

Abstract
A phenotypic screen of a compound library for antiparasitic activity on Trypanosoma brucei, the causative agent of Human African Trypanosomiasis (HAT), led to the identification of N-(2-aminoethyl)-N-phenyl benzamides as a starting point for hit-to-lead medicinal chemistry. Eighty two analogues were prepared, which led to the identification of a set of highly potent N-(2-aminoethyl)-N-benzyloxyphenyl benzamides with the most potent compound 73 having an in vitro EC50 = 0.001 μM. The compounds displayed drug-like properties when tested in a number of in vitro assays. Compound 73 was orally bioavailable and displayed good plasma and brain exposure in mice, cured 2 out of 3 mice infected with Trypanosoma brucei in acute model when dosed orally at 50 mg/kg once per day for 4 days. Given its potent antiparasitic properties and its ease of synthesis, compound 73 represents a potential lead for the development of drug to treat Human African Trypanosomiasis.
Keywords: Human African Trypanosomiasis, “Sleeping Sickness”, Trypanosoma brucei inhibitor, Hit-to-lead optimization
1. Introduction
Human African Trypanosomiasis (HAT, Sleeping Sickness) occurs in 36 sub-Saharan Africa countries where biting tse-tse flies transmit the disease. The etiologic agent, Trypanosoma brucei, is a flagellated protozoan parasite that disseminates through the body during the early hemolymphatic stage and eventually enters the central nervous system to cause late-stage disease. Symptoms of late-stage HAT include sleep disturbance, cognitive dysfunction, coma, and death. Unless diagnosed and treated during the early stage, drugs must cross the blood-brain barrier to be effective. As a result, treatment options are severely limited for late-stage disease and consist of nifurtimox-eflornithine combination treatment (NECT) or the arsenical drug, melarsoprol.1,2 NECT is expensive and requires intravenous administration of the eflornithine component. Melarsoprol causes severe side effects including fatalities in 3–10% of patients. Two drugs are still being studied in clinical trials, fexinidazole3 and SCYX-7158.4 New drugs that are safer and simpler to use (preferably by oral administration) are urgently needed to address this pernicious disease.
Despite modern advances in chemistry, genomics, and high-throughput screening technology, antiparasitic drug discovery remains an immense challenge.5–7 Debates are ongoing whether target-based versus cell-based (“phenotypic”) screening are preferable for identifying novel drug classes.8,9 With respect to developing new drugs for HAT, we considered the additional requirement for needing compounds with brain permeability properties. The vast majority of small molecule (>98%) do not cross the blood-brain barrier.10 As a result, we felt that it was advantageous to use a cell-based screening strategy that would identify membrane permeable small molecules and provide a broad diversity of chemical scaffolds from which brain-permeable compounds might be identified.
A high throughput screen of a library of 700,000 compounds for growth inhibitory activity against Trypanosoma brucei was conducted as previously described.11 The 1035 confirmed and selective hits could be grouped into 115 distinct scaffolds. We have previously described a series of substituted 2-phenylimidazopyridines derived from this high throughput screening that was optimized by medicinal chemistry to result in compounds showing curative activity in the murine model of acute T. brucei infection.11 Other hits from the screening were evaluated for their potential to be further developed based on selectivity (parasite vs. mammalian cells), chemical tractability, and compliance with Lipinski rules. One of these hits, compound 1 (GNF-00-0394-8224-1), became the object of a hit-to-lead medicinal chemistry project and is described herein.
2. Results and discussion
2.1. Properties of lead compound (1)
Lead compound 1 was selected from the available hits based on drug-like features including low MW of 363.6, clogP of 3.48, H-bond donors of 1, H-bond acceptors of 2. Additional measurements from biological assays are shown in Table 1. It had good activity on T. brucei cells with selectivity over mammalian cells of >30-fold. It resisted metabolism in mouse liver microsomes with t½ > 60 min. Importantly, it showed excellent permeability into brain tissue following intraperitoneal injection in mice (Supporting information, Fig. S1), a necessary attribute for treating late-stage trypanosomiasis. As a hit compound, the one disadvantage is fairly potent activity on CYP3A4 enzyme with an IC50 of 0.074 μM (average of 2 independent assays). The CYP3A4 activity was determined to be attributable to the primary amine which was also necessary for the antiparasitic activity (discussed below). In the literature, other benzamides with activity against T. brucei are reported but with no primary amino group and completely different SAR profile.12–15
Table 1.
Properties of the original hit compound (1) from high-throughput screen.
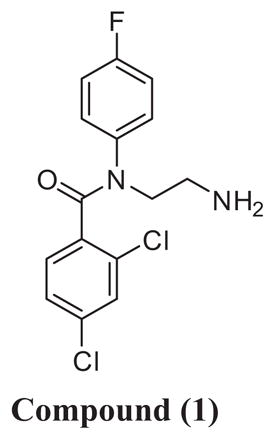 Compound (1) | |
|---|---|
| MW | 363.6 |
| cLogP | 3.48 |
| T. brucei brucei EC50 (μM)a | 1.21 |
| HepG2 cells CC50 (μM)b | 40.0 |
| CRL-8150 CC50 (μM)c | 30.0 |
| Mouse liver microsome t1/2 (min)d | >60 |
| CYP3A4 IC50 (μM)e | 0.074 |
Concentration of compound required to inhibit growth by 50% (EC50) of T. brucei brucei strain BF427.
Concentration of compound required to inhibit growth by 50% (CC50) of mammalian cell lines human hepatocytes (HepG2) and human lymphoblasts (CRL-8150) respectively.
Time required by liver microsomes (mouse) to reduce the amount of compound by half.
Concentration of compound required to inhibit by 50% (IC50) of human cytochrome P450 (3YP3A4 isoform) enzyme.
2.2. Synthesis of 1 and its analogues
The N-(2-aminoethyl)-N-phenyl benzamide derivatives (2–44) and compound 1 were synthesized starting from corresponding commercial anilines 84 by reductive alkylation with N-Boc-2-aminoacetaldehyde (Scheme 1). Benzoylation of amine 85 and subsequent removal of the Boc protecting group with hydrochloric acid in dioxane gave final compounds purified by flash chromatography or HPLC.
Scheme 1.

Synthesis of N-(2-aminoethyl)-N-phenyl-benzamidesa (1–44). aReagents and conditions: (a) N-Boc-2-aminoacetaldehyde, NaCNBH3, chloroform, rt; (b) R2COCl, DIPEA, dichloromethane, 4 °C to rt; (c) 4 M HCl in dioxane, rt.
Scheme 2 shows the synthetic route to make N-(2-aminoethyl)-N-benzyloxyphenyl benzamides analogues. The compounds were synthesized in four steps starting with alkylation of 4-nitrophenol (87) with alkyl or benzyl bromides (86) (Scheme 2, condition (a)). A second step – reduction of the aromatic nitro group was performed in two different conditions depending on presence of halogens in the aromatic ring (Scheme 2, condition (b)). The reduction of molecule containing aromatic halogens (chlorine) was performed with activated zinc/copper pair in aqueous ammonium chloride to prevent dehalogenation observed with use of palladium catalyst. The third step – an N-alkylation of anilines 88 by reductive amination with N-Boc-2-aminoacetaldehyde (Scheme 2, condition (c)). Final step is benzoylation of amine 89 and subsequent removal of the Boc protecting group under room temperature with hydrochloric acid in dioxane that yield final compounds 64–83 as hydrochloric salt (Scheme 2, conditions (d, e)). All final compounds were purified by HPLC.
Scheme 2.

Synthesis of N-(2-aminoethyl)-N-benzyloxyphenyl-benzamidesa (64–83). aReagents and conditions: (a) Acetone, 60 °C, K2CO3;(b) Pt/C, H2, EtOAc, rt or Zn/Cu, aq. NH4Cl/diethylether, rt; (c) N-Boc-2-aminoacetaldehyde, Na(OAc)3BH, chloroform, rt; (d) R2COCl, DIPEA, dichloromethane, 4 °C to rt; (e) 4 M HCl in dioxane, rt.
2.3. Structure–activity studies of analogues of (1)
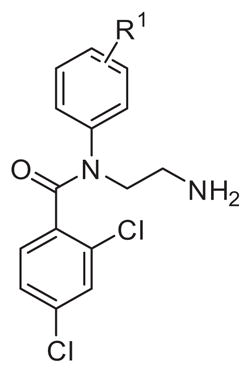
2.3.1. Substitutions at the aniline ring (R1)
The biochemical target of action for compound 1 is not known, thus an unbiased approach was taken to investigate different substitutions at varying parts of the molecule. First, changes to the para-fluoro substituent (R1 position) were investigated (Table 2). Replacement with chlorine (2) or bromine (3) led to a 4-fold enhancement of potency. Trifluoromethyl (4) methyl (5) and phenyl (6) derivatives retained potency, while isopropyl (7) was less active in comparison to 1. Replacement of fluorine (1) by methoxy (8), nitro (9) or amino (10) group eliminated activity on T. brucei cells.
Table 2.
SAR optimization of site R1 of N-(2-aminoethyl)-N-phenyl-2.4-dichlorobenzamides.*
| |||
|---|---|---|---|
| Compound | R1 | EC50 (μM)a | CC50 (μM)b |
| 1 | 4-F | 1.21 | 30.0c, 40.0d |
| 2 | 4-Cl | 0.31 | 12.5 c, 14.2d |
| 3 | 4-Br | 0.33 | 11.5c,11.0d |
| 4 | 4-CF3 | 1.05 | 13.2 e |
| 5 | 4-CH3 | 1.52 | 24.9c |
| 6 | 4-Ph | 1.83 | |
| 7 | 4-CH(CH3)2 | 3.89 | |
| 8 | 4-OCH3 | >6 | 56.1e |
| 9 | 4-NO2 | >10 | |
| 10 | 4-NH2 | >10 | 11.0c |
| 11 | 3-Cl | 3.05 | |
| 12 | 2,4-Di-Cl | 0.51 | |
| 13 | 3,4-Di-Cl | 1.68 | |
| 14 | 3-Cl-4F | 2.72 | |
| 15 | 4-Cl-3-OCH3 | 4.52 | |
| 16 | 4-(4-Cl-Ph-O) | 0.17 | |
| 17 | 4-(3-Cl-Ph-O) | 0.28 | |
| 18 | 4-(2-Cl-Ph-O) | 3.60 | |
| 19 | 3-(4-Cl-Ph-O) | 2.13 | |
All compounds prepared as HCl salts.
Concentration of compound required to inhibit growth by 50% (EC50) of T. brucei brucei strain BF427.
Concentration of compound required to inhibit growth by 50% (CC50) of mammalian cell lines.
Human lymphoblasts (CRL-8155).
Human hepatocytes (HepG2).
Rat myoblasts (L6).
To investigate the influence of substitution position in aromatic ring on activity, the meta-chloro substituted analogue 11 was prepared and showed 10-fold reduction in potency over corresponding para-chloro (2) derivative, indicating the importance of para-position (R1) for activity. From 2,4-di-chloro (12), 3,4-dichloro (13), 4-fluoro-3-chloro (14) and 4-chloro-3-methoxy (15) substituted analogues, only 2,4-dichloro (12) derivative has more than 2-fold improved activity with respect to 1 and just small reduction of potency with regards to 2.
Substitution at para-position with the bigger and more lipophilic 4-chlorophenoxy (16) or 3-chlorophenoxy (17) group led to enhanced antiparasitic activity by 7-fold and 4-fold respectively, while 2-chlorophenoxy (18) derivative resulted in reduced potency. Moving 4-chlorophenoxy substituent to the meta-position (19) drastically reduced the potency indicating importance of 4-substitution position for antiparasitic activity with no obvious size limitation of substituents in this preferred substitution position.
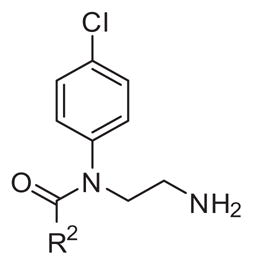
2.3.2. Substitutions at the benzoyl position (R2)
The influence of varying the benzoyl part of the molecule (R2) on antiparasitic activity was assessed while keeping the 4-chlorophenyl substituent unchanged (Table 3). First, the unsubstituted benzoyl derivative 20 is less active than all other compound in this series. Among isomers of mono-substituted benzoyl derivatives the most active are the 2-substituted benzoyl analogues. The 2-chlorobenzoyl derivative 23 is 3 fold more active than 3-chloro (22) and almost 8-fold more active than corresponding 4-chlorobenzoyl derivative 21. 2-Methyl derivative 24 is 14-fold more active than 4-methyl derivative 25 and 2-methoxy analogue 26 is at least 4-times more potent than corresponding 4-methoxy (27). The 2-chloro (23) and 2-methyl (24) analogues are equipotent while 2-methoxy derivative 26 is at least 5 times less active compere to 23, 24. In the case of mono-fluoro isomers 28, 29, 30 there is no difference in activity indicating that substituents larger than F are needed to influence potency favorably. Among the dichloroisomers (2, 31–34) 2,4-dichloro (2) and 2,3-dichloro (31) are the most active and equally potent with 2-chloro derivative 23.
Table 3.
SAR optimization of benzoyl site R2 of N-(2-aminoethyl)-N-4-Cl-phenyl-benzamides.*
| |||
|---|---|---|---|
| Compound | R2 | EC50 (μM)a | CC50 (μM)b |
| 20 | Ph | 13.00 | |
| 21 | 4-Cl-Ph | 3.12 | |
| 22 | 3-Cl-Ph | 1.20 | |
| 23 | 2-Cl-Ph | 0.44 | >50c, >50d |
| 24 | 2-CH3 | 0.52 | |
| 25 | 4-CH3 | 7.00 | |
| 26 | 2-OCH3 | 2.51 | |
| 27 | 4-OCH3 | >10 | |
| 28 | 4-F-Ph | 4.01 | |
| 29 | 3-F-Ph | 4.03 | |
| 30 | 2-F-Ph | 3.90 | 104.1e |
| 31 | 2,3-Di-Cl | 0.31 | |
| 32 | 2,5-Di-Cl | 0.60 | |
| 33 | 2,6-Di-Cl | 0.78 | >50c,>50d |
| 34 | 3,5-Di-Cl | 1.08 | |
| 35 | 4-Cl-3-NO2 | 2.90 | |
| 36 | 2-Cl-5-NO2 | 1.20 | |
| 37 | 2,4,6-Tri-Cl | 0.23 | 8.5c, 21.5d |
| 38 | 2,4-Di-CH3 | 0.97 | |
| 39 | 2,4-Di-OCH3 | 4.82 | |
| 40 | 2,4-Di-F | 1.24 | |
| 41 | 2,3,4,5,6-Penta-F | 0.54 | |
| 42 | 2-CF3 | 0.33 | |
| 43 | 2-CF3-4-F | 0.18 | 42.3c, 37.7d |
| 44 | 2-CF3-3-Pyr | 0.59 | |
All compounds prepared as HCl salts.
Concentration of compound required to inhibit growth by 50% (EC50) of T. brucei brucei strain BF427. Pentamidine isethionate was used as control with EC50 = 0.0021 ± 0.00001 μM.
Concentration of compound required to inhibit growth by 50% (CC50) of mammalian cell lines.
Human lymphoblasts (CRL-8155).
Human hepatocytes (HepG2).
Rat myoblasts (L6).
4-Chloro-3-nitro analogue 35 retains the potency of monochloro derivative 21, while 2-chloro-5-nitro compound 36 is 3 times less potent than corresponding 2-chloro analogue 23. 2,4,6-tri-Chlorobenzoyl derivative 37 is the most active from all chlorobenzoyl derivatives. No increase in activity was observed comparing the 2,4-dimethyl (38) and 2,4-dimethoxy (39) derivatives with corresponding mono-ortho-substituted analogues. Interestingly pentafluoro derivative 41 showed 26-fold improvement in activity compared to 20 and the 2,4-difluorobenzoyl compound 40 was at least 3-fold more active than 2-floro (30) or 4-fluoro (28) derivatives. Making a bigger ortho-benzoyl substituent like 2-trifluoromethyl derivative 42 improves the potency and the most active analogue in this SAR series was 2-trifluoromethyl-4-flurobenzoyl derivative 43. In order to make the compounds more water soluble the 2-trifluoromethyl-pyridinoyl compound 44 was made and it showed a high potency against T. brucei parasites (EC50 = 0.59 μM). Selected compounds were tested for growth inhibition activity on mammalian cells and were observed to have low toxicity (Table 3).
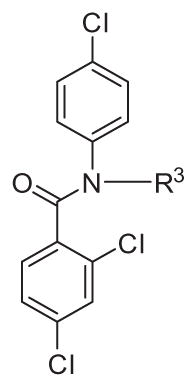
2.3.3. Substitutions at the ethylamino position (R3)
To investigate the SAR of ethylamino group (R3), we synthesized compounds derivatives of 2 (Table 4). Removing amino group at position R3 (45) as well as acylation (46) and dimethylation (47) of amino group eliminated anti-T. brucei activity. The IC50 of compound 45 on CYP3A4 was 11.9 μM (>100-fold weaker than compound 2 with CYP3A4 EC50 = 0.070 μM) indicating that the CYP450 inhibitory activity was due to the free amino group.
Table 4.
SAR optimization of ethylamino site R3 of N-4-Cl-phenyl-2,4-benzoylmides.
| ||
|---|---|---|
| Compound | R3 | EC50 a (μM) |
| 45 | CH2CH3 | >10 |
| 46 | CH2CH2NHC(O)CH3 | >10 |
| 47 | CH2CH2N(CH3)2 | >10 |
| 48 | CH2CH2N(CH2)4O | >10 |
| 49 | CH2CH2OH | >10 |
| 50* | CH2CH2CH2NH2 | 2.90 |
| 51* | CH2CH(CH3)NH2 | 1.05 |
| 52* | CH(CH3)CH2NH2 | 3.56 |
| 53 | CH2-2-furane | >10 |
| 54 | CH2-2-thiophene | >10 |
| 55* | CH2-2-imidazole | >10 |
| 56* | CH2-5-imidazole | >10 |
| 57* | 3-Pirrolidine | 4.47 |
| 58* | CH2CH2NHCH3 | 1.65 |
| 59 | CH2CH2C(O)NH2 | >10 |
| 60 | CH2CN | >10 |
| 61* | 3-Piperidine | >10 |
| 62* | 4-Piperidine | 4.11 |
| 63* | 3-Pyridine | >10 |
Compounds prepared as HCl salts.
Concentration of compound required to inhibit growth by 50% (EC50) of T. brucei brucei strain BF427. Pentamidine isethionate was used as control with EC50 = 0.0021 ± 0.00001 μM.
The morpholino analogue 48 was also inactive in the T. brucei EC50 assay. Changing NH2 group to OH led to an inactive alcohol 49. Elongation of the alkyl chain to propylamine (50) resulted in 10-fold loss of activity with almost the same gain in activity for propyl chain isomers (51, 52). Methylene-heterocycle derivatives 53–56 were made to keep the three C—C bond distance between heteroatoms the same as in compound 2, however, all of these compounds 53–56 were inactive. 3-Pyrrolidine derivative 57 showed low activity with EC50 of 4.5 μM. Monomethylation of the amino group (58) decreased activity 5-fold compared to 2. The carbonyl amide (59) and CN (60) derivatives were inactive. Among piperidine isomers, the 4-piperidine derivative 62 was at ~2.5 fold more active than 3-piperidine (61) but 10-fold less active than compound 2. The 3-pyridine analogue 63 was inactive. Based on this SAR the unprotected, unsubstituted ethylamino group is indispensable for antiparasitic activity.
2.3.4. Substitutions at the aniline ring (R1) – round 2
After the first round of optimization leading to analogues 15 and 42 with EC50 (T.b.b.) = 0.17 μM and 0.18 μM respectively, we performed a second round of optimization on site R1 (Table 5).
Table 5.
N-(2-Aminoethyl)-N-benzyloxyphenyl-benzamides.*
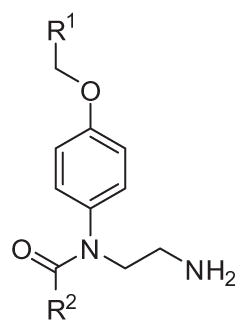
| ||||
|---|---|---|---|---|
| Compound | R1 | R2 | EC50 (μM)a | CC50 (μM)b |
| 64 | 4-Chlorophenyl | 2,4-Dichlorophenyl | 0.031 | 2.00c, 2.00d |
| 65 | ” | 2-Trifluoromethyl-4-fluorophenyl | 0.007 | 3.67c, 7.53d |
| 66 | ” | 2-Trifluoromethyphenyl | 0.007 | 3.08c, 5.72d |
| 67 | ” | 2-Chloro-3-pyridyl | 0.030 | 11.46c, 15.22d |
| 68 | ” | 2-Trifluoromethyl-3-pyridyl | 0.003 | 8.01c, 10.22d |
| 69 | 4-Isopropylphenyl | 2,4-Dichlorophenyl | 0.005 | 3.10c, 3.19d |
| 70 | ” | 2-Trifluoromethyl-4-fluorophenyl | 0.002 | 2.83c, 3.66d |
| 71 | ” | 2-Trifluoromethyphenyl | 0.003 | 2.22c, 3.99d |
| 72 | ” | 2-Chloro-3-pyridyl | 0.002 | 4.03c, 10.53d |
| 73 | ” | 2-Trifluoromethyl-3-pyridyl | 0.001 | 2.03c, 3.83d |
| 74 | ” | 2-Nitrophenyl | 0.001 | 3.03c |
| 75 | ” | 2-Bromophenyl | 0.001 | 1.91c |
| 76 | 3,5-dimethylphenyl | 2,4-Dichlorophenyl | 0.020 | 2.45c, 4.10d |
| 77 | ” | 2-Trifluoromethyl-4-fluorophenyl | 0.005 | 2.24c, 4.53d |
| 78 | ” | 2-Trifluoromethyphenyl | 0.010 | 2.94c, 3.94d |
| 79 | ” | 2-Chloro-3-pyridyl | 0.015 | 5.12c, 8.38d |
| 80 | ” | 2-Trifluoromethyl-3-pyridyl | 0.003 | 4.74c, 6.46d |
| 81 | 3-Chlorophenyl | 2,4-Dichlorophenyl | 0.043 | 2.38c, 3.78d |
| 82 | 4-tert-Buthylphenyl | 2,4-Dichlorophenyl | 0.005 | 2.12c, 3.83d |
| 83 | 4-Ethylphenyl | 2,4-Dichlorophenyl | 0.015 | 2.59c, 5.60d |
All compounds prepared as HCl salts.
Concentration of compound required to inhibit growth by 50% (EC50) of T. brucei brucei strain BF427. Pentamidine isethionate was used as control with EC50 = 0.0021 ± 0.00001 μM.
Concentration of compound required to inhibit growth by 50% (CC50) of mammalian cell lines.
Human lymphoblasts (CRL-8155).
Human hepatocytes (HepG2).
It was found that 4-chlorobenzyloxy derivatives substantially increased activity with analogue 64 having an EC50 of 0.031 μM. Switching to 2-trifluoromethyl-4-fluorobenzoyl and 2-trifluoromethyl derivatives 65, 66 further improve potency to single digit nanomolar EC50 values. From nicotinoyl derivatives the 2-trifluoromethylnicotinoyl (68) was 10-fold more active than 2-chloronicotinoyl (67) even though both are very active with EC50 of 0.003 μM and 0.03 μM, respectively. Changing the position of the chlorine substituent in the chlorobenzoxy part to 3-chlorobenzoxy (81) resulted in comparable potency to 4-chlorobenzoxy analogue 64. Introduction of an isopropyl group at position 4 further improved potency. The most active compounds 69–75, containing a 4-isopropylbenzyloxy moiety, had EC50 values in the low nanomolar range. Interestingly, the 2-nitorobenzoyl (74) and 2-bromobenzoyl (75) compounds were as active as 2-trifluoromethylnicotinoyl derivative 73, each with EC50 values of 0.001 μM. The 2-trifluoromethyl-4-fluorobenzoyl (70) and 2-trifluoromethylbenzoyl (71) derivatives had EC50 values of 0.002 and 0.003 μM, respectively. 3,5-Dimethyl analogues 78–80 showed slight reduction in potency compared to corresponding 4-isopropyl derivatives but still retained low nanomolar antiparasitic activity with EC50 of 0.003 μM for the most active 2-trifluoromethylnicotinoyl analogue 80. Switching to 4-terbutyl (82) or 4-ethylbenzyloxy (83) derivatives retained the potency. The second-generation SAR effort around aniline ring R1 shows that oxybenzyl moiety with the lipophilic substituent in aromatic ring (chloro, methyl, isopropyl, tert-butyl) at 4 and/or 3 position(s) significantly improves in-vitro antiparasitic activity of lead compounds.
3. Biological studies
3.1. Pharmacological testing
The analogues with the highest potency against T. brucei cultures were subjected to additional pharmacological profiling (Table 6). Eight out of 10 compounds that were tested showed half-life time >60 min in human liver microsome incubations. This may be related to strong potency against CYP450 enzymes as indicated by inhibition of CYP3A4 in the range of 0.003–0.179 μM. Pharmacokinetics in mice were assessed by oral gavage dosing at 50 mg/kg and sampling plasma at intervals out to 8 h. The maximum plasma concentration (Cmax) was generally in the range of 1–2 μM, with the exceptions of 65 and 76 having Cmax values of 4.4 and 5.9 μM, respectively.
Table 6.
Antiparasitic activity, pharmacological data and efficacy of selected compounds.
| Comp. | EC50 (μM) a
|
CYP3A4 IC50 (μM)b | Liver microsome T1/2 (min)c | Brain/plasma ratiod | Cma × (μM)e | AUC ± SEM (min*μM)f | Acute cure rate (mice)g | |
|---|---|---|---|---|---|---|---|---|
| T. brucei | T. rhod | |||||||
| 64 | 0.031 | 0.005 | 0.179 | 35 (mouse), >60 (human) | 0.42 ± 0.06 | 0.53 ± 0.23 | 205.05 ± 91.48 | 4/5 |
| 65 | 0.007 | 0.010 | >60 (mouse), >60 (human) | 0.38 ± 0.05 | 4.39 ± 0.56 | 1560.83 ± 364.67 | 2/3 | |
| 66 | 0.007 | >60 (human) | 0.88 ± 0.35 | 1.67 ± 0.41 | 616.97 ± 146.07 | 1/3 | ||
| 69 | 0.005 | 0.019 | 0.015 | >60 (human) | 0.32± 0.07 | 1.49 ± 0.36 | 1260.70 ± 128.72 | 2/3 |
| 70 | 0.002 | 52 (human) | 0.22 ± 0.10 | 1.78 ± 0.31 | 665.63 ± 204.69 | 1/3 | ||
| 72 | 0.002 | >60 (human), >60 (mouse) | 0.16 ± 0.09 | 1.89 ± 0.28 | 735.07 ± 72.62 | 2/3 | ||
| 73 | 0.001 | 0.002 | 0.144 | >60 (human) | 0.23 ± 0.19 | 1.00 ± 0.77 | 334.63 ± 236.47 | 2/3h |
| 76 | 0.020 | 0.003 | >60 (human) | 0.18 ± 0.09 | 5.90 ± 1.31 | 2032.63 ± 689.16 | 1/3 | |
| 78 | 0.010 | >60 (human) | 0.96 ± 0.24 | 1.01 ± 0.41 | 390.43 ± 155.95 | 1/2 | ||
| 79 | 0.015 | 39 (human) | 0.06 ± 0.01 | 2.07 ± 1.52 | 653.33 ± 432.23 | 2/3 | ||
Concentration of compound required to inhibit growth by 50% (EC50) of T. brucei brucei strain BF427 and T. brucei rhodesiense strain STIB900. Pentamidine isethionate was used as control with EC50 = 0.0021 ± 0.00001 μM.
Concentration of compound required to inhibit by 50% (IC50) of human cytochrome P450 (3YP3A4 isoform) enzyme.
Time required by liver microsomes (mouse, human) to reduce the amount of compound by half.
Ratio of compound concentration in brain to concentration in plasma 1 h after intraperitoneal injection of 5 mg/kg (values are the mean of three mice, n = 3).
Maximum concentration of compound in blood by oral admission (values are the mean of three mice, n = 3).
Area under the curve of concentration of compound in blood over the time (values are the mean of three mice, n = 3).
Ratio of number of cured mice over total number of mice in each compound experiment after 60 days post infection.
The parasitemia relapse (in one mice) was observed 31 days after treatment.
These two compounds also had the highest area under the curve (AUC0–8 h) measurements (Table 6). Penetration into brain tissue was assessed by comparing brain to plasma concentrations of compounds at 60 min following a single intraperitoneal injection at 5 mg/kg. Compounds 64, 65, 66, 69, 78 shows high CNS exposure with the brain-to-plasma concentration ratios >0.3.
3.2. Efficacy studies in mice
Given the favorable pharmacological properties and antiparasitic activity, ten compounds were selected for efficacy testing in the mouse model of acute HAT infection using the STIB900 strain of T. brucei rhodesiense. Mice were treated for 4 days beginning 48 hours post infection at 50 mg/kg once per day by oral gavage. Parasitemia was followed out to 60 days. Compounds 64, 65, 69, 72, 73, 79 had cure rates of 67–80% while compounds 66, 70, 76, 78 showed partial cure rates of 33% (Table 6). The highest parasitemia rebound time (parasitemia free period) was observed for compound 73 which was 31 days post infection (see Supporting information, Fig. S3).
Selected compounds were tested in a murine model of late-stage HAT. Mice were injected with the TREU667 strain that was allowed to establish infection for 21 days. Dosing with high concentration and long duration was chosen as a proof of concept to see if the full cure could be achieved. Mice were treated twice a day at 50 mg/kg by oral gavage from day 21 to 34 (14 days). Diminazene aceturate single dose of 10 mg/kg on day 21 was used as one of the controls. Diminazene does not cross the blood-brain barrier. It causes temporary clearance of parasitemia which later relapses, most likely from parasites leaving the brain and returning to the bloodstream. Compounds 76 and 65 showed partial suppression during treatment whereas compounds 72 and 73 led to complete suppression during the treatment phase, however, all mice relapsed with parasites in the blood. The longest parasite free (blood) time (12 days post-treatment/46 days post infection) was observed for treatment with compound 65 (Table 7).
Table 7.
Late-stage efficacy model in mice. The data indicate the number of mice free of parasitemia/number of mice in the group.
| Day post-infection | 0 | 21–34 | 34 | 35 | 36 | 37 | 38 | 39 | 40 | 41–42 | 43–45 | 46–49 | 50–51 | 52–180 |
| Day post-treatment | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7–8 | 9–11 | 12–15 | 16–17 | 18–180 | ||
| Vehicle | Infected | DOSING | 0/4 | – | – | – | – | – | – | – | – | – | – | – |
| Diminazene day 21a | Infected | DOSING | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 1/3 | 0/3 | – |
| 65b | Infected | DOSING | 4/4 | 4/4 | 4/4 | 2/4 | 1/4 | 1/4 | 1/4 | 1/4 | 1/4 | 0/4 | – | – |
| 72b | Infected | DOSING | 5/5 | 5/5 | 5/5 | 4/5 | 3/5 | 2/5 | 1/5 | 0/5 | – | – | – | – |
| 73b | Infected | DOSING | 5/5 | 5/5 | 5/5 | 4/5 | 4/5 | 3/5 | 0/5 | – | – | – | – | – |
| 76b | Infected | DOSING | 4/4 | 3/4 | 3/4 | 3/4 | 3/4 | 2/4 | 1/4 | 0/4 | – | – | – | – |
| SCYX-7158c | Infected | DOSING | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
Single intraperitoneal dose of diminazene aceturate at 10 mg/kg in water on day 21 (clear parasites from blood and peripheries but not from the brain).
Mice were injected with the T. brucei brucei (strain TREU667) that was allowed to establish infection for 21 days. Mice were treated at 50 mg/kg twice a day by oral gavage from day 21 to 34 (14 days).
Control oxaborole compound, SCYX-7158, developed by Anacor Pharmaceuticals.
This indicated that the compounds did not fully suppress parasites in the periphery or were unable to clear parasites from the central nervous system and they re-established infection in the blood. The control compound, SCYX-7158, cured all mice at the same dose (50 mg/kg PO twice per day).
3.3. Washout experiments
Although N-(2-aminoethyl)-N-benzyloxyphenyl benzamide compounds demonstrated very low EC50 values on cultured T. brucei and reasonably good pharmacokinetic profiles in mice, they were only successful at giving partial cures in the acute infection model. Therefore, washout experiments were performed to evaluate the time and concentrations of compounds required to kill T. brucei cultures. The compounds in this series required relatively long exposures at high concentrations compared to the clinical drug pentamidine (Table 8).
Table 8.
Washout experiments with T. brucei.a
| Compound | 24H | 48H | 72H | 96H |
|---|---|---|---|---|
| Pentamidine | 4X | 4X | 1X | 1X |
| 64 | 32X | 16X | 16X | 16X |
| 69 | 16X | 32X | 16X | 32X |
| 72 | 16X | 8X | 8X | 4X |
| 79 | 32X | 64X | 64X | 16X |
| 66 | 128X | 128X | 64X | 64X |
The values indicate the concentration of compound relative to the 48-h EC50 and the duration of exposure that resulted in no outgrowth of cells after another 10 days of observation in media without compound.
For example, parasites incubated in vitro with compound 66 for 96 h required a concentration of 64-times the EC50 to completely kill the culture. This contrasted with pentamidine which killed all the parasites at 4-times the EC50 after only 24 h exposure. Thus, it appears that parasites are able to rebound after exposures to compounds well above the EC50 values.
4. Conclusions
Eighty two analogues of compound 1 were synthesized to optimize anti-trypanosomal activity and pharmacological properties. Several compounds with EC50 values as low as 0.001–0.002 μM were identified. They demonstrated reasonably good oral bioavailability and plasma exposures, and had good penetration into brain tissue. However, in murine efficacy models of HAT infection, the compounds showed only partial cures or suppression. In vitro washout studies suggested that the compounds completely eliminate parasites only with long exposure at concentrations many fold above EC50 values and this probably is responsible for the suboptimal results in the efficacy experiments.
5. Experimental section
5.1. Chemistry
All starting materials were purchased from various chemical vendors and used without further purification unless noted. Thin-layer chromatography was performed on Merck Silica Gel 60 F254 pre-coated plates. Column chromatography was conducted under medium pressure on silica (Cleanert Silica (40–60 μm)) from Agela Technologies. 1H NMR spectra were recorded on a Bruker AV-300 or AV-500 spectrometers. Chemical shifts were referenced with respect to the residual solvents signals. Electrospray (ESI) mass spectra were obtained on Bruker Esquire Ion Trap Mass Spectrometer. All target compounds were purified by Varian semi-preparative HPLC (Varian PrepStar, model 218, column YMC ODS-A, 100x20 mm, 5 μm, flow: 10 mL/min, UV detector at : 218 nm and 254 nm) with mobile phase 1 (water: methanol, gradient 50%–80% methanol over 15 min) or mobile phase 2 (water (0.01% HCl): methanol, gradient 15%–50% methanol over 15 min). All final compounds are judged to be >95% pure by HPLC (UV at 254 nm and 218 nm).
5.1.1. General synthesis of compounds 2–19
Compounds 2–19 were synthesized by reductive alkylation of commercially available anilines with N-Boc-2-aminoacetaldehyde and benzoylation with 2,4-dichlorobenzoyl chloride with subsequent removal of Boc-protecting group with 4 N HCl in dioxane. Compound precipitated from hexane/ dioxane reaction mixture as HCl salt.
5.1.1.1
N-(2-Aminoethyl)-2,4-dichloro-N-(4-chlorophenyl)benzamide (2). was prepared from 4-chloroaniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (65 mg, 84%); 1H NMR (300 MHz, CDCl3): δ 7.23 (1H, bs), 7.18 (2H, d, J = 9 Hz), 7.08 (2H, d, J = 9 Hz), 7.07 (2H, bs), 3.96 (2H, t, J = 9 Hz), 2.94 (2H, t, J = 9 Hz); ESI MS m/z 343.4 (M+H)+, 326.2 (M−NH3+H)+.
5.1.1.2
N-(2-Aminoethyl)-N-(4-bromophenyl)-2,4-dichlorobenzamide hydrochloride (3). was prepared from 4-bromoaniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (100 mg, 80%); 1H NMR (300 MHz, CDCl3): δ 8.55 (3H, bs), 7.90 (1H, d, J = 9 Hz), 7.26 (2H, d, J = 9 Hz), 7.19 (2H, d, J = 9 Hz), 7.12 (1H, d, J = 3 Hz), 7.04 (1H, dd, J = 9 Hz, J = 3 Hz), 4.27 (2H, bs), 3.34 (2H, bs); ESI MS m/z 387.5 (M+H)+, 370.7 (M−NH3+H)+.
5.1.1.3
N-(2-Aminoethyl)-2,4-dichloro-N-[4-(trifluoromethyl)phenyl] benzamide hydrochloride (4). was prepared from 4-trifluoroaniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (38 mg, 50%); 1H NMR (300 MHz, CDCl3): δ 8.68 (3H, bs), 7.98 (1H, d, J = 9 Hz), 7.55–7.35 (4H, bs), 7.12 (1H, s), 7.06 (1H, d, J = 8 Hz), 7.12 (1H, d, J = 3 Hz), 4.31 (2H, bs), 3.34 (2H, bs); ESI MS m/z 377.3 (M+H)+, 360.7 (M−NH3+H)+.
5.1.1.4
N-(2-Aminoethyl)-2,4-dichloro-N-(4-methylphenyl)benzamide hydrochloride (5). was prepared from p-toluidine, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (65 mg, 90%); 1H NMR (300 MHz, CDCl3): δ 8.55 (3H, bs), 7.87 (1H, bs), 7.14 (2H, bs), 7.07 (1H, s), 7.00 (1H, s), 6.92 (2H, d, J = 6 Hz), 4.25 (2H, bs), 3.32 (2H, bs), 2.21 (3H, s); ESI MS m/z 323.7 (M+H)+, 306.8 (M−NH3+H)+.
5.1.1.5
N-(2-Aminoethyl)-2,4-dichloro-N-(4-phenylphenyl)benzamide (6). was prepared from 4-phenylaniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (17 mg, 45%); 1H NMR (300 MHz, CDCl3): δ 8.71 (3H, bs), 8.03 (1H, d, J = 9 Hz), 7.50–7.30 (9H, bs), 7.10 (2H, bs), 4.37 (2H, bs), 3.43 (2H, bs); ESI MS m/z 385.3 (M+H)+, 368.6 (M−NH3+H)+.
5.1.1.6
N-(2-Aminoethyl)-2,4-dichloro-N-[4-(propan-2-yl)phenyl] benzamide hydrochloride (7). was prepared from 4-isopropylaniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (75 mg, 83%); 1H NMR (300 MHz, d6-DMSO): δ 8.15 (3H, bs), 7.62 (1H, d, J = 9 Hz), 7.49 (1H, d, J = 2 Hz), 7.33 (1H, dd, J = 9 Hz, J = 2 Hz), 7.26 (2H, d, J = 9 Hz), 7.15 (2H, d, J = 9 Hz), 4.04 (2H, bs), 2.99 (2H, bs), 2.80 (1H, m), 1.11 (3H, s), 1.08 (3H, s); ESI MS m/z 351.3 (M+H)+, 334.2 (M−NH3+H)+.
5.1.1.7
N-(2-Aminoethyl)-2,4-dichloro-N-(4-methoxyphenyl)benzamide hydrochloride (8). was prepared from p-anisidine, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (60 mg, 80%); 1H NMR (300 MHz, CDCl3): δ 8.56 (3H, bs), 7.90 (1H, d, J = 9 Hz), 7.22 (2H, d, J = 9 Hz), 7.09 (1H, s), 7.02 (1H, d, J = 9 Hz), 6.63 (2H, d, J = 9 Hz), 4.25 (2H, bs), 3.70 (3H, s), 3.33 (2H, bs); ESI MS m/z 339.6 (M+H)+, 322.3 (M−NH3+H)+.
5.1.1.8
N-(2-Aminoethyl)-2,4-dichloro-N-(4-nitrophenyl)benzamide hydrochloride (9). was prepared from 4-nitroaniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (20 mg, 77%); 1H NMR (300 MHz, d6-DMSO): δ 8.25 (3H, bs), 8.16 (2H, d, J = 9 Hz), 7.81 (1H, d, J = 9 Hz), 7.67 (2H, d, J = 9 Hz), 7.51 (1H, bs), 7.43 (1H, d, J = 9 Hz), 4.16 (2H, bs), 2.99 (2H, bs); ESI MS m/z 354.8 (M+H)+, 337.2 (M−NH3+H)+.
5.1.1.9
N-(2-Aminoethyl)-N-(4-aminophenyl)-2,4-dichlorobenzamide (10). was prepared from 9 by reduction with SnCl2 in ethyl acetate (10 mg, 77%); 1H NMR (300 MHz, CDCl3): δ 7.21 (1H, d, J = 2 Hz), 7.02 (2H, d, J = 2 Hz), 6.88 (2H, d, J = 9 Hz), 6.45 (2H, d, J = 9 Hz), 3.90 (2H, t, J = 9 Hz), 3,67 (2H, bs), 2.91 (2H, t, J = 9 Hz); ESI MS m/z 324.5 (M+H)+, 307.4 (M−NH3+H)+.
5.1.1.10
N-(2-Aminoethyl)-2,4-dichloro-N-(3-chlorophenyl)benzamide hydrochloride (11). was prepared from 3-chloroaniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (50 mg, 74%); 1H NMR (300 MHz, d6-DMSO): δ 8.20 (3H, bs), 7.74 (1H, d, J = 9 Hz), 7.58 (1H, s), 7.51 (1H, d, J = 2 Hz), 7.45–7.25 (4H, m), 4.10 (2H, bs), 3.00 (2H, m); ESI MS m/z 343.3 (M +H)+, 326.9 (M−NH3+H)+.
5.1.1.11
N-(2-Aminoethyl)-2,4-dichloro-N-(2,4-dichlorophenyl)benzamide (12). was prepared from 2.4-dichloroaniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (75 mg, 98%); 1H NMR (300 MHz, d6-DMSO): δ 8.16 (3H, bs), 7.72 (1H, d, J = 2 Hz), 7.69 (1H, d, J = 9 Hz), 7.57 (1H, d, J = 2 Hz), 7.52 (1H, d, J = 9 Hz), 7.43 (1H,dd, J = 9 Hz, J = 2 Hz), 7.34 (1H, dd, J = 9 Hz, J = 2 Hz), 4.59 (2H,bs), 3.05 (2H, bs); ESI MS m/z 377.3 (M+H)+, 360.7 (M−NH3+H)+.
5.1.1.12
N-(2-Aminoethyl)-2,4-dichloro-N-(3,4-dichlorophenyl)benzamide hydrochloride (13). was prepared from 2.4-dichloroaniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (78 mg, 57%); 1H NMR (300 MHz, d6-DMSO): δ 8.24 (3H, bs), 7.95–7.75 (2H, bs), 7.65–7.35 (4H, bs), 4.11 (2H,bs), 3.00 (2H, bs). ESI MS m/z 377.5 (M+H)+, 360.4 (M−NH3+H)+.
5.1.1.13
N-(2-Aminoethyl)-2,4-dichloro-N-(3-chloro-4-fluorophenyl) benzamide hydrochloride (14). was prepared from 3-chloro-4-fluoro-aniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (74 mg, 54%); 1H NMR (300 MHz, d6-DMSO): δ 8.27 (3H, bs), 7.82 (2H, d, J = 9 Hz), 7.55–7.30 (4H, bs), 4.09 (2H,bs), 3.01 (2H, bs); ESI MS m/z 361.3 (M+H)+, 344.4 (M−NH3+H)+.
5.1.1.14
N-(2-Aminoethyl)-2,4-dichloro-N-(4-chloro-3-methoxyphenyl) benzamide hydrochloride (15). was prepared from 4-chloro-3-methoxy-aniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (27 mg, 63%); 1H NMR (300 MHz, d6-DMSO): δ 8.20 (3H, bs), 7.79 (1H, d, J = 9 Hz), 7.50 (1H, d, J = 2 Hz), 7.42–7.26 (3H, bm), 6.96 (1H, dd, J = 9 Hz, J = 2 Hz), 4.10 (2H, bs), 3.76 (3H, s), 3.01 (2H, bs); ESI MS m/z 373.2 (M+H)+, 356.4 (M−NH3+H)+.
5.1.1.15
N-(2-Aminoethyl)-2,4-dichloro-N-[4-(4-chlorophenoxy)phenyl] benzamide hydrochloride (16). was prepared from 4-(4-chlorophenoxyl)-aniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride as HCl salt (35 mg, 79%); 1H NMR (300 MHz, d6-DMSO): δ 8.17 (3H, bs), 7.66 (1H, d, J = 9 Hz), 7.51 (1H, d, J = 3 Hz), 7.43–7.36 (5H, bm), 6.92 (2H, d, J = 9 Hz), 6.91 (2H, d, J = 9 Hz), 4.07 (2H, bs), 3.01 (2H, bs). ESI MS m/z 435.2 (M +H)+, 418.4 (M−NH3+H)+.
5.1.1.16
N-(2-Aminoethyl)-2,4-dichloro-N-[4-(3-chlorophenoxy)phenyl] benzamide hydrochloride (17). was prepared from 4-(3-chlorophenoxy)-aniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride. Purified by HPLC as HCl salt (100 mg, 75%); 1H NMR (500 MHz, d6-DMSO): δ 8.11 (3H, bs), 7.63 (1H, d, J = 9 Hz), 7.51 (1H, d, J = 2 Hz), 7.45–7.35(4H, bm), 7.20 (1H, d, J = 9 Hz), 6.98 (2H, d, J = 9 Hz), 6.90–6.80 (2H, bm), 4.08 (2H, bs), 3.03 (2H, t, J = 8 Hz); ESI MS m/z: 435.4 (M+H)+, 418.5 (M−NH3+H)+.
5.1.1.17
N-(2-Aminoethyl)-2,4-dichloro-N-[4-(2-chlorophenoxy)phenyl] benzamide hydrochloride (18). was prepared from 4-(2-chlorophenoxy)aniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride. Purified by HPLC as HCl salt (52 mg, 51%); 1H NMR (300 MHz, d6-DMSO): δ 8.14 (3H, bs), 7.63 (1H, d, J = 9 Hz), 7.58 (1H, dd, J = 9 Hz, J = 2 Hz), 7.51 (1H, d, J = 2 Hz), 7.40–7.30(4H, bm), 7.24 (1H, dd, J = 9 Hz, J = 2 Hz), 6.96 (1H, dd, J = 9 Hz, J = 2 Hz), 6.83 (2H, d, J = 9 Hz), 4.06 (2H, bs), 3.02 (2H, bs); ESI MS m/z: 435.5 (M+H)+, 418.2 (M−NH3+H)+.
5.1.1.18
N-(2-Aminoethyl)-2,4-dichloro-N-[3-(4-chlorophenoxy)phenyl] benzamide hydrochloride (19). was prepared from 3-(4-chlorophenoxy)aniline, N-Boc-2-aminoacetaldehyde and 2,4-dichlorobenzoyl chloride. Purified by HPLC as HCl salt (18 mg, 90%); 1 H NMR (300 MHz, d6-DMSO): δ 8.19 (3H, bs), 7.63 (1H, d, J = 9 Hz), 7.56 (1H, d, J = 2 Hz), 7.45–7.30 (4H, bm), 7.23 (1H, dd, J = 9 Hz, J = 2 Hz), 7.08 (1H, bs), 6.94 (1H, dd, J = 9 Hz, J = 2 Hz), 6.73 (2H, d, J = 9 Hz), 4.09 (2H, bs), 3.00 (2H, bs); ESI MS m/z: 435.4 (M+H)+, 418.7 (M−NH3+H)+.
5.1.2. General synthesis of compounds 20–44
Compounds 20–44 were synthesized by benzoylation of tert-butyl N-{2-[(4-chlorophenyl)amino]ethyl}carbamate (obtained by reductive amination of N-Boc-2-aminoacetaldehyde with 4-chloroaniline) with corresponding benzoyl chlorides and subsequent removal of Boc-protecting group by HCl. All compounds precipitated from Boc-removal reaction mixture as HCl salt.
5.1.2.1. N-(2-Aminoethyl)-N-(4-chlorophenyl)benzamide hydrochloride (20)
Compound 20 was prepared by reaction of amine with benzoyl chloride and subsequent removal of Boc-protecting group by HCl (16 mg, 53%); 1H NMR (300 MHz, CDCl3): δ 8.65 (3H, bs), 7.35 (2H, bs), 7.20–6.95 (7H, bs), 4.33 (2H, bs), 3.34 (2H, bs); ESI MS m/z: 275.2 (M+H)+, 258.7 (M−NH3+H)+.
5.1.2.2. N-(2-Aminoethyl)-4-chloro-N-(4-chlorophenyl)benzamide hydrochloride (21)
Compound 21 was prepared by reaction of amine with 4-chlorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (35 mg, 92%); 1H NMR (300 MHz, d6-DMSO): δ 8.16 (3H, bs), 7.37 (4H, s), 7.32 (4H, s), 4.07 (2H, t, J = 6 Hz), 2.95 (2H, m); ESI MS m/z: 309.1 (M+H)+, 292.3 (M−NH3+H)+.
5.1.2.3. N-(2-Aminoethyl)-3-chloro-N-(4-chlorophenyl)benzamide hydrochloride (22)
Compound 22 was prepared by reaction of amine with 3-chlorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (35 mg, 91%); 1H NMR (300 MHz, d6-DMSO): δ 8.17 (3H, bs), 7.45–7.30 (6H, m), 7.27–7.18 (2H, m), 4.06 (2H, t, J = 6 Hz), 2.95 (2H, m); ESI MS m/z: 309.3 (M+H)+, 292.5 (M−NH3+H)+.
5.1.2.4. N-(2-Aminoethyl)-2-chloro-N-(4-chlorophenyl)benzamide hydrochloride (23)
Compound 23 was prepared by reaction of amine with 2-chlorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (30 mg, 79%); 1H NMR (300 MHz, d6-DMSO): δ 8.14 (3H, bs), 7.60 (1H, m), 7.40–7.32 (4H, m), 7.28–7.23 (3H, m), 4.07 (2H, bt), 3.00 (2H, bs); ESI MS m/z 309.1 (M +H)+, 292.2 (M−NH3+H)+.
5.1.2.5. N-(2-Aminoethyl)-N-(4-chlorophenyl)-2-methylbenzamide hydrochloride (24)
Compound 24 was prepared by reaction of amine with o-toluoyl chloride and subsequent removal of Boc-protecting group by HCl (19 mg, 51%); 1H NMR (300 MHz, d6-DMSO): δ 8.16 (3H, bs), 7.32–7.01 (8H, m), 4.07 (2H, bs), 2.98 (2H, bs), 2.24 (3H,s); ESI MS m/z 289.0 (M+H)+.
5.1.2.6. N-(2-Aminoethyl)-N-(4-chlorophenyl)-4-methylbenzamide hydrochloride (25)
Compound 25 was prepared by reaction of amine with p-toluoyl chloride and subsequent removal of Boc-protecting group by HCl (27 mg, 93%); 1H NMR (300 MHz, d6-DMSO): δ 8.10 (3H, bs), 7.39–7.30 (4H, m), 7.19 (2H, d, J = 9 Hz), 7.04 (2H, d, J = 9 Hz), 4.06 (2H, t, J = 9 Hz), 2.96 (2H, m), 2.23 (3H, s); ESI MS m/z 289.1 (M+H)+, 272.2 (M−NH3+H)+.
5.1.2.7. N-(2-Aminoethyl)-N-(4-chlorophenyl)-2-methoxybenzamide hydrochloride (26)
Compound 26 was prepared by reaction of amine with 2-methoxybenzoyl chloride and subsequent removal of Boc-protecting group by HCl (29 mg, 87%); 1H NMR (500 MHz, d6-DMSO): δ 8.11 (3H, bs), 7.38 (1H, d, J = 9 Hz), 7.30–7.15 (5H, m), 6.85 (1H, m), 6.75 (1H, d, J = 9 Hz), 4.04 (2H, bs), 3.56 (3H, s), 2.94 (2H, bs); ESI MS m/z 305.0 (M+H)+, 288.0 (M−NH3+H)+.
5.1.2.8. N-(2-Aminoethyl)-N-(4-chlorophenyl)-4-methoxybenzamide hydrochloride (27)
Compound 27 was prepared by reaction of amine with 4-methoxybenzoyl chloride and subsequent removal of Boc-protecting group by HCl (25 mg, 68%); 1H NMR (300 MHz, d6-DMSO): δ 8.14 (3H, bs), 7.39–7.30 (4H, m), 7.24 (2H, d, J = 9 Hz), 6.77 (2H, d, J = 9 Hz), 4.05 (2H, t, J = 9 Hz), 3.70 (3H, s), 2.94 (2H, m); ESI MS m/z 305.2 (M+H)+, 288.1 (M−NH3+H)+.
5.1.2.9. N-(2-Aminoethyl)-N-(4-chlorophenyl)-4-fluorobenzamide hydrochloride (28)
Compound 28 was prepared by reaction of amine with 4-fluorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (30 mg, 83%); 1H NMR (300 MHz, d6-DMSO): δ 8.13 (3H, bs), 7.40–7.34 (6H, m), 7.09 (2H, t, J = 9 Hz), 4.07 (2H, t, J = 6 Hz), 2.95 (2H, m); ESI MS m/z 293.4 (M +H)+, 276.7 (M−NH3+H)+.
5.1.2.10. N-(2-Aminoethyl)-N-(4-chlorophenyl)-3-fluorobenzamide hydrochloride (29)
Compound 29 was prepared by reaction of amine with 3-fluorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (33 mg, 92%); 1H NMR (300 MHz, d6-DMSO): δ 8.13 (3H, bs), 7.38 (4H, s), 7.30–7.08 (4H, m), 4.06 (2H, t, J = 6 Hz), 2.95 (2H, m); ESI MS m/z 293.2 (M+H)+, 276.2 (M−NH3+H)+.
5.1.2.11. N-(2-Aminoethyl)-N-(4-chlorophenyl)-2-fluorobenzamide hydrochloride (30)
Compound 30 was prepared by reaction of amine with 2-fluorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (31 mg, 86%); 1H NMR (300 MHz, d6-DMSO): δ 8.10 (3H, bs), 7.53 (1H, t, J = 9 Hz), 7.34 (5H, bs), 7.13 (1H, t, J = 6 Hz), 7.02 (1H, t, J = 6 Hz), 4.08 (2H, t, J = 6 Hz), 2.96 (2H, bs); ESI MS m/z 293.1 (M+H)+, 276.0 (M−NH3+H)+
5.1.2.12. N-(2-Aminoethyl)-2,3-dichloro-N-(4-chlorophenyl)benzamide hydrochloride (31)
Compound 31 was prepared by reaction of amine with 2,3-dichlorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (43 mg, 77%); 1H NMR (300 MHz, d6-DMSO): δ 8.22 (3H, bs), 7.65 (1H, d, J = 9 Hz), 7.50 (1H, d, J = 9 Hz), 7.45–7.35 (4H, m), 7.27 (1H, t, J = 9 Hz), 4.07 (2H, bs), 3.00 (2H, bs); ESI MS m/z 343.4 (M+H)+, 326.5 (M−NH3+H)+.
5.1.2.13. N-(2-Aminoethyl)-2,5-dichloro-N-(4-chlorophenyl)benzamide hydrochloride (32)
Compound 32 was prepared by reaction of amine with 2,5-dichlorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (48 mg, 85%); 1H NMR (300 MHz, d6-DMSO): δ 8.21 (3H, bs), 7.90 (1H, d, J = 2 Hz), 7.47–7.29 (5H, m), 4.08 (2H, bs), 3.00 (2H, bs); ESI MS m/z 343.2 (M +H)+, 326.3 (M−NH3+H)+.
5.1.2.14. N-(2-Aminoethyl)-2,6-dichloro-N-(4-chlorophenyl)benzamide hydrochloride (33)
Compound 33 was prepared by reaction of amine with 2,6-dichlorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (45 mg, 80%); 1H NMR (300 MHz, d6-DMSO): δ 8.18 (3H, bs), 7.40–7.25 (7H, m), 4.06 (2H, t, J = 9 Hz), 3.04 (2H, bs); ESI MS m/z 343.3 (M+H)+, 326.5 (M−NH3+H)+.
5.1.2.15. N-(2-Aminoethyl)-3,5-dichloro-N-(4-chlorophenyl)benzamide hydrochloride (34)
Compound 34 was prepared by reaction of amine with 3,5-dichlorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (48 mg, 86%); 1H NMR (300 MHz, d6-DMSO): δ 8.15 (3H, bs), 7.55 (1H, s), 7.43 (4H, s), 7.39 (2H, s), 4.05 (2H, t, J = 9 Hz), 2.96 (2H, bs); ESI MS m/z 343.6 (M+H)+, 326.8 (M−NH3+H)+.
5.1.2.16. N-(2-Aminoethyl)-4-chloro-N-(4-chlorophenyl)-3-nitrobenzamide hydrochloride (35)
Compound 35 was prepared by reaction of amine with 4-chloro-3-nitrobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (26 mg, 62%); 1H NMR (300 MHz, d6-DMSO): δ 8.23 (3H, bs), 8.10 (1H, bs), 7.68 (1H, d, J = 9 Hz), 7.56 (1H, d, J = 9 Hz) 7.48–7.40 (4H, m), 4.09 (2H, t, J = 9 Hz), 2.96 (2H, bs); ESI MS m/z 354.4 (M+H)+, 337.5 (M−NH3+H)+.
5.1.2.17. N-(2-Aminoethyl)-2-chloro-N-(4-chlorophenyl)-5-nitrobenzamide hydrochloride (36)
Compound 36 was prepared by reaction of amine with 2-chloro-5-nitrobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (27 mg, 63%); 1H NMR (300 MHz, d6-DMSO): δ 8.71 (1H, d, J = 2 Hz), 8.17 (3H, bs), 8.09 (1H, dd, J = 9 Hz, J = 2 Hz), 7.59 (1H, d, J = 9 Hz), 7.47 (2H, d, J = 9 Hz) 7.37 (2H, d, J = 9 Hz), 4.13 (2H, bs), 3.00 (2H, bs); ESI MS m/z 354.3 (M+H)+, 337.2 (M−NH3+H)+.
5.1.2.18. N-(2-Aminoethyl)-2,4,6-trichloro-N-(4-chlorophenyl)benzamide hydrochloride (37)
Compound 37 was prepared by reaction of amine with 2,4,6-trichlorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (35 mg, 76%); 1H NMR (300 MHz, CDCl3): δ 7.30–7.10 (6H, m), 4.09 (4H, bs), 3.14 (2H, bs); ESI MS m/z 377.4 (M+H)+, 360.6 (M−NH3+H)+.
5.1.2.19. N-(2-Aminoethyl)-N-(4-chlorophenyl)-2,4-dimethylbenzamide hydrochloride (38)
Compound 38 was prepared by reaction of amine with 2,4-dimethylbenzoyl chloride and subsequent removal of Boc-protecting group by HCl (14 mg, 38%); 1H NMR (300 MHz, d6-DMSO): δ 8.09 (3H, bs), 7.39–7.32 (4H, m), 7.20 (2H, d, J = 9 Hz), 7.03 (2H, d, J = 9 Hz), 4.05 (2H, t, J = 9 Hz), 2.97 (2H, m), 2.25 (3H, s), 2.23 (3H, s); ESI MS m/z 303.5 (M+H)+, 286.7 (M−NH3+H)+.
5.1.2.20. N-(2-Aminoethyl)-N-(4-chlorophenyl)-2,4-dimethoxybenzamide hydrochloride (39)
Compound 39 was prepared by reaction of amine with 2,4-dimethoxylbenzoyl chloride and subsequent removal of Boc-protecting group by HCl (26 mg, 65%); 1H NMR (500 MHz, d6-DMSO): δ 8.13 (3H, bs), 7.32–7.29 (3H, m), 7.23 (2H, d, J = 9 Hz), 6.45 (1H, d, J = 2 Hz), 6.28 (1H, bs), 4.02 (2H, bs), 3.69 (3H, s), 3.52 (3H, s), 2.91 (2H, bs); ESI MS m/z 335.6 (M+H)+, 318.7 (M−NH3+H)+.
5.1.2.21. N-(2-Aminoethyl)-N-(4-chlorophenyl)-2,4-difluorobenzamide hydrochloride (40)
Compound 40 was prepared by reaction of amine with 2,4-difluorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (32 mg, 94%); 1H NMR (500 MHz, d6-DMSO): δ 8.22 (3H, bs), 7.67 (1H, m), 7.42–7.37 (4H, bs), 7.08 (2H, m), 4.09 (2H, bs), 2.95 (2H, bs); ESI MS m/z 311.0 (M+H)+, 294.2 (M−NH3+H)+.
5.1.2.22. N-(2-Aminoethyl)-N-(4-chlorophenyl)-2,3,4,5,6-pentafluorobenzamide hydrochloride (41)
Compound 41 was prepared by reaction of amine with 2,3,4,5,6-pentafluobenzoyl chloride and subsequent removal of Boc-protecting group by HCl (15 mg, 38%); 1H NMR (300 MHz, d6-DMSO): δ 8.21 (3H, bs), 7.46 (2H, d, J = 9 Hz), 7.37 (2H, d, J = 9 Hz), 4.11 (2H, t, J = 9 Hz), 3.00 (2H, bs); ESI MS m/z 365.2 (M+H)+, 348.4 (M−NH3+H)+.
5.1.2.23. N-(2-aminoethyl)-N-(4-chlorophenyl)-2-(trifluoromethyl) benzamide hydrochloride (42)
Compound 42 was prepared by reaction of amine with 2-trifluoromethylbenzoyl chloride and subsequent removal of Boc-protecting group by HCl. Product was purified by HPLC (76 mg, 92%); 1H NMR (300 MHz, CDCl3): δ 8.55 (3H, bs), 7.85 (1H, m), 7.43 (1H, m), 7.28 (2H, d, J = 4 Hz), 7.18 (2H, d, J = 9 Hz), 7.04 (2H, d, J = 9 Hz), 4.21 (2H, bs), 3.34 (2H, bs); ESI MS m/z 343.0 (M+H)+, 326.1 (M−NH3+H)+.
5.1.2.24. N-(2-Aminoethyl)-N-(4-chlorophenyl)-4-fluoro-2-(trifluoromethyl) benzamide hydrochloride (43)
Compound 43 was prepared by reaction of amine with 2-trifluoromethyl-4-fluorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl. Product was purified by HPLC (76 mg, 87%); 1H NMR (300 MHz, CDCl3): δ 8.53 (3H, bs), 7.96 (1H, m), 7.20 (2H, d, J = 9 Hz), 7.15 (1H, d, J = 2 Hz), 7.10 (2H, d, J = 9 Hz), 6.98 (1H, dt, J = 9 Hz, J = 2 Hz), 4.13 (2H, bs), 3.34 (2H, bs); ESI MS m/z 361.2 (M+H)+, 344.5 (M−NH3+H)+.
5.1.2.25. N-(2-Aminoethyl)-N-(4-chlorophenyl)-2-(trifluoromethyl) pyridine-3-carboxamide hydrochloride (44)
Compound 44 was prepared by reaction of amine with 2-trifluoromethyl-4-fluorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl. Product was purified by HPLC (44 mg, 89%); 1H NMR (500 MHz, d6-DMSO): δ 8.63 (1H, d, J = 5 Hz), 8.29 (1H, d, J = 10 Hz), 8.19 (3H, bs), 7.63 (1H, m), 7.42–7.35 (4H, m), 4.08 (2H, bs), 3.02 (2H, bs); ESI MS m/z 344.3 (M+H)+, 327.7 (M−NH3+H)+.
5.1.3. General synthesis of compounds 45–63
Compounds 45–63 were synthesized by reductive alkylation of 4-chloroaniline with corresponding aldehydes (or alkylation with alkyl bromides) with following benzoylation with 2,4-dichlorobenzoyl chloride and subsequent removal of Boc-protecting group.
5.1.3.1. 2,4-Dichloro-N-ethyl-N-(4-fluorophenyl)benzamide (45)
Compound 45 was prepared by reaction of N-ethyl-N-4-fluorophenyl amine with 2,4-dichlorobenzoyl chloride and subsequent removal of Boc-protecting group by HCl. Product was purified by flash chromatography on silica gel with dichloromethane as mobile phase (63 mg, 27%); 1H NMR (300 MHz, CDCl3): δ 7.22 (1H, dd, J = 2 Hz, J = 0.6 Hz), 7.11–7.00 (4H, m), 6.92 (2H, d, J = 9 Hz), 3.94 (2H, q, J = 9 Hz), 1.23 (3H, t, J = 9 Hz); ESI MS m/z 313.3 (M+H)+.
5.1.3.2. N-{2-[N-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)formamido] ethyl}acetamide (46)
Compound 46 was prepared by acylation of 2 with acetic acid anhydride. Product was purified by flash chromatography on silica gel with dichloromethane; methanol 10:1 as mobile phase (4 mg, 16%); 1H NMR (300 MHz, CDCl3): δ 7.53 (1H, d, J = 9 Hz), 7.45–7.40 (3H, m), 7.30 (1H, dd, J = 2 Hz, J = 9 Hz), 7.20 (2H, d, J = 9 Hz), 3.95 (2H, t, J = 6 Hz), 3.62 (2H, m), 1.84 (3H, s); ESI MS m/z 385.2 (M+H)+.
5.1.3.3. 2,4-Dichloro-N-(4-chlorophenyl)-N-[2-(dimethylamino)ethyl] benzamide (47)
Compound 47 was prepared by alkylation of 4-chloroaniline with N,N-dimethyl-N-chloroethyl amine and follow up benzoylation with 2,4-dichlorobenzoyl chloride. Product was purified by flash chromatography on silica gel with dichloromethane; methanol 10:1 as mobile phase (50 mg, 67%); 1H NMR (300 MHz, CDCl3): δ 7.25–7.00 (7H, m), 3.99 (2H, t, J = 6 Hz), 2.55 (2H, t, J = 6 Hz), 2.28 (6H, s); ESI MS m/z 371.1 (M+H)+.
5.1.3.4. 2,4-Dichloro-N-(4-chlorophenyl)-N-[2-(morpholin-4-yl)ethyl] benzamide (48)
Compound 48 was prepared by alkylation of 4-chloroaniline with N-chloroethylmorpholine and follow up benzoylation with 2,4-dichlorobenzoyl chloride. Product was purified by flash chromatography on silica gel with dichloromethane; methanol 10:0.5 as mobile phase (65 mg, 20%); 1H NMR (300 MHz, CDCl3): δ 7.25–7.00 (7H, m), 4.04 (2H, t, J = 6 Hz), 3.70 (4H, bs), 2.55 (2H, t, J = 6 Hz), 2.49 (4H, bs); ESI MS m/z 413.2 (M +H)+.
5.1.3.5. 2,4-Dichloro-N-(4-chlorophenyl)-N-(2-hydroxyethyl)benzamide (49)
Compound 49 was prepared by alkylation of 4-chloroaniline with tert-buthyldimethylsilil protected glycol aldehyde and follow up benzoylation with 2,4-dichlorobenzoyl chloride and subsequent deprotection with HCl. Product was purified by flash chromatography on silica gel with dichloromethane as mobile phase (130 mg, 77%); 1H NMR (300 MHz, d6-DMSO): δ 7.23 (1H, d, J = 9 Hz), 7.15 (2H, d, J = 9 Hz), 7.11–7.06 (3H, m), 4.07 (2H, t, J = 6 Hz), 3.88 (2H, m). ESI MS m/z 344.5 (M+H)+.
5.1.3.6. N-(3-Aminopropyl)-2,4-dichloro-N-(4-chlorophenyl)benzamide (50)
Compound 50 was prepared by alkylation of 4-chloroaniline with 2-N-Boc-aminopropanal and follow up benzoylation with 2,4-dichlorobenzoyl chloride and subsequent deprotection with HCl. Product precipitated from reaction mixture as HCl salt (75 mg, 90%); 1H NMR (300 MHz, CDCl3): δ 7.20–7.00 (7H, m), 3.97 (2H, t, J = 6 Hz), 3.15 (2H, m), 2.02 (2H, t, J = 6 Hz); ESI MS m/z 377.3 (M+H)+, 360.7 (M−NH3+H)+.
5.1.3.7. N-(2-Aminopropyl)-2,4-dichloro-N-(4-chlorophenyl)benzamide hydrochloride (51)
Compound 51 was prepared by alkylation of 4-chloroaniline with 2-methyl-2-N-Boc-aminopropanal and follow up benzoylation with 2,4-dichlorobenzoyl chloride and subsequent deprotection with HCl. Product precipitated from reaction mixture as HCl salt (100 mg, 85%); 1H NMR (300 MHz, CDCl3): δ 8.67 (3H, bs), 7.95 (1H, d, J = 9 Hz), 7.30 (2H, d, J = 9 Hz), 7.13 (2H, d, J = 9 Hz), 7.13 (1H, bs), 7.07 (1H, dd, J = 2 Hz, J = 9 Hz), 4.63 (1H, bs), 3.66 (2H, bs), 1.43,1.45 (3H, s); ESI MS m/z 357.2 (M+H)+, 340.3 (M−NH3+H)+.
5.1.3.8. N-(1-Aminopropan-2-yl)-2,4-dichloro-N-(4-chlorophenyl) benzamide hydrochloride (52)
Compound 52 was prepared by alkylation of 4-chloroaniline with N-Boc-aminopropanone and follow up benzoylation with 2,4-dichlorobenzoyl chloride and subsequent deprotection with HCl. Product precipitated from reaction mixture as HCl salt (90 mg, 46%); 1H NMR (300 MHz, CDCl3): δ 8.66 (3H, bs), 7.81 (1H, bs), 7.31 (2H, bs), 7.18–7.10 (3H, m), 6.95 (1H, dd, J = 2 Hz, J = 9 Hz), 5.25 (1H, bs), 3.26 (2H, bs), 1.14 (3H, s); ESI MS m/z 357.3 (M+H)+, 340.5 (M−NH3+H)+.
5.1.3.9. 2,4-Dichloro-N-(4-chlorophenyl)-N-(furan-2-ylmethyl)benzamide (53)
Compound 53 was prepared by alkylation of 4-chloroaniline with furfural and follow up benzoylation with 2,4-dichlorobenzoyl chloride. Product was purified by flash chromatography on silica gel with hexane : dichloromethane 10:7 as mobile phase (150 mg, 80%); 1H NMR (300 MHz, CDCl3): δ 7.37 (1H, m), 7.22 (1H, t, J = 2 Hz,), 7.13 (2H, d, J = 9 Hz), 7.06 (2H, bs), 6.94 (2H, d, J = 9 Hz), 6.31 (1H, m), 6.28 (1H, m), 5.04 (2H, s); ESI MS m/z 381.0 (M+H)+.
5.1.3.10. 2,4-Dichloro-N-(4-chlorophenyl)-N-(thiophen-2-ylmethyl) benzamide (54)
Compound 54 was prepared by alkylation of 4-chloroaniline with 2-thiophen aldehyde and follow up benzoylation with 2,4-dichlorobenzoyl chloride. Product was purified by flash chromatography on silica gel with hexane : dichloromethane 10:7 as mobile phase (171 mg, 86%); 1H NMR (300 MHz, CDCl3): δ 7.30–7.20 (2H, m), 7.11 (2H, d, J = 9 Hz), 7.05 (2H, bs), 6.90 (2H, d, J = 9 Hz), 6.95–6.85(2H, m), 5.19 (2H, s); ESI MS m/z 396.1 (M+H)+.
5.1.3.11. 2,4-Dichloro-N-(4-chlorophenyl)-N-(1H-imidazol-2-ylmethyl) benzamide (55)
Compound 55 was prepared by alkylation of 4-chloroaniline with 2-carboxoimidazole aldehyde and follow up benzoylation with 2,4-dichlorobenzoyl chloride. Product was purified by HPLC (37 mg, 20%); 1H NMR (300MHz, d6-DMSO): δ 11.99 (1H, s), 7.60–7.20 (7H, m), 7.05 (1H,bs), 6.81 (1H, bs), 5.03 (2H, s); ESI MS m/z 380.2 (M+H)+.
5.1.3.12. 2,4-Dichloro-N-(4-chlorophenyl)-N-(1H-imidazol-4-ylmethyl)benzamide (56)
Compound 56 was prepared by alkylation of 4-chloroaniline with 5-carboxoimidazole aldehyde and follow up benzoylation with 2,4-dichlorobenzoyl chloride. Product was purified by HPLC (45 mg, 24%); 1H NMR (300 MHz, d6-DMSO): δ 11.94 (1H, bs), 7.55 (1H, s), 7.40–7.15 (7H, m), 6.95 (1H, s), 4.93 (2H, s). ESI MS m/z 380.8 (M+H)+.
5.1.3.13. 2,4-Dichloro-N-(4-chlorophenyl)-N-(pyrrolidin-3-yl)benzamide hydrochloride (57)
Compound 57 was prepared by alkylation of 4-chloroaniline with N-Boc-3-pirrolidinone and follow up benzoylation with 2,4-dichlorobenzoyl chloride and subsequent removal of Boc group by HCl. Product was purified by HPLC (86 mg, 53%); 1H NMR (300 MHz, d6-DMSO): δ 9.35 (1H, s), 9.16 (1H,s), 7.55–7.30 (7H, m), 4.96 (1H, m), 3.60 (1H, bs), 3.23(3H, bs), 2.26 (1H, m), 1.87 (1H, m); ESI MS m/z 369.5 (M+H)+.
5.1.3.14. 2,4-Dichloro-N-(4-chlorophenyl)-N-[2-(methylamino)ethyl] benzamide hydrochloride (58)
Compound 58 was prepared by alkylation of 4-chloroaniline with N-Boc-N-methylaminoacetaldehyde and follow up benzoylation with 2,4-dichlorobenzoyl chloride and subsequent removal of Boc group by HCl. Product was purified by HPLC (36 mg, 51%); 1H NMR (300 MHz, d6-DMSO): δ 9.03 (2H, bs), 7.79 (1H, d, J = 9 Hz), 7.50–7.30 (6H, m), 4.13 (2H, bs), 3.09 (2H, t, J = 6 Hz), 2.59 (3H, s); ESI MS m/z 357.4 (M+H)+, 326.4 (M−CH3NH2)+.
5.1.3.15. 2-[N-(4-Chlorophenyl)-1-(2,4-dichlorophenyl)formamido] acetamide (59)
Compound 59 was prepared by alkylation of 4-chloroaniline with chloroacetamide and follow up benzoylation with 2,4-dichlorobenzoyl chloride. Product was purified by HPLC (64 mg, 60%); 1H NMR (300 MHz, CDCl3): δ 7.27 (1H, bs), 7.21–7.10 (6H, m), 4.48 (2H, s); ESI MS m/z 357.0 (M+H)+, 340.2 (M−NH3+H)+.
5.1.3.16
2,4-Dichloro-N-(4-chlorophenyl)-N-(cyanomethyl)benzamide (60). was prepared by alkylation of 4-chloroaniline with paraformaldehyde in presence of potassium cyanide and follow up benzoylation with 2,4-dichlorobenzoyl chloride. Product was purified by HPLC (150 mg, 38%); 1H NMR (300 MHz, CDCl3): δ 7.30–7.15 (3H, bs), 7.20–7.10 (4H, m), 4.76 (2H, s); ESI MS m/z 339.2 (M+H)+.
5.1.3.17. 2,4-Dichloro-N-(4-chlorophenyl)-N-(piperidin-3-yl)benzamide hydrochloride (61)
Compound 61 was prepared by alkylation of 4-chloroaniline with 3-N-Boc-piperidone and follow up benzoylation with 2,4-dichlorobenzoyl chloride and subsequent removal of Boc protecting group with HCl. Product was purified by HPLC (95 mg, 58%); 1H NMR (300 MHz, CDCl3): δ 9.96 (1H, bs), 9.78 (1H, bs), 7.38 (1H, d, J = 9 Hz), 7.25–7.10 (5H, m), 7.03 (1H, d, J = 9 Hz), 5.24 (1H, bs), 4.24 (1H, d, J = 9 Hz), 3.63 (1H, d, J = 9 Hz), 2.87 (1H, bs), 2.67 (1H, bs), 2.17 (2H, m), 1.98 (1H, m), 1.56 (1H, m); ESI MS m/z 383.6 (M+H)+.
5.1.3.18. 2,4-Dichloro-N-(4-chlorophenyl)-N-(piperidin-4-yl)benzamide hydrochloride (62)
Compound 62 was prepared by alkylation of 4-chloroaniline with 4-N-Boc-piperidone and follow up benzoylation with 2,4-dichlorobenzoyl chloride and subsequent removal of Boc protecting group with HCl. Product was purified by HPLC (120 mg, 57%); 1H NMR (300 MHz, CDCl3): δ 9.65 (1H, bs), 9.37 (1H, bs), 7.25–7.15 (3H, m), 7.10–6.97 (4H, m), 4.88 (1H, t, J = 12 Hz), 3.52 (2H, d, J = 12 Hz), 2.99 (2H, bs), 2.13 (2H, d, J = 12 Hz), 2.00–1.70 (2H, m); ESI MS m/z 383.7 (M+H)+.
5.1.3.19. 2,4-Dichloro-N-(4-chlorophenyl)-N-(pyridin-3-yl)benzamide (63)
Compound 63 was prepared by arylation of 3-aminopyridine with 4-chlorophenylboronic acid in presence of cupper catalyst, follow up benzoylation with 2,4-dichlorobenzoyl chloride. Product was purified by HPLC (16 mg, 29%); 1H NMR (300 MHz, CDCl3): δ 8.47 (2H, bs), 7.50–7.00 (8H, m); ESI MS m/z 377.1 (M+H)+.
5.1.4. General synthesis of compounds 64–83
5.1.4.1. Representative synthesis of 73
5.1.4.1.1. Alkylation of nitrophenol with benzylbromide
To the solution of 4-nitrophenol (1.39 g, 10 mmol) and 4-isopropylbenzylbromide (2.13 g, 10 mmol) in acetone (20 mL) a K2CO3 (4.1 g, 30 mmol) was added and the resulting mixture was stirred at 60 °C overnight in round bottom flask with reverse condenser. The progress of the reaction was monitored by TLC (Hexanes: DCM = 2:1). The mixture was cooled to RT and filtrated. Solvent was removed in rotovap. Residue was re-suspended in chloroform and filtrated trough Celite-545. Solvent removed and the residue dried in vacuum. Obtained 2.6 g (96%) of off white solid. Used for next step without further purification.
1H NMR (300 MHz, CDCl3): δ 8.21 (2H, d, J = 9 Hz), 7.35 (2H, d, J = 9.0 Hz), 7.28 (2H, d, J = 9.0 Hz), 7.03 (2H, d, J = 9 Hz), 5.21 (2H, s), 2.94 (1H, m), 1.28 (3H, s), 1.25 (3H, s).
5.1.4.1.2. Reduction of nitro group
Method A
A round bottom flask was loaded with a solution of 1-(4-nitrophenoxymethyl)-4-(propan-2-yl)benzene 2.6 g (9.6 mmol) in 50 mL of EtOAc and 200 mg of 5% Pt/C, flushed with nitrogen and subsequently with hydrogen gas. The mixture was stirred under hydrogen atmosphere (hydrogen filled balloon) at RT for 6 h. Reaction was monitored by TLC (Hexanes: DCM = 1:1). After reaction was completed the mixture was filtered through Celite-545. Solvent was removed and residue dried in vacuum to afford 2.08 g (96%) of off-white solid. It was used without father purification in next step.
Method B
To solution of 0.5 g of CuCl2 H2O in diluted (aprox. 1%) HCl a zinc granules (5 g, 20 mesh) was added. Elevation of gas was observed and the blue colored solution become colorless. The residual granular zinc was washed with water (2 × 50 mL) and loaded with 50 mL of saturated ammonium chloride solution. A solution of corresponding nitrobenzene (7.58 mmol) in 50 mL of diethyl ether was added to activated zinc in aq. ammonium chloride and mixture was stirred vigorously at rt overnight. Progress of the reaction was monitored by TLC (DCM). After no starting compound was observed by TCL ammonium hydroxide was added (10 mL) and the resulting mixture was extracted with Et2O (2 × 25 mL). The aqueous layer was removed and the organic layer washed with sat. aq. NaHCO3 aq. (30 mL), H2O (30 mL), brine (30 mL) then dried over Na2SO4 and concentrated in vacuo. The crude solid was purified by column chromatography (CH2Cl2) to afford corresponding amine in 95% yield.
1H NMR (300 MHz, CDCl3): δ 7.36 (2H, d, J = 9 Hz), 7.24 (2H, d, J = 9.0 Hz), 6.83 (2H, d, J = 9.0 Hz), 6.65 (2H, d, J = 9 Hz), 4.96 (2H, s), 3.34 (2H, bs) 2.92 (1H, m), 1.28 (3H, s), 1.25 (3H, s)
5.1.4.1.3. Reductive amination with N-Boc-2-aminoacetaldehyde
A solution of 4-{[4-(propan-2-yl)benzyl]oxy}aniline 1.2 g, 4.9 mmol and N-Boc-2-aminoacetaldehyde 0.638 g, 4.0 mmol in 15 mL chloroform, molecular sieves 3 Å 0.5 g, was stirred at RT for 2 h. Sodium triacetoxyborohydride 1.27 g 6.0 mmol was added portion wise and resulting mixture stirred at RT overnight. After completion of the reaction, it was diluted with 10 mL of CH2Cl2 and filtered. The filtrate was washed with water, sat. aq NaHCO3 and brine, the organic layer dried over Na2SO4, filtered, and concentrated. The crude product was purified by HPLC (Varian PrepStar, model 218, column YMC ODS-A, 100 × 20 mm, 5 μm, flow: 10 mL/min, detector: 218 nm and 254 nm, solvent: water: methanol, gradient 50%–80% methanol over 15 min). Fractions containing product (at 10–12 min) were collected, solvent evaporated in speedvac with no heat to give 440 mg (29% yield) of white solid product.
1H NMR (300 MHz, CDCl3): δ 7.35 (2H, d, J = 9 Hz), 7.23 (2H, d, J = 9.0 Hz), 6.85 (2H, d, J = 9.0 Hz), 6.57 (2H, d, J = 9 Hz), 4.95 (2H, s), 4.80 (1H, bs), 3.66 (1H, bs), 3.35 (2H, m) 3.21 (2H, t, J = 6 Hz), 2.91 (1H, m), 1.45 (9H, s), 1.26 (3H, s), 1.24 (3H, s).
5.1.4.1.4. Benzoylation and removal of Boc-group
To the ice both cooled solution of tert-butyl N-{2-[(4-{[4-(propan-2-yl)phenyl] methoxy}phenyl)amino]ethyl}carbamate 115 mg (0.3 mmol) and DIPEA 58 mg (0.45 mmol) in 3 mL DCM a 2-trifluoromethyl nicotinoyl chloride 75 mg (0.36 mmol) was added and the reaction mixture stirred at RT for 2 h. Progress of the reaction was monitored by TLC (CH2Cl2:MeOH = 10:0.1). Reaction mixture was cooled down in ice both and 0.5 mL of 4 M HCl in Dioxane was added. Reaction stirred at RT for 4 h, solvent removed, residue dissolved in methanol and load to HPLC (Varian PrepStar, model 218, column YMC ODS-A, 100 × 20 mm, 5 μm, flow: 10 mL/min, detector: 218 nm and 254 nm, solvent: water (0.01% HCl): methanol, gradient 15%–50% methanol over 15 min). Fractions with the product peak were collected. Solvent was removed in speedvac to give 80 mg (54% yield) of final compound 73 as HCl salt.
1H NMR (300 MHz, CDCl3): δ 8.57 (3H, bs), 7.54 (1H, bm), 7.29–7.16 (7H, m), 6.72 (2H, d, J = 9 Hz), 4.85 (2H, s), 3.32 (2H, bs), 2.90 (1H, m), 1.25 (3H, s), 1.23 (3H, s). 1 H NMR (300 MHz, d6-DMSO): δ 8.60 (1H, d, J = 2 Hz), 8.26 (1H, d, J = 9 Hz), 8.22 (3H, bs), 7.60 (1H, dd, J = 9 Hz), 7.32–7.18 (6H, m), 6.90 (2H, d, J = 9 Hz), 4.93 (2H, s), 4.04 (2H, bs), 2.99 (2H, bm), 2.87 (1H, m), 1.19 (3H, s), 1.17 (3H, s). ESI MS m/z 458.2 (M+H)+, 441.6 (M−NH3+H)+.
5.1.4.2. N-(2-Aminoethyl)-2,4-dichloro-N-{4-[(4-chlorophenyl)methoxy] phenyl}benzamide hydrochloride (64)
Compound 64 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 4-chlorobenzyl bromide, using reduction Method B, and benzoylation with 2,4-dichlorobenzoyl chloride. Final product purified by HPLC as HCl salt (116 mg, 28%). 1H NMR (300 MHz, d6-DMSO): δ 8.15 (3H, bs), 7.65 (1H, d, J = 6 Hz), 7.49 (1H, d, J = 2 Hz), 7.43 (3H, bm), 7.35 (1H, dd, J = 2 Hz, J = 6 Hz), 7.30 (2H, d, J = 9 Hz), 6.90 (2H, d, J = 9 Hz), 5.01 (2H, s), 4.02 (2H, bs), 2.97 (2H, bs); ESI MS m/z 448.1 (M+H)+, 432.1 (M−NH3+H)+.
5.1.4.3. N-(2-Aminoethyl)-N-{4-[(4-chlorophenyl)methoxy]phenyl}-4-fluoro-2-(trifluoromethyl)benzamide hydrochloride (65)
Compound 65 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 4-chlorobenzyl bromide, using reduction Method B, and benzoylation with 2-trifluoromethyl-4-fluorobenzoyl chloride. Final product purified by HPLC as HCl salt (36 mg, 75%). HPLC 100 area % (254 nm); 1H NMR (300 MHz, CDCl3): δ 8.54 (3H, bs), 7.97 (1H, m), 7.39–7.23 (4H, m), 7.15 (2H, d, J = 6 Hz), 7.10 (1H, dd, J = 3 Hz, J = 9 Hz), 6.96 (1H, bm), 6.70 (2H, d, J = 9.0 Hz), 4.86 (2H, s), 4.16 (2H, bs), 3.33 (2H, bt); ESI MS m/ z: 467.3 (M+H)+, 450.4 (M−NH3+H)+.
5.1.4.4. N-(2-Aminoethyl)-N-{4-[(4-chlorophenyl)methoxy]phenyl}-2-(trifluoromethyl)benzamide hydrochloride (66)
Compound 66 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 4-chlorobenzyl bromide, using reduction Method B, and benzoylation with 2-trifluoromethylbenzoyl chloride. Final product purified by HPLC as HCl salt (35 mg, 69%). HPLC 100 area % (254 nm); 1H NMR (300 MHz, CDCl3): δ 8.58 (3H, bs), 7.84 (1H, d, J = 9 Hz), 7.42 (1H, d, J = 9H) 7.34–7.20 (6H, m), 7.15 (2H, d, J = 9 Hz), 6.67 (2H, d, J = 9 Hz), 4.85 (2H, s), 4.19 (2H, bs), 3.33 (2H, bt); ESI MS m/z: 449.2 (M+H)+, 432.3 (M−NH3+H)+.
5.1.4.5. N-(2-Aminoethyl)-2-chloro-N-{4-[(4-chlorophenyl)methoxy] phenyl}pyridine-3-carboxamide dihydrochloride (67)
Compound 67 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 4-chlorobenzyl bromide, using reduction Method B, and benzoylation with 2-chloronicotynoyl chloride. Final product purified by HPLC as HCl salt (47 mg, 75%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, d6-DMSO): δ 8.25 (1H, dd, J = 2 Hz, J = 8 Hz) 8.24 (3H, bs), 8.16 (1H, dd, J = 2 Hz, J = 8 Hz), 7.42 (4H, bs), 7.36 (2H, d, J = 9 Hz), 7.36 (1H, m), 6.90 (2H, d, J = 9 Hz), 5.01 (2H, s), 4.05 (2H, bs), 3.00 (2H, bm); ESI MS m/z: 416.4 (M+H)+, 399.9 (M−NH3+H)+.
5.1.4.6. N-(2-Aminoethyl)-N-{4-[(4-chlorophenyl)methoxy]phenyl}-2-(trifluoromethyl)pyridine-3-carboxamide hydrochloride (68)
Compound 68 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 4-chlorobenzyl bromide, using reduction Method B, and benzoylation with 2-trifluoromethylnicotynoyl chloride. Final product purified by HPLC as HCl salt (52 mg, 76%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, d6-DMSO): δ 8.60 (1H, d, J = 4 Hz), 8.30 (1H, d, J = 8 Hz), 8.29 (3H, bs), 7.60 (1H, m), 7.41 (4H, bs), 7.27 (2H, d, J = 9 Hz), 6.90 (2H, d, J = 9 Hz), 4.99 (2H, s), 4.04 (2H, bs), 3.00 (2H, bm); ESI MS m/z: 450.5 (M +H)+, 434.2 (M−NH3+H)+.
5.1.4.7. N-(2-Aminoethyl)-2,4-dichloro-N-(4-{[4-(propan-2-yl)phenyl] methoxy}phenyl)benzamide hydrochloride (69)
Compound 69 was prepared in the same way as 73 using 2,4-dichlorolbenzoyl chloride for benzoylation. Final product was purified by HPLC as HCl salt (80 mg, 54%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, CDCl3): δ 8.51 (3H, bs), 7.93 (1H, d, J = 9 Hz), 7.32–7.19 (6H, m), 7.10 (1H, d, J = 2 Hz), 7.04(1H, dd, J = 2 Hz, J = 9 Hz), 6.72 (2H, d, J = 9.0 Hz), 4.87 (2H, s), 4.23 (2H, bs), 3.33(2H,bt), 2.91 (1H, m), 1.25 (3H, s), 1.23 (3H, s); ESI MS m/z 457.5 (M+H)+, 440.1 (M-NH3+H)+.
5.1.4.8. N-(2-Aminoethyl)-4-fluoro-N-(4-{[4-(propan-2-yl)phenyl] methoxy}phenyl)-2-(trifluoromethyl)benzamide hydrochloride (70)
Compound 70 was prepared in the same way as 73 using 2-trifluoromethyl-4-fluorobenzoyl chloride for benzoylation. Final product was purified by HPLC as HCl salt (47 mg, 71%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, CDCl3): δ 8.55 (3H, bs), 7.98 (1H, m), 7.30–7.18 (4H, m), 7.15 (2H, d, J = 9 Hz), 7.10 (1H, dd, J = 3 Hz, J = 9 Hz), 6.97 (1H, bm), 6.72 (2H, d, J = 9.0 Hz), 4.86 (2H, s), 4.20 (2H, bs), 3.33 (2H, bt), 2.90 (1H, m), 1.25 (3H, s), 1.23 (3H, s); ESI MS m/z: 475.2 (M+H)+, 458.5 (M−NH3+H)+.
5.1.4.9. N-(2-Aminoethyl)-N-(4-{[4-(propan-2-yl)phenyl]methoxy} phenyl)-2-(trifluoromethyl)benzamide hydrochloride (71)
Compound 71 was prepared in the same way as 73 using 2-trifluorobenzoyl chloride for benzoylation. Final product was purified by HPLC as HCl salt (42 mg, 66%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, CDCl3): δ 8.59 (3H, bs), 7.87 (1H, d, J = 9 Hz), 7.42 (1H, d, J = 9H) 7.34–7.18 (6H, m), 7.15 (2H, d, J = 9 Hz), 6.68 (2H, d, J = 9 Hz), 4.83 (2H, s), 4.19 (2H, bs), 3.33 (2H, bt), 2.90 (1H, m), 1.24 (3H, s), 1.22 (3H, s); ESI MS m/z: 457.3 (M+H)+, 440.8 (M−NH3+H)+.
5.1.4.10. N-(2-Aminoethyl)-2-chloro-N-(4-{[4-(propan-2-yl)phenyl] methoxy}phenyl)pyridine-3-carboxamide hydrochloride (72)
Compound 72 was prepared in the same way as 73 using 2-chloronicotynoyl chloride for benzoylation. Final product was purified by HPLC as HCl salt (60 mg, 92%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, d6-DMSO): δ 8.25 (1H, dd, J = 2 Hz, J = 8 Hz) 8.24 (3H, bs), 8.17 (1H, dd, J = 2 Hz, J = 9 Hz), 7.40–7.20 (7H, m), 6.90 (2H, d, J = 9 Hz), 4.94 (2H, s), 4.05 (2H, bs), 2.99 (2H, bm), 2.87 (1H, m), 1.19 (3H, s), 1.17 (3H,s); ESI MS m/z: 424.3 (M+H)+, 407.5 (M−NH3+H)+.
5.1.4.11. N-(2-Aminoethyl)-2-nitro-N-(4-{[4-(propan-2-yl)phenyl] methoxy}phenyl)benzamide hydrochloride (74)
Compound 74 was prepared in the same way as 73 using 2-nitrobenzoyl chloride for benzoylation. Final product was purified by HPLC as HCl salt (15 mg, 41%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, CDCl3): δ 8.58 (3H, bs), 8.12 (1H, d, J = 7 Hz), 7.82 (1H, d, J = 9 Hz), 7.49 (1H, dd, J = 9H) 7.25–7.10 (7H, m), 6.64 (2H, d, J = 9 Hz), 4.81 (2H, s), 4.28 (2H, bs), 3.39 (2H, bs), 2.89 (1H, m), 1.24 (3H, s), 1.22 (3H, s); ESI MS m/z: 434.4 (M+H)+, 417.2 (M−NH3+H)+.
5.1.4.12. N-(2-Aminoethyl)-2-bromo-N-(4-{[4-(propan-2-yl)phenyl] methoxy}phenyl)benzamide hydrochloride (75)
Compound 75 was prepared in the same way as 73 using 2-bromobenzoyl chloride for benzoylation. Final product was purified by HPLC as HCl salt (39 mg, 60%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, CDCl3): δ 8.63 (3H, bs), 7.85 (1H, d, J = 4 Hz), 7.30–7.17 (7H, m), 7.10 (1H, dd, J = 4 Hz), 6.95 (1H, dd, J = 4 Hz), 6.68 (2H, d, J = 6 Hz), 4.84 (2H, s), 3.35 (2H, bs), 2.90 (1H, m), 1.24 (3H, s), 1.23 (3H, s); ESI MS m/z: 467.4 (M+H)+, 450.4 (M−NH3+H)+.
5.1.4.13. N-(2-Aminoethyl)-2,4-dichloro-N-{4-[(3,5-dimethylphenyl) methoxy]phenyl}benzamide hydrochloride (76)
Compound 76 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 3.5-dimethylbenzyl bromide, using reduction Method A, and benzoylation with 2,4-dichlorobenzoyl chloride. Final product purified by HPLC as HCl salt (57 mg, 74%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, CDCl3): δ 8.57 (3H, bs), 7.91 (1H, d, J = 9 Hz), 7.23 (2H, d, J = 9 Hz), 7.08 (1H, d, J = 2 Hz), 7.03 (1H, dd, J = 2 Hz, J = 9 Hz), 6.96 (3H, bm), 6.71 (2H, d, J = 9.0 Hz), 4.82 (2H, s), 4.24 (2H, bs), 3.35(2H, bt), 2.30 (6H, s); ESI MS m/z 443.5 (M +H)+, 426.7 (M−NH3+H+)+.
5.1.4.14. N-(2-Aminoethyl)-N-{4-[(3,5-dimethylphenyl)methoxy]phenyl}-4-fluoro-2-(trifluoromethyl)benzamide hydrochloride (77)
Compound 77 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 3.5-dimethylbenzyl bromide, using reduction Method A, and benzoylation with 2-trifluoromethyl-4-fluorobenzoyl chloride. Final product purified by HPLC as HCl salt (17 mg, 63%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, CDCl3): δ 8.56 (3H, bs), 7.98 (1H, dd, J = 6 Hz), 7.15 (2H, d, J = 9 Hz), 7.10 (1H, dd, J = 2 Hz, J = 9 Hz), 7.05–6.90 (4H, bs), 6.72 (2H, d, J = 9 Hz), 4.82 (2H, s), 4.20 (2H, bs), 3.34 (2H, bs), 2.29 (6H, s); ESI MS m/z 461.3 (M+H)+, 444.4 (M−NH3+H)+.
5.1.4.15. N-(2-Aminoethyl)-N-{4-[(3,5-dimethylphenyl)methoxy]phenyl}-2-(trifluoromethyl)benzamide hydrochloride (78)
Compound 78 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 3.5-dimethylbenzyl bromide, using reduction Method A, and benzoylation with 2-trifluoromethylbenzoyl chloride. Final product purified by HPLC as HCl salt (25 mg, 40%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, CDCl3): δ 8.58 (3H, bs), 7.87 (1H, d, J = 9 Hz), 7.40 (1H, d, J = 9 Hz), 7.33–7.19 (2H, m), 7.15 (2H, d, J = 9 Hz), 6.94 (3H, bs),6.70 (2H, d, J = 9 Hz), 4.79 (2H, s), 4.15 (2H, bs), 3.34 (2H, bs), 2.29 (6H, s); ESI MS m/z 443.2 (M +H)+, 426.4 (M−NH3+H)+.
5.1.4.16. N-(2-Aminoethyl)-2-chloro-N-{4-[(3,5-dimethylphenyl) methoxy]phenyl}pyridine-3-carboxamide dihydrochloride (79)
Compound 79 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 3.5-dimethylbenzyl bromide, using reduction Method A, and benzoylation with 2-chloronicotynoyl chloride. Final product purified by HPLC as HCl salt (40 mg, 75%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, d6-DMSO): δ 8.26 (1H, dd, J = 2 Hz, J = 6 Hz), 8.22 (3H, bs), 8.15 (1H, dd, J = 2 Hz, J = 9 Hz), 7.34 (2H, d, J = 9 Hz), 7.34 (1H, m), 7.00–6.92 (3H, bs), 6.88 (2H, d, J = 9 Hz), 4.90 (2H, s), 4.05 (2H, bs), 2.99 (2H, bs), 2.24 (6H, s); ESI MS m/z 410.3 (M+H)+, 393.5 (M−NH3+H)+.
5.1.4.17. N-(2-Aminoethyl)-N-{4-[(3,5-dimethylphenyl)methoxy]phenyl}-2-(trifluoromethyl)pyridine-3-carboxamide hydrochloride (80)
Compound 80 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 3,5-dimethylbenzyl bromide, using reduction Method A, and benzoylation with 2-trifluoromethylnicotynoyl chloride. Final product purified by HPLC as HCl salt (45 mg, 67%). HPLC 100 area% (254 nm); 1H NMR (300 MHz, d6-DMSO): δ 8.61 (1H, d, J = 2 Hz), 8.30–8.20 (4H, bs), 7.60 (1H, dd, J = 9 Hz), 7.25 (2H, d, J = 9 Hz), 6.96 (2H, bs), 6.94 (1H, bs), 6.90 (2H, d, J = 9 Hz), 4.90 (2H, s), 4.04 (2H, bs), 3.00 (2H, bs), 2.24 (6H, s); ESI MS m/z 444.3 (M+H)+, 428.1 (M−NH3+H)+.
5.1.4.18. N-(2-Aminoethyl)-2,4-dichloro-N-{4-[(3-chlorophenyl) methoxy]phenyl}benzamide hydrochloride (81)
Compound 81 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 3-chlorobenzyl bromide, using reduction Method B, and benzoylation with 2,4-dichlorobenzoyl chloride. Final product purified by HPLC as HCl salt (45 mg, 18%). 1H NMR (300 MHz, d6-DMSO): δ 8.24 (3H, bs), 7.69 (1H, d, J = 6 Hz), 7.47 (1H, d, J = 2 Hz), 7.40–7.30 (6H, bm), 6.90 (2H, d, J = 9 Hz), 5.03 (2H, s), 4.03 (2H, bs), 2.96 (2H, bs); ESI MS m/z 448.3 (M+H)+, 432.4 (M−NH3+H)+.
5.1.4.19. N-(2-Aminoethyl)-N-{4-[(4-tert-butylphenyl)methoxy]phenyl}-2,4-dichlorobenzamide hydrochloride (82)
Compound 82 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 4-tert-butylbenzyl bromide, using reduction Method A, and benzoylation with 2,4 dichlorobenzoyl chloride. Final product purified by HPLC as HCl salt (35 mg, 53%). 1H NMR (300 MHz, CDCl3): δ 7.40 (2H, d, J = 9 Hz), 7.30 (2H, d, J = 9 Hz), 7.23 (1H, bs), 7.10–7.00 (4H, bs), 6.78 (2H, d, J = 9 Hz), 4.91 (2H, s), 4.00 (2H, t, J = 6 Hz), 3.43(2H, m), 1.44 (9H, s); ESI MS m/z 471.5 (M+H)+, 454.8 (M−NH3+H)+.
5.1.4.20. N-(2-Aminoethyl)-2,4-dichloro-N-{4-[(4-ethylphenyl)methoxy] phenyl}benzamide hydrochloride (83)
Compound 83 was prepared in the same way as 73 starting with alkylation of p-nitrophenol with 4-ethylbenzyl bromide, using reduction Method A, and benzoylation with 2,4 dichlorobenzoyl chloride. Final product purified by HPLC as HCl salt (95 mg, 73%). 1H NMR (300 MHz, CDCl3): δ 8.58 (3H, bs), 7.92 (2H, d, J = 9 Hz), 7.30–7.15 (6H, m), 7.09 (1H, d, J = 2 Hz), 7.03 (1H, d, J = 9 Hz), 6.70 (2H, d, J = 9 Hz), 4.87 (2H, s), 4.24 (2H, bs), 3.34(2H, bs), 2.64 (2H, q, J = 9 Hz), 1.24 (3H, t, J = 9 Hz); ESI MS m/z 443.3 (M+H)+, 426.6 (M−NH3+H)+.
5.2. T. brucei growth inhibition assay
Compounds were tested for antitrypanosomal activity against T. brucei brucei (strain BF427) or against T. rhodesiense (strain STIB900) in HMI-9 media as previously described.16 Cells were tested in triplicate against serial dilutions of compounds along with a Pentamidine isethionate (Sigma-Aldrich, St. Louis, MO) control and quantified with AlamarBlue (ThermoFisher Scientific, Waltham, MA) at 48H.17 EC50 values were calculated by non-linear regression using software by the Collaborative Drug Database (Burlingame, CA. www.collaborativedrug.com).
5.3. Cytotoxicity on mammalian cells
Compounds were assayed for cytotoxicity against CRL-8155 (human lymphoblasts) and HepG2 cells (human hepatocellular carcinoma) as previously described.18 Cells were exposed to serial dilutions of compounds for 48 hours and toxicity was quantified using AlamarBlue (ThermoFisher Scientific, Waltham, MA).18 Compounds were assayed in quadruplicate and EC50 values were calculated non-linear regression using software by the Collaborative Drug Database (Burlingame, CA. www.collaborativedrug.com).
5.4. In vitro liver microsome assays
Pooled liver microsomes from mouse, cow, and human sources were purchased from BD Biosciences (San Jose, CA). Microsome reactions were incubated at 37 °C with 0.16 M KH2PO4, 1mM NADPH, 0.5 mg/mL of microsomes, and 1.5 μM of test compounds. Reactions were quenched at time points (0, 5, 10, 15, 30, 60 min) with the addition of 4X the sample volume of 100% acetonitrile and processed using liquid chromatography/tandem mass spectrometry analysis.
5.5. CYP3A4 inhibition assay
Inhibition of human cytochrome P450 (3YP3A4 isoform) was determined with a commercial kit (BD Biosciences) according the manufacturer’s instructions.
5.6. Mouse oral pharmacokinetics
Compounds were orally administered to mice at a concentration of 50 mg/kg in 5% DMSO, 3% EtOH, 7% Tween80 and 0.9% saline. Three mice were used per group, and 40 μL of blood was collected from the tail at 0.5, 1, 2, 4, 6 and 8 h post dose into heparinized capillary tubes. Plasma was separated via centrifugation and extracted with acetonitrile and compound concentrations were measured by liquid chromatography/tandem mass spectrometry.
5.7. Brain:plasma compound concentration measurements in mice
Each test compound was administered at 5 mg/kg in 5% DMSO, 3% EtOH, 7% Tween80 and 0.9% saline by intraperitoneal injection to 3 mice.11 At 1 h post injection, blood was collected and the brain was removed and homogenized in acetonitrile. Levels of compound in the blood and brain were determined via liquid chromatography/ tandem mass spectrometry. Calculations of brain concentrations accounted for 3% volume/weight of blood in the brain.
5.8. Acute efficacy studies in mice
Experiments were carried out using the standard operating procedure used by WHO screening centers and done in compliance with the University of Washington IACUC approved protocol.19 Groups of 3 female Swiss-Webster mice (ND4 outbred, ages 6–8 weeks) were in infected on day 0 with 1 × 104 T. brucei rhodesiense (strain STIB900) parasites. 50 mg/kg of compound was administered orally in 5% DMSO, 7% Tween80, 3% EtOH and 0.9% saline in a 200 μL volume twice a day at 12 h intervals from day 2 to day 5, for a total of 8 doses. Parasitemia was monitored via microscopic analysis of tail blood for 60 days post infection, or until parasites were detected.
5.9. Chronic efficacy
Again, the experiments followed standard operating procedure used by WHO screening centers and done in compliance with the University of Washington IACUC approved protocol.19 Groups 5 mice were infected with T. brucei brucei (strain TREU667) at Day 0 to establish a chronic infection. Treatment began on day 21 post infection, and mice received 50 mg/kg test compound orally in 5% DMSO, 7% Tween 80, 3% EtOH and 0.9% saline in a 200 μL volume twice a day for 14 days (a total of 28 doses). A control group received vehicle with no compound and another control group received a single intraperitoneal dose of diminazene aceturate at 10 mg/kg in water on day 21. Post dosing, parasitemia was monitored via microscopic examination of tail blood slides until 6 months post infection. Mice were removed from the experiment once parasites were detected in the blood.
5.10. Washout studies
T. brucei brucei (strain 427) cells were plated at 1 × 104 cells/ well on round-bottom 96-well tissue culture treated plates using a separate plate for each time point. At specified times (24H, 48H, 72H, 96H), the plates were centrifuged at 1209 RCF, washed three times with IMDM media (ThermoFisher Scientific, Waltham, MA) equilibrated to 37 °C and 5% CO2, and resuspended in HMI-9 media with 10% fetal bovine serum and 1% penicillin/streptomycin. 20 Plates were then incubated at 37 °C and 5% CO2 and monitored via light microscopy for parasite growth. Positive outgrowth was determined by detecting live and expanding parasites via light microscopy by 10 days post washout.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Richard Glynne, Frantisek Supek, Arnab Chatterjee, Advait Nagle and The Genomics Institute of the Novartis Research Foundation for conducting the high-throughput screen leading to the identification of compound 1. We also acknowledge helpful advice from Richard Tidwell and Donald Patrick from the University of North Carolina. The research was supported by grants from the National Institutes of Health (AI106850, AI054384, and AI070218).
A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2016.11.019.
References
- 1.Priotto G, Kasparian S, Mutombo W, et al. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, noninferiority trial. Lancet. 2009;374:56–64. doi: 10.1016/S0140-6736(09)61117-X. [DOI] [PubMed] [Google Scholar]
- 2.Bisser S, N’Siesi F-X, Lejon V, et al. Equivalence trial of Melarsoprol and Nifurtimox monotherapy and combination therapy for the treatment of Second-Stage Trypanosoma brucei gambiense Sleeping Sickness. J Infect Dis. 2007;195:322–329. doi: 10.1086/510534. [DOI] [PubMed] [Google Scholar]
- 3.New Oral Drug Candidate for African Sleeping Sickness; DNDi press release [Geneva, Switzerland and Kinshasa, Democratic Republic of the Congo – 6 December 2012]. http://www.dndi.org/media-centre/press-releases/354-media-centre/pressreleases/1505-new-oral-drug-candidate-hat.html.
- 4.Pivotal Clinical Trial to Begin for First Oral Drug Candidate Specifically Developed for Sleeping Sickness; DNDi press release [Basel, Switzerland – 8 September 2015]. http://www.dndi.org/2015/media-centre/press-releases/prscyx-7158/.
- 5.Payne D, Gwynn M, Holmes D, Pompliano D. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 6.Munos B. Lessons from 60 years of pharmaceutical innovation. Nat Rev Drug Discov. 2009;8:959–968. doi: 10.1038/nrd2961. [DOI] [PubMed] [Google Scholar]
- 7.Nagle A, Khare S, Kumar A, et al. Recent developments in drug discovery for Leishmaniasis and Human African Trypanosomiasis. Chem Rev. 2014;114:11305–11347. doi: 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swinney D, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 9.Eder J, Sedrani R, Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat Rev Drug Discov. 2014;13:577–587. doi: 10.1038/nrd4336. [DOI] [PubMed] [Google Scholar]
- 10.Pardridge W. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tatipaka HB, Gillespie JR, Chatterjee AK, et al. Substituted 2-phenylimidazopyridines: a new class of drug leads for human African trypanosomiasis. J Med Chem. 2014;57:828–835. doi: 10.1021/jm401178t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrins L, Gazdik M, Rahmani R, et al. Pyridyl benzamides as a novel class of potent inhibitors for the kinetoplastid Trypanosoma brucei. J Med Chem. 2014;57:6393–6402. doi: 10.1021/jm500191u. [DOI] [PubMed] [Google Scholar]
- 13.Rahmani R, Ban K, Jones A, et al. 6-Arylpyrazine-2-carboxamides: a new core for Trypanosoma brucei inhibitors. J Med Chem. 2015;58:6753–6765. doi: 10.1021/acs.jmedchem.5b00438. [DOI] [PubMed] [Google Scholar]
- 14.Hwang J, Smithson D, Holbrook G, et al. Optimization of the electrophile of chloronitrobenzamide leads active against Trypanosoma brucei. Bioorg Med Chem Lett. 2013;23:4127–4131. doi: 10.1016/j.bmcl.2013.05.049. [DOI] [PubMed] [Google Scholar]
- 15.Hwang J, Smithson D, Zhu F, et al. Optimization of chloronitrobenzamides (CNBs) as therapeutic leads for Human African Trypanosomiasis (HAT) J Med Chem. 2013;56:2850–2860. doi: 10.1021/jm301687p. [DOI] [PubMed] [Google Scholar]
- 16.Shibata S, Gillespie JR, Kelley AM, et al. Selective inhibitors of methionyl-tRNA synthetase have potent activity against Trypanosoma brucei infection in mice. Antimicrob Agents Chemother. 2011;55:1982–1989. doi: 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raz B, Iten M, Grether-Buhler Y, Kaminsky R, Brun R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 1997;68:139–147. doi: 10.1016/s0001-706x(97)00079-x. [DOI] [PubMed] [Google Scholar]
- 18.Shibata S, Gillespie JR, Ranade RM, et al. Urea-based inhibitors of Trypanosoma brucei methionyl-tRNA synthetase: selectivity and in vivo characterization. J Med Chem. 2012;55:6342–6351. doi: 10.1021/jm300303e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dardonville C, Barrett MP, Brun R, Kaiser M, Tanious F, Wilson WD. DNA binding affinity of bisguanidine and bis(2-aminoimidazoline) derivatives with in vivo antitrypanosomal activity. J Med Chem. 2006;49:3748–3752. doi: 10.1021/jm060295c. [DOI] [PubMed] [Google Scholar]
- 20.Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol. 1989;75:985–989. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.