

Abstract
Aldosterone elicits rapid physiological responses in target tissues such as the distal nephron through the stimulation of cell signaling cascades. We identified protein kinase D (PKD1) as an early signaling response to aldosterone treatment in the M1-cortical collecting duct (M1-CCD) cell line. PKD1 activation was blocked by the PKC inhibitor chelerythrine chloride and by rottlerin, a specific inhibitor of PKCδ. The activation of PKCδ and PKCε coincided with PKD1 activation and while a complex was formed between PKD1 and PKCε after aldosterone treatment, there was a concurrent reduction in PKD1 association with PKCδ. A stable PKD1 knockdown M1-CCD-derrived clone was developed in which PKD1 expression was 90% suppressed by gene silencing with a PKD1-specific siRNA. The effect of aldosterone treatment on the subcellular distribution of enhanced cyan fluorescent protein (eCFP)-tagged epithelial sodium channel (ENaC) subunits in wild type (WT) and PKD1 suppressed cells was examined using confocal microscopy. In an untreated confluent monolayer of M1-CCD cells, α, β, and γ ENaC subunits were evenly distributed throughout the cytoplasm of WT and PKD1-suppressed cells. After 2 min treatment, aldosterone stimulated the localization of each of the ENaC subunits to discrete regions within the cytoplasm of WT cells. The translocation of eCFP-ENaC subunits in WT cells was inhibited by rottlerin and the mineralocorticoid receptor (MR) antagonist spironolactone. No subcellular translocation of eCFP-ENaC subunits was observed in PKD1-suppressed cells treated with aldosterone. These data demonstrate the involvement of a novel MR/PKCδ /PKD1 signaling cascade in the earliest ENaC subunit intracellular trafficking events that follow aldosterone treatment.
ALDOSTERONE PROMOTES Na+ absorption and K+ secretion in target tissues such as the epithelia of the distal colon and nephron (reviewed in Ref. 1). The osmotic movement of water concurrent with Na+ absorption means that the net effect of aldosterone release is to increase extracellular fluid volume and consequently raise blood pressure (2). The classical mechanism of aldosterone action involves binding of aldosterone to the cytoplasmic mineralocorticoid receptor (MR), which functions as a ligand-dependent transcription factor. The earliest investigation of rapid physiological responses to aldosterone described the hormone effects on Na+ and K+ excretion into the urine within 5 min after its intraarterial administration (3). Increased activity of the epithelial sodium channel (ENaC) is the rate-limiting step in promoting Na+ reabsorption across the apical membrane of epithelial cells in the aldosterone-sensitive distal nephron. The distal convoluted tubule and the connecting tubule comprise the major sites for Na+ conservation in the renal tubule; however, the principal cells of the cortical collecting duct (CCD) also contribute significantly to this process (4, 5, 6). It is generally accepted that the aldosterone-induced increase in cell surface ENaC expression and channel open probability is regulated in two stages by: 1) promoting the trafficking and stabilization of pre-expressed ENaC subunits at the apical cell membrane and 2) through the MR-dependent regulation of ENaC subunit gene transcription.
ENaC subunits are stabilized in the cell membrane to enhance channel density after aldosterone treatment through suppression of channel degradation by the proteasome. This suppression occurs through the phosphorylation and inactivation of the E3 ubiquitin-protein ligase Nedd4-2 by serum glucocorticoid-stimulated kinase (SGK)-1 (7, 8). Increased expression of SGK-1 in response to aldosterone treatment has been the cornerstone for understanding the regulation of ENaC cell surface expression for many years. However, the transcriptional regulation of signaling intermediates such as SGK-1 cannot account for the most rapid stimulation of ENaC activity by aldosterone. An increase in the amiloride-sensitive ENaC current within 2 min after aldosterone treatment has been observed in isolated rabbit principal CCD cells (9). The rapid signaling processes that promoted ENaC activity in this model have yet to be elucidated; however, a rapid increase in ENaC activity through elevated apical membrane channel density has been linked to rapid vesicle trafficking processes coupled to activation of the RhoA small GTPase (10).
We previously demonstrated that aldosterone rapidly stimulates the activation of a novel protein kinase, protein kinase D (PKD1) in the M1-CCD cell line, and this activation is mediated through the MR-dependent trans-activation of the epidermal growth factor receptor (11). The PKD family of diacylglycerol-stimulated serine/threonine protein kinases comprises three known isoforms: PKD1/PKCμ, PKD2, and PKD3/PKCν (as reviewed in Ref. 12), which are classified as a subgroup of the calcium/calmodulin-dependent protein kinase family (13). PKD1 is implicated in crucial cellular processes including proliferation and apoptosis through its association with other signaling intermediates including ERK1/2 and c-Jun N-terminal kinase/MAPK (reviewed in Ref. 12). PKD1 has also been implicated in intracellular trafficking by regulating the fission of vesicles from the trans-Golgi network (TGN) and the direct phosphorylation of the TGN-associated kinase phosphatidylinositol (PI)-4 kinase III-β (14, 15, 16). The RhoA-dependent rapid membrane translocation of ENaC is coupled to vesicle trafficking through PI-4P5K (10), and the ENaC subunits interact directly with phosphatidylinositide signaling intermediates (17).
In this study, we elucidate the molecular mechanism of PKD1 activation and its role in regulating the rapid translocation of preexpressed ENaC subunits in CCD cells after treatment with aldosterone. This is an entirely novel mechanism by which aldosterone modulates the subcellular distribution of ENaC within minutes of treatment and presents a new paradigm for the rapid action of aldosterone in renal collecting duct cells.
RESULTS
Aldosterone Stimulates PKD1 Activation via a PKC-Dependent Pathway
We have previously demonstrated that aldosterone stimulates the concentration-dependent activation of PKD1 in M1-CCD cells (11). The activation of PKD1 in different experimental systems requires the phosphorylation of its activation loop residues Ser744 and Ser748 PKC isoforms (18). PKD1 then undergoes autophosphorylation at residue Ser916, which is believed to stabilize PKD1 activation. Autophosphorylation at residue Ser916 was used as a measure of PKD1 kinase activity. To determine whether PKC activation was involved in the aldosterone-induced activation of PKD1, serum-starved cells were treated with the potent and general PKC inhibitor chelerythrine chloride (10 μm) for 30 min before aldosterone treatment (IC50 PKC = 0.66 μm). Aldosterone (10 nm) induced a 3-fold increase in PKD1 autophosphorylation at Ser916 indicative of kinase activation within 5 min of hormone treatment. PKD1 autophosphorylation was inhibited by chelerythrine chloride, implicating PKC in the activation pathway (Fig. 1A). In other experimental systems, novel PKC (nPKC) rather than classical/conventional isoforms are the most prevalent in PKD regulation. The role of the nPKC isoform PKCδ in the activation of PKD1 in response to aldosterone was examined by preincubating the cells with the specific pharmacological inhibitor rottlerin (IC50 PKCδ = 3–5 μm). Rottlerin is widely used as a PKCδ-specific inhibitor; however, questions have recently arisen regarding the absolute specificity of rottlerin and whether it can also antagonize tyrosine kinase activity (19). M1-CCD cells were preincubated with rottlerin (20 μm) for 30 min before aldosterone treatment. Rottlerin blocked the activation of PKD1 in response to aldosterone, which normally occurred 5 min after addition of the hormone (Fig. 1B). This indicates a role for novel, calcium-independent PKC isoforms in PKD1 regulation in CCD cells treated with aldosterone. Rottlerin can also inhibit PKCε and PKCη activity at higher concentrations (IC50 PKCε = 80–100 μm); however, the amount of dimethylsulfoxide vehicle required to achieve such concentrations in this experimental model also stimulated the activation of PKD1 without the addition of aldosterone (data not shown).
Fig. 1.
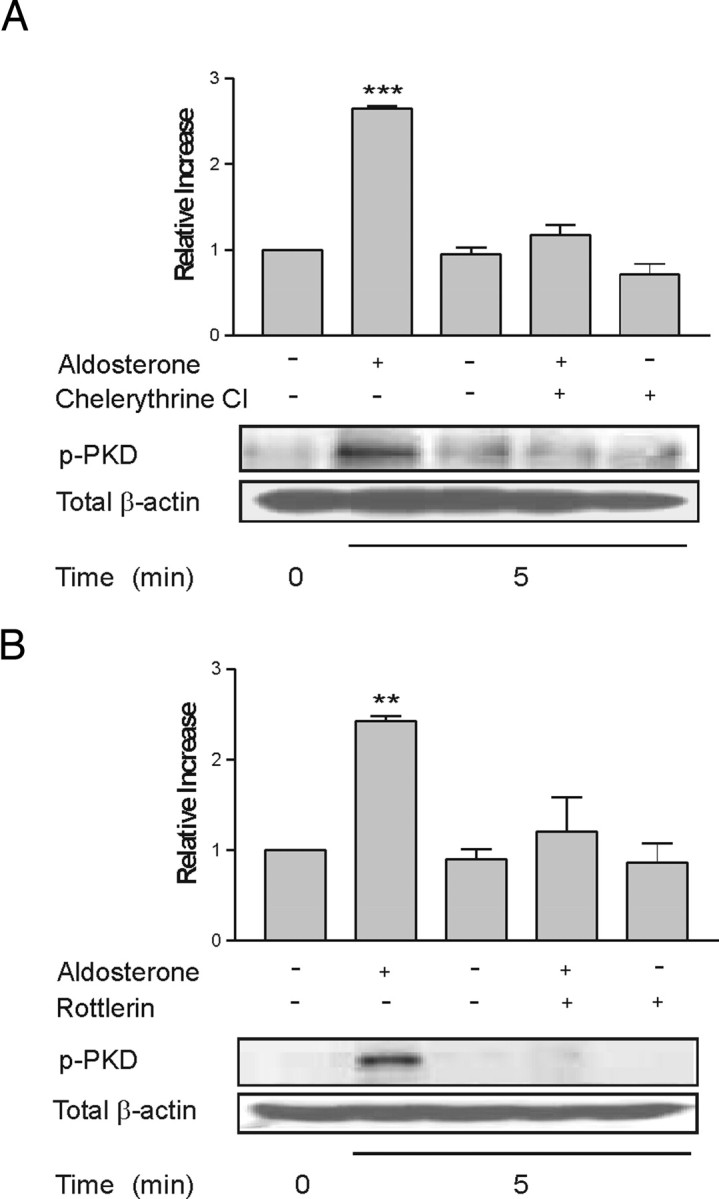
Aldosterone Induces PKD1 Activation via a PKC-Dependent Pathway
PKC isoform involvement in PKD1 activation was determined using specific inhibitors. M1-CCD cells were treated with the general PKC inhibitor chelerythrine chloride (A) at 10 μm for 30 min or the PKCδ antagonist rottlerin (B) at 20 μm for 30 min before the addition of aldosterone (10 nm). The effect of these inhibitors on aldosterone-induced PKD1 autophosphorylation was determined by Western blotting using a phospho-Ser916 site-specific antibody. Values represent the means of four separate experiments **, P < 0.01; ***, P < 0.001 vs. control.
Aldosterone Rapidly Stimulates PKCδ and PKCε Activation in M1-CCD Cells
PKC is known to play a role in aldosterone-mediated rapid signaling events and physiological responses. The levels of PKC isoform activation can be followed through changes in phosphorylation states at key amino acid residues. PKCδ and PKCε undergo autophosphorylation at Ser643 and Ser729, respectively, after activation; therefore, the level of phosphorylation at these residues can be used as a measure of the catalytic activity of the kinases. Both PKCδ and PKCε demonstrated low levels of autophosphorylation in resting M1-CCD cells. After aldosterone treatment, there was a 2- to 3-fold increase in phosphorylation at the respective autophosphorylation residues above the basal level at 2 min and again at 30 min for PKCδ (Fig. 2A) and PKCε (Fig. 2B). PKCδ and PKCε are thus strongly activated in response to aldosterone in M1-CCD cells with temporal kinetics similar to that previously described for PKD1 activation (11).
Fig. 2.
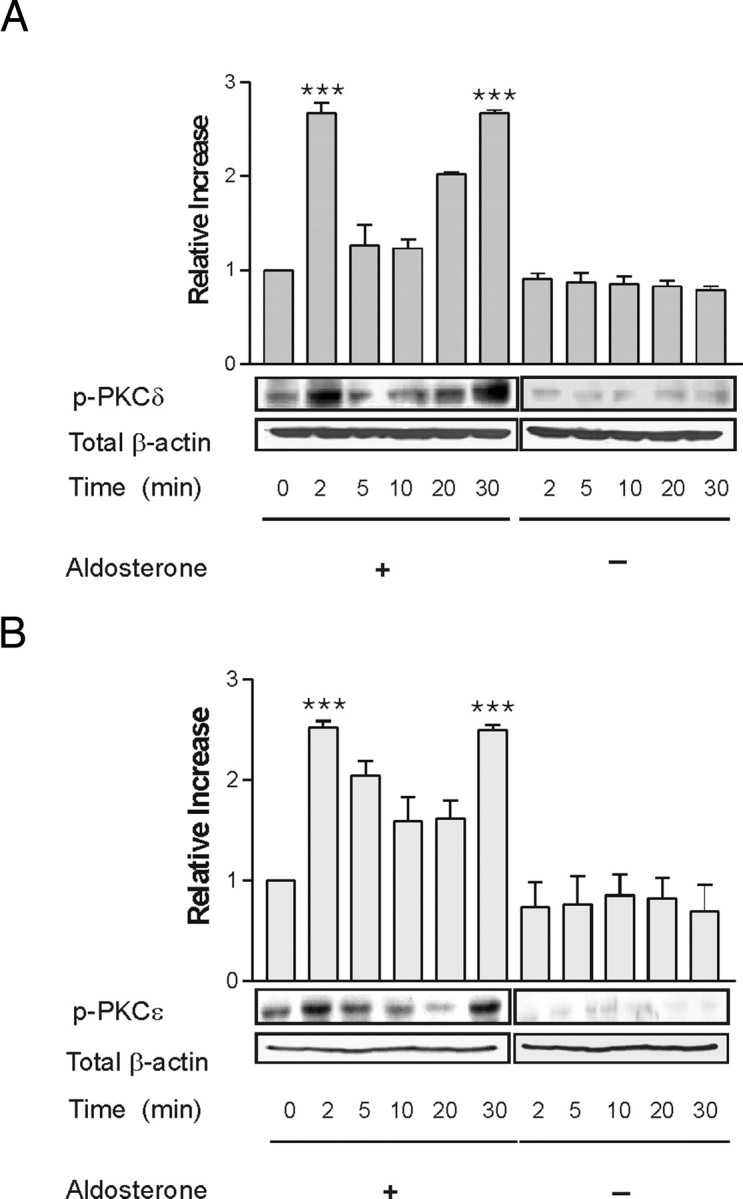
Aldosterone Induces Activation of nPKC Isoforms
M1-CCD cells were treated with aldosterone (10 nm), and autophosphorylation of nPKC isoforms was determined by Western blotting over a time course of 30 min. Autophosphorylation of PKCδ was determined using a phospho-Ser643-specific antibody (A), and autophosphorylation of PKCε was determined using a phospho-Ser729-specific antibody (B). Values represent the means of six separate experiments. ***, P < 0.001 vs. control.
Aldosterone Promotes the Association between PKD1 and PKCε
The activation of PKD1 through the stimulation of G protein-coupled receptor (GPCR) agonists is mediated by nPKC isoforms, most often PKCε. Because aldosterone synchronously stimulated the activation of the nPKC isoforms PKCδ and PKCε with PKD1 activation in M1-CCD cells, we examined whether physical association of these kinases occurred after aldosterone treatment. PKD1 was immunoprecipitated using a total PKD1 antibody from serum-starved cells at 0, 2, and 5 min after aldosterone treatment. The isolated proteins were Western blotted and probed with an antibody specific for PKCε. There was only weak association between PKD1 and PKCε in untreated cells and for up to 2 min after hormone treatment; however, after 5 min treatment, a stable complex formed between these two kinases that could be immunoprecipitated (Fig. 3A). Immunoprecipitation of PKCε from these cells using a total PKCε antibody followed by Western blotting for total PKD1 confirmed the formation of a stable complex between these two kinases within 5 min of aldosterone treatment (Fig. 3B). To investigate whether PKCδ also formed a complex with PKD1 in aldosterone-treated cells, PKD1 was immunoprecipitated from lysates 0, 5, and 15 min after aldosterone treatment using a total PKD1 antibody. The immunoprecipitates were analyzed by Western blotting using a PKCδ-specific antibody. PKCδ was found to be already associated with PKD1 in a stable complex in the M1-CCD cells before treatment with aldosterone. At 5 and 15 min after aldosterone treatment there was a significant reduction in the physical association between these two kinases (Fig. 3C). Commercially available anti-PKCδ antibodies are not recommended for immunoprecipitation, and consequently the inverse immunoprecipitation was not performed.
Fig. 3.
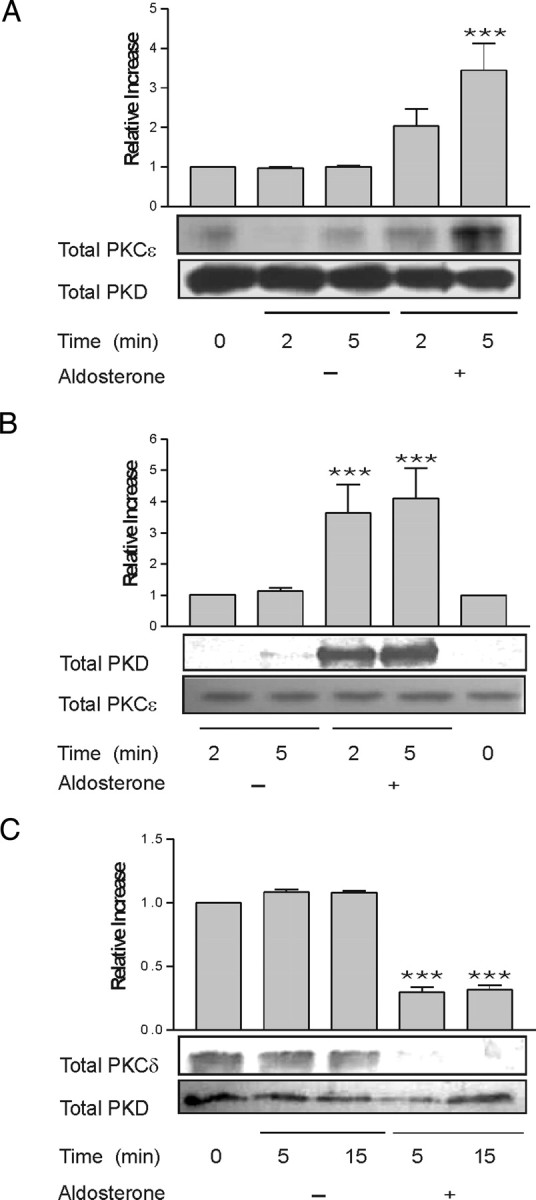
Aldosterone Promotes the Association of PKD1 with PKCε and Dissociation from PKCδ
The interaction between PKD1 and PKCε was investigated by co-immunoprecipitation. Lysates were prepared from M1-CCD cells treated with aldosterone (10 nm) for 0, 2, or 5 min. PKD1 was immunoprecipitated from the lysates using a total PKD1 antibody coupled to protein G-Sepharose. A, Immunoprecipitated proteins were separated by SDS-PAGE, Western blotted, and probed using a total PKCε antibody. Values represent the means of five separate experiments. ***, P < 0.001 vs. control. B, The reverse immunoprecipitation was performed using total PKCε antibody and then Western blotting with total PKD1. Values represent the means of four separate experiments. ***, P < 0.001 vs. control. C, The association between PKD1 and PKCδ was determined by immunoprecipitating PKD1 from lysates using a total PKD1 antibody and then Western blotting with total PKCδ antibody. Values represent the means of four separate experiments. ***, P < 0.001 vs. control. The immunocomplexes from each experiment were also reprobed with the same antibody used in the immunoprecipitation.
Aldosterone Promotes Rapid ENaC Translocation
The subcellular relocalization of ENaC subunits is a crucial stage in the increased stimulation of ENaC activity in response to aldosterone. PKD1 has been implicated in regulating subcellular trafficking, and the involvement of PKD1 in the early trafficking of ENaC subunits in response to aldosterone was investigated. The translocation of each of the ENaC subunits after treatment with aldosterone was analyzed by confocal microscopy, using plasmids expressing enhanced cyan fluorescent protein (eCFP)-tagged α, β, and γ ENaC subunit fusion proteins (Fig. 4). After 48 h growth in the presence of serum and 5 h under serum-free conditions to mitigate the effects of serum steroids, each of the eCFP-tagged ENaC subunits were found to be distributed uniformly throughout the cytoplasm of the cells. After aldosterone treatment for 2 min, a large portion of each of the eGFP-ENaC subunit fusion proteins became localized to a small number of discrete, densely staining areas within the cytoplasm of each cell.
Fig. 4.

Aldosterone Rapidly Promotes Redistribution of ENaC Subunits
M1-CCD cells were transfected with plasmids expressing ENaCα, ENaCβ, or ENaCγ as eCFP fusion proteins (green). The subcellular distribution of each of the ENaC subunits was examined by confocal microscopy in untreated cell monolayers and cells treated with aldosterone (10 nm) for 2 min. Cells were counterstained with TRITC-labeled phalloidin to detect polymerized actin (red), and single (XY) focal plane images through the center of the cells are shown. Small dense accumulations of each of the ENaC subunits were observed within 2 min of aldosterone treatment (white arrows). M1-CCD cells were transfected with a plasmid expressing ENaCα as an eCFP fusion protein (green). The subcellular distribution of ENaCα was examined by confocal microscopy in cell monolayers pretreated with spironolactone (20 μm) or rottlerin (20 μm) for 30 min in advance of aldosterone (10 nm) treatment for 2 min. Cells were counterstained with TRITC-labeled phalloidin to detect polymerized actin (red), and single (XY) focal plane images through the center of the cells are shown. Small dense accumulations of ENaCα were observed within 2 min of aldosterone treatment (white arrows), which were absent in cells pretreated with spironolactone or rottlerin.
The activation of aldosterone-induced rapid signaling responses is at least to some degree initiated through the binding of aldosterone to MR. This has been demonstrated through the effectiveness of MR-specific pharmacological antagonists in inhibiting a rapid nongenomic response to aldosterone. The MR antagonists spironolactone and RU28318 completely abolish aldosterone-induced PKD1 activation 5 min after hormone treatment (11). Preincubation of M1-CCD cells with the MR antagonist spironolactone for 10 min before aldosterone treatment inhibited the translocation of each of the eCFP-ENaC subunits to the discrete cytoplasmic sites (Fig. 4). The eCFP-ENaC subunits remained uniformly distributed in the cytoplasm of the spironolactone-treated M1-CCD cells. PKD1 is activated through the phosphorylation of its activation loop Ser residues by nPKCs. We demonstrated that preincubation with the PKCδ-specific antagonist rottlerin inhibited the aldosterone-induced activation of PKD1 (Fig. 1B). Rottlerin preincubation (20 μm, 30 min) of M1-CCD cells also abolished the aldosterone-induced rapid translocation of eCFP-tagged ENaCα in response to aldosterone (Fig. 4). These data confirm that MR and PKCδ are intermediate transducers in the activation of PKD1 by aldosterone to promote early ENaC trafficking events.
PKD1 Knockdown in M1-CCD Cells
Currently, the investigation of PKD1-dependent signaling pathways relies upon the use of nonspecific pharmacological inhibitors or the use of overexpressed dominant-negative mutants of PKD1. Recent descriptions of small interfering RNA (siRNA) models for identifying and studying PKD1 downstream effectors suggest that this important kinase can be suppressed in its expression while retaining cell viability (20, 21). This suggestion is further strengthened by the recent description of an avian cell line that is viable even after PKD1 gene knockout (22). To investigate the role of PKD1 in transducing the physiological effects stimulated by aldosterone in the CCD, a stable PKD1 knockdown cell line was developed. Three potential siRNA target sites within the murine PKD1 gene were identified, and expression cassettes expressing these siRNAs were individually transfected into M1-CCD cells. Western blotting using a PKD1-specific antibody revealed different degrees of effectiveness for each of the cassettes in suppressing PKD1 expression (Fig. 5A). Expression cassette 3 was found to be most effective in suppressing PKD expression by the M1-CCD cells, and this expression cassette was subcloned into the pSEC-Neo expression vector. A stable M1-CCD clone was derived through G418 selection where PKD1 protein expression was suppressed 80–90% as compared with nontransfected and empty vector stably transfected M1-CCD cells (Fig. 5B).
Fig. 5.

Development of a siRNA-Based PKD1 Suppression Model
A, The efficacy for each of the expression cassettes (oligo1, oligo2, and oligo3) to suppress PKD1 expression in the M1-CCD cell line was investigated by Western blotting. M1-CCD cells were transfected with equivalent amounts (50 ng) of each of the PKD1 siRNA expression cassettes or a GAPDH kit control. Lysates were prepared and Western blotted using a PKD1-specific antibody. A β-actin-specific antibody was used as a loading control. B, The efficacy of PKD1 suppression in a stable cell line knockdown (KD) harboring oligo3 siRNA expression cassette ligated into pSEC-Neo was compared with untransfected wild-type (WT) or empty vector control (C) transfected cells. Lysates were prepared and Western blotted using a PKD1-specific antibody. A β-actin-specific antibody was used as a loading control.
Suppression of PKD1 Expression Does Not Prevent Epithelial Polarization
Membrane transporters are specifically distributed into the apical or basolateral cell membrane after cell polarization. The Na+/K+ ATPase is located in the basolateral membrane of polarized CCD epithelial cells (23). To confirm that the M1-CCD cells formed a polarized epithelial monolayer when cultured on a glass surface, cells were seeded into chamber slides and allowed to reach 100% confluency and probed for Na+/K+ ATPase localization. The cells were fixed with paraformaldehyde and permeabilized. Immunofluorescent staining was performed to determine the subcellular distribution of Na+/K+ ATPase, and the cells were also stained with tetramethylrhodamine isothiocyanate (TRITC)-labeled phalloidin to detect polymerized actin. Confocal microcopy was performed to generate Z-stack sections through the monolayer.
The subcellular distribution of polymerized actin within the cells was consistent with the formation of a polarized epithelium. Actin stress fibers and focal adhesion were observed in the sections nearest the basal surface of the cells, and a dense ring of polymerized actin was observed in each cell below the apical cell membrane. This actin collar is consistent with the formation of tight junctions between the cells of the monolayer. The Na+/K+ ATPase was located sparsely in the cytoplasm of the M1-CCD cells, but the Na+/K+ ATPase was located at a high density in the basolateral membrane. The Na+/K+ ATPase was absent from the apical membrane, demonstrating that the M1-CCD cells had formed a polarized epithelium on the glass surface (Fig. 6).
Fig. 6.

Polarization of M1-CCD Cells on Glass
Wild-type M1-CCD cells (A) and M1-CCD cells harboring a plasmid expressing PKD1-specific siRNA (B) were grown on a glass surface until a confluent monolayer was established. The polymerized actin structure of the cells was stained using TRITC-labeled phalloidin (red), and the distribution of the Na+/K+ ATPase pump within the cells was determined by confocal immunofluorescence using a specific monoclonal antibody and detected using an antimouse Alexa 488 conjugate (green). The pump was located only at the basolateral surface of both cell lines.
The PKD1 knockdown M1-CCD cells were similarly examined to establish whether the loss of PKD1 expression affected their capacity to form a polarized epithelium. Confocal microscopy revealed that there was no significant difference in the distribution of polymerized actin or the Na+/K+ ATPase in PKD1-suppressed M1-CCD cells as compared with the wild-type cells (Fig. 6). Focal adhesions and polymerized actin rings were still observed, and the pump was located in the basolateral membrane but absent from the apical membrane. These results indicate that PKD1 suppression does not affect cell polarization in this experimental system.
Suppression of PKD1 Expression Does Not Affect the Genomic Response to Aldosterone
The genomic response to aldosterone is detected in all tissues expressing MR. The up-regulation of SGK-1 is the earliest transcriptional and translational effect elicited by aldosterone in the cells of the distal nephron. Changes in SGK-1 protein expression can be observed within 20 min of aldosterone treatment (24). The effect of suppressing PKD1 expression in the M1-CCD cells on the MR-mediated genomic response to aldosterone was determined by investigating the expression of SGK-1. We observed a 4- to 5-fold increase in SGK-1 expression above basal levels in wild-type M1-CCD cells treated with aldosterone (10 nm) compared with untreated or vehicle-treated cells after 30 min (Fig. 7). A similar level of SGK-1 protein induction (3.5- to 4-fold) was observed in cells stably suppressed in PKD1 expression. These results demonstrate that PKD1 suppression did not significantly affect the earliest phases of aldosterone-induced gene expression.
Fig. 7.
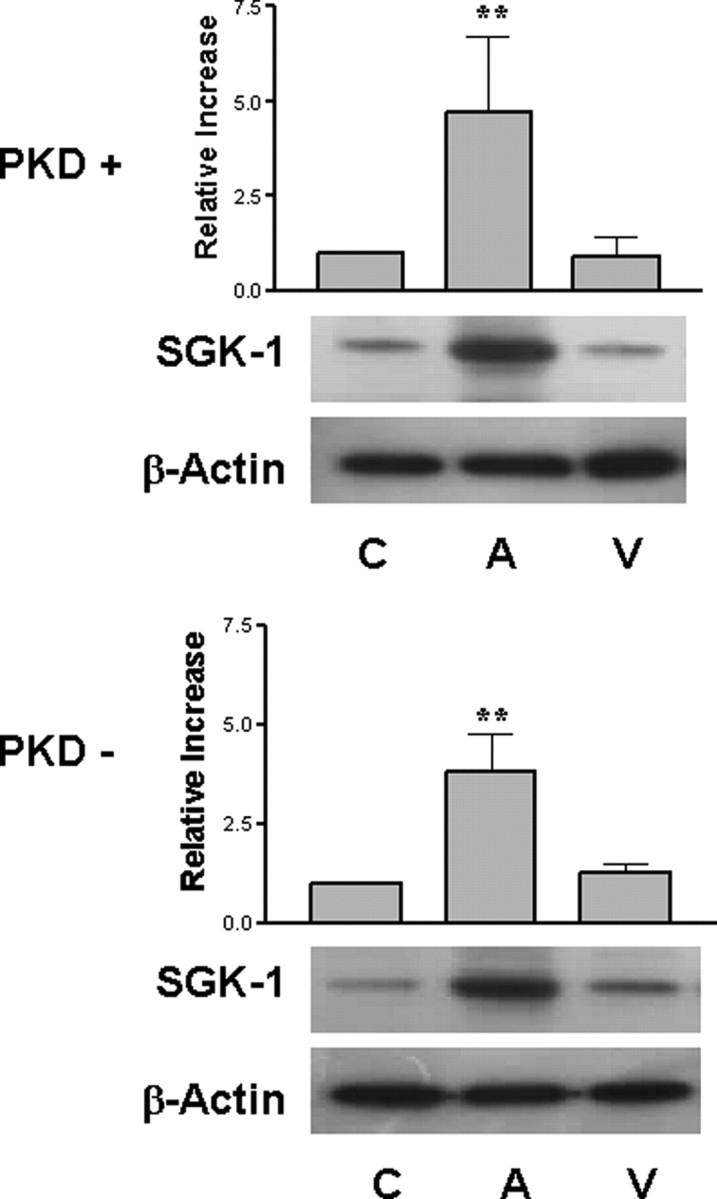
SGK-1 Induction by Aldosterone Is Not Sensitive to PKD1 Suppression
Lysates were prepared from wild-type M1-CCD cells (PKD+) and M1-CCD cells suppressed in PKD1 expression (PKD−) either treated with aldosterone (10 nm) for 30 min (A), vehicle treated for 30 min (V), or not treated (C). Equivalent amounts were separated on SDS-PAGE gel (80 μg protein) and then Western blotted and probed using a SGK-1-specific antibody. Values represent the means of four separate experiments. **, P < 0.01. Expression of β-actin was used as a control for gel loading.
Suppression of PKD1 Expression Affects Early Aldosterone-Induced ENaC Translocation
PKD isoforms are implicated in the regulation of vesicle fission from the TGN to facilitate protein trafficking from the Golgi to the cell membrane. We thus postulate that PKD1 activation may be involved in the rapid translocation of eCFP-ENaC subunits observed within 2 min of aldosterone treatment in M1-CCD cells. This hypothesis was tested in wild-type M1 cells and in the PKD1 knockdown M1-CCD clone. The cells were cotransfected with the eCFP-ENaCα expression plasmid, and the distribution of the fusion protein was examined by confocal microscopy. In contrast to wild-type M1 cells, the PKD1-suppressed cells transfected with the eCFP ENaCα-eCFP plasmid showed no redistribution of the ENaCα subunit in response to aldosterone within 2 min, confirming that PKD1 activation is a prerequisite for the early translocation of ENaC (Fig. 8).
Fig. 8.

Suppression of PKD1 Expression Blocks ENaCα Redistribution
Wild-type M1-CCD cells and cells stably suppressed in their PKD1 expression were transfected with a plasmid expressing ENaCα as an eCFP fusion protein (green). The subcellular distribution of ENaCα was examined by confocal microscopy in untreated cell monolayers and cells treated with aldosterone (10 nm) for 2 min. Cells were counterstained with TRITC-labeled phalloidin to detect polymerized actin (red), and single (XY) focal plane images through the center of the cells are shown. Small dense accumulations of ENaCα were observed within 2 min of aldosterone treatment in wild-type (white arrows) but not in PKD1-suppressed cells.
PKCε in PKD1 Activation and ENaC Translocation
We demonstrated that PKCε interacted with PKD1 after aldosterone treatment (Fig. 3A). In view of the problems with pharmacologically inhibiting PKCε activity to investigate the role of this kinase in the aldosterone-stimulated responses, siRNA was used to suppress PKCε expression. M1-CCD cells were transfected with an On-Target Plus siRNA Smart Pool (Dharmacon, Lafayette, CO) specific for murine PKCε, or cells were mock transfected. PKCε expression was investigated 72 h after transfection. The PKCε-specific siRNA suppressed PKCε expression in the M1-CCD cells by 90% compared with mock-transfected cells (Fig. 9). The activation of PKD1 in aldosterone-treated M1-CCD cells was compared with PKD1 activation in cells transfected with the PKCε-specific siRNA. Aldosterone (10 nm) stimulated a 3-fold increase in PKD1 autophosphorylation 5 min after treatment (Fig. 9). PKD1 activation was not activated above the vehicle response in mock-transfected cells. These data confirm the role of PKCε in mediating aldosterone-induced PKD1 activation in aldosterone-treated M1-CCD cells.
Fig. 9.

PKD Activation and PKDε siRNA
M1-CCD cells (PKCε+) or M1-CCD cells transfected with PKCε-specific siRNA (PKCε−) were grown in serum-free medium for 24 h and then treated with aldosterone (+) or vehicle only (−) for 5 min or left untreated. Lysates were prepared and analyzed by Western blot with a total PKCε polyclonal antibody, a PKD1 anti-phospho-Ser916-specific polyclonal antibody, or a total PKD1 polyclonal antibody. The data shown are representative of three independent experiments.
The stable knockdown of PKD1 expression inhibited the aldosterone-induced ENaC subunit intracellular translocation. Also, the suppression of PKCε expression inhibited the activation of PKD1 in response to aldosterone. It would therefore be expected that suppression of PKCε expression would also antagonize the rapid translocation of ENaC subunits in response to aldosterone. M1-CCD cells were separately transfected with each of the ENaC subunits, in combination with or without the PKCε-specific siRNA pool. The subcellular distribution of each of the ENaC subunits in +/− PKCε-specific siRNA transfected cells was compared before and after aldosterone treatment. We found that the suppression of PKCε expression using siRNA resulted in abnormal distribution of each of the ENaC subunits in the cytoplasm of M1-CCD cells. Instead of the uniform distribution observed in nontransfected cells, the ENaC subunits were restricted to densely stained areas within the cytoplasm of PKCε (−) cells that also colocalized with the actin-specific phalloidin-TRITC stain (Fig. 10). This observation suggested that PKCε plays an important role in the subcellular distribution of all three ENaC subunits under basal conditions even without aldosterone treatment and that the role of PKCε in subcellular trafficking is more complex than solely modulating PKD1 activity.
Fig. 10.

ENaC Subunit Localization Is Modulated by PKCε
M1-CCD cells were cotransfected with plasmids expressing each of the ENaCβ subunits (α, β, and γ) as an eCFP fusion protein (green) and PKCε-specific siRNA. After 48 h propagation in full medium, the transfected cells were examined by confocal microscopy. Cells were counterstained with TRITC-labeled phalloidin to detect polymerized actin (red), and single (XY) focal plane images through the center of the cells are shown of each fluor separated and as a merged image.
DISCUSSION
The distal nephron is a major site for hormonal regulation of Na+ efflux from the body. Na+ is reabsorbed at the luminal or apical surface of the principal epithelial cells from the renal ultrafiltrate through the ENaC and the Na+/Cl− cotransporter. Na+ is transported across the basolateral membrane of the epithelial cells and into the blood by the Na+/K+ ATPase, which also maintains a gradient for apical Na+ uptake. The membrane transporters implicated directly and indirectly in these processes are regulated by aldosterone at the transcriptional level over a number of hours after treatment (1). Some transporters are also activated rapidly, within a few minutes through the stimulation of cell signaling cascades that modulate the activity and subcellular distribution of pre-expressed transporter proteins. Transmembrane Na+/H+ exchanger activity was also identified as responding rapidly to aldosterone in the MDCK canine and M1-CCD murine renal cell lines (25).
The level of apical ENaC activity at the surface of principal cells of the CCD is the rate-limiting factor for Na+ reabsorption in the distal nephron. However, a role for the earliest rapid signaling responses stimulated by aldosterone in the CCD in promoting Na+ transport has been unclear. This is essentially because detection of increased ENaC activity in cell culture models through the measurement of short circuit current (ISC), occurs much later than the measurable effect of aldosterone on the ionic composition of the renal ultrafiltrate would predict (3). Recent work on the ENaC-related acid-sensing ion channel suggests that functional ENaC is a heterotrimer (1xα, 1xβ, 1xγ) (26) that assembles in the endoplasmic reticulum where it undergoes N-linked glycosylation. The channel undergoes further posttranslational modification on its passage through the Golgi; this includes furin cleavage of α- and γ-subunits and the concurrent substitution of a high-mannose glycosylation pattern for a complex one. The glycosylation and proteolytic processing of the ENaC channel are prerequisite for its full activity; however, partially processed or immature ENaC subunits are also associated with the cell membrane (27). Under high Na+ conditions ENaC is found in vesicles throughout the cytoplasm of CCD cells; however, Na+ depletion or aldosterone exposure results in translocation of the ENaC vesicular pool to the apical cell membrane well in advance of ENaC subunit transcriptional changes (28). This redistribution of pre-expressed ENaC has been linked to changes in SGK-1 expression within 1 h of aldosterone treatment. The earliest changes in SGK-1 expression using a near physiological concentration of aldosterone (10 nm) was observed 30 min after hormone treatment (29). In contrast, the earliest changes in ENaC activity are detected within 5 min of aldosterone treatment (3, 9) and must depend upon signaling events not coupled to increased SGK-1 availability.
We have shown that PKD1 is activated within 5 min in response to aldosterone in the M1-CCD cell line (11), and PKD is activated in response to diverse stimuli either at the cell membrane or at the TGN (reviewed in Ref. 30). PKD activity is stimulated by phosphorylation at two key residues within its activation loop (Ser744 and Ser748) by nPKC isoforms PKCδ, PKCε, and PKCη. Activation of PKD at the TGN results in its persistence at this organelle rather than shuttling to the nucleus, which is observed in response to cell membrane-associated PKD1 activation. PKD1 has been implicated in intracellular trafficking by regulating the fission of vesicles from the TGN (14, 15, 16). The precise mechanism is unclear; however, the interaction of PKD with the TGN-associated kinase PI-4 kinase III-β is believed to be significant (16).
The biphasic activation of PKCδ and PKCε coincided with the peaks of PKD1 autophosphorylation previously described, and these observations suggest that PKD1 activation is mediated by nPKC isoforms. PKCδ has been implicated in the activation of PKD by different agonists through pharmacological inhibition or expression knockdown using siRNA (20, 31, 32, 33, 34, 35, 36). However, an increased physical association in the form of a complex that can be immunoprecipitated has been demonstrated in only two instances (20, 35). In the M1-CCD cells, PKCδ was found in a complex with PKD1 in resting cells, an association that was relaxed after PKD1 activation by aldosterone. This observation suggests that the interaction between PKD and nPKCs may be different in individual experimental models. PKCε and PKCη are emerging as the most frequent activating kinases for PKD1 in different cell types in response to different agonists. PKCε directly phosphorylates and activates PKD1 in response to thrombin (35), bombesin, and phorbol 12,13-dibutyrate (37). The activation of PKD1 requires the formation of a complex with PKCε such as that observed in this study. PKCδ and PKCε can act synergistically to induce and stabilize their coactivation through trans-phosphorylation (38). PKCε phosphorylates its own activation loop residues and those of PKCδ, whereas PKCδ phosphorylates the hydrophobic motif sites of both PKC isoforms (38). This may also occur in response to aldosterone. The spironolactone-sensitive, rapid activation of PKCε by aldosterone in cardiomyocytes has been ascribed a physiological significance through the up-regulation of Na+/K+-ATPase activity (39, 40). The induction of hypertrophy in cardiomyocytes by GPCR agonists such as angiotensin II has been linked to the formation of a PKCε-PKD1 signaling complex at the Z-discs of these cells (41). We have previously reported cyclical activation of PKD1 as demonstrated by Ser916 autophosphorylation in M1-CCD cells (11). This response has also been observed in cardiomyocytes but not in cardiac fibroblasts after aldosterone treatment (42). Aldosterone-induced proliferation of cardiomyocytes was inhibited by Gö6976, an inhibitor of PKC/PKD activity, suggesting a role for this signaling cascade in aldosterone-induced cardiac hypertrophy. The induction of PKD1 activity in aldosterone-responsive tissues may prove to be an important aspect of the physiological responses stimulated by this hormone and also contribute to its pathological effects in target tissues.
PKD family isoforms have roles in establishing the structure of the Golgi and regulating TGN to membrane trafficking (43). PKD2 and PKD3 kinase-dead mutants caused TGN tabulation and inhibited TGN to membrane vesicle transport in a nonpolarized cell monolayer. In the same study, PKD2 was implicated in basolateral membrane-specific vesicle trafficking in a polarized epithelium (43). PKD activation had been regarded as a consequence of stimuli at the cell membrane, initiated through GPCRs or receptor tyrosine kinases. The activation of PKD through Golgi-associated heterotrimeric G proteins, specifically those with Gβ1γ2 and Gβ3γ2 subunits as distinct from membrane-associated receptors allows for organelle-targeted PKD activation resulting in fragmentation of the Golgi and the release of transport vesicles (44). This mechanism allows molecules leaving the Golgi to activate single or multiple PKD isoforms through Golgi-specific receptors and G proteins to direct the delivery of vesicle cargoes to the appropriate surface of a polarized cell (44). In this investigation, the formation of membrane-bound structures that were rich in ENaC subunits was observed within 5 min of treatment with aldosterone. The existence of an endoplasmic reticulum-Golgi intermediate compartment has been proposed as the initial site for post-endoplasmic reticulum protein sorting (45). The reported structure of the endoplasmic reticulum-Golgi intermediate compartment complex is consistent with the subcellular distribution of ENaC subunits observed within 2 min of aldosterone treatment. Data presented here demonstrate that aldosterone promotes the rapid transport of preexpressed ENaC subunits to membrane-bound structures within minutes of treatment. ENaC translocation is dependent on a PKD1-coupled signaling cascade that may potentiate or synergize with the delayed effects of aldosterone on ENaC subunit trafficking through SGK-1. PKD1 thus plays a role in pre-Golgi trafficking of ENaC in addition to its role as a regulator of the TGN.
MATERIALS AND METHODS
Cell Culture and Reagents
The M1-CCD cell line (CRL-2038; American Type Culture Collection, Manassas, VA) is derived from renal CCD microdissected from a mouse transgenic for the early region of SV40 virus [strain Tg(SV40E) Bri7] (46). M1-CCD cells were routinely grown in 75-cm2 polystyrene culture flasks containing 1:1 DMEM and Ham’s F-12 medium (DMEM/F-12) with fetal bovine serum (10%), supplemented with l-glutamine (2 mm), penicillin (100 U/ml), streptomycin (100 μg/ml), and dexamethasone (5 μm). Cultures were maintained in an atmosphere of 70% humidity, 5% CO2 at 37 °C. Medium was changed every 2–3 d, and cells were subcultured by trypsinization before they became confluent. For experimental purposes, cells were propagated in 6- or 10-cm polystyrene dishes or in eight-well glass chamber slides (Nunc, Roskilde, Denmark) as required, and experiments were performed once a confluent monolayer had been established. Cells were maintained in serum and steroid-free medium for either 5 or 24 h as indicated before antagonist or hormone treatment. M1-CCD cells were treated with spironolactone (10 μm) for the specific inhibition of the MR (Calbiochem, La Jolla, CA) and rottlerin (20 μm) for the specific inhibition of PKCδ (Sigma-Aldrich, Dublin, Ireland) for the times indicated. Aldosterone (Steraloids, Newport, RI) was prepared as a 50 μm stock in ethanol and then further diluted in culture medium so that the final concentration was 10 nm. All other chemical reagents used in this investigation were purchased from Sigma-Aldrich unless otherwise indicated.
Development of PKD1 siRNA Knockdown Cell Line
Three potential siRNA target sequences (sequence 1: bases 885–905, 5′-AATTCGCAGTCATACATCGGACG-3′; sequence 2: bases 2201–2221, 5′-AATTACTCAGATACTAGTGGCTT-3′; and sequence 3: bases 2705–2725, 5′-AATTGGAGAACGCTATATTACCC-3′) were identified within the open reading frame encoding the murine PKD1 mRNA (European Molecular Biology Laboratory, Heidelberg, Germany). The specificity of these sequences for PKD1 was confirmed by BLAST database search analysis. Oligonucleotides were designed on the basis of these sequences to facilitate the synthesis of DNA expression cassettes that expressed the respective siRNA under the control of the murine U6 promoter using the Silencer Express system according to the manufacturer’s directions (Ambion, Austin, TX). The expression cassettes were independently transfected into subconfluent M1-CCD cells grown on 6-cm dishes using Oligofectamine (Invitrogen, Carlsbad, CA). The efficacy of each of the expression cassettes in suppressing PKD1 expression was verified after 72 h by Western blotting using a PKD1-specific antibody. Dishes of cells were washed with ice-cold PBS (pH 7.4) and lysed on ice in chilled lysis buffer [20 mm Tris-HCl (pH 7.6), 250 mm NaCl, 3 mm EGTA, 3 mm EDTA, 1 mm dithiothreitol (DTT), 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 mm NaF, 1 mm PMSF, and 0.5% Nonidet P-40] at the time points indicated. The protein concentration of the lysates was determined and standardized by Bradford Assay (Bio-Rad, Hercules, CA).
Expression cassettes that effectively suppressed PKD1 expression were subcloned between the EcoR1 and HindIII sites of the pSEC-Neo mammalian expression vector (Ambion). Recombinant plasmids were transfected into M1-CCD cells grown on 10-cm dishes using Lipofectamine 2000 (Invitrogen). After 24 h, the medium was substituted with medium containing G418, the synthetic homolog of neomycin (200 μg/ml), and this medium was changed every 3 d over a period of 2 wk. Neomycin-resistant cells harboring the plasmid survived in the selective medium, and colonies of recombinant cells were isolated by localized trypsinization. Cell numbers were amplified in selective medium, and all experiments using the PKD1 stable knockdown cell line were performed in the presence of G418 (200 μg/ml). Continued suppression of PKD1 expression in the knockdown cell line was verified by Western blotting using the PKD1-specific antibody.
PKCε siRNA Inhibition
M1-CCD cells were seeded at 80% confluency in 10-cm dishes or eight-well chamber slides. A murine PKCε-specific On Target plus Smart pool of four inhibitory siRNA duplexes, was synthesized by Dharmacon with sense sequences as follows: duplex 5, GAGGACGAUUGUUCGAAUUU; duplex 6, GGAGUGAUCUACAGGGAUUUU; duplex 7, CGGAAACACCCUUAUCUAAUU; and duplex 8, GGUAGACGCCAGAGGAAUUUU. The siRNA pool (28 pmol per 10-cm plate or 5 pmol per chamber slide well) was transfected into the M1-CCD cells using Lipofectamine 2000 according to manufacturer-specified protocols. The cells were allowed to recover after transfection, and PKCε suppression was allowed to establish over the time periods indicated.
SDS-PAGE and Western Blotting
Cell lysates were prepared in chilled lysis buffer, cleared by centrifugation, and then dissolved in 2× Laemmli sample buffer (Sigma-Aldrich). Proteins were separated by SDS-PAGE on 10% polyacrylamide gels and then electrotransferred onto polyvinylidene difluoride membranes and probed with the specified primary antibodies diluted in 5% BSA in Tris-buffered saline (TBS) [50 mm Tris-HCl (pH 7.5), 150 mm NaCl] according to the supplier’s instructions. Bound antibodies were detected using antimouse or antirabbit horseradish peroxidase secondary conjugates. Antibodies were obtained from Abcam (Cambridge, UK) (Na+/K+ ATPase and PKCε phospho-Ser729); BD Biosciences (San Diego, CA) (total PKCδ and total PKCε), Cell Signaling Technology (Beverly, MA) (SGK-1, PKCδ phospho-Ser643, total PKD1, and PKD1 phospho-Ser916), or Sigma-Aldrich (β-actin, antimouse IgG horseradish peroxidase secondary conjugate, and antirabbit IgG horseradish peroxidase secondary conjugate). Labeled bands were visualized by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ). Exposed films were digitally photographed, and band density was measured using GeneSnap image analysis software (Synoptics, Cambridge, UK).
Immunoprecipitations
The formation of stable complexes between PKD1 and the PKC isoforms was determined using immunoprecipitation. Cleared lysates prepared as above in lysis buffer without DTT were incubated with the primary antibody indicated. Immunocomplexes were recovered by incubation with protein G-Sepharose (Amersham Biosciences) and isolated by centrifugation. The immunocomplexes were washed five times with lysis buffer without DTT and then subjected to SDS-PAGE and Western blotting as described above.
Confocal Microscopy
M1-CCD cells were routinely grown in eight-well chamber slides (Nunc). The establishment of a polarized epithelial monolayer by the cells when grown in this way was established by immunofluorescence using a Na+/K+ ATPase-specific antibody. The monolayer was washed with ice-cold PBS and then fixed with 4% freshly prepared paraformaldehyde in PBS, and the cell membranes were disrupted with 0.2% Triton X-100 in TBS. Nonspecific binding of antibodies and fluorescent conjugates was blocked using 2% fish skin gelatin (Sigma-Aldrich) and 1% fetal bovine serum in TBS. Effective polarization of the epithelium was demonstrated using an antibody specific for the Na+/K+ ATPase (located in the basolateral membrane). Bound antibody was detected according to manufacturer’s directions using a rabbit antimouse Alexa 488 conjugate (Invitrogen). Cells were counterstained with TRITC-labeled phalloidin (Invitrogen) for the detection of polymerized actin. Cells were mounted in Vectashield (Vector Laboratories, Burlingame, CA) and examined using an LSM 510 Meta confocal microscope (Zeiss, Oberkochen, Germany). Excitation wavelengths for Alexa Fluor 488 and TRITC were 488 and 543 nm, respectively.
Plasmids expressing eCFP N-terminally tagged murine α, β, and γ ENaC subunits were a gift from Prof. J. D. Stockand (University of Texas, San Antonio, TX). M1-CCD or M1-CCD PKD siRNA cells were grown in eight-well chamber slides for 24 h and then transfected when at 80% confluency with the individual eCFP-ENaC subunit expression plasmids using Lipofectamine 2000 for 6 h. The cells were grown for 48 h in the presence of 10% serum to achieve 100% confluency and then maintained in serum-free medium without steroids for an additional 5 h. Cells were subjected to agonist and antagonist treatments for the times indicated, fixed, and permeabilized, and nonspecific binding was blocked as above. Transfected cells were counterstained with TRITC-labeled phalloidin for the detection of polymerized actin. Cells were mounted in Vectashield and examined using an LSM 510 Meta confocal microscope. Images were captured at ×40 magnification with ×8 zoom. The excitation wavelengths used for eCFP, Alexa Fluor 594, and TRITC were 488, 543, and 543 nm, respectively. Scans were performed at 1-μm interval depths through the fixed cells and merged, or single fluor images are presented as single XY planes through the center of the cells or combined scans of the XY, XZ, and YZ planes as indicated.
Statistical Analysis
Densitometric values are presented graphically as the means of at least four independent experiments + sem with one representative Western blot. The ANOVA of densitometry values relative to untreated controls was performed using Dunnett’s modified t test on one-way ANOVA data, and P < 0.05 was treated as significant.
Acknowledgments
The eCFP-ENaC expression plasmids used in this investigation were a gift from Professor J. D. Stockand, University of Texas.
NURSA Molecule Pages:
Ligands: Aldosterone | Spironolactone.
Footnotes
This work was supported by The Higher Education Authority of Ireland (HEA) under the Program for Research in Third Level Institutions (PRTLI) Cycle 3 and by a Wellcome Trust program grant to B.J.H. (060809/Z/00).
Disclosure Statement: The authors have nothing to declare.
First Published Online January 17, 2008
Abbreviations: CCD, Cortical collecting duct; DTT, dithiothreitol; eCFP, enhanced cyan fluorescent protein; ENaC, epithelial sodium channel; GPCR, G protein-coupled receptor; MR, mineralocorticoid receptor; nPKC, novel PKC; PI, phosphatidylinositol; PKD1, protein kinase D; SGK, serum glucocorticoid-stimulated kinase; siRNA, small interfering RNA; TBS, Tris-buffered saline; TGN, trans-Golgi network; TRITC, tetramethylrhodamine isothiocyanate.
References
- 1.Booth RE, Johnson JP, Stockand JD 2002. Aldosterone. Adv Physiol Educ 26:8–20 [DOI] [PubMed] [Google Scholar]
- 2.Pan YJ, Young DB 1982. Experimental aldosterone hypertension in the dog. Hypertension 4:279–287 [DOI] [PubMed] [Google Scholar]
- 3.Ganong WF, Mulrow PJ 1958. Rate of change in sodium and potassium excretion after injection of aldosterone into the aorta and renal artery of the dog. Am J Physiol 195:337–342 [DOI] [PubMed] [Google Scholar]
- 4.Loffing J, Zecevic M, Feraille E, Kaissling B, Asher C, Rossier BC, Firestone GL, Pearce D, Verrey F 2001. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am J Physiol Renal Physiol 280:F675–F682 [DOI] [PubMed]
- 5.Loffing J, Summa V, Zecevic M, Verrey F 2001. Mediators of aldosterone action in the renal tubule. Curr Opin Nephrol Hypertens 10:667–675 [DOI] [PubMed] [Google Scholar]
- 6.Ronzaud C, Loffing J, Bleich M, Gretz N, Grone HJ, Schutz G, Berger S 2007. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol 18:1679–1687 [DOI] [PubMed] [Google Scholar]
- 7.Snyder PM, Olson DR, Thomas BC 2002. Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem 277:5–8 [DOI] [PubMed] [Google Scholar]
- 8.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O 2001. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J 20:7052–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou ZH, Bubien JK 2001. Nongenomic regulation of ENaC by aldosterone. Am J Physiol Cell Physiol 281:C1118–C1130 [DOI] [PubMed]
- 10.Pochynyuk O, Medina J, Gamper N, Genth H, Stockand JD, Staruschenko A 2006. Rapid translocation and insertion of the epithelial Na+ channel in response to RhoA signaling. J Biol Chem 281:26520–26527 [DOI] [PubMed] [Google Scholar]
- 11.McEneaney V, Harvey BJ, Thomas W 2007. Aldosterone rapidly activates protein kinase D via a mineralocorticoid receptor/EGFR trans-activation pathway in the M1 kidney CCD cell line. J Steroid Biochem Mol Biol 107:180–190 [DOI] [PubMed] [Google Scholar]
- 12.Rozengurt E, Rey O, Waldron RT 2005. Protein kinase D signaling. J Biol Chem 280:13205–13208 [DOI] [PubMed] [Google Scholar]
- 13.Hanks SK 2003. Genomic analysis of the eukaryotic protein kinase superfamily: a perspective. Genome Biol 4:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baron CL, Malhotra V 2002. Role of diacylglycerol in PKD recruitment to the TGN and protein transport to the plasma membrane. Science 295:325–328 [DOI] [PubMed] [Google Scholar]
- 15.Maeda Y, Beznoussenko GV, Van Lint J, Mironov AA, Malhotra V 2001. Recruitment of protein kinase D to the trans-Golgi network via the first cysteine-rich domain. Embo J 20:5982–5990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hausser A, Storz P, Martens S, Link G, Toker A, Pfizenmaier K 2005. Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIβ at the Golgi complex. Nat Cell Biol 7:880–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pochynyuk O, Tong Q, Staruschenko A, Stockand JD 2007. Binding and direct activation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. J Physiol 580(Pt 2):365–372 [DOI] [PMC free article] [PubMed]
- 18.Waldron RT, Rey O, Iglesias T, Tugal T, Cantrell D, Rozengurt E 2001. Activation loop Ser744 and Ser748 in protein kinase D are transphosphorylated in vivo. J Biol Chem 276:32606–32615 [DOI] [PubMed] [Google Scholar]
- 19.Soltoff SP 2007. Rottlerin: an inappropriate and ineffective inhibitor of PKCδ. Trends Pharmacol Sci 28:453–458 [DOI] [PubMed] [Google Scholar]
- 20.Storz P, Toker A 2003. Protein kinase D mediates a stress-induced NF-κB activation and survival pathway. EMBO J 22:109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hausser A, Link G, Bamberg L, Burzlaff A, Lutz S, Pfizenmaier K, Johannes FJ 2002. Structural requirements for localization and activation of protein kinase Cμ (PKCμ) at the Golgi compartment. J Cell Biol 156:65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matthews SA, Liu P, Spitaler M, Olson EN, McKinsey TA, Cantrell DA, Scharenberg AM 2006. Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol Cell Biol 26:1569–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verrey F 2001. Sodium reabsorption in aldosterone-sensitive distal nephron: news and contributions from genetically engineered animals. Curr Opin Nephrol Hypertens 10:39–47 [DOI] [PubMed] [Google Scholar]
- 24.Naray-Fejes-Toth A, Canessa C, Cleaveland ES, Aldrich G, Fejes-Toth G 1999. sgk is an aldosterone-induced kinase in the renal collecting duct. Effects on epithelial Na+ channels. J Biol Chem 274:16973–16978 [DOI] [PubMed] [Google Scholar]
- 25.Oberleithner H, Weigt M, Westphale HJ, Wang W 1987. Aldosterone activates Na+/H+ exchange and raises cytoplasmic pH in target cells of the amphibian kidney. Proc Natl Acad Sci USA 84:1464–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jasti J, Furukawa H, Gonzales EB, Gouaux E 2007. Structure of acid-sensing ion channel 1 at 1.9 Å resolution and low pH. Nature 449:316–323 [DOI] [PubMed] [Google Scholar]
- 27.Hughey RP, Bruns JB, Kinlough CL, Kleyman TR 2004. Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J Biol Chem 279:48491–48494 [DOI] [PubMed] [Google Scholar]
- 28.Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA 1999. Aldosterone-mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. J Clin Invest 104:R19–R23 [DOI] [PMC free article] [PubMed]
- 29.Naray-Fejes-Toth A, Fejes-Toth G 2000. The sgk, an aldosterone-induced gene in mineralocorticoid target cells, regulates the epithelial sodium channel. Kidney Int 57:1290–1294 [DOI] [PubMed] [Google Scholar]
- 30.Ghanekar Y, Lowe M 2005. Protein kinase D: activation for Golgi carrier formation. Trends Cell Biol 15:511–514 [DOI] [PubMed] [Google Scholar]
- 31.Song J, Li J, Lulla A, Evers BM, Chung DH 2006. Protein kinase D protects against oxidative stress-induced intestinal epithelial cell injury via Rho/ROK/PKC-δ pathway activation. Am J Physiol Cell Physiol 290:C1469–C1476 [DOI] [PMC free article] [PubMed]
- 32.Li J, O’Connor KL, Hellmich MR, Greeley Jr GH, Townsend Jr CM, Evers BM 2004. The role of protein kinase D in neurotensin secretion mediated by protein kinase C-α/-δ and Rho/Rho kinase. J Biol Chem 279:28466–28474 [DOI] [PubMed] [Google Scholar]
- 33.Tinsley JH, Teasdale NR, Yuan SY 2004. Involvement of PKCδ and PKD in pulmonary microvascular endothelial cell hyperpermeability. Am J Physiol Cell Physiol 286:C105–C111 [DOI] [PubMed]
- 34.Britt KL, Findlay JK 2002. Estrogen actions in the ovary revisited. J Endocrinol 175:269–276 [DOI] [PubMed] [Google Scholar]
- 35.Tan M, Xu X, Ohba M, Ogawa W, Cui MZ 2003. Thrombin rapidly induces protein kinase D phosphorylation, and protein kinase Cδ mediates the activation. J Biol Chem 278:2824-2828 [DOI] [PubMed] [Google Scholar]
- 36.Bradford MD, Soltoff SP 2002. P2X7 receptors activate protein kinase D and p42/p44 mitogen-activated protein kinase (MAPK) downstream of protein kinase C. Biochem J 366:745–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rey O, Reeve Jr JR, Zhukova E, Sinnett-Smith J, Rozengurt E 2004. G protein-coupled receptor-mediated phosphorylation of the activation loop of protein kinase D: dependence on plasma membrane translocation and protein kinase Cε. J Biol Chem 279:34361–34372 [DOI] [PubMed] [Google Scholar]
- 38.Rybin VO, Sabri A, Short J, Braz JC, Molkentin JD, Steinberg SF 2003. Cross-regulation of novel protein kinase C (PKC) isoform function in cardiomyocytes. Role of PKCε in activation loop phosphorylations and PKCδ in hydrophobic motif phosphorylations. J Biol Chem 278:14555–14564 [DOI] [PubMed] [Google Scholar]
- 39.Mihailidou AS, Mardini M, Funder JW 2004. Rapid, nongenomic effects of aldosterone in the heart mediated by ε protein kinase C. Endocrinology 145:773–780 [DOI] [PubMed] [Google Scholar]
- 40.Mihailidou AS, Bundgaard H, Mardini M, Hansen PS, Kjeldsen K, Rasmussen HH 2000. Hyperaldosteronemia in rabbits inhibits the cardiac sarcolemmal Na+-K+ pump. Circ Res 86:37–42 [DOI] [PubMed] [Google Scholar]
- 41.Iwata M, Maturana A, Hoshijima M, Tatematsu K, Okajima T, Vandenheede JR, Van Lint J, Tanizawa K, Kuroda S 2005. PKCε-PKD1 signaling complex at Z-discs plays a pivotal role in the cardiac hypertrophy induced by G-protein coupling receptor agonists. Biochem Biophys Res Commun 327:1105–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsybouleva N, Zhang L, Chen S, Patel R, Lutucuta S, Nemoto S, DeFreitas G, Entman M, Carabello BA, Roberts R, Marian AJ 2004. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation 109:1284–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yeaman C, Ayala MI, Wright JR, Bard F, Bossard C, Ang A, Maeda Y, Seufferlein T, Mellman I, Nelson WJ, Malhotra V 2004. Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat Cell Biol 6:106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diaz Anel AM, Malhotra V 2005. PKCη is required for β1γ2/β3γ2- and PKD-mediated transport to the cell surface and the organization of the Golgi apparatus. J Cell Biol 169:83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Appenzeller-Herzog C, Hauri HP 2006. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci 119:2173–2183 [DOI] [PubMed] [Google Scholar]
- 46.Stoos BA, Naray-Fejes-Toth A, Carretero OA, Ito S, Fejes-Toth G 1991. Characterization of a mouse cortical collecting duct cell line. Kidney Int 39:1168–1175 [DOI] [PubMed] [Google Scholar]