005).
We also examined the effects of PMA on the metalloprotease gene MMP-3, which is normally not expressed in untreated skin but is induced in dermal monocytic cells by PMA (17). The robust induction of MMP-3 that we observed in WT skin treated with PMA was significantly reduced in K5-GR-TR line 298 and K5-GR mice (1.6-fold). Pretreatment with Dex further repressed transcription of MMP-3, ranging from 2- to 5-fold in the different genotypes (Fig. 5, MMP-3). Finally, we evaluated cycD1 mRNA levels as a marker of PMA-induced proliferation (16), and although we observed a weak repression of this gene as a consequence of GR-TR and GR expression, these differences were not statistically significant.
GR Transrepression Abrogates the PMA-Induced Activation of JNK at Early Time Points and Reduces NF-κB Binding at Later Stages
MAPKs are crucial mediators of keratinocyte function (18), and GR negatively modulates these proteins in different physiopathological settings (19). We thus examined whether the expression and/or activation of ERK and JNK played a role in the altered cutaneous responses of K5-GR-TR transgenic mice to PMA. Total protein and phosphorylated isoforms of ERK and JNK were checked by immunoblotting using whole-cell extracts from adult K5-GR-TR and WT skin, quantitated, and expressed as relative MAPK activity (Fig. 6). In WT mice, PMA induced phospho- (P-)ERK activation at 2 h, which remained high up to 8 h. GR-TR decreased the basal rate of ERK phosphorylation, although the kinetics of PMA induction was similar to that for WT mice (Fig. 6, A and B). Notably, PMA caused a robust and transient JNK activation (10- to 12-fold) that was abrogated in K5-GR-TR skin. It is also important that GR-TR augmented constitutive basal c-Jun levels (Fig. 6A), which is in agreement with previous work reporting that Dex induces up-regulation of c-jun through DNA-binding-independent mechanisms (17). Surprisingly, c-Fos was similarly increased by GR-TR expression. Remarkably, and contrary to PMA-induced accumulation of Jun and Fos proteins in WT skin, we did not observe any increase in these AP-1 proteins in K5-GR-TR PMA-treated skin (Fig. 6A).
Fig. 6.

GR-TR Abrogates the PMA-Induced Activation of JNK at Early Time Points
A, Immunoblotting using whole-cell extracts from adult K5-GR-TR and WT dorsal skin that were either untreated or treated with PMA at the indicated times to check the expression of ERK, P-ERK, JNK, P-JNK, c-Jun, and c-Fos. B, Quantitation of the relative levels of P-ERK/ERK and P-JNK/JNK was determined in at least three individuals of each genotype.
We also evaluated the implication of NF-κB activation in the GR-TR-mediated effects on the cutaneous responses to acute and chronic PMA (Fig. 7). At early times, PMA induced NF-κB binding to a similar extent in both WT and transgenic skin. NF-κB complexes in adult skin consisted of p50/p65 and p50/p50 dimers, as determined by supershift assays (Fig. 7A, right panel). After 1 wk of PMA exposure, NF-κB binding was reduced in transgenic skin as compared with WT, thus indicating that the interference of GR with the binding of NF-κB occurs at later stages (Fig. 7A; see Discussion). The observed differences in NF-κB binding were not due to changes in the amount and/or subcellular localization of p65 or inhibitor of κB, α (IκBα), as shown by immunoblotting using skin fractionated extracts from transgenic and WT littermates (Fig. 7B). Moreover, the expression of either WT GR or GR-TR in keratinocytes did not transcriptionally induce ikba (Fig. 7C), ruling out that up-regulation of IκBα is necessary for GR-mediated inhibition of NF-κB activity in keratinocytes. Overall, our data indicate that GR transrepression antagonizes the PMA-induced activation of both JNK and NF-κB, although these interactions occur with different kinetics.
Fig. 7.

GR-TR Reduces NF-κB Activation at Later Stages
A, EMSA showing NF-κB-binding activity of total skin extracts from WT and transgenic mice that were either untreated or PMA-treated for the indicated times. The composition of the NF-κB complexes (p50/p65 and p50/p50 dimers) was determined by supershift assays (right panel). PI, Preimmune serum. Free probe is indicated at the bottom of the gel. B, Cytoplasmic and nuclear extracts were prepared from adult K5-GR-TR and WT skin to examine the expression of p65 and ΙκBα by using specific antibodies. C, RT-PCR analysis showing ikba transcript levels in skin from adult WT, K5-GR, and K5-GR-TR mice.
DISCUSSION
Role of GR-TR in Epithelial Development
We have generated a novel transgenic mouse model that expresses the transrepression function of GR in an attempt to better understand the mechanisms of GR action in the physiopathology of the epidermis, cutaneous appendages, and other stratified epithelia. It is important to note that, similar to other published dimerization mutants, the P493R, A494S mutant used in our study is not globally deficient in transactivation because these mutants were competent for GRE binding or transcriptional enhancement of some genes, such as PNMT (20, 21). This implies that the mechanisms through which GR-TR can regulate keratinocyte responses include not only transrepression but also transactivation of genes with a more complex GRE configuration, as previously reported for other GC-target genes (20, 21).
Our previous work demonstrated that overexpression of the WT form of GR (K5-GR mice) caused numerous congenital malformations of ectodermal-derived tissues, including the epidermis, HFs, eyes, teeth, and exocrine glands (7, 8, 9). One of the aims of this work was to decipher whether the transrepression function of GR was sufficient to produce abnormal epithelial development in these tissues. The fact that both transgenic mice, K5-GR and K5-GR-TR, use the same k5 regulatory sequences, along with our data indicating that both transgenes are expressed at similar protein levels allows us to compare the impact of GR vs. GR-TR in keratinocytes in vivo (7) (Fig. 1 and data not shown).
A major finding of the current study is that GR-mediated transrepression is sufficient to impair the development of eyelid and cornea epithelia (Fig. 2), because K5-GR-TR mice showed an ocular phenotype indistinguishable from K5-GR mice (8). Interestingly, the characterization of several knockout mouse models has allowed the identification of at least two signaling pathways in the control of eyelid closure (reviewed in Ref. 22). One pathway involves the transduction of TGF-β and activin signals by the MAPK kinase kinase 1 (MEKK1)/JNK/c-Jun pathway, and the second pathway involves TGF-α/epidermal growth factor receptor (EGFR)/ERK signaling. Previous reports have shown that both JNK1−/−/JNK2−/− and c-jun conditional knockout in epidermis (c-jun-Δep) display an EOB phenotype (13, 14, 15). In c-jun-Δep mice, the ocular phenotype was ultimately due to reduced EGFR expression; however, in JNK-deficient embryos, decreased EGFR signaling despite normal levels of EGFR caused ocular anomalies. Remarkably, we did not find changes in JNK/c-Jun activities in K5-GR-TR eyelids (Fig. 2C), which indicates that JNK-dependent phosphorylation of c-Jun is not sufficient to mediate normal eyelid closure and that alternate pathways regulate the process of eyelid fusion. We examined EGFR expression and activity in the leading edge of 16.5-d post conception (16.5 dpc) eyelids in HT, HM, and WT littermates, but our results were not conclusive because the expression of these markers was very weak in both transgenic and WT embryos (not shown). Overall, our findings suggest that GR elicits the EOB phenotype by additional pathways that remain to be defined.
This study also raises another interesting issue, namely, that the mechanisms by which GR mediates epithelial morphogenesis are tissue specific. Supporting this hypothesis, the transrepression function of GR was sufficient to induce dysmorphogenesis in some epithelia (eyelid and cornea), whereas both transactivation- and transrepression-dependent functions were seemingly required for abnormal development in other tissues (developing epidermis and HF, teeth, and secretory Meibomian glands) (Figs. 2 and 3 and data not shown) (9). Moreover, the mechanisms governing GR action in distinct epithelia differ in embryos vs. adult individuals. The failure of GR to produce skin anomalies during embryonic development contrasts with the epidermal atrophy and reduced HF number observed in adult transgenic mice (Figs. 3 and 4).
Antiproliferative vs. Antiinflammatory Effects of GR: Requirement of Transrepression and Transactivation
Our results in the two independent K5-GR-TR lines studied were highly reproducible, thus ruling out insertion-dependent effects of the transgene. Furthermore, the observed phenotypes correlated with transgene dosage, as demonstrated by a comparison between lines 298 and 306 as well as the severity of defects observed in HM vs. HT mice of line 298 (Fig. 4, E–H). The use of K5-GR-TR and K5-GR mice to analyze the proliferative and inflammatory responses to PMA in vivo has allowed us to compare the requirements of transrepression and transactivation functions in these processes (Fig. 5).
Our data show that the antiproliferative actions exerted by GR-TR and GR in keratinocytes were very similar in acute and chronic PMA processes (Fig. 4). We here demonstrate that the antiproliferative effects of K5-GR-TR in PMA-treated keratinocytes rely on the repression of both JNK and NF-κB activities and do not require GRE-dependent activation (Figs. 4, 6, and 7), in agreement with previous reports where this mutant was tested in several cell lines (11, 23). Our data showed that GR-TR inhibited PMA-induced NF-κB binding after 1 wk of PMA treatment and not at early time points (Fig. 7A). However, given that GR-mediated inhibition of NF-κB activity does not necessarily rely on reduced NF-κB binding to DNA, it is plausible that GR-TR may exert a dual effect by repressing gene transcription without affecting DNA binding at early times and reducing DNA binding at later stages of PMA treatment.
Because GR negatively regulated JNK activity after PMA treatment, and given that JNK positively regulates AP-1 function through phosphorylation of c-Jun, AP-1 activity would be expected to be diminished in the skin of K5-GR-TR mice. Accordingly, we detected reduced transcription of the AP-1 target gene MMP-3 (Fig. 5), which is regulated in an antagonistic manner by phorbol esters and GCs (17). Additionally, we found that k6β mRNA, the inducible isoform of k6 that also contains AP-1 sites in its promoter, was strongly reduced in PMA-treated skin of K5-GR-TR mice (Fig. 4D). It has been described that GR represses K6 through GR/AP-1 negative cross-talk as well as through binding of GR monomers to nGREs at its promoter (24). In our model, both mechanisms could account for the observed repression of K6 and support the role of the transrepression function of GR in the inhibition of keratinocyte proliferation. On the other hand, our results indicate that GR-TR represses AP-1-dependent transcription induced by PMA, despite detection of increased levels of Jun and Fos proteins in untreated skin (Fig. 6A).
The antiinflammatory effects of GR-TR and GR differed, as demonstrated primarily by reduction of dermal infiltrates (Fig. 4G) and, more importantly, by the differential repression of several proinflammatory genes (Fig. 5). Although previous reports claim that GR protein interactions are sufficient to mediate GC antiinflammatory effects (3), we found only a weak reduction of inflammatory infiltrates in the dermis of transgenic K5-GR-TR upon exposure to PMA relative to the response observed in WT littermates, whereas this reduction was statistically significant in K5-GR mice (Fig. 4). Moreover, the addition of Dex reduced dermal cellularity to a similar extent in all genotypes, including WT mice, thus suggesting that the effect of this ligand was mediated by the endogenous GR. In Fig. 5, we show that GR-TR and GR differentially repressed several proinflammatory genes, with IL-1β and MMP-3 being the most affected. The fold repression of IL-1β and MMP-3 after PMA treatment (4 and 48 h) was equivalent for K5-GR-TR line 298 and K5-GR but less pronounced in the K5-GR-TR line 306 (Fig. 5), in agreement with transgene expression levels (Fig. 1 and data not shown). However, the induction of IL-6 and TNF-α genes by PMA was significantly reduced in K5-GR but not K5-GR-TR skin, as compared with WT littermates. Very recently, it has been shown that in contact allergy, GR requires GRE-dependent transcription for the suppression of allergic inflammation specifically in macrophages (25). This report demonstrated that GR dimerization is required in macrophages for the repression of certain cytokine genes (e.g. IL-1β and MCP-1) but not for others such as TNF-α (26). Our results indicate that transactivation also plays a role in the GR-mediated transrepression of certain proinflammatory genes in vivo. The situation could be even more complex, because it has been reported that the up-regulation of MAPK phosphatase-1 (MKP-1) by GCs contributed to their antiinflammatory effects in a cutaneous air pouch model (26, 27, 28). However, in other models of inflammation, both MKP-1-dependent and -independent components of the antiinflammatory action of GCs may coexist in a single cell type (28). We examined MKP-1 at the mRNA and protein level and found no differences among K5-GR-TR, K5-GR, and WT mice in the absence or presence of Dex (not shown). Thus, our results indicate that the antiproliferative action exerted in vivo by GR in keratinocytes does not involve MKP-1 (data not shown).
The generation of selective GR agonists has provided valuable information regarding the dissociation between transrepression and transactivation; however, it is still unclear which mechanisms are responsible for the adverse effects elicited by GCs (1, 4, 10). A previous study evaluating the side effects of the selective GR agonist compound ZK 216348 demonstrated that after prolonged treatment, skin thinning was less pronounced as compared with the classical GC prednisone, thus suggesting that transactivation may be involved in skin atrophy (29). In K5-GR-TR adult mice, the expression of the transgene inhibited growth in epidermal keratinocytes as well as HF density, similar to the skin atrophy and occasional alopecia observed after prolonged treatments with GC analogs in human patients (10). However, GR-induced skin atrophy does not necessarily use the same molecular mechanisms implicated in the antiproliferative actions mediated by GR in response to PMA exposure. The generation and functional characterization of K5-GR-TR mice demonstrates that the antiproliferative effects of GR are mediated solely through its transrepression function, specifically by impairing JNK/AP-1 and NF-κB signaling pathways, whereas the GR-mediated antiinflammatory actions appear to use both transrepression and transactivation mechanisms. These findings may have implications for understanding the therapeutic potential of WT GR, which functions as a potent tumor suppressor in skin tumorigenesis (30, 31). Given that both sustained hyperplasia and persistent inflammation are hallmarks of tumor formation (32), K5-GR-TR mice represent a powerful tool to decipher the contribution of inhibiting proliferation and inflammation in the prevention and/or treatment of epithelial skin tumors.
MATERIALS AND METHODS
Generation of Transgenic Mice
The cDNA encoding the rat GR mutant, which contains a double mutation (P493R, A494S) rendering a GR defective in transactivation but effective in transrepression (11), was placed into a cassette containing the 5.2-kb bovine keratin 5 (bk5) regulatory sequences, the 5′ β-globin intron 2, and the 3′ polyadenylation sequences (33). The transgene construct, denominated K5-GR-TR, was excised from the plasmid vector with NotI, isolated from agarose gels, purified by Elutip columns (Schleicher and Schuell, Dassel, Germany), adjusted to a final concentration of 2 mg/ml, and microinjected into C57BL/6J × DBA/2J F2 mouse embryos, as previously described (34). Founder mice and progeny were identified by PCR analysis of tail genomic DNA using specific primers that recognize either mouse GR (endogenous) or rat GR (transgene).
Transgenic mice from two independent K5-GR-TR HT lines (298 and 306) were analyzed to rule out insertion-dependent effects. Results of both lines are shown in Figs. 4 and 5. For the other experiments, only results from line 298 are illustrated. Routinely, the mating schedule was programmed by intercrosses of HT mice with WT mice, thus obtaining 50% of transgenic and 50% of nontransgenic progeny, the latter being used as controls. In addition, HM mice of line 298 were generated, although this line was difficult to maintain because HM females had difficulties in lactating their litters. The total number of control and K5-GR-TR transgenic mice examined was 270. Of these, 78 were late embryos and newborn mice, and 192 were adult mice. K5-GR heterozygous mice (line 285) have been previously reported (7).
Animal Handling and Treatments
Mice were housed in microisolated boxes in which the air is filtered in both directions, allowing maximal isolation. Animal experimentation was always conducted with accepted standards of humane animal care in our registered animal facility (CV-46007, Centro de Investigación Príncipe Felipe CIPF). Experiments were performed in accordance with the Principles of Laboratory Animal Care (National Institutes of Health Publication no. 85-23, revised 1985) and with the current Spanish and European normative that governs research with animals (Real Decreto 1201/2005, B.O.E. no. 252, October 10, 2005, and Convenio Europeo 1-2-3, March 18, 1986).
Embryos were obtained by cesarean derivation at 18.5 dpc (the morning of the day that the vaginal plug was seen was considered as 0.5 dpc). In all experiments performed in adult mice, female mice in the resting stage of the hair cycle (8–10 wk of age) were used. Dorsal skins were shaved 48 h before the treatments. At the indicated times, skin was excised and either homogenized to obtain RNA and protein or fixed for immunohistochemical analysis. At least three or four animals per experimental group were examined. In the control group, animals were treated with vehicle (acetone) only.
PMA and Dex (both from Sigma Chemical Co., St. Louis, MO) were applied topically at the indicated doses and times. PMA was given at a unique dose of 8 μg/mouse in the acute treatments (2–48 h) and repeatedly applied every other day at 2 μg/mouse in the chronic treatments for 1–3 wk. Dex (8 μg/mouse in 200 μl) was topically applied 24 h before PMA treatment.
Antibodies
The antibodies used included rabbit polyclonal antibodies to GR (sc-1004), ERK (sc-154), JNK (sc-474), P-c-jun (sc-822), p65 (sc-372), IκBα (sc-371), and c-Fos (sc-52) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies against c-jun (no. 9165), P-ERK (Thr202/Tyr204) (no. 4376), and P-JNK (Thr183/Tyr185) (no. 9251) were purchased from Cell Signaling Technology Inc. (Beverley, MA). An antibody against actin (A-2066; Sigma) was used for loading control. Secondary peroxidase-conjugated antirabbit antibody was from Amersham (Aylesbury, UK), and secondary peroxidase-conjugated antimouse antibody and biotin-conjugated antirabbit or antimouse antibodies were from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA).
Antibodies against K5 (PRB-160P), K6 (PRB-169P), and K10 (PRB-159P) were from Covance (Babco, Berkeley, CA).
Transfection Studies
The PB cell line (mouse adult nontransformed keratinocytes) was grown in DMEM (BioWhitakker, Inc., Walkersville, MD) supplemented with 10% fetal calf serum (BioWhitakker). Transient transfection assays were done by the calcium phosphate method using either MMTV-CAT or 8x-κB-CAT reporter plasmids along with expression vectors encoding for empty vector (pcDNA3), K5-GR, or K5-GR-TR (5 μg/each). To determine MMTV-CAT activity, PB cells were incubated with charcoal-stripped serum overnight to deplete endogenous GCs and then treated with either vehicle or Dex (1 μm) for 6 h. In transfections experiments using 8x-κB-CAT reporter, PB cells were treated with either vehicle or TNF-α (10 ng/ml) for 4 h. CAT activity was assayed by the CAT ELISA kit (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer’s recommendations. Three independent experiments were performed in triplicate and mean value ± sd estimated. Statistical significance was analyzed by Student’s t test.
Histological and Immunohistochemical Analysis
Whole embryos, newborn mice, and skin samples were paraformaldehyde or ethanol fixed and embedded in paraffin. Consecutive 4-μm-thick sections were obtained and routinely stained with hematoxylin and eosin. Briefly, for immunostaining, paraffin sections were dewaxed, microwaved in 10 mm citrate solution, and blocked with 1% BSA. Slides were then incubated with the primary antibody for at least 1 h, washed three times with PBS, and then incubated with conjugated secondary antibodies for 1 h. Finally, the reaction was visualized with the avidin-biotin complex (ABC) kit from Dako (Vectastain Elite; Vector Laboratories, Inc., Burlingame, CA) using diaminobenzidine as chromogenic substrate for peroxidase. Slides were mounted and analyzed by light microscopy (Leica DM RXA2), and microphotographs were taken at the indicated magnification.
BrdU Labeling
Epithelial cell proliferation was measured by ip injection of BrdU (130 mg/g body weight; Roche, Indianapolis, IN) 1 h before animals were killed. BrdU incorporation was detected by immunohistochemistry of paraffin-embedded sections using a mouse anti-BrdU monoclonal antibody (Roche, no. 1170376). The number of BrdU-positive cells and the number of total cells were determined in at least 6 mm of interfollicular epithelium in each sample. Data reflect the average from five mice of each genotype (WT, K5-GR-TR line 298 HT, K5-GR-TR line 298 HM, K5-GR-TR line 306, and K5-GR line 285). Error bars indicate sd. Statistical significance was determined by Student’s t test comparing the PMA response (acute and chronic) of each genotype relative to PMA-treated WT mice. Statistical differences of PMA plus Dex vs. PMA-treated WT are also shown.
Morphometric Analysis
Quantitation of the epidermal width, dermal cellularity, and average number of HFs was performed in dorsal skin sections stained with hematoxylin and eosin. At least 10 individual fields of 1 mm per slide were counted using the software MetaMorph (Premier Offline 7.0; Molecular Devices, Downingtown, PA). Experiments were performed in at least five individuals of each genotype, and differences were assessed by using the t test, with statistical significance when P < 0.05.
RNA Preparation and Semiquantitative RT-PCR
Total RNA was isolated from back skin of newborn and adult transgenic and control mice by using Trizol reagent (Invitrogen, Molecular Probes, Eugene, OR), following manufacturer’s recommendations. Reverse transcription was performed by using 1 μg RNA and oligo-dT (Fermentas Inc., Burlington, Canada) followed by PCR using specific oligonucleotides for k6β (forward, 5′-CACCATCAAATACACCACCAGCG-3′; reverse, 5′-AAGCAGCCAAAAAGAGAAGCGAG-3′), TNF-α (forward, 5′-ATG AGC ACA GAA AGC ATG ATC-3′; reverse, 5′-TAC AGG CTT GTC ACT CGA ATT-3′), IL-6 (forward, 5′-GAC AAA GCC AGA GTC CTT CAG AGA G-3′; reverse, 5′-CTA GGT TTG CCG AGT AGA TCT C-3′), IL-1β (forward, 5′-ATG GCA ACT GTT CCT GAA CTC ACC T-3′; reverse, 5′-CAG GAC AGG TAT AGA TTC TTT CCT TT-3′), cyclin D1 (forward, 5′-CAT CAA GTG TGACCC GGA CTG-3′; reverse, 5′-CCT CCT CCT CAG TGG CCT TG-3′), MMP-3 (forward, 5′-TGT ACC CAG TCT ACA AGT CCT CCA C-3′; reverse, 5′-CTG CGA AGA TCC ACT GAA GAA GTA G-3′), ikba (forward, 5′-CGC CCA AGC ACC CGG ATA CAG C-3′; reverse, 5′-TGG GGT CAG TCA CTC GAA GCA CAA-3′), and β-actin (forward, 5′-CCA CCA GAC AAC ACT GTG TTG GCA T-3′; reverse, 5′-AGA GGT ATC CTG ACC CTG AAG TAC C-3′). To achieve semiquantitative amplification, 25 cycles were used; bands corresponding to specific PCR products were scanned by using ImageQuant (Amersham), quantitated with Quantity One Software (Bio-Rad, Hercules, CA), and values were estimated relative to β-actin levels. Experiments were performed in at least three individuals of each genotype, and differences were assessed by using the t test, with statistical significance when P < 0.05.
Immunoblotting
Whole-cell extracts and cytoplasmic and nuclear extracts (20 μg) were prepared as previously described (30), boiled in Laemmli buffer, and separated on 10% SDS-PAGE gels, then transferred to nitrocellulose filters (Hybond ECL; Amersham). Filters were blocked with 5% nonfat dry milk in PBS/0.1% Tween 20 at 4 C overnight, washed three times in PBS/0.1% Tween 20, and incubated with the indicated antibodies. After washing, membranes were incubated with a peroxidase-conjugated secondary antibody (Amersham), washed again, and analyzed using the enhanced chemiluminescence method (ECL; Amersham), according to the manufacturer’s recommendations. The membranes were stained with Ponceau S to verify equal protein loading and transfer.
Specific bands were scanned by using ImageQuant (Amersham), quantitated with Quantity One Software (Bio-Rad) and graph plotted. Values for control samples (basal protein levels in WT skin) were arbitrarily set as 1, and other values are expressed as relative to WT. Experiments were performed in at least three individuals of each genotype and differences were assessed by using the t test, with statistical significance when P < 0.05.
EMSA
EMSA were performed by incubating total cell extracts from mouse skin with a labeled oligonucleotide corresponding to a palindromic κB site previously described (30). Complexes were separated on 5.5% native polyacrylamide gels in 0.25× Tris-borate-EDTA buffer, dried, and exposed to Hyperfilm-MP (Amersham) at −70 C.
Acknowledgments
We are grateful to D. Burks for helpful comments on this manuscript.
NURSA Molecule Pages:
Ligands: Dexamethasone;
Nuclear Receptors: GR.
Footnotes
This work was supported by Grants SAF2005-00412 from the Spanish Ministerio de Educación y Ciencia and Fundación Areces (050507070007).
Disclosure Statement: The authors have nothing to disclose.
First Published Online January 3, 2008
Abbreviations: AP-1, Activator protein-1; BrdU, bromodeoxyuridine; Dex, dexamethasone; dpc, days post conception; EGFR, epidermal growth factor receptor; EOB, eyelid opening at birth; GC, glucocorticoid; GR, glucocorticoid receptor; HF, hair follicle; HM, homozygous; HT, heterozygous; IκBα, inhibitor of κB, α; JNK, c-Jun N-terminal kinase; K6, keratin 6; MKP-1, MAPK phosphatase-1; NF-κB, nuclear factor-κB; P-, phospho-; PMA, phorbol 12-myristate 13-acetate; WT, wild type.
References
- 1.Rhen T, Cidlowski JA 2005. Antiinflammatory action of glucocorticoids: new mechanisms for old drugs. N Engl J Med 353:1711–1723 [DOI] [PubMed] [Google Scholar]
- 2.Cole TJ, Blendy AP, Monaghan K, Schmid W, Aguzzi A, Fantuzzi G, Hummler E, Unsicker K, Schütz G 1995. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev 9:1608–1621 [DOI] [PubMed] [Google Scholar]
- 3.Reichardt HM, Tuckermann JP, Göttlicher M, Vujic M, Weih F, Angel P, Herrlich P, Schütz G 2001. Represion of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J 20:7168–7173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buttgereit F, Burmester G, Lipworth BJ 2005. Optimised glucocorticoid therapy: the sharpening of an old spear. Lancet 365:801–803 [DOI] [PubMed] [Google Scholar]
- 5.Seckl JR, Nyirenda MJ, Walker BR, Chapman KE 1999. Glucocorticoids and fetal programming. Biochem Soc Trans 27:74–78 [DOI] [PubMed] [Google Scholar]
- 6.Segre JA 2006. Epidermal barrier formation and recovery in skin disorders. J Clin Invest 116 1150–1158 [DOI] [PMC free article] [PubMed]
- 7.Pérez P, Page A, Bravo A, Del Rio M, Gimenez-Conti I, Budunova IV, Slaga TJ, Jorcano JL 2001. Altered skin development and impaired proliferative and inflammatory responses in transgenic mice overexpressing the glucocorticoid receptor. FASEB J 15:2030–2032 [DOI] [PubMed] [Google Scholar]
- 8.Cascallana JL, Bravo A, Page A, Budunova I, Slaga TJ, Jorcano JL, Pérez P 2003. Disruption of eyelid and cornea development by targeted overexpression of the glucocorticoid receptor. Int J Dev Biol 47:59–64 [PubMed] [Google Scholar]
- 9.Cascallana JL, Bravo A, Donet E, Leis H, Lara MF, Paramio JM, Jorcano JL, Pérez P 2005. Ectoderm-targeted overexpression of the glucocorticoid receptor induces hypohidrotic ectodermal dysplasia. Endocrinology 146:2629–2638 [DOI] [PubMed] [Google Scholar]
- 10.Schäcke H, Döcke W, Asadullah K 2002. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 96:23–43 [DOI] [PubMed] [Google Scholar]
- 11.Helmberg A, Auphan N, Caelles C, Karin M 1995. Glucocorticoid-induced apoptosis of human leukemic cells is caused by the repressive function of the glucocorticoid receptor. EMBO J 14:452–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Addison WH, How HW 1999. The development of the eyelids of the albino rat, until the completion of disjunction. Am J Anat 29:1–31 [Google Scholar]
- 13.Li G, Gustafson-Brown C, Hanks SK, Nason K, Arbeit JM, Pogliano K, Wisdom RM, Johnson RS 2003. c-Jun is essential for organization of the epidermal leading edge. Dev Cell 4:865–877 [DOI] [PubMed] [Google Scholar]
- 14.Zenz R, Scheuch H, Martin P, Frank C, Eferl R, Kenner L, Sibilia M, Wagner EF 2003. c-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Dev Cell 4:879–889 [DOI] [PubMed] [Google Scholar]
- 15.Weston CR, Wong A, Hall JP, Goad MEP, Flavell RA, Davis RJ 2004. The c-Jun NH2-terminal kinase is essential for epidermal growth factor expression during epidermal morphogenesis. Proc Natl Acad Sci USA 101:14114–14119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodríguez-Puebla ML, Robles AI, Johnson DG, LaCava M 1998. Synchronized proliferation induced by 12-O-tetradecanoylphorbol-13-acetate treatment of mouse skin: an in vivo model for cell cycle regulation. Cell Growth Differ 9:31–39 [PubMed] [Google Scholar]
- 17.Tuckermann JP, Reichardt HM, Arribas R, Richter KH, Schütz G, Angel P 1999. The DNA-binding-independent function of the glucocorticoid receptor mediates repression of AP-1-dependent genes in skin. J Cell Biol 147:1365–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozanne BW, Spence HJ, McGarry LC, Hennigan RF 2007. Transcription factors control invasion: AP-1 the first among equals. Oncogene 26:1–10 [DOI] [PubMed] [Google Scholar]
- 19.De Bosscher K, Vanden Berghe W, Haegeman G 2003. The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 24:488–522 [DOI] [PubMed] [Google Scholar]
- 20.Adams M, Meijer OC, Wang J, Bhargava A, Pearce D 2003. Homodimerization of the glucocorticoid receptor is not essential for response element binding: activation of the phenylethanolamine N-methyltransferase gene by dimerization-defective mutants. Mol Endocrinol 17:2583–2592 [DOI] [PubMed] [Google Scholar]
- 21.Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Ha CM, Darimont BD, Garabedian MJ, Yamamoto KR 2003. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci USA 100:13845–138450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia Y, Karin M 2004. The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol 14:94–101 [DOI] [PubMed] [Google Scholar]
- 23.Bladh LG, Lidén J, Pazirandeh A, Rafter I, Dahlman-Wright K, Nilsson S, Okret S 2005. Identification of target genes involved in the antiproliferative effect of glucocorticoids reveals a role for nuclear factor-κB repression. Mol Endocrinol 19:632–643 [DOI] [PubMed] [Google Scholar]
- 24.Radoja N, Komine M, Jho SH, Blumenberg M, Tomic-Canic M 2000. Novel mechanism of steroid action in skin through glucocorticoid receptor monomers. Mol Cell Biol 20:4328–4339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tuckermann JP, Kleiman A, Moriggl R, Spanbroek R, Neumann A, Illing A, Clausen BE, Stride B, Förster I, Habenicht AJR, Reichardt HM, Tronche F, Schmid W, Schütz G 2007. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest 117:1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberman AC, Druker J, Perone MJ, Arzt E 2007. Glucocorticoids in the regulation of transcription factors that control cytokine synthesis. Cytokine Growth Factor Rev 18:45–56 [DOI] [PubMed] [Google Scholar]
- 27.Owens DM, Keyse SM 2007. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 26:3203–3213 [DOI] [PubMed] [Google Scholar]
- 28.Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, Saklatvala J, Clark AR 2006. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med 203:1883–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schäcke H, Schottelius A, Döcke WD, Strehlke P, Jaroch S, Schmees N, Rehwinkel H, Hennekes H, Asadullah K 2004. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA 101:227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Budunova IV, Kowalczyk D, Pérez, P, Yao YJ, Jorcano JL, Slaga TJ 2003. Glucocorticoid receptor functions as a potent suppressor of mouse skin carcinogenesis. Oncogene 22:3279–3287 [DOI] [PubMed] [Google Scholar]
- 31.Leis H, Page A, Ramirez A, Bravo A, Segrelles C, Paramio J, Barettino D, Jorcano JL, Pérez P 2004. Glucocorticoid receptor counteracts tumorigenic activity of Akt in skin through interference with the phosphatidylinositol 3-kinase signaling pathway. Mol Endocrinol 18:303–311 [DOI] [PubMed] [Google Scholar]
- 32.Mueller MM 2006. Inflammation in epithelial skin tumours: old stories and new ideas. Eur J Cancer 42:735–744 [DOI] [PubMed] [Google Scholar]
- 33.Ramírez A, Bravo A, Jorcano JL, Vidal MA 1994. Sequences 5′ of the bovine keratin 5 gene direct tissue- and cell-type specific expression of a lacZ gene in the adult and during development. Differentiation 58:53–64 [DOI] [PubMed] [Google Scholar]
- 34.Hogan B, Beddington R, Costantini F, Lacy E 1997. Manipulating the mouse embryo. New York: Cold Spring Harbor Press