

Abstract
Somatostatin (SS) is a widely distributed polypeptide that exerts inhibitory effects on hormone secretion and cell proliferation by interacting with five different receptors (SST1-SST5). β-Arrestins have been implicated in regulating SST internalization, but the structural domains mediating this effect are largely unknown. The aim of this study was to characterize the intracellular mechanisms responsible for internalization of human SST5 in the rat pituitary cell line GH3 and to identify the SST5 structural domains involved in this process. To this purpose we evaluated, by fluorescence microscopy and biochemical assay, the ability of wild-type, progressive C-terminal truncated and third cytoplasmatic loop mutants SST5-DsRed to associate with β-arrestin-enhanced green fluorescent protein and to internalize under SS28 stimulation. The truncated mutants were comparable to the wild-type receptor with respect to recruitment of β-arrestin-2 and internalization, whereas the third loop mutants R240W, S242A, and T247A showed the abolishment or reduction of arrestin association and a significant reduction of receptor internalization (14.4%, 29%, and 30.9% vs. 52.4% of wild type) and serine phosphorylation upon SS28 stimulation. Moreover, we evaluated the ability of simultaneous mutation of these three residues (R240, S242, and T247) and C-terminal truncated receptors to internalize. The progressive truncation of the C-terminal tail resulted in a progressive increased internalization (21.6%, 36.7%, and 41%, respectively) with respect to the full-length total third-loop mutant (15%). In conclusion, our results indicate the SST5 third intracellular loop as an important mediator of β-arrestin/receptor interaction and receptor internalization, whereas they suggest that residues 328–347 within the C terminus may play an inhibitory role in receptor internalization.
SOMATOSTATIN (SS) PHYSIOLOGICALLY regulates several biological functions, including inhibition of endocrine and exocrine secretion, modulation of neurotransmission, motor, and cognitive functions, inhibition of intestinal motility, absorption of nutrient and ions, and vascular contractility (reviewed in Refs. 1, 2, 3). SS is also an important hormonal regulator of cell proliferation and differentiation (4, 5). These actions are mediated by a family of five specific G protein-coupled receptors (GPCRs) termed SST1–5, with important differences in tissue distribution, coupling to second messengers, affinity for SS and its analogs, and intracellular trafficking (1, 2, 3). These receptors couple to Gi/Go proteins and generate a complex series of intracellular signals, the inhibition of adenylyl cyclase and the reduction of intracellular cAMP levels being common to the five receptors.
The high density of SST, in particular SST2 and 5, on human neuroendocrine tumors has been used clinically to treat patients harboring GH- or TSH-secreting pituitary adenomas as well as islet cell or carcinoid tumors with specific SS analogs (6).
A common property of most GPCRs is their ability to decrease receptor responsiveness in the continued presence of agonist (7, 8, 9). This regulation involves a coordinated series of events that include phosphorylation of the receptor by G protein-coupled receptor kinases, recruitment of β-arrestins, signal termination (desensitization), and receptor targeting to clathrin-coated pits for endocytosis (10). Although the first identified sites of receptor phosphorylation required for arrestin binding were localized in the C terminus of prototypical GPCRs, such as rhodopsin and β2-adrenergic receptor, recent studies indicate that relevant phosphorylation sites can be localized anywhere on the intracellular surface of the receptor (11, 12, 13, 14).
It was previously shown that agonist activation of SSTs is followed by receptor desensitization and internalization (15, 16, 17, 18, 19, 20, 21). In particular, a previous study reported that the five rat SSTs transfected in human embryonic kidney 293 cells differ in their patterns of β-arrestin mobilization and endosomal sorting (22), SST2A showing stable colocalization with β-arrestin in the same endocytic vescicles, SST3 and SST5 showing transient translocation with β-arrestin to the plasma membrane, and SST1 and SST4 β-arrestin showing independent trafficking.
Because SS is the hypothalamic peptide that physiologically inhibits GH secretion, the aim of this study was to investigate the molecular determinants mediating the interaction of the human SST5 with β-arrestin and subsequent internalization after stimulation with SS28, the natural peptide with highest affinity for SST5, in a pituitary cell line.
RESULTS
Internalization Properties of Wild-Type SST5 in GH3 Cells
Human SST5 gene expression in GH3 cells was first characterized by RT-PCR analysis in transfected cells. Native GH3 cells did not express SST5, whereas GH3 cells transiently transfected with SST5-DsRed2 expressed SST5 mRNA and protein (data not shown). To investigate subcellular localization of the expressed protein, cells were analyzed by fluorescence microscopy 48 h after transfection. As shown in Fig. 1A, SST5-DsRed2 appears to be localized at the plasma membrane. The functional activity of the wild-type SST5-DsRed2 receptor was confirmed by examining its ability to inhibit adenylyl cyclase and reduce intracellular levels of cAMP (51 ± 3% reduction of forskolin-stimulated intracellular cAMP after 30 min incubation with 100 nm SS28). To study wild-type SST5 internalization after agonist stimulation, GH3 cells were transiently transfected with SST5-DsRed2 and incubated in the absence or presence of saturating concentration of 1 μm SS28 (21, 22), the natural peptide with highest affinity for SST5. As shown in Fig. 1A, in unstimulated cells, wild-type SST5 was clearly visible at the cell surface. After 30 min incubation the receptor was localized within intracellular vescicles, distributed throughout the cytoplasm. The membrane to intracellular fluorescence ratio (fR) after 30 min of stimulation resulted in 47.6% of control with a resulting internalization of 52.4% in SS28-stimulated cells with respect to untreated cells (Table 1). The same results were observed after 60 min of incubation (data not shown).
Fig. 1.
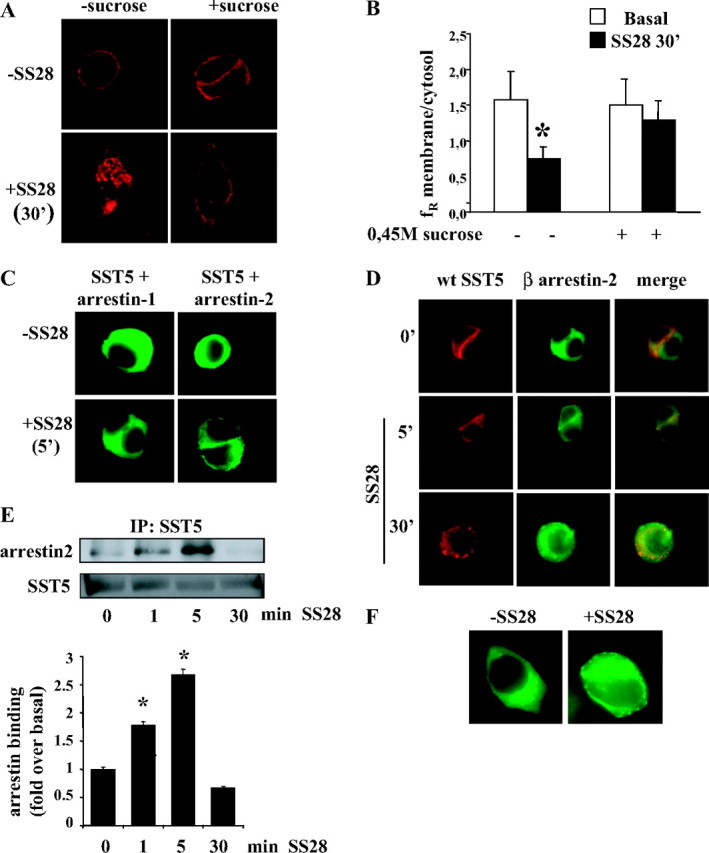
Effects of SS28 Treatment on the Wild-Type SST5 (Red) Internalization and Arrestin (Green) Mobilization
A, Representative images of GH3 cells transiently transfected with wild-type SST5. Cells were preincubated 48 h after transfection in the absence or presence of 0.45 m sucrose for 30 min at 37 C and then stimulated with the agonist. The figure shows SST5 distribution in the absence of the agonist and after 30 min of 1 μm SS28 incubation. The results shown are representative images of five individual experiments. B, SST5 internalization was evaluated using ImagePro-Plus 6.0 software. Fluorescence density mean in two separate regions corresponding to the plasma membrane and to the cytoplasm was densitometrically determined. Mean membrane to intracellular fluorescence ratio (fR) was then calculated. For each group, at least 30 cells from three independent transfections were analyzed by three independent investigators, and the mean value was used for the graph. *, P < 0.01 vs. basal. C, Translocation of β-arrestins to the plasma membrane. GH3 cells were transiently cotransfected with wild-type SST5 and either β-arrestin-1 or β-arrestin-2. SS28 (1 μm) was added to the culture medium for 5 min. Cells were then fixed and examined by fluorescence microscopy. Shown are representative images from one of at least five independent experiments. The figure shows translocation of β-arrestin-2 but not β-arrestin-1 to the plasma membrane. D, Internalization of wild-type SST5 and β-arrestin-2 trafficking. GH3 cells transiently cotransfected with SST5-DsRed and β-arrestin-2-GFP were incubated with 1 μm SS28 for 5 and 30 min. Fixed cells were analyzed by fluorescence microscopy. The figure shows representative images from one of at least five individual experiments. The figure shows translocation of β-arrestin-2 to the plasma membrane in the middle panel and colocalization of SST5-DsRed with β-arrestin-2 in the right panel. E, Time course of the association of wild-type SST5 with β-arrestin-2. GH3 cells transiently cotransfected with wild-type SST5 and β-arrestin-2 were washed and incubated at 37 C with 1 μm SS28 for the indicated times before cross-linking. Lysates were immunoprecipitated (IP) with SST5 antibody, and the presence of β-arrestin-2 and SST5 was detected using anti-GFP and anti-SST5 antibody, respectively (upper panel). The figure shows a representative experiment and the analysis of the data obtained from five separate experiments performed with the image analysis program NIH ImageJ (lower panel). β-Arrestin-2 binding is expressed as the fold increase relative to unstimulated cells and represents mean ± se. *, P < 0.05 vs. 0 min. F, Translocation of β-arrestin-2 to the plasma membrane induced by wild-type, unmodified SST5. GH3 cells were transiently cotransfected with wild-type, unmodified SST5 and β-arrestin-2-EGFP. SS28 (1 μm) was added to the culture medium for 5 min. Cells were then fixed and examined by fluorescence microscopy. Shown are representative images from one of three independent experiments. The figure shows the redistribution of β-arrestin-2 from the cytoplasm to the cell membrane induced by SS28 stimulation. wt, Wild type.
Table 1.
Quantitative Analysis of Internalization of Wild-Type and Mutated SST5 Receptor
| FR 0 min | FR 30 min | % FR | % Internalization | P | |
|---|---|---|---|---|---|
| Wild type | 1.577 ± 0.391 | 0.750 ± 0.163 | 47.56 | 52.44 | |
| Wild type + sucrose | 1.487 ± 0.257 | 1.328 ± 0.315 | 89.30 | 10.70 | 0.00001 |
| D347 | 1.510 ± 0.365 | 0.900 ± 0.212 | 59.62 | 40.38 | 0.04141 |
| D338 | 1.316 ± 0.210 | 0.685 ± 0.171 | 52.06 | 47.94 | 0.32311 |
| D328 | 1.392 ± 0.290 | 0.579 ± 0.187 | 41.63 | 58.37 | 0.02041 |
| S242A | 1.463 ± 0.185 | 1.039 ± 0.171 | 71.01 | 28.99 | 0.00011 |
| E243A | 1.322 ± 0.156 | 0.764 ± 0.222 | 57.80 | 42.20 | 0.05741 |
| T247A | 1.452 ± 0.147 | 1.003 ± 0.202 | 69.07 | 30.93 | 0.00461 |
| R240W | 1.403 ± 0.169 | 1.201 ± 0.210 | 85.60 | 14.40 | 0.00001 |
| RST | 1.414 ± 0.224 | 1.202 ± 0.206 | 85.01 | 14.99 | 0.00001 |
| RST-347 | 1.386 ± 0.182 | 1.087 ± 0.182 | 78.43 | 21.57 | 0.0348b |
| RST-338 | 1.413 ± 0.207 | 0.895 ± 0.257 | 63.32 | 36.68 | 0.0306b |
| RST-328 | 1.344 ± 0.132 | 0.792 ± 0.067 | 58.97 | 41.03 | 0.0013b |
Percent of receptor internalization was calculated from the stimulated to basal fluorescence ratio (fR). For each group, at least 30 cells from three independent transfections were analyzed by three independent investigators, and the mean value was used for the graphs.
P vs. wt; bP vs. correspondent deleted mutant.
GPCR endocytosis commonly occurs via clathrin-coated pits after ligand activation (25, 26). Hypertonic sucrose is a commonly used agent that destabilizes clathrin-coated pits and inhibits clathrin-dependent endocytosis. To verify the molecular mechanism of SST5 internalization, transfected cells were preincubated with 0.45 m sucrose for 30 min and then incubated without or with SS28 for 30 min (Fig. 1A). After this manipulation SST5 remained at the cell membrane in cells incubated with SS28, with a fR of 89.3% of control, resulting in 10.7% internalization (Fig. 1B and Table 1). This result supports the involvement of clathrin-coated pits in SS28-mediated SST5 internalization.
SST5 Associates with β-Arrestin-2, But Not with β-Arrestin-1, after 5 min of SS28 Stimulation
Arrestins participate in receptor internalization by binding the components of the endocytotic machinery of the clathrin-coated pit. To visualize the translocation of β-arrestins to the plasma membrane, we cotransfected GH3 cells with wild-type SST5 and either β-arrestin-1 or -2 conjugated to EGFP. The results are presented in Fig. 1C. In the absence of the agonist, both β-arrestin isoforms were uniformly distributed throughout the cytoplasm. A 5-min SS28 stimulation induced a rapid redistribution of β-arrestin-2 from the cytoplasm to the cell membrane, where it appears as punctate fluorescence clusters, whereas no change in β-arrestin-1 distribution was detected. Figure 1D shows merge imaging indicating a colocalization of β-arrestin-2 with wild-type SST5 after 5 min of agonist incubation. After 30 min of SS28 exposure, β-arrestin-2 redistributed to the cytoplasm, whereas endocytic vesicles contained only the receptor.
A biochemical assay was used to confirm colocalization studies and to measure the amount of SST5-arrestin complexes. GH3 cells cotransfected with wild-type SST5 and β-arrestin-1 or -2, after incubation with SS28, were treated with dithiobis (succinimidylpropionate) (DSP), a cell-permeable, cleavable cross-linking agent, and SST5 receptors were immunoprecipitated from cell lysates. The cross-linked complexes were dissociated by incubation of the immunoprecipitates with sodium dodecyl sulfate (SDS) sample buffer containing reducing agents and analyzed by Western blotting. The formation of SST5-arrestin complexes was quantified by detecting the presence of arrestin in immunoprecipitates. The presence of equal amounts of receptor was confirmed by reprobing with anti-SST5 antibody. Figure 1E shows that SS28-induced β-arrestin-2 association was slightly increased above basal level at 1 min and was maximal at 5 min (2.7-fold over basal). The complexes were completely dissociated after 30 min stimulation. No significant change in β-arrestin-1 level after 1 min, 5 min, or 30 min stimulation was observed (data not shown).
These data demonstrated that after SS28 stimulation SST5 associated with β-arrestin-2 but not with β-arrestin-1. The cross-linking/coimmunoprecipitation assay showed that the SST5-β-arrestin-2 interaction was transient and the complexes rapidly dissociated.
To verify that DsRed2 protein fused to the C terminus of the receptor did not alter receptor-arrestin interaction, we transiently transfected GH3 cells with wild-type, unmodified SST5 (23) and β-arrestin-2. Fluorescence microscopy analysis of β-arrestin mobilization revealed that 5 min of SS28 stimulation induced the redistribution of β-arrestin-2 from the cytoplasm to the cell membrane, similarly to that obtained with GH3 cells transfected with the receptor fused to DsRed2 protein (Fig. 1F).
SST5 C-Tail Deletion Mutants Still Internalize and Associate with β-Arrestin-2
For many GPCRs, formation of receptor-arrestin complexes involves phosphorylation of serines and threonines in the C-terminal tail of the receptor. This region of SST5 contains 55 residues. Of the three serines and three threonines present within this region, NetPhos 2.0 server indicated only Thr361 as a potential phosphorylation target (score = 0.659; threshold > 0.5). To identify SST5 structural domains involved in β-arrestin interaction and receptor internalization, we evaluated the effects of progressive truncations of the SST5 C-terminal tail (Fig. 2).
Fig. 2.

Schematic Representation of Wild-Type and Mutant SST5 Receptors
The third intracellular loop (A) and the C-terminal domain (B) are shown in detail to illustrate the structure of the different mutant receptors that were constructed. A, Residues in the third intracellular loop mutated to Ala (Ser242, Glu243, and Thr247) and residue Arg240 mutated to Trp are highlighted in black. B, The figure shows progressive deletions of the C-terminal tail introduced by PCR to create truncated receptors with variable length C-tail (D347, D338, and D328). Potential phosphorylation sites (Ser and Thr residues) in the C-terminal domain are highlighted in black. Of these, NetPhos 2.0 server indicated only Thr361 as a potential phosphorylation target (score = 0.659; threshold > 0.5). C, Schematic representation of the model proposed for SST5 arrestin binding and internalization. Under basal conditions, C-terminal tail of wild-type SST5 is associated with the third cytoplasmic loop. SS28 binding promotes receptor activation and phosphorylation of residues within the third cytoplasmic loop, allowing these two domains to disassociate and arrestin to bind to the receptor. Arrestin binding targets the receptor to clathrin-coated pits for endocytosis. wt, Wild type. The P within a circle indicates receptor phosphorylation.
Each truncated construct (D347, D338, and D328) transiently cotransfected in GH3 cells with β-arrestin-2 localized at the plasma membrane in the absence of the agonist (Fig. 3A). The basal expression level at the plasma membrane of the wild-type and different truncated receptors was similar in transiently transfected cells, as indicated by Western blot analysis of SST5 immunoprecipitation performed on plasma membrane cell extracts (Fig. 3B, lower panel). D338 and D328 receptors, but not D347, were partially internalized upon 5 min of stimulation. A 30-min agonist exposure induced evident internalization of D338 and D328, whereas the truncation at position 347 attenuated receptor internalization (Fig. 3A). As shown in Fig. 3B, 30 min of SS28 stimulation induced a progressive reduction of fR that corresponded to progressive deletions (Table 1). Internalization of D328 mutant (58.4%) was significantly increased (P = 0.02) with respect to wild-type SST5 (52.4%), suggesting that a short or absent C-terminal tail could enhance the internalization process.
Fig. 3.

Internalization Properties and Arrestin Binding of SST5 Deletion Mutants
A, Internalization of progressive deleted SST5 mutants and β-arrestin-2 trafficking. GH3 cells cotransfected with β-arrestin-2 and SST5 with progressive deletions of the C-terminal tail were incubated with 1 μm SS28 for 5 and 30 min. Fixed cells were analyzed by fluorescence microscopy. Representative images from one of at least five individual experiments are shown. Each truncated construct (D347, D338, and D328) transiently cotransfected in GH3 cells with β-arrestin-2 localized at the plasma membrane in the absence of the agonist. The figures show translocation of β-arrestin-2 to the plasma membrane at 5 min of SS28 stimulation (“β-arr-2” panel) and colocalization of SST5 with β-arrestin-2 (“merge” panel). A 30-min agonist exposure induced evident internalization of D338 and D328 whereas the truncation at position 347 attenuated receptor internalization. B, Internalization of SST5-truncated mutants was evaluated using ImagePro-Plus 6.0 software. SS28 stimulation for 30 min induced a progressive reduction of fR that corresponded to progressive deletions. Mean membrane to intracellular fluorescence ratio (fR) was calculated. For each group, at least 30 cells from three independent transfections were analyzed, and the mean value was used for the graph. §, P < 0.05 vs. wild-type SST5 + SS28 30′. *, P < 0.01 vs. basal. The lower panel shows a representative Western blot of wild-type and deleted SST5 immunoprecipitated from cell membrane extracts of transiently transfected cells in basal conditions. C, Time course of the association of D328 SST5 with β-arrestin-2. GH3 cells transiently cotransfected with D328 SST5 and β-arrestin-2 were washed and incubated at 37 C with 1 μm SS28 for the indicated times before cross-linking. Lysates were immunoprecipitated (IP) with SST5 antibody, and the presence of β-arrestin-2 and SST5 was detected using anti-GFP and anti-SST5 antibody, respectively (upper panel). Representative images from at least five individual experiments are shown. The analysis of the data was performed with the image analysis program NIH ImageJ (lower panel). β-Arrestin-2 binding is expressed as the fold increase relative to unstimulated cells and represents mean ± se. *, P < 0.05 vs. 0 min. arr2, Arrestin-2; wt, wild type.
Fluorescence microscopy analysis of β-arrestin mobilization revealed that, after 5 min of SS28 stimulation, the truncation mutants induced a translocation of β-arrestin-2 to the plasma membrane where it colocalized with the receptors (Fig. 3A).
Cross-linking/coimmunoprecipitation assay (Fig. 3C) showed that the amount of β-arrestin-2 coimmunoprecipitated with D328 receptor was slightly increased at 1 min and was considerably increased above basal levels at 5 min of stimulation (2.69-fold over basal). No association of β-arrestin-1 to D328 SST5 was detected (data not shown).
SST5 Third Loop Mutants Are Associated with Reduced Internalization and Phosphorylation after SS28 Stimulation
To identify a region of SST5 that mediates agonist-induced internalization, we focused our studies on the third intracellular loop, which contains two predicted sites of phosphorylation (Ser242, P = 0.994; and Thr247, P = 0.461). The presence of a glutamic acid residue (Glu243) in close proximity is predicted to enhance their phosphorylation by a G protein-coupled receptor kinase (7) and to increase the affinity for β-arrestin. We created three-point mutant receptors by site-directed mutagenesis in which these three residues were replaced by Ala (S242A, E243A, and T247A mutants). We also tested a naturally occurring SST5 third loop mutant (R240W) previously found in one acromegalic patient resistant to SS analogs (23). As shown in Fig. 4, A and B, mutant receptors were correctly targeted to the plasma membrane. Average data from multiple experiments in which the internalization was quantified are shown in Table 1 and Fig. 4B. The mutation of Ser242 or Thr247 to Ala strongly reduced SST5 internalization (29.0% and 30.9%, respectively, with respect to 52.4% of wild type), whereas E243A mutant showed lower (42.2%), but not significant (P = 0.057), internalization with respect to wild-type SST5 after 30 min of agonist incubation. Both mutation of Arg240 to Trp and the simultaneous mutation of R240, S242, and T247 in the total third-loop mutant [R240, S242, and T247 (RST)] almost completely abolished internalization (14.4% and 15.0%, respectively). To exclude differences in the level of expression at the plasma membrane, we performed immunoprecipitation followed by Western blot analysis on plasma membrane cell extracts. Western blots showed comparable expression levels of the wild type and different mutated receptors at the cell surface (Fig. 4B, lower panel).
Fig. 4.
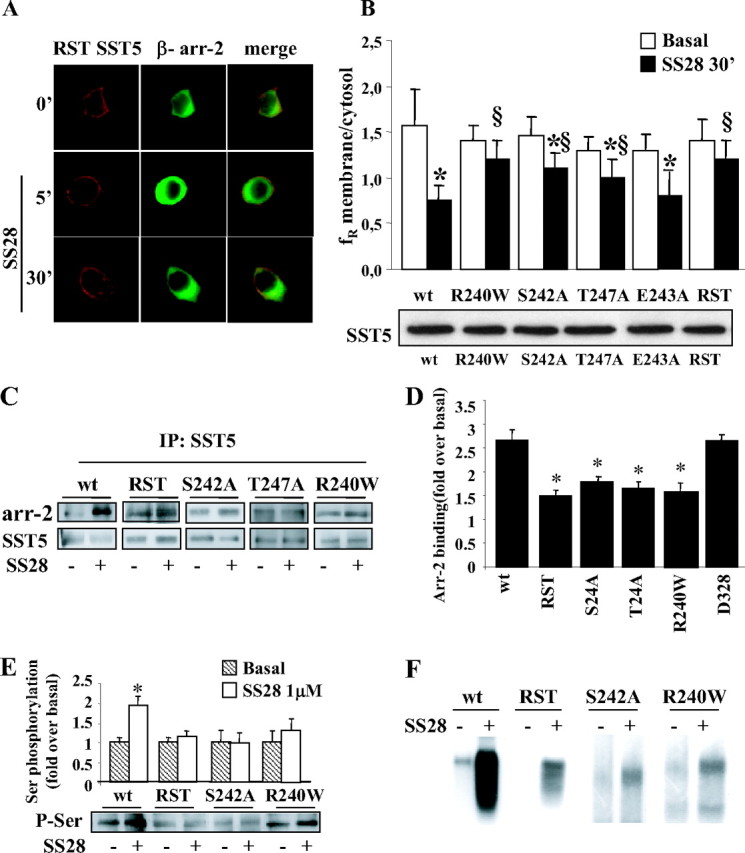
Effects of Introducing Mutations within the Third Cytoplasmic Loop of SST5 (Red) on β-Arrestin-2 (Green) Translocation and Receptor Internalization
A, GH3 cells transiently cotransfected with β-arrestin-2 and SST5 with simultaneous mutations of R240, S242, and T247 (RST) were incubated with SS28 1 μm for 5 and 30 min. We created three-point mutant receptors by site-directed mutagenesis in which these three residues were replaced by Ala (S242A, E243A, and T247A mutants). We also tested a naturally occurring SST5 third-loop mutant (R240W) previously found in one acromegalic patient resistant to SS analogs. As shown here for RST mutant as a representative example, mutant receptors were correctly targeted to the plasma membrane. No β-arrestin translocation was observed after 5 min SS28 incubation and, accordingly, RST mutant internalization was almost completely abolished. Fixed cells were analyzed by fluorescence microscopy. The figure shows representative images from one of at least five individual experiments. B, SST5 third-loop mutants (R240W, S242A, T247A, E243A, and RST) internalization was evaluated using ImagePro-Plus 6.0 software. Mean membrane to intracellular fluorescence ratio (fR) was calculated. For each group, at least 30 cells from three independent transfections were analyzed and the mean value was used for the graph. *, P < 0.01 vs. basal; §, P < 0.01 vs. wild-type SST5 + SS28 30′. The lower panel shows a representative Western blot of wild-type and third-loop mutated SST5 receptors immunoprecipitated from cell membrane extract in basal conditions. C, Association of third-loop mutants SST5 with β-arrestin-2. GH3 cells transiently cotransfected with wild-type, RST, S242A, T247A, or R240W SST5 and β-arrestin-2 were washed and incubated at 37 C with 1 μm SS28 for 5 min before cross-linking. Lysates were immunoprecipitated (IP) with SST5 antibody, and the presence of β-arrestin-2 and SST5 was detected using anti-GFP and anti-SST5 antibody, respectively. The figure shows a representative experiment. D, The figure compares the association of β-arrestin-2 to wild-type, third loop-mutated, and D328-deleted SST5 receptors. The analysis of the data obtained from five separate experiments was performed with the image analysis program NIH ImageJ. β-Arrestin-2 binding is expressed as the fold increase relative to unstimulated cells and represents mean ± se. *, P < 0.05 vs. wild type. E, Analysis of wild-type and mutated SST5 phosphorylation. GH3 cells transiently transfected with wild type or mutated SST5 were incubated with SS28 1 μm for 5 min. SST5 was immunoprecipitated from cell lysates, and Western blot analysis was performed using antiphosphoserine antibody (lower panel). Loading of equal amounts of receptor proteins in each lane was confirmed with anti-SST5 antibody. The upper panel shows the analysis of the data obtained from five separate experiments performed with the image analysis program NIH ImageJ and represents mean ± se. *, P < 0.05 vs. basal. The level of agonist-induced serine phosphorylation, expressed as the fold increase relative to unstimulated cells, was reduced for RST (1.15-fold over basal), S242A (0.93-fold over basal), and R240W (1.38-fold over basal) receptor with respect to wild-type SST5 (1.93-fold over basal). F, Autoradiogram of SDS-PAGE analysis of immunoprecipitates from whole-cell phosphorylation assays. GH3 cells were transiently transfected with the appropriate plasmids for 48 h, labeled with [32P]orthophosphate, and incubated with 1 μm SS28 for 5 min. SST5 was immunoprecipitated, and the samples were subjected to SDS-PAGE followed by autoradiography. A representative experiment is shown. arr-2, arrestin-2; wt, wild type; P-Ser, phosphoserine.
By fluorescence microscopy, no β-arrestin translocation to RST, R240W, S242A, T247A, and E243A SST5 was observed after 5 min of SS28 incubation (Fig. 4A).
To measure the arrestin association by the cross-linking/coimmunoprecipitation assay, transfected cells were stimulated with SS28 for 5 min followed by DSP cross-linking, immunoprecipitation, and Western blotting (Fig. 4C). When we transfected cells with RST SST5 and β-arrestin-2, the amount of arrestin in the immunoprecipitates after 5 min of stimulation was 1.5-fold over basal, suggesting a strongly reduced arrestin association for the RST mutant with respect to wild-type SST5 (2.7-fold over basal). Reduced arrestin association was also observed with S242A, T247A, and R240W mutants (Fig. 4, C and D).
To test whether mutation of the RST motif affects the phosphorylation of SST5, GH3 cells transiently transfected with wild-type or mutated SST5 were incubated with 1 μm SS28 for 5 min. SST5 was immunoprecipitated from cell lysates, and Western blot analysis was performed using antiphosphoserine or antiphosphothreonine antibodies. Loading of equal amounts of receptor proteins in each lane was confirmed with anti-SST5 antibody. The level of SST5 serine phosphorylation after 5 min of SS28 stimulation was reduced for RST, S242A, and R240W mutant receptors with respect to wild type (Fig. 4E). No significant difference of threonine phosphorylation was measured after agonist stimulation of SST5 in the presence or absence of mutations. These data suggest that, in our system, the residue Ser242, but not Thr247, is a target for phosphorylation. To further confirm these results, we measured the incorporation of radioactive phosphate by whole-cell phosphorylation assay (Fig. 4F). We observed a marked increase in phosphorylation of wild-type SST5 after incubation with SS28, whereas in cells expressing RST, S242A, and R240W mutant receptors agonist-induced receptor phosphorylation was reduced with respect to wild type.
Third-Loop Mutated and C-Tail-Deleted SST5
To determine whether mutations of the third loop would reduce internalization if these mutations were created within the context of the truncated constructs, we evaluated the ability of the total third-loop mutant (RST) and C-terminal truncated receptors to internalize. As shown in Table 1 and Fig. 5, A and B, progressive truncation of the C-terminal tail resulted in a progressive increased internalization (21.6%, 36.7%, and 41.0%, respectively) with respect to full-length RST mutant, which however remained reduced with respect to wild-type or deleted receptor.
Fig. 5.
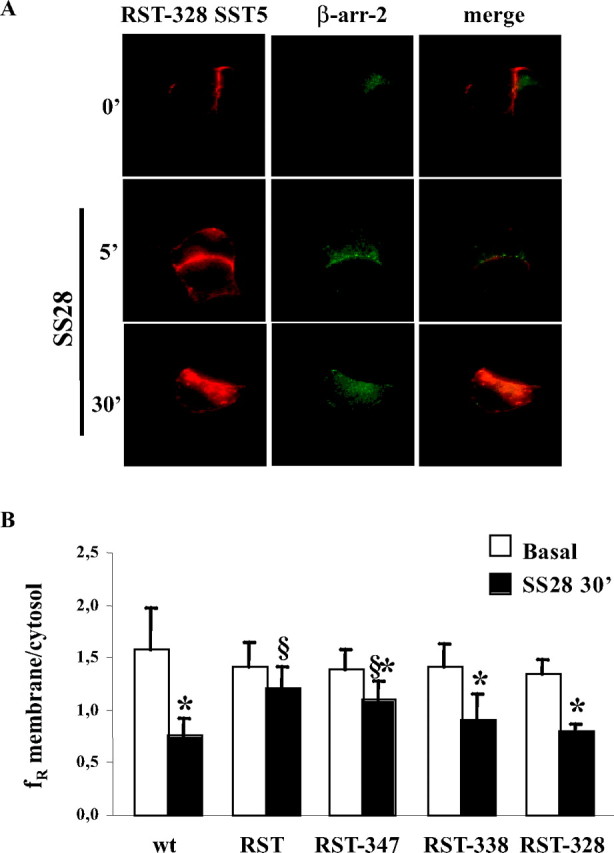
Determination Whether Mutations Would Reduce Internalization
To determine whether mutations of the third loop would reduce internalization if these mutations were created within the context of the truncated constructs, we evaluated the ability of SST5 with simultaneous mutations of RST and C-terminal truncated receptors to internalize. A, GH3 cells transiently cotransfected with β-arrestin-2 and C-terminal tail truncation (D328) on third loop-mutated RST SST5 were incubated with 1 μm SS28 for 5 and 30 min. Fixed cells were analyzed by fluorescence microscopy. The figure shows representative images from one of at least five individual experiments. B, Effect of C-terminal tail truncations on third loop-mutated RST-SST5 internalization. The figure shows that progressive truncation of the C-terminal tail resulted in a progressive increased internalization with respect to full-length RST mutant, which, however, remained reduced with respect to wild-type or deleted receptor. Quantitative analysis was performed by ImagePro-Plus 6.0 software. For each group, at least 30 cells from three independent transfections were analyzed, and the mean value was used for the graph. *, P < 0.01 vs. basal; §, P < 0.01 vs. wild type SST5 + SS28 30′. arr-2, Arrestin-2; wt, wild type.
Biochemical Analysis of SST5 Endocytosis Using Cleavable Biotin
To confirm SST5 internalization data, a highly sensitive biochemical assay based upon cleavable biotin was used. SST5 receptors on the cell surface were biotinylated with a cleavable reagent. After SS28 stimulation, glutathione, a membrane-impermeant reducing agent, was used to cleave biotin of cell surface proteins, whereas internalized receptors were inaccessible to glutathione (28). As shown in Fig. 6A, by this approach we demonstrated that 1 μm SS28 induces 47.8% internalization of wild-type SST5 after 30 min of stimulation, whereas only 14.9% of the surface population of RST receptors was internalized in the same conditions. The amount of internalized, glutathione-resistant D347, D338, and D328 SST5 was, respectively, 42.3%, 52.7% and 63.8%. These results are in complete agreement with the data obtained with fluorescence microscopy (Fig. 6B).
Fig. 6.
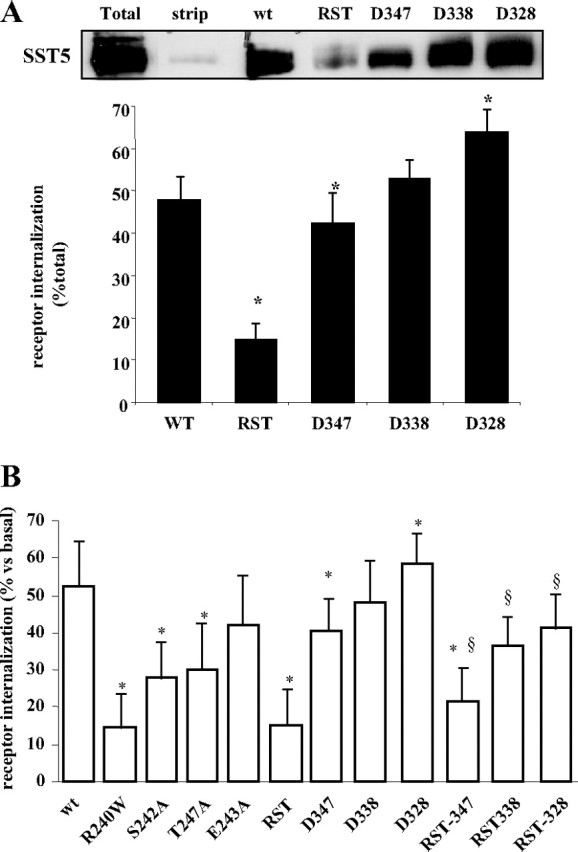
Effect of Third-Loop Mutations and Progressive C-Terminal Tail Truncation on SST5 Internalization
SST5 internalization was evaluated either by fluorescence microscopy and biochemical assay. A, Internalization of wild-type and mutated receptor was determined biochemically using cell surface biotinylation assay. The upper panel shows a representative experiment. Lane 1 (total) shows the total amount of SST5 receptor biotinylated at the cell surface; lane 2 (strip) indicates the efficiency of biotin cleavage by gluthatione from SST5 receptors present in the plasma membrane. Internalized receptors were detected by their resistance to biotin cleavage after incubation of biotinylated cells with 1 μm SS28 for 30 min at 37 C. The analysis of the data obtained from five separate experiments was performed with the image analysis program NIH ImageJ (lower panel). Receptor internalization was expressed as a percentage relative to the total amount of biotinylated receptor present without cleavage, and the mean value (±se) was used for the graph. *, P < 0.05 vs. wild type. B, The figure shows the summary of % internalization of wild-type compared with mutated SST5 receptors, by fluorescence microscopy. Images of GH3 cells transiently cotrasfected with β-arrestin-2 and SST5 were processed using Image ProPlus 6.0 software, and the amount of internalized receptor after agonist stimulation was analyzed in single cells. Percent of receptor internalization was calculated from the stimulated (30 min; 1 μm SS28) to basal fR ratio. *, P < 0.05 vs. wt; §, P < 0.05 vs. correspondent deleted mutant. wt, Wild type.
DISCUSSION
In the present study, we characterized the structural domains of human SST5 involved in internalization and β-arrestin recruitment processes in a pituitary model, using both biochemical and fluorescence microscopy approaches, and demonstrated that the third intracellular loop is crucial for agonist-dependent receptor internalization and β-arrestin interaction. As demonstrated in other cell systems (15, 16, 17, 18, 19, 20, 21), wild-type SST5 functionally expressed at the plasma membrane in GH3 cells efficiently internalized upon the stimulation with SS28, the natural peptide with highest affinity for SST5 (52.4% internalization). Moreover, SST5 internalization was inhibited by hypertonic sucrose, a classical inhibitor of clathrin-mediated endocytosis, demonstrating that SST5 trafficking and signaling are regulated by clathrin-dependent pathways. We also observed that SS28 induced the recruitment of β-arrestin-2, but not β-arrestin-1, to the plasma membrane where it colocalized with SST5. The arrestin-receptor complexes dissociated at or near the cell membrane and, after prolonged agonist exposure, SST5 internalized in endocytotic vesicles, whereas arrestin redistributed in the cytoplasm. The molecular mechanism of arrestin-receptor interaction is based on the presence of two sensor sites within the arrestin protein: an activation sensor that binds receptor elements that change conformation upon activation, and a phosphate sensor that binds receptor-attached phosphates or negatively charged residues, independently from the sequence context (29). Because, for most GPCRs, the first phosphorylation sites relevant for arrestin binding are localized either in the C terminus or the third intracellular loop, we concentrated on these two regions to define SST5 structural domains involved in receptor internalization and binding to β-arrestin-2. Using a series of C-terminal tail-truncated receptors, we demonstrated that a short or absent C-terminal tail did not reduce, but even enhanced, the internalization process. In fact, the deleted mutants, D347, D338, and D328, retained the ability to interact with β-arrestin and to internalize. Moreover, internalization efficiency increased with the length of the deleted region. Indeed, the mutant with the shorter C-tail, i.e. D328 mutant, internalized more efficiently than the wild-type receptor, suggesting that residues 328–347 within the SST5 C terminus play an inhibitory role in receptor internalization, as previously shown for a very small subset of GPCRs (11, 14, 30). Conversely, the observation that the D347 mutant showed reduced internalization with respect to full-length SST5 suggests that the segment corresponding to amino acids 347–364 contains positive molecular signals for internalization. Interestingly, deletion of the last 17 residues occurring in D347 mutant abolished the only phosphorylation site (threonine 361) predicted by NetPhos 2.0 server.
These data are in contrast with the only study on SST5 structure-function relationship available in the literature (16). In fact, it has been previously reported that the C-tail was critical for SST5 internalization, coupling with adenylyl cyclase and desensitization in Chinese hamster ovary K1 cells (16). Admittedly, the different cellular host system used for receptor expression might account for the different results. Furthermore, because in that study internalization was investigated by measuring radioligand, the results might reflect trafficking and processing of the radioligand but not of the receptor itself.
To identify third-loop residues involved in arrestin interaction, we created three-point mutant receptors in which serines, threonines, or acidic residues were replaced by Ala (S242A, E243A, and T247A). We also tested a naturally occurring SST5 third-loop mutant (R240W) previously detected in our laboratory in one acromegalic patient resistant to SS analogs (23). No β-arrestin translocation (by fluorescence microscopy) or a strongly reduced arrestin association (by cross-linking-coimmunoprecipitation assay) were detectable after SS28 stimulation of S242A, T247A, and R240W, whereas E243A partially retained the ability to associate with arrestin. Accordingly, the E243A mutant showed slightly lower internalization, suggesting that the role of this acidic residue is not crucial for arrestin binding. By contrast, Ser242 mutation to Ala caused a significant decrease in receptor internalization (29.0% with respect to 52.4% of wild type), indicating that the phosphorylation of this residue is essential to activate arrestin phosphate sensor. This hypothesis has been further confirmed by the finding of reduced phosphorylation of the RST, R240W, and S242A mutants by immunoprecipitation experiments using antiphosphoserine antibody and whole-cell phosphorylation assay. Surprisingly, R240W mutant showed a dramatic loss of internalization (14.4%) that was comparable with that observed with the simultaneous mutation of the three residues, Arg, Ser, and Thr, within the third loop (RST). Although mutation of Arg240 to Trp is predicted by NetPhos 2.0 server to abolish Ser242 phosphorylation, to explain the more dramatic reduction of internalization induced by this mutant in comparison with Ser242 mutation, we hypothesize that Arg240 residue, highly conserved in all five SS subtypes, might also be directly involved in receptor binding by the activation sensor of arrestin. The location of the regions that bind arrestin activation sensor has not been fully characterized in any GPCR. However, numerous studies indicate that arrestins can bind in vitro to specific GPCR fragments located within the third intracellular loop, which do not become phosphorylated upon agonist activation (31, 32).
Our data suggest a model for SST5 internalization comparable with that hypothesized for dopamine D1 receptor (Fig. 1) (11). Under basal conditions, the C terminus of D1 is in close association with the third cytoplasmic loop. Receptor activation promotes a sequential phosphorylation of residues, first on the C-terminal tail and then within the third cytoplasmic loop, allowing these two domains to disassociate and arrestin to bind to the activated third loop. Our results indicate that the third intracellular loop of wild-type SST5 relieves an intrinsic inhibitory effect of the C-terminal tail on arrestin binding and receptor internalization. Accordingly, these processes are facilitated by the truncation of the C-terminal tail as indicated by the observation that D328 mutant internalized more efficiently than wild-type SST5. If the third loop is damaged (R240W, S242A, and T247A mutants), the interaction of this region with β-arrestin might be sterically prevented by the C terminus. To test whether the role of the third intracellular loop is to remove the C-tail or to directly bind arrestin, we evaluated the effect of multiple third-loop mutations (RST) within the context of the truncated constructs. In the absence of the C-terminal domain, the mutated receptor showed an increased internalization (41.0%) with respect to full-length RST mutant (15.0%), suggesting that the third cytoplasmic loop of SST5 is essential to remove the C-tail inhibition.
In conclusion, our results indicate SST5 third intracellular loop as a crucial mediator of β-arrestin binding and receptor internalization, whereas they suggest that residues 328–347 within the C terminus may play an inhibitory role in receptor internalization. Whether the first and second intracellular loops are also involved in β-arrestin interaction remains to be investigated.
MATERIALS AND METHODS
Materials
GH3 cells (ATCC CCL-82.1) were purchased from American Type Culture Collection (Manassas, VA). Ham F10 medium, fetal calf serum, l-glutamine, penicillin, streptomycin, T4 DNA ligase, and AccuPrime proofreading polymerase were from Invitrogen (Carlsbad, CA). Restriction enzymes were from New England Biolabs (Beverly, MA). Transfection reagent Jet PEI was from Polyplus Transfection (San Marcos, CA). The plasmid encoding β-arrestin-1-EGFP and pDsRed2-N1 expression vector were kindly provided by Dr. Bodduluri Haribabu (University of Louisville Health Sciences, Louisville, KY). FluoSave mounting medium was from Calbiochem (San Diego, CA). SS28, sucrose, paraformaldehyde, and antiphosphoserine antibody were from Sigma-Aldrich (St. Louis, MO). Protein A/G Plus-Agarose, anti-SST5 (H-54) and antiphosphothreonine antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibiotin, horseradish peroxidase-linked antibody was from Cell Signaling Technology (Danvers, MA). DSP and Sulfo-NHS-SS-Biotin were from Pierce Chemical Co. (Rockford, IL).
Plasmids and Constructs
To generate the wild-type (full-length and truncated) and the mutated forms, human wild-type SST5 cDNA was amplified starting from the previous cassette construct DNA as template (23) and subcloned into AgeI/HindIII multiple cloning site of pDsRed2-N1 expression vector. Point mutations were introduced into SST5 by PCR-based mutagenesis replacing serine 242, glutamic acid 243, and threonine 247 with alanine (Fig. 2). The naturally occurring mutant R240W was amplified starting from the previous cassette construct DNA as previously reported (23). A total third-loop mutant (RST) was created by replacing serine 242 and theonine 247 with alanine using R240W mutant as a template. Progressive deletion mutants of the SST5 C-terminal tail were created by PCR using wild-type or mutated SST5 as templates (Fig. 2). To construct fusions to the N terminus of DsRed2, the target genes were cloned into pDsRed2-N1 in frame with the DsRed2 coding sequence, with no intervening in-frame stop codons. Human β-arrestin-2 cDNA was amplified by RT-PCR using human pituitary mRNA as a template and subcloned into the SacI/SalI multiple cloning site of the expression vector encoding β-arrestin-1-EGFP. Primers were designed to avoid changes in the reading frame, with no intervening in-frame stop codons, to construct fusion to the N terminus of EGFP. The primers used for the construction of different mutant and wild-type receptors and β-arrestin-2 are shown in Table 2. The sequence of all constructs was verified by dideoxynucleotide sequencing.
Table 2.
Oligonucleotide Primers Used in the Creation of Wild-Type SST5, C-Tail Deletion Mutants, Third Loop Mutants, and β-Arrestin-2
| Oligo | Sequence (5′–3′) |
|---|---|
| SST5 forward | TGTATAAGCTTGCCATGGAGCCCCTGTTCC |
| SST5 reverse | GCCTAACCGGTGCCAGCTTGCTGGTCTGCA |
| D347 reverse | CGCATACCGGTGCCGTGGCCTCCTGCTGCT |
| D338 reverse | CCGATACCGGTGCGTCTGGACGCGGCTCCG |
| D328 reverse | GCGATACCGGTGCCTTGGCACCAGAGCCCT |
| S242A junction forward | CTGCGTGCGGCGGCGCGCGGAGCGGAAGGTGAC |
| S242A junction reverse | GTCACCTTCCGCTCCGCGCGCCGCCGCACGCAG |
| E243A junction forward | TGCGGCGGCGCTCGGCGCGGAAGGTGACGCG |
| E243A junction reverse | CGCGTCACCTTCCGCGCCGAGCGCCGCCGCA |
| T247A junction forward | CGGAGCGGAAGGTGGCGCGCATGGTGTTG |
| T247A junction reverse | CAAGACCATGCGCGCCACCTTCCGCTCCG |
| R240W/S242A junction forward | TGCGTGCGGTGGCGCGCGGAGCGGAAGGTG |
| R240W/S242A junction reverse | CACCTTCCGCGCCGCGCGCCACCGCACGCA |
| BARR2 forward | TGTATGAGCTCGCCATGGGGGAGAAACCCG |
| BARR2 reverse | TGTATGTCGACGCCGCAGAGTTGATCATCA |
Cell Culture and Transfection
Rat pituitary GH3 cells were grown in Ham’s F10 medium supplemented with 10% fetal calf serum, 2 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37 C in a humidified atmosphere of 95% air-5% CO2. Chinese hamster ovary K1 cells were cultured in αMEM containing 10% fetal calf serum at 37 C in a humidified atmosphere of 95% air-5% CO2. Transient transfections of β-arrestin and SST5 were performed using JetPEI according to the instruction of the manufacturer. Western blot analysis was performed to control the expression level of SST5-DsRed2 in transiently transfected cells. Immunoblotting was performed using a 1:1000 dilution of the SST5(H-54) antibody and revealed by a chemiluminescent detection system.
To confirm the functional activity of the wild-type SST5-DsRed2 receptor, intracellular cAMP was measured by enzymatic immunoassay (Amersham Pharmacia Biotech, Piscataway, NJ) as previously reported (23).
Fluorescence Microscopy
GH3 cells were transiently cotransfected with β-arrestin-EGFP and SST5-DsRed2. Cells were treated 48 h after transfection with saturating concentration of SS28 (1 μm) for the indicated times, fixed with 4% paraformaldehyde for 20 min at room temperature, and washed several times in PBS. Coverslips were mounted on glass slides with FluoSave mounting medium for fluorescence microscopy examination. All images were collected using Leica DM IRE 2 (Leica Corp., Deerfield, IL) and processed with Image ProPlus 6.0 software.
Quantitation of Internalization
SST5 internalization was evaluated either by fluorescence microscopy and biochemical analysis. Images of GH3 cells transiently cotransfected with β-arrestin and SST5 were processed using Image ProPlus 6.0 software. The amount of internalized receptor after agonist stimulation was analyzed in single cells. The fluorescence density mean (F) in two separate regions corresponding to the plasma membrane and to the entire intracellular area were densitometrically determined. Mean membrane to intracellular fluorescence ratio (fR) was then calculated according to the following equation:
fR = [F(membrane) − F(background)]/[F(citosol) − F(background)]
For each group, at least 30 cells from three independent transfections were analyzed by three independent investigators, and the mean value was used for the graphs.
Biochemical analysis of receptor endocytosis was performed using the cleavable biotin method (28). Transiently transfected GH3 cells were washed three times with ice-cold PBS, and the cell surface was biotinylated with 500 μg/ml cleavable sulfo-NHS-S-S-biotin for 30 min at 4 C. Unreacted biotin was quenched and removed by three washes with cold Tris-buffered saline-10 mm glycine. Biotinylated cells were incubated in prewarmed medium with or without SS28 1 μm at 37 C for 30 min, and then cells were chilled on ice to stop endocytosis. To release the biotin label from proteins at the cell surface, cells were washed twice with cold glutathione strip buffer (50 mm glutathione, 75 mm NaCl, 75 mm NaOH, 10% fetal bovine serum in H2O) at 4 C for 20 min. Excess glutathione was then quenched for 30 min at 4 C in iodoacetamide buffer (50 mm iodoacetamide; 1% BSA; in PBS, pH 7.4). Cells were lysed with 0.5 ml lysis buffer in the presence of protease inhibitors.
For immunoprecipitation, 60 μg of total cellular protein was incubated with 2 μg of SST5(H-54) antibody for 1 h at 4 C. The resuspended volume of protein A/G Plus-Agarose (20 μl) was then added, and tubes were incubated for 90 min at 4 C on a rotating device. The pellet was washed with PBS with 0.05% BSA and PBS, and resuspended in 30 μl of PBS with 0.1% SDS for elution. Eluted proteins were resolved by SDS-PAGE under reducing or nonreducing conditions and transferred to a nitrocellulose filter. To detect biotinylated proteins, 1:500 dilution of antibiotin, horseradish peroxidase-linked antibody was used. The presence of equal amounts of receptor in the immunoprecipitates was confirmed by stripping and reprobing with anti-SST5 antibody (1:2000) and antirabbit secondary antibody covalently coupled to horseradish peroxidase (1:2000). The resulting bands were evaluated with the image analysis program NIH ImageJ. Experiments were repeated at least five times.
Quantitation of SST5-Arrestin Interaction
The immunoprecipitation method was modified to include a cross-linking step as recently described (33, 34) for coimmunoprecipitation of SST5 and β-arrestin. GH3 cells transiently cotransfected with SST5 and β-arrestin were washed three times with 0.15 m NaCl, 20 mm HEPES, pH 7.4, and incubated with or without 1 μm SS28 at 37 C for the indicated times. At the end of this incubation, 2 mm solution of DSP was added, and the cells were incubated 30 min at room temperature, washed with PBS containing 50 mm Tris-HCl (pH 7.4), and lysed in 0.5 ml lysis buffer. Total cellular protein (100 μg) was incubated with 2 μg of SST5(H-54) antibody, and immunoprecipitation of SST5 was performed as described above. The cross-linked complexes were dissociated by incubation of the immunoprecipitates with SDS sample buffer containing reducing agents and analyzed by Western blotting. β-Arrestin was visualized with anti-GFP (green fluorescent protein) antibody (1:1000) and antimouse secondary antibody covalently coupled to horseradish peroxidase. The presence of equal amounts of receptor in the immunoprecipitates was confirmed by stripping and reprobing with anti-SST5 antibody (1:2000). The amount of arrestin expressed was determined using 30 μg of the whole-cell lysate containing equivalent amounts of protein.
Analysis of Protein Phosphorylation
The phosphorylation sites in wild-type and mutant SST5 were predicted using NetPhos 2.0 Server (http://www.cbs.dtu.dk/services/NetPhos/). The NetPhos 2.0 server produces neural network predictions for serine, threonine, and tyrosine phosphorylation sites in eukaryotic proteins (24).
For the phosphorylation assay, GH3 cells were transiently transfected with wild-type or mutated SST5 for 48 h, incubated with 1 μm SS28 for the indicated times, and lysed with 0.5 ml lysis buffer in the presence of protease inhibitors. SST5 was immunoprecipitated with 2 μg of SST5 antibody as described above.
Western blot analysis was performed using antiphosphoserine (1:800) and antiphospho-threonine antibody (1:400), and loading of equal amounts of receptor proteins in each lane was confirmed by reprobing with anti-SST5 antibody (1:2000).
To further confirm these results, we performed a whole-cell phosphorylation assay (27). GH3 cells were transiently transfected with the appropriate plasmids for 48 h, washed with serum- and phosphate-free medium, and then labeled with 200 μCi/ml carrier-free [32P]orthophosphate for 60 min at 37 C. Labeled cells were incubated with 1 μm SS28 for 5 min, washed with ice-cold PBS, and lysed with radioimmune precipitation buffer (50 mm Tris-HCl, pH 7.4; 150 mm NaCl; 5 mm EDTA; 10 mm NaF; 10 mm disodium pyrophosphate; 1% Nonidet P40; 0.5% sodium deoxycholate; 0.1% SDS; 0.2 mm phenylmethylsulfonylfluoride; 10 μg/ml leupeptin; 1 μg/ml pepstatin A; 1 μg/ml aprotinin; and 10 μg/ml bacitracin). SST5 was immunoprecipitated with 2 μg of SST5 antibody as described above. The samples were subjected to SDS-PAGE followed by autoradiography. Loading of equal amounts of receptor proteins in each lane was confirmed by Western blot analysis.
Statistical Analysis
The results are expressed as the mean ± sd. A paired two-tailed Student’s t test was used to detect the significance between two series of data. P < 0.05 was accepted as statistically significant.
Footnotes
This work was partially supported by Associazione Italiana per la Ricerca sul Cancro (Milan), PRIN 2006060982_002 (to A.S.), and Ricerca Corrente Funds of Fondazione Ospedale Maggiore Policlinico Mangiagalli e Regina Elena Instituto di Ricovero e Cura a Carattere Scientifico (Milan).
Author Disclosure: The authors have nothing to disclose.
First Published Online December 20, 2007
E.P. and G.M. contributed equally to this work and should both be considered first authors.
Abbreviations: DSP, Dithiobis (succinimidylpropionate); EGFP, enhanced GFP; GFP, green fluorescent protein; GPCR, G protein-coupled receptor; RST, R240, S242, and T247 mutant; SDS, sodium dodecyl sulfate; SS, somatostatin.
References
- 1.Patel YC 1999. Somatostatin and its receptors family. Front Neuroendocrinol 20:157–198 [DOI] [PubMed] [Google Scholar]
- 2.Moller LN, Stidsen CE, Hartmann B, Holst JJ 2003. Somatostatin receptors. Biochim Biophys Acta 1616:1–84 [DOI] [PubMed] [Google Scholar]
- 3.Reisine T, Bell G 1995. Molecular biology of somatostatin receptor. Endocr Rev 16:427–442 [DOI] [PubMed] [Google Scholar]
- 4.Weckbecker G, Liu R, Tolcsvai L, Bruns C 1992. Antiproliferative effects of the somatostatin analogue octreotide (SMS 201–995) on ZR-75–1 human breast cancer cells in vivo and in vitro. Cancer Res 52:4973–4978 [PubMed] [Google Scholar]
- 5.Douziech N, Calvo E, Coulombe Z, Muradia G, Bastien J, Aubin RA, Lajas A, Morrisset J 1999. Inhibitory and stimulatory effects of somatostatin on two human pancreatic cancer cell lines: a primary role for tyrosine phosphatase SHP-1. Endocrinology 140:765–777 [DOI] [PubMed] [Google Scholar]
- 6.Farooqi S, Bevan JS, Sheppard MC, Wass JAH 1999. The therapeutic value of somatostatin and its analogues. Pituitary 2:79–88 [DOI] [PubMed] [Google Scholar]
- 7.Ferguson SS 2001. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signalling. Pharmacol Rev 53:1–24 [PubMed] [Google Scholar]
- 8.Pierce KL, Lefkowitz RJ 2001. Classical and new roles of β-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci 2:727–733 [DOI] [PubMed] [Google Scholar]
- 9.Kohout TA, Lefkowitz RJ 2003. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol 63:9–18 [DOI] [PubMed] [Google Scholar]
- 10.Luttrell LM, Lefkowitz RJ 2002. The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci 115:455–465 [DOI] [PubMed] [Google Scholar]
- 11.Kim OJ, Gardner BR, Williams DB, Marinec PS, Cabrera DM, Peters JD 2004. The role of phosphorylation in D1 dopamine receptor desensitization: evidence for a novel mechanism of arrestin association. J Biol Chem 279:7999–8010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhaskaran RS, Min L, Krishnamurthy H, Ascoli M 2003. Studies with chimeras of the gonadotropin receptors reveal the importance of third intracellular loop threonines on the formation of the receptor/nonvisual arrestin complex. Biochemistry 42:13950–13959 [DOI] [PubMed] [Google Scholar]
- 13.Celver J, Xu M, Jin W, Lowe J, Chavkin C 2004. Distinct domains of the μ-opioid receptor control uncoupling and internalization. Mol Pharmacol 65:528–537 [DOI] [PubMed] [Google Scholar]
- 14.Kishi H, Krishnamurthy H, Galet C, Bhaskaran RS, Ascoli M 2002. Identification of a short linear sequence in the C-terminal tail of the rat follitropin receptor that modulates arrestin-3 binding in a phosphorylation-independent fashion. J Biol Chem 277:21939–21946 [DOI] [PubMed] [Google Scholar]
- 15.Hukovic N, Panetta R, Kumar U, Patel YC 1996. Agonist-dependent regulation of cloned human somatostatin receptor types 1–5 (hSSTR1–5): subtype selective internalization or upregulation. Endocrinology 137:4046–4049 [DOI] [PubMed] [Google Scholar]
- 16.Hukovic N, Panetta R, Kumar U, Rocheville M, Patel YC 1998. The cytoplasmic tail of the human somatostatin receptor type 5 is crucial for interaction with adenylyl cyclase and in mediating desensitization and internalization. J Biol Chem 273:21416–21422 [DOI] [PubMed] [Google Scholar]
- 17.Hukovic N, Rocheville M, Kumar U, Sasi R, Khare S, Patel YC 1999. Agonist-dependent up-regulation of human somatostatin receptor type 1 requires molecular signals in the cytoplasmic C-tail. J Biol Chem 274:24550–24558 [DOI] [PubMed] [Google Scholar]
- 18.Roosterman D, Roth A, Kreienkamp HJ, Richter D, Meyerhof W 1997. Distinct agonist-mediated endocytosis of cloned rat somatostatin receptor subtypes expressed in insulinoma cells. J Neuroendocrinol 9:741–751 [DOI] [PubMed] [Google Scholar]
- 19.Roth A, Kreienkamp HJ, Nehring RB, Roosterman D, Meyerhof W, Richter D 1997. Endocytosis of the rat somatostatin receptors: subtype discrimination, ligand specificity, and delineation of carboxy-terminal positive and negative sequence motifs. DNA Cell Biol 16:111–119 [DOI] [PubMed] [Google Scholar]
- 20.Stroh T, Jackson AC, Sarret P, Dal Farra C, Vincent JP, Kreienkamp HJ, Mazella J, Beaudet A 2000. Intracellular dynamics of SST5 receptors in transfected COS-7 cells: maintenance of cell surface receptors during ligand-induced endocytosis. Endocrinology 141:354–365 [DOI] [PubMed] [Google Scholar]
- 21.Cescato R, Schulz S, Waser B, Eltschinger V, Rivier JE, Wester H-J, Culler M, Ginj M, Liu Q, Schonbrunn A, Reubi JC 2006. Internalization of sst2, sst3, and sst5 receptors: effects of somatostatin agonist and antagonist. J Nucl Med 47:502–511 [PubMed] [Google Scholar]
- 22.Tulipano G, Stumm R, Pfeiffer M, Kreienkamp H-J, Hollt V, Schulz S 2004. Differential β-arrestin trafficking and endosomal sorting of somatostatin receptor subtypes. J Biol Chem 279:21374–21382 [DOI] [PubMed] [Google Scholar]
- 23.Ballarè E, Persani L, Lania AG, Filopanti M, Giammona E, Corbetta S, Mantovani S, Arosio M, Beck-Peccoz P, Faglia G, Spada A 2001. Mutation of somatostatin receptor type 5 in an acromegalic patient resistant to somatostatin analog treatment. J Clin Endocrinol Metab 86:3809–3814 [DOI] [PubMed] [Google Scholar]
- 24.Blom N, Gammeltoft S, Brunak S 1999. Sequence- and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294:1351–1362 [DOI] [PubMed] [Google Scholar]
- 25.Ferguson SS, Zhang J, Barak LS, Caron MG 1998. Molecular mechanisms of G-protein coupled receptors desensitization and resensitization. Life Sci 62:1561–1565 [DOI] [PubMed] [Google Scholar]
- 26.Le Roy C, Wrana JL 2005. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol 6:112–126 [DOI] [PubMed] [Google Scholar]
- 27.Galliera E, Jala VR, Trent JO, Bonecchi R, Signorelli P, Lefkowitz RJ, Mantovani A, Locati M, Haribabu B 2004. β-Arrestin-dependent constitutive internalization of the human chemokine decoy receptor D6. J Biol Chem 279:25590–25597 [DOI] [PubMed] [Google Scholar]
- 28.Cao TT, Mays RW, von Zastrow M 1998. Regulated endocytosis of G-protein-coupled receptors by a biochemically and functionally distinct subpopulation of clathrin-coated pits. J Biol Chem 38:24592–24602 [DOI] [PubMed] [Google Scholar]
- 29.Gurevich VV, Gurevich EV 2006. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol Ther 110:465–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whistler JL, Tsao P, Von Zastrow M 2001. A phosphorylation-regulated brake mechanism controls the initial endocytosis of opioid receptors but is not required for post-endocytic sorting to lysosomes. J Biol Chem 276:34331–34338 [DOI] [PubMed] [Google Scholar]
- 31.Cheng Z-J, Zhao J, Sun Y, Hu W, Wu Y-L, Cen B, Wu GX, Pei G 2000. β-arrestin differentially regulates the chemokine receptor CXCR4-mediated signaling and receptor internalization, and this implicates multiple interaction sites between β-arrestin and CXCR4. J Biol Chem 275:2479–2485 [DOI] [PubMed] [Google Scholar]
- 32.Cen B, Xiong Y, Ma L, Pei G 2001. Direct and differential interaction of β-arrestins with the intracellular domains of different opioid receptors. Mol Pharmacol 59:758–764 [DOI] [PubMed] [Google Scholar]
- 33.Min L, Galet C, Ascoli M 2002. The association of arrestin-3 with the human lutropin/choriogonadotropin receptor depends mostly on receptor activation rather than on receptor phosphorylation. J Biol Chem 277:702–710 [DOI] [PubMed] [Google Scholar]
- 34.Qian H, Pipolo L, Thomas WG 2001. Association of β-arrestin 1 with the type 1A angiotensin II receptor involves phosphorylation of the receptor carboxyl terminus and correlates with receptor internalization. Mol Endocrinol 15:1706–1719 [DOI] [PubMed] [Google Scholar]