

Abstract
An increased level of glucocorticoid may be related to the pathophysiology of depressive disorder. The involvement of brain-derived neurotrophic factor (BDNF) in the antidepressive effect has also been suggested; however, the possible influence of glucocorticoid on the action of BDNF in the developing central nervous system has not been elucidated. In this study, we investigated the effect of glucocorticoid (dexamethasone, DEX) on synaptic maturation and function enhanced by BDNF in early developing hippocampal neurons. In the immature stage, BDNF increased the outgrowth of dendrites and the expression of synaptic proteins including glutamate receptors and presynaptic proteins. Pretreatment with DEX significantly inhibited the BDNF-dependent up-regulation of both dendritic outgrowth and synaptic proteins. In the more mature stage, the BDNF-reinforced postsynaptic Ca2+ influx was decreased by DEX. BDNF-enhanced presynaptic glutamate release was also suppressed. RU486, a glucocorticoid receptor antagonist, canceled the DEX-dependent blocking effect on the action of BDNF. After down-regulation of glucocorticoid receptor by small interfering RNA application, no inhibitory effect of DEX on the BDNF-increased synaptic proteins was observed. Interestingly, the BDNF-activated MAPK/ERK pathway, which is an essential intracellular signaling pathway for the BDNF-increased synaptic proteins, was reduced by DEX. These results suggest that BDNF-mediated synaptic maturation is disturbed after neurons are exposed to high-level glucocorticoid in their development stage.
GLUCOCORTICOID IS A STRESS hormone that is regulated by the hypothalamic-pituitary-adrenal axis including the hippocampus (1). This hormone is needed to cope with stressful life events and regulates the expression of many target genes at the cellular level (2). However, failure in the control of glucocorticoid homeostasis is thought to be closely related to the symptoms of depressive disorder (3). We previously reported that acutely elevated glucocorticoid levels did not return to the basal level in depressive patients (4). Interestingly, increased glucocorticoid is reported to influence learning and memory in humans (5). Depressive patients’ deficits in cognition, learning, and memory have also been reviewed (6, 7). These studies suggest that sustained elevation of glucocorticoid is involved in the pathophysiology of depression.
Many studies using in vivo and in vitro systems in animals have reported that excessive glucocorticoid causes neuronal damage. Cognitive or learning deficits in glucocorticoid-administered animals occur (8, 9). As expected, chronic stress and administration of glucocorticoid result in dendritic atrophy of hippocampal neurons (10, 11, 12, 13). In medial prefrontal cortex, the dendrite atrophy induced by daily 10-min restraint stress for 1 wk was reported (14). In addition, infants that received maternal separation stress during postnatal d 2–14 (P2–P14) exhibited decreased mossy fiber in the hippocampus (15). On the other hand, an acute function of the glucocorticoid receptor (GR) is also shown, which appeared within a few minutes. GR prolongs an acute N-methyl-d-aspartate receptor (NMDA receptor)-mediated increase in intracellular Ca2+ in hippocampal neurons (16). GR function is involved in a potentiation of the response to NMDA in dopamine-sensitive neurons in the ventral tegmental area (17). Taking these findings together, clarifying the action of glucocorticoid/GR signaling is important for understanding the symptoms of depressive disorder.
Recent reports suggest that the action of growth factors including neurotrophin is involved in depressive disorder. In particular, the decrease in brain-derived neurotrophic factor (BDNF) may be related to the pathophysiology of mental disorders. For example, BDNF expression is low in the brains of suicide victims with depressive disorder (18). A reduction in the concentration of serum BDNF was observed in depressive patients (19). In animal studies, the down-regulation of BDNF in the hippocampus of glucocorticoid-administered or stressed rats was confirmed (20, 21). Thus, down-regulation of BDNF expression may result in depressive disorder. As further evidence, the mechanism by which antidepressants exert the antidepressive effect also supports the importance of BDNF. The increase of BDNF level is thought to be a mechanism by which the antidepressive effect is exerted. Chronic treatment with antidepressants up-regulates the mRNA of BDNF in rats (22). We recently reported that antidepressants potentiated the BDNF function; that is, antidepressants reinforce BDNF-stimulated glutamate release via enhancing phospholipase Cγ/Ca2+ (PLCγ) intracellular signaling (23), suggesting that the reinforcement of BDNF-stimulated intracellular signaling is also involved in the antidepressive effect.
BDNF is a neurotrophic factor that promotes neuronal differentiation, survival, and plasticity in the peripheral nervous system and central nervous system (CNS) (24). In CNS, BDNF elicits long-term potentiation, which is related to synaptic plasticity (25). We previously reported that BDNF enhanced depolarization-induced release of glutamate (26) and potentiated the spontaneous Ca2+ oscillations in cultured cortical neurons (27), suggesting that BDNF strengthens the activity of neural networks. In developing CNS neurons, BDNF promotes neurite outgrowth (28) and increases the synaptic proteins including synapsin I, synaptophysin, and synaptotagmin (26, 29). Therefore, BDNF plays an important role in establishing synaptic connections and neuronal maturation.
Glucocorticoid is also reported to influence maturation of the CNS in animal experiments (30, 31, 32, 33); however, the cellular mechanism underlying glucocorticoid action in neuronal development is largely unknown. Therefore, we focused on the cross-talk between BDNF and the glucocorticoid action in vitro system. There are two receptors for glucocorticoid: GR and mineralocorticoid receptor (MR) (34). GR is suggested to be related to the stress response because it has a lower affinity for glucocorticoid than MR. In the present study, we used dexamethasone (DEX), which is a selective agonist for GR (35), as an excessive glucocorticoid exposure model. We found that BDNF-increased neurite outgrowth and synaptic proteins in developing hippocampal neurons were suppressed by pretreatment with DEX. The down-regulation of BDNF-increased pre- and postsynaptic function was still observed in the more mature stage. Interestingly, DEX inhibited the BDNF-activated MAPK signaling, a required signaling for the BDNF-increased synaptic proteins and functions. Our present findings demonstrate that BDNF-reinforced synaptic maturation is disturbed in neurons by exposure to high-level glucocorticoid in their development stage.
RESULTS
BDNF-Enhanced Outgrowth of Neurites Was Suppressed by DEX Pretreatment
To investigate the influence of glucocorticoid (DEX) on the maturation of synaptic connections and functions, in which BDNF exerts important effects, we first focused on the change of structure of young hippocampal neurons. In our cultures, the glutamic acid decarboxylase (GAD)-positive γ-aminobutyric acid (GABA)-ergic neurons were about 10% (Fig. 1A, yellow in b); thus, the other microtubule-associated protein 2 (MAP2)-positive cells are expected to be glutamatergic neurons (Fig. 1A, a and b). As illustrated in Fig. 1B, DEX (final 10 μm) was applied at 1 d in vitro (DIV1) before the addition of BDNF (100 ng/ml, at DIV2). Forty-eight hours (at DIV4) after BDNF addition, the neurite outgrowth was determined. As shown in Fig. 1C, BDNF tends to enhance neurite outgrowth of GABAergic (MAP2/GAD-double-positive cells) neurons (Fig. 1C, a–c and d–f), and pretreatment with DEX slightly inhibited the effect of BDNF (Fig. 1C, d–f and g–i), although significance was not observed. Sole DEX treatment had no effects (Fig. 1C, a–c and j–l). The thickness of neurites seemed to be increased by BDNF; however, the number of primary neurites per cell for each experimental condition revealed no significant change (Fig. 1C, m). Next, the effect of DEX on glutamatergic neurons was examined. BDNF increased the number of neurites of glutamatergic (MAP2/glutamate-double-positive) neurons (Fig. 1D, a–c and d–f). Interestingly, DEX pretreatment blocked the BDNF-enhanced outgrowth (Fig. 1D, d–f and g–i). Sole DEX treatment exerted no influence (Fig. 1D, a–c and j–l). Quantitative data show the inhibitory effect of DEX on the BDNF-increased glutamatergic neurites (Fig. 1D, m). The number of neurites of the MAP2-positive but GAD-negative neurons, most of which were considered to be glutamatergic neurons, was also examined [none, 4.08 ± 1.44 (n = 76 cells); BDNF, 7.46 ± 2.84 (n = 82); BDNF plus DEX, 4.64 ± 2.20 (n = 105); DEX, 3.54 ± 1.38 (n = 97)] and confirmed the inhibitory effect of DEX. We confirmed that treatment with DEX and/or BDNF did not affect cell survival at DIV4 (supplemental Fig. 1, published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). These results suggest that DEX suppresses the BDNF-enhanced neurite outgrowth in early developing hippocampal neurons.
Fig. 1.
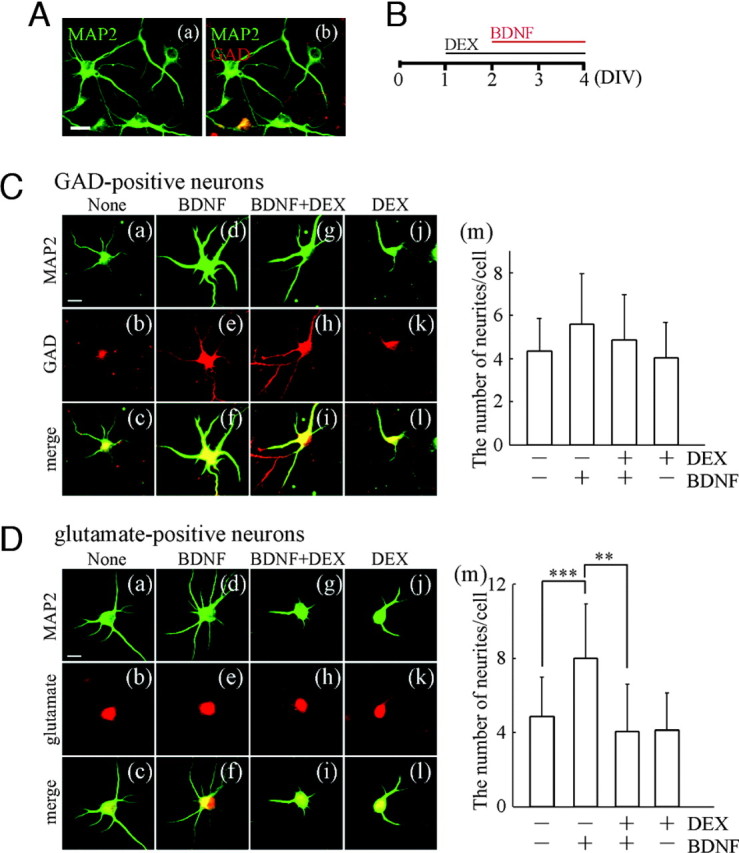
BDNF-Enhanced Outgrowth of Neurites Was Suppressed in the Presence of DEX
A, Cultured hippocampal neurons at DIV4 were immunostained with anti-MAP2 (green, neuronal dendritic marker) and anti-GAD (red, GABAergic neuronal marker) antibodies. a, MAP2 image; b, MAP2 and GAD merged image. Approximately 10% of MAP2-positive cells were GAD-positive GABAergic neurons in our cultures. Bar, 20 μm. B, Experimental schedule of BDNF and DEX application for immunostaining. DEX was applied at DIV1, and then BDNF was also applied at DIV2 in the presence of DEX. Forty-eight hours after BDNF addition, the cultured cells were fixed for immunostaining. C, Influence of DEX treatment on neurite outgrowth of GABAergic neurons. a, d, g, and j, The neurites were revealed by immunostaining with anti-MAP2 antibody; b, e, h, and k, GABAergic neurons were determined by staining with anti-GAD antibody; c, f, i, and l, merged images; d–f, BDNF has a slight tendency to increase the number of neurites compared with control (a–c) (none), although significance was not observed; g–i, DEX pretreatment before BDNF application; j–l, sole DEX treatment. Bar, 20 μm. m, The number of neurites per cell was counted. Data represent mean ± sd (none, n = 60; BDNF, n = 58; BDNF plus DEX, n = 73; DEX, n = 48). The n indicates the number of cells. D, Effect of DEX on neurite outgrowth of glutamatergic neurons. MAP2-positive (a, d, g, and j) and glutamate-positive (b, e, h, and k) glutamatergic neurons are shown. c, f, i, and l, Merged images; d–f, BDNF increased the number of neurites compared with control (a–c); g–i, DEX significantly suppressed the BDNF effect; j–l, sole DEX had no influence compared with control. Bar, 20 μm. m, Quantification indicates that the number of glutamatergic neurites was increased by BDNF, and the increase was suppressed by DEX. Data represent mean ± sd (none, n = 200; BDNF, n = 200; BDNF plus DEX, n = 200; DEX, n = 200). The n indicates the number of cells. Reproducibility was confirmed by three independent cultures. Statistical significance was determined by ANOVA. ***, P < 0.001; **, P < 0.01.
BDNF Did Not Increase Expression of Synaptic Proteins in the Presence of DEX
The change of neurite outgrowth might be associated with synaptic connection. Thus, expressions of pre- and postsynaptic proteins were examined. As expected, the application of BDNF dramatically increased various synaptic proteins including NMDA receptor 2A (NR2A), NR2B, glutamate receptor 1 (GluR1), synapsin I, and synaptosome-associated protein 25 (SNAP25). DEX pretreatment (final 0.1–100 μm) inhibited the effect of BDNF in a dose-dependent manner (Fig. 2A). Quantification after Western blotting was performed (Fig. 2B). The expression of postsynaptic proteins (glutamate receptor subunits), such as NR2A (Fig. 2B, a), NR2B (Fig. 2B, b), and GluR1 (Fig. 2B, c), were up-regulated by BDNF. Presynaptic proteins, synapsin I (Fig. 2B, d), SNAP25 (Fig. 2B, e), were also increased by BDNF. The level of class III β-tubulin (TUJ1, a neuronal marker) is shown as a control (Fig. 2B, f). The increases of these synaptic proteins caused by BDNF were inhibited by DEX in a dose-dependent manner (Fig. 2B). As depicted in supplemental Fig. 2A (published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org), DEX application alone seemed to have a tendency to reduce the level of synaptic proteins compared with control; however, quantification performed with the result of DEX (10 μm) showed no significance (supplemental Fig. 2B). To quantify Western blotting, normalization using response to exogenous BDNF application was performed because control protein levels are unstable due to their low expression in immature neurons and because endogenous expression of BDNF remains unaltered in response to 72 h exposure of DEX (supplemental Fig. 2C).
Fig. 2.

BDNF-Increased Expression of Synaptic Proteins Was Inhibited by Pretreatment with DEX
A, Dose-dependent effect of DEX on the BDNF-increased synaptic proteins. BDNF dramatically increased pre- and postsynaptic proteins at DIV4 (48 h after BDNF application, see legend to Fig. 1B). BDNF-increased synaptic proteins were inhibited by DEX in a dose-dependent manner. B, Quantitative analysis revealed that BDNF increased NR2A (a), NR2B (b), GluR1 (c), synapsin I (d), and SNAP25 (e). DEX pretreatment inhibited the BDNF effect. f, Expression of TUJ1 (class III β-tubulin), a neuronal marker, was not changed. Data represent mean ± sd (n = 6, from six independent cultures). Data were normalized to a level in BDNF stimulation without DEX pretreatment. Statistical significance was confirmed by Kruskal-Wallis test and Mann-Whitney U test. *, P < 0.05 (control vs. BDNF); #, P < 0.05 (BDNF vs. BDNF plus DEX).
DEX Decreased the BDNF-Enhanced Postsynaptic Function in More Mature Neurons
The inhibitory effect of DEX on the actions of BDNF in developing hippocampal neurons may result in a decrease in synaptic function of a more mature stage. Thus, to examine postsynaptic function, we performed Ca2+ imaging analysis at DIV7 (5 d after BDNF application). Fluo-3-filled cell images and time-course analysis of the BDNF-potentiated Ca2+ increase triggered by glutamate (final 1 μm) are shown (Fig. 3, A and B). Glutamate is expected to evoke the Ca2+ influx through ionotropic glutamate receptors. As shown, an increase in intracellular Ca2+ concentration stimulated by glutamate was potentiated by BDNF. However, DEX pretreatment (1 μm) reduced the BDNF-potentiated effect (Fig. 3, A and B). Quantification of the level of increased Ca2+ was conducted (Fig. 3C). Stimulation by high KCl (final 50 mm) was also tested. The BDNF-dependent enhancement in the Ca2+ increase stimulated by high KCl was blocked by DEX (Fig. 3D). Sole DEX treatment has an inhibitory effect compared with control, implying the involvement of influence of DEX on voltage-sensitive ion channels (Ca2+, K+ channels, etc.) in case of KCl stimulation. These results suggest that the inhibitory effect of DEX on BDNF-increased postsynaptic proteins in developing neurons results in a decrease in postsynaptic function of a more mature stage.
Fig. 3.
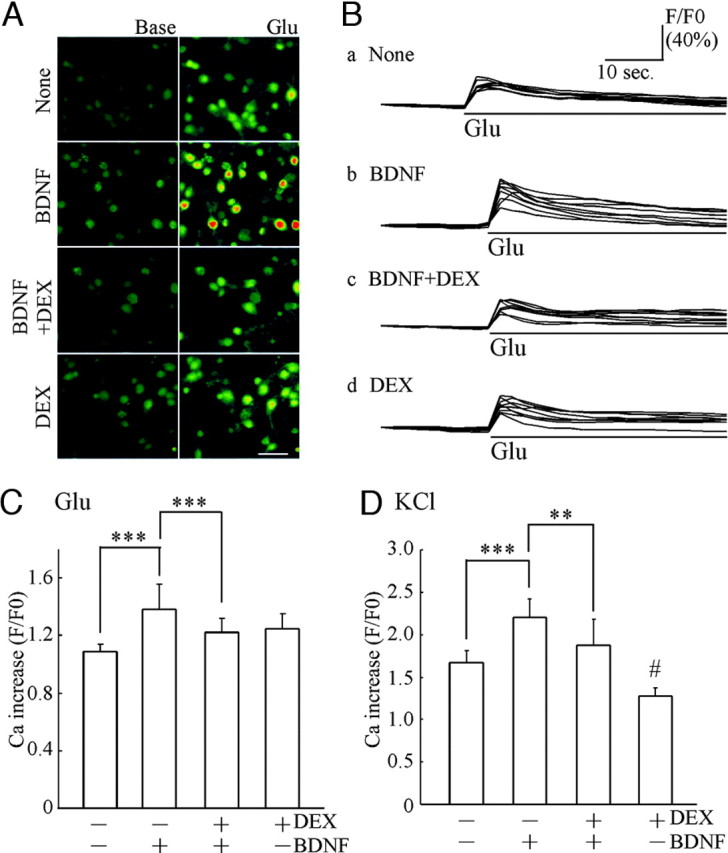
DEX Decreased the BDNF-Potentiated Ca2+ Increase Triggered by Glutamate or High KCl Stimulation
A, Intracellular Ca2+ mobilization was monitored after Fluo-3 loading. Left, Basal intensity of fluorescence in Fluo-3-filled cells for each experimental condition; right, images after glutamate (final 1 μm) stimulation. These images were obtained 4 sec before (Base) and after glutamate stimulation (Glu), respectively. Bar, 50 μm. B, Time-course analysis of intracellular Ca2+ before and after glutamate addition. Each trace indicates the representative intensity of fluorescence from nine neurons in untreated cultures (none) (a) and cultures pretreated with BDNF (b), BDNF plus DEX (c), and DEX (d). Bars indicate exposure time to glutamate. Data are shown as a ratio (F/F0; intensity after stimulation/ basal intensity before stimulation). C, Quantitative analysis of Ca2+ increase induced by glutamate (1 μm). Enhancement of intracellular Ca2+ increase was observed in BDNF-treated cultures. Pretreatment of DEX (1 μm) inhibited the BDNF-potentiated Ca2+ increase. Data represent mean ± sd. Fluorescent intensity was measured from cells in a sister culture (n is the number of cells selected randomly): none, n = 33; BDNF, n = 46; BDNF plus DEX, n = 48; DEX, n = 49. ***, P < 0.001 (Kruskal-Wallis test and Mann-Whitney U test). The Ca2+ imaging analysis was performed at DIV7 (5 d after BDNF application). D, KCl-induced Ca2+ increase was also potentiated by BDNF and inhibited by DEX. High potassium (high KCl solution, final 50 mm) was applied to induce cell depolarization. Data represent mean ± sd. None, n = 9; BDNF, n = 13; BDNF plus DEX, n = 8; DEX, n = 11. ***, P < 0.001; **, P < 0.01; #, P < 0.05 (none vs. DEX), Kruskal-Wallis test and Mann-Whitney U test.
DEX Decreased the BDNF-Enhanced Presynaptic Function in More Mature Neurons
We then examined the presynaptic function in mature neurons after BDNF treatment with or without DEX pretreatment during the development stage. Because changes in the expression of presynaptic proteins occurred (see Fig. 2), first, the number of presynaptic sites was estimated. The presynaptic site of neurons at DIV16 was determined after long maintenance. DEX was applied at DIV1 before the addition of BDNF (DIV2). Two weeks (DIV16) after BDNF addition, the neurons were fixed for immunostaining. Representative images of immunostaining with anti-synapsin I antibody indicated that BDNF increased the number of synapsin I-positive presynaptic sites (Fig. 4A, a–d; b and d are magnified images). DEX inhibited the BDNF effect (Fig. 4A, c–e, f; d and f are magnified). A significant reduction was observed after sole DEX treatment compared with control (Fig. 4A, a, b, and g, h; b and h are magnified). Quantification indicated that the BDNF-dependent increase of presynaptic sites was completely abolished by DEX exposure (Fig. 4A, i). Next, to clarify the function of individual presynaptic sites, exocytosis imaging analysis with FM1-43 dye was carried out. The intensity of FM1-43 fluorescence in presynaptic terminals was reduced after stimulation with high KCl (Fig. 4B, a and b). Although BDNF enhanced the elimination of the FM-43 fluorescence (Fig. 4B, c), DEX had little effect on the BDNF-enhanced elimination, implying that DEX did not affect the function of individual presynaptic sites. Thus, we measured the amount of released glutamate in hippocampal cultures after depolarization because the decrease in the number of synaptic sites might result in reduction of the total amount of released neurotransmitter. The basal levels of glutamate (before KCl stimulation) are shown as white bars in Fig. 4C. KCl stimulation rapidly induced glutamate release, and BDNF potentiated the depolarization-evoked release (Fig. 4C, black bars). Markedly, DEX inhibited the BDNF-potentiated glutamate release in a dose-dependent manner. These results suggest that the inhibition of BDNF-potentiated presynaptic glutamate release in DEX-pretreated cultures reflects a decrease in the number of presynaptic sites.
Fig. 4.

BDNF Failed to Increase the Number of Presynaptic Sites and the Release of Glutamate after DEX Pretreatment
A, The number of presynaptic sites was quantified after immunostaining with anti-synapsin I antibody. Top, Representative images from untreated cultures (none) (a) and cultures pretreated with BDNF (c), BDNF plus DEX (e), and DEX (g). Bar, 20 μm. Bottom, High-magnification images of dendrites from untreated cells (none) (b) and cells pretreated with BDNF (d), BDNF plus DEX (f), and DEX (h). Bar, 10 μm. i, Quantification of the number of synapsin I-positive presynaptic sites per dendritic shaft (50 μm). Data represent mean ± sd. None, n = 39; BDNF, n = 40; BDNF plus DEX, n = 46; DEX, n = 42. The n is the number of dendritic shafts selected randomly from cultured neurons for each experimental condition. BDNF increased the synapsin I-positive presynaptic number, and DEX treatment canceled the BDNF effect. Neurons at DIV16 were fixed after long maintenance in the presence or absence of BDNF or DEX, respectively. ***, P < 0.001, ANOVA. B, Effect of DEX on BDNF-potentiated presynaptic exocytotic activity. FM-43 images of before (a) and after (b) KCl (50 mm) stimulation. FM-dye signal revealed the presynaptic sites (arrowheads). The exocytotic activity in presynaptic sites was determined by elimination of the fluorescence. c, Quantification of reduction of FM-dye fluorescence. BDNF enhanced the elimination of FM-dye fluorescence. DEX had no influence. Data represent mean ± sd. The n (= 30) is monitored fluorescence dots in neurons for each experimental condition. The presynaptic sites of DIV7 neurons were monitored. Bar, 5 μm. ***, P < 0.001, Kruskal-Wallis test and Mann-Whitney U test. C, Amount of glutamate released from hippocampal cultures. White bars indicate the basal release of glutamate without stimulation. Black bars show the glutamate release after KCl stimulation. The reduction caused by DEX (0.1–10 μm) in the BDNF-potentiated glutamate release was observed. The glutamate release in DIV7 cultures (5 d after BDNF application) was measured and indicated as relative release compared with basal release in control (without DEX and BDNF). Data represent mean ± sd (n = 4). The n indicates the number of wells for each experimental condition on a plate. *, P < 0.05 (control vs. BDNF); #, P < 0.05 (BDNF vs. BDNF plus DEX), Kruskal-Wallis test and Mann-Whitney U test.
DEX Showed No Inhibitory Effect in the Presence of GR Antagonist RU486
To investigate the mechanisms underlying the DEX-decreased action of BDNF, the possible involvement of GR was examined. Exposure of glucocorticoid is suggested to reduce GR protein expression (36). Consistently, application of DEX (10 μm) to hippocampal cultures at DIV1 prevented developmental increase of GR expression (Fig. 5A, a). On the other hand, MR expression was not changed (Fig. 5A, a). The down-regulation of GR after DEX addition was observed in a dose-dependent manner (Fig. 5A, b). In contrast, DEX did not affect the MR expression at any concentration of DEX (Fig. 5A, b). Quantification of GR expression after DEX exposure for 24 h was conducted (Fig. 5A, c). Furthermore, we tested the effect of RU486, an antagonist of GR. Levels of pre- and postsynaptic proteins including NR2A, NR2B, GluR1, synapsin I, and SNAP25 were examined (Fig. 5B, a). RU486 canceled the DEX-dependent decrease in BDNF-increased synaptic proteins. As a control, TUJ1 is also shown. The BDNF-enhanced Ca2+ increase was decreased in DEX-pretreated cultures; however, no reduction in Ca2+ increase was observed in coapplication of DEX with RU486 (Fig. 5B, b). As expected, RU486 blocked the DEX-dependent decrease in the BDNF-enhanced glutamate release (Fig. 5B, c). Sole DEX treatment slightly reduced glutamate release, implying the possibility that a change in the activity of voltage-sensitive ion channels is involved.
Fig. 5.

Inhibitory Effect of DEX Was through GR
A, DEX inhibited increase in GR expression during in vitro maturation. a, GR and MR expression after DEX application at DIV2. No increase in GR expression was observed by DEX (10 μm) exposure for 24–48 h. MR expression was intact. b, DEX inhibited the increase in GR expression in a dose-dependent manner. DEX (0.01–10 μm) pretreatment was carried out at DIV1 for 24 h. MR expression was also shown. c, Quantitative analysis of GR expression 24 h after DEX (1.0 or 10 μm) application. Data represent mean ± sd; n = 4, from separate cultures. *, P < 0.05, Kruskal-Wallis test and Mann-Whitney U test. B, RU486, a GR antagonist, prevented DEX-dependent decrease in BDNF-dependent biological effects. a, RU486 (RU) reversed the DEX-dependent decrease in BDNF-increased synaptic proteins. Levels of pre- and postsynaptic proteins including NR2A, NR2B, GluR1, synapsin I, and SNAP25 are shown. As a control, TUJ1 is also detected. RU486 (1.0 μm) was applied 20 min before DEX (10 μm) addition. Twenty-four hours later, BDNF was added. b, RU486 prevented DEX-dependent decrease in BDNF-potentiated postsynaptic Ca2+ increase. RU486 (1.0 μm) was applied 20 min before DEX (1.0 μm) addition. Twenty-four hours later, BDNF was added. Imaging analysis was performed at DIV7 (5 d after BDNF application). Sole RU486 treatment had no effect. None, n = 57; BDNF, n = 147; BDNF plus DEX, n = 152; BDNF plus DEX plus RU486, n = 210; DEX only, n = 61; RU486 only, n = 133. Fluo-3 fluorescent intensity was measured from cells in a sister culture (n is the number of cells selected randomly). Data represent mean ± sd. ***, P < 0.001, ANOVA. c, RU486 reversed the DEX-dependent reduction in glutamate release stimulated by KCl. RU486, DEX, and BDNF were applied as described in b. Data represent mean ± sd; n = 4. The n indicates the number of wells for each experimental condition on a plate. *, P < 0.05; #, P < 0.05 (none vs. DEX), Kruskal-Wallis test and Mann-Whitney U test.
To further clarify the involvement of GR, the effect of small interfering RNA (siRNA) for knockdown of GR was examined. The siRNA transfection was performed at DIV1. The levels of GR and MR at DIV2 indicated the GR-specific reduction after siRNA application (Fig. 6A, a–c). The expression levels of TUJ1 were not altered (Fig. 6A, a). After the GR knockdown by siRNA, DEX treatment before BDNF addition was carried out. As shown in Fig. 6B, DEX failed to decrease the BDNF-increased synaptic proteins after down-regulation of GR caused by siRNA. The levels of proteins at DIV5 are shown. DEX-dependent inhibition on the BDNF-increased NR2A, NR2B, GluR1, synapsin I, SNAP25, and synaptotagmin did not occur in GR-siRNA-transfected cultures, suggesting that GR function is involved in the inhibitory effect of DEX.
Fig. 6.

DEX Did Not Exert Inhibitory Effect on the BDNF-Increased Synaptic Proteins after Down-Regulation of Endogenous GR by siRNA
A, a, The level of endogenous GR was decreased after GR-siRNA transfection. Scramble (control) siRNA had no effect. MR and TUJ1 are shown as a negative control. The levels of GR, MR, and TUJ1 were detected at DIV2. SiRNA transfection was performed at DIV1. b and c, Quantification of GR and MR expression at DIV2. Data represent mean ± sd; n = 4. Significant reduction was confirmed by Student’s t test. ***, P < 0.001. B, DEX had no inhibitory effect on the BDNF-increased synaptic proteins after GR-siRNA transfection. The levels of proteins at DIV5 are shown. DEX-dependent inhibition on the BDNF-increased NR2A, NR2B, GluR1, synapsin I, SNAP25, and synaptotagmin did not occur in GR-siRNA-transfected cultures. SiRNA transfection was performed at DIV1, and then, DEX was applied at DIV2 followed by BDNF addition at DIV3. GR and TUJ1 were also shown.
DEX Decreased the BDNF-Increased Synaptic Proteins via Reducing Activation of the MAPK Pathway Stimulated by BDNF
To further determine what kind of mechanism is involved in DEX effects, we focused on the intracellular signaling. We previously reported that the activation of MAPK/ERK stimulated by BDNF is essential for BDNF-increased synaptic proteins in cultured cortical neurons (26). Thus, we tested the effect of the MAPK pathway inhibitor U0126 in the present system. U0126 (final 10 μm) was applied 20 min before adding BDNF at DIV2, and then samples were collected at DIV4 (Fig. 7A). U0126 suppressed the phosphorylation of ERK1/2 [activated ERK1/2 form, phosphorylated ERK1/2 (pERK1/2)]. The BDNF-dependent increase in the expression of synaptic proteins, including NR2A, NR2B, GluR1, synapsin I, and synaptotagmin, was prevented by U0126. Quantification of levels of pERK1/2 and synaptic proteins (48 h after BDNF application) was conducted (Tables 1 and 2, respectively). As shown in Table 1 and supplemental Fig. 3, we detected the BDNF-stimulated ERK1/2 activation at least 24 h, 48 h, and 5 d after BDNF addition, although the more highly activated ERK1/2 at 6 or 12 h after BDNF stimulation was observed (see Fig. 7B). These results suggest that the up-regulation of these synaptic proteins was through activation of the MAPK pathway triggered by BDNF. Because DEX suppressed the BDNF-increased synaptic proteins, it was possible that DEX inhibited the BDNF-stimulated ERK1/2. Thus, we next examined the effect of DEX on the BDNF-stimulated ERK1/2. Six hours after BDNF application, significant activation of ERK1/2 was observed; however, DEX suppressed the ERK1/2 activation (Fig. 7B, a). The levels of activated ERK1/2 after the application of BDNF with or without DEX pretreatment were determined (Fig. 7B, b). The ERK1/2 activation at 12 h after BDNF stimulation and the suppression of ERK1/2 activity caused by DEX was significant (Fig. 7B, c and d). We examined another pathway activated by TrkB (BDNF receptor), i.e. the phosphatidylinositol 3-kinase pathway. However, DEX pretreatment did not affect the activation of Akt (phosphorylated Akt, pAkt), a component of the phosphatidylinositol 3-kinase pathway, stimulated by BDNF (Fig. 7B, a and c). Total expression of Akt or ERK1/2 was not influenced by DEX (Fig. 7B, a and c). Finally, involvement of the MAPK pathway in BDNF-enhanced synaptic function was examined. U0126 completely blocked the BDNF-potentiated Ca2+ increase triggered by glutamate (Fig. 7C). The BDNF-enhanced glutamate release was also inhibited by U0126 (Fig. 7D). These results suggest that activation of the MAPK pathway is necessary for the BDNF-potentiated synaptic function and that the exposure to a high level of glucocorticoid suppressed the BDNF action through inhibition of the MAPK pathway.
Fig. 7.

Inhibitory Effect of DEX on the Action of BDNF Was via Reducing Activation of the MAPK Pathway
A, U0126 (a specific inhibitor of MAPK kinase, an upstream molecule of MAPK/ERK) suppressed the phosphorylation of ERK1/2 (pERK1/2) and the increases of synaptic proteins induced by BDNF. BDNF-up-regulated NR2A, NR2B, GluR1, synapsin I, and synaptotagmin was prevented by U0126. No change of total ERK1/2 was observed. U0126 (final 10 μm) was applied 20 min before BDNF addition. BDNF was added at DIV2. Forty-eight hours later, samples for immunoblotting were collected. B, BDNF-stimulated ERK activation was decreased by DEX (10 μm) pretreatment. a, Levels of pERK1/2, ERK1/2, pAkt, and Akt at 6 h after BDNF treatment are shown. DEX was applied at DIV1, and then BDNF was added at DIV2. The ERK1/2 activation was suppressed by DEX, whereas the Akt activation was intact. b, Quantitative analysis of activated ERK1/2 at 6 h was performed. Data represent mean ± sd; n = 4. *, P < 0.05 vs. basal pERK1/2 in untreated cells (without BDNF or DEX); #, P < 0.05 vs. BDNF-stimulated pERK1/2 without DEX pretreatment, Kruskal-Wallis test and Mann-Whitney U test. c, Activation of ERK1/2 and Akt at 12 h after BDNF application. The activated ERK1/2 by BDNF was still observed, and DEX inhibited the BDNF-activated ERK1/2. DEX did not affect the Akt activation by BDNF. d, Quantitative analysis of activated ERK1/2 at 12 h after BDNF application. Data represent mean ± sd; n = 4. *, P < 0.05 vs. basal pERK1/2 in untreated cells (without BDNF or DEX); #, P < 0.05 vs. BDNF-stimulated pERK1/2 without DEX pretreatment, Kruskal-Wallis test and Mann-Whitney U test. C, BDNF-potentiated Ca2+ increase required activation of the MAPK pathway. U0126 canceled the BDNF-potentiated Ca2+ increase. U0126 was applied 20 min before BDNF addition at DIV2. Ca2+ imaging was carried out at DIV7 (5 d after BDNF application). None, n = 50; BDNF, n = 50; BDNF plus DEX, n = 50; DEX, n = 50. The n indicates the number of cells. Data represent mean ± sd. ***, P < 0.001, ANOVA. D, BDNF-potentiated glutamate release was blocked by U0126. U0126 and BDNF were applied as indicated in C; n = 4. The n indicates the number of wells for each experimental condition on a plate. *, P < 0.05, Kruskal-Wallis test and Mann-Whitney U test.
Table 1.
Effect of U0126 on the Activation of ERK1/2 Stimulated by BDNF
| None | BDNF | BDNF + U0126 | U0126 | |
|---|---|---|---|---|
| pERK1 | 0.26 ± 0.14 | 1.001 | 0.39 ± 0.322 | 0.18 ± 0.15 |
| pERK2 | 0.37 ± 0.21 | 1.001 | 0.51 ± 0.272 | 0.21 ± 0.16 |
| ERK1 | 0.90 ± 0.12 | 1.00 | 1.13 ± 0.32 | 1.28 ± 0.47 |
| ERK2 | 0.90 ± 0.12 | 1.00 | 0.97 ± 0.52 | 1.00 ± 0.18 |
Each band was quantified by densitometry after Western blot analysis. U0126 and BDNF were applied as indicated in Fig. 7A. To quantify Western blot, normalization using the response to exogenous BDNF application was performed. Data represent the mean ± sd (n = 5).
P < 0.01 (none vs. BDNF), Kruskal-Wallis test and Mann-Whitney U test.
P < 0.01 (BDNF vs. BDNF plus U0126), Kruskal-Wallis test and Mann-Whitney U test.
Table 2.
Effect of U0126 on the BDNF-Increased Synaptic Proteins
| None | BDNF | BDNF + U0126 | U0126 | |
|---|---|---|---|---|
| NR2A | 0.28 ± 0.09 | 1.001 | 0.52 ± 0.222 | 0.29 ± 0.13 |
| NR2B | 0.38 ± 0.14 | 1.001 | 0.65 ± 0.152 | 0.45 ± 0.12 |
| GluR1 | 0.41 ± 0.29 | 1.001 | 0.68 ± 0.162 | 0.23 ± 0.21 |
| Synapsin I | 0.67 ± 0.25 | 1.001 | 0.65 ± 0.162 | 0.54 ± 0.19 |
| TUJ1 | 0.94 ± 0.16 | 1.00 | 1.12 ± 0.13 | 1.06 ± 0.14 |
U0126 and BDNF were applied as indicated in Fig. 7A. To quantify Western blotting, normalization using the response to exogenous BDNF application was performed. BDNF-induced increases in NR2A, NR2B, GluR1, and synapsin I were prevented by U0126. As a control, TUJ1 is also shown. Data represent the mean ± sd (n = 5).
P < 0.01 (none vs. BDNF), Kruskal-Wallis test and Mann-Whitney U test.
P < 0.01 (BDNF vs. BDNF plus U0126), Kruskal-Wallis test and Mann-Whitney U test.
DISCUSSION
In the present study, we found that DEX disturbed BDNF-mediated synaptic maturation in developing hippocampal neurons. Pretreatment with DEX inhibited BDNF-reinforced neurite outgrowth and synaptic protein expression in the early development stage. In the more mature stage, the inhibitory effect of DEX on synaptic function increased by BDNF was also observed. Interestingly, the reduction in the BDNF-activated MAPK/ERK pathway, a required intracellular signaling for BDNF-increased synaptic proteins, was observed after DEX exposure. Thus, it is possible that BDNF-mediated synaptic maturation is suppressed via reducing activation of the MAPK pathway when young neurons are exposed to high-level glucocorticoid during their development stage.
Morphological change of mature neurons seems to depend on the duration of stress hormone exposure. Chronic stress and administration of glucocorticoid resulted in dendritic atrophy of hippocampal neurons (10, 11, 12, 13). Dendrite atrophy of the medial prefrontal cortex of adult rat was also observed after mild and short stress (14). Immature neurons might be more sensitive to stressful stimulation than mature neurons. Indeed, maternal separated infants during P2–P14 exhibited increase of anxiety, learning deficits, and decreased mossy fiber in the CA3 region of the hippocampus after they grew up (15). In our system, the number of neurites was estimated 3 d after starting DEX exposure with or without BDNF addition. Sole DEX exposure did not alter the number of neurites, although the inhibitory effect on the BDNF-increased neurite outgrowth was significant. The 3-d exposure to DEX may be a relatively shorter duration of exposure. If young neurons are exposed for a longer period, significant dendrite atrophy may be induced by sole DEX exposure.
BDNF is a key molecule involved in many neuronal aspects of developing and mature neurons. In particular, BDNF promotes the outgrowth of neurites and increases the expression of synaptic proteins, which are required for establishing synaptic connections or functions during the development stage (24). In hippocampal slice cultures prepared from P7 rats, treatment with BDNF for 48 h increased synaptic vesicle proteins, such as synaptotagmin, synaptophysin, and synaptobrevin (29). Exercise-induced increase in the expression of synapsin I and synaptophysin in the hippocampus has been reported, and this increase in synaptic protein expression depended on BDNF (37). In dissociated cortical neurons, we recently reported that chronic treatment with BDNF enhanced the expression of synapsin I, synaptotagmin, and synaptophysin (26). These results suggest that BDNF is an essential molecule for establishing the basal machinery of synaptic function and plasticity in CNS neurons. Consistently, in the present study, BDNF significantly increased the expression of presynaptic proteins including synapsin I, synaptotagmin, and SNAP25 in cultured hippocampal neurons (Fig. 2 and supplemental Fig. 2). In the exocytotic release of neurotransmitter, synaptic vesicle-associated proteins (synapsin I, synaptotagmin, synaptobrevin, synaptophysin, etc.) and plasma membrane-associated proteins (syntaxin, SNAP25, etc.) are important (38). Indeed, after BDNF application during the in vitro maturation period, cultured hippocampal neurons at the mature stage showed more synapsin I-positive presynaptic sites compared with that in the absence of BDNF (Fig. 4A). However, cultured neurons after coapplication with DEX failed to increase synaptic protein (Fig. 2) and synapsin I-positive sites (Fig. 4A). To our knowledge, there is no report that DEX suppresses the expression of presynaptic proteins in an in vitro system. Indeed, although sole DEX application at high dose seemed to reduce synaptic proteins compared with control, quantification showed no significance (supplemental Fig. 2). After long exposure, sole DEX treatment decreased the number of synapsin I-positive sites compared with control. Because BDNF expression gradually increases in the development stage (39), the reduction in the number of presynaptic sites by DEX alone might result from inhibition of the endogenous BDNF expression. In our cultures, significant reduction in endogenous BDNF after 72 h exposure of DEX was not observed (supplemental Fig. 2C), suggesting no involvement of endogenous BDNF in synaptic protein expression at DIV4. In the case of a decrease in the number of synapsin I-positive sites after long DEX exposure, down-regulation of endogenous BDNF may be involved in the effect of sole DEX addition. Furthermore, it is possible that the change in the efficiency of secretion of endogenous BDNF after DEX exposure is involved in the effects of sole DEX treatment, although further study is required.
Application of DEX before BDNF addition significantly inhibited the BDNF-up-regulated synaptic proteins, whereas sole DEX treatment had little influence compared with the control, suggesting that glucocorticoid may have a more severe effect on the BDNF-dependent biological effects compared with that in the control condition. Interestingly, DEX affected BDNF-induced neurite outgrowth, especially of glutamatergic neurons (Fig. 1). BDNF is important not only for excitatory but also for inhibitory neurons (40). Indeed, BDNF tended to enhance the neurite outgrowth of both glutamatergic and GABAergic neurons in our cultures. However, inhibition by DEX on the BDNF-enhanced neurite outgrowth of GABAergic neurons was weak, implying that glutamatergic neurons have a higher sensitivity to glucocorticoid.
The inhibitory action of DEX on the BDNF-increased synaptic proteins in the early development stage may result in down-regulation of synaptic function in the mature stage. Exposure of neurons to DEX early in culture reduced the BDNF-enhanced presynaptic activity and glutamate release (Fig. 4C). One possibility is that the activity of individual synapses is down-regulated. However, DEX had no significant influence on exocytotic efficiency in individual synaptic sites (Fig. 4B, FM imaging). As shown in Fig. 4A, immunostaining with anti-synapsin I antibody showed a decrease in the number of synaptic sites. Taking these findings together, it is possible that the number of synaptic connections is decreased.
Generally, if inputs from presynapses are reduced for a long time, the sensitivity of postsynapses will increase plastically to compensate for the reduction of the presynaptic inputs (41). However, DEX decreased both pre- and postsynaptic functions in the present study. BDNF is produced and secreted in a neuronal activity-dependent manner (42, 43, 44); thus, continuous weak synaptic activity could result in the reduction of BDNF level as is observed in depressive disorder (19). The down-regulation of BDNF itself or function caused by glucocorticoid exposure in the immature period might make a neuronal system vulnerable to various stresses. Glucocorticoid exposure or stress application during the early postnatal days is suggested to influence later life (15, 30). DEX-injected rats at P1 show a reduction of long-term potentiation in the CA1 region of the hippocampus, and reduction in NR2B level in adulthood occurs (31). Maternal deprivation stress to neonatal (P9) rats suppresses the expression of mRNA of BDNF, NR2A, and NR2B in the hippocampus and prefrontal cortex when they become adults (32). These studies, including our results, support the idea that glucocorticoid exposure during the development of neurons results in inhibition of network establishment (33).
Long-term exposure of glucocorticoid is suggested to reduce GR protein (36). Consistently, DEX-dependent suppression of a developmental increase in the GR expression was confirmed (Fig. 5A). In our system, to reveal the marked inhibitory action of DEX, the high dose of DEX is required, although the dose dependency of DEX in its inhibitory action was observed (Fig. 2). Thus, to clarify the involvement of GR, the siRNA for GR knockdown was applied. We found that DEX failed to inhibit the BDNF-increased synaptic proteins after siRNA application (Fig. 6). Moreover, RU486, a GR antagonist, reversed the inhibitory effect of DEX (Fig. 5B). These results suggest that DEX exerts its inhibitory effect through GR. The high dose required for the present study may be a result of nonspecific binding to extracellular molecules derived from serum in media for plating of cells.
BDNF binds to the TrkB receptor and activates various signaling pathways, including the MAPK and phosphatidylinositol 3-kinase pathways (45). In our study, DEX treatment had no effect on activation of the phosphatidylinositol 3-kinase pathway (Fig. 7B), which is important for neuronal survival (46, 47). Consistently, DEX did not have any effect on neuronal viability (supplemental Fig. 1). Therefore, DEX might inhibit the MAPK pathway, especially. MAPK signaling plays multiple roles in neurons. We recently reported that 17β-estradiol protects cortical neurons against oxidative stress-induced cell death through reduction in the activity of MAPK (48). Interestingly, it was reported that an early phase of the MAPK activation contributed to the cellular adaptive response, but the late phase of the signal activation exerted a toxic response to oxidative stress in the HT22 mouse hippocampal cell line (49). With regard to synaptic function, we recently reported that long-lasting (over 9–24 h) activation of the MAPK pathway is important for BDNF-increased presynaptic protein levels and glutamate release (26). In hippocampal neurons, the suppression of BDNF-stimulated MAPK activation was observed in DEX-treated cultures (Fig. 7). Moreover, BDNF-induced increases in synaptic proteins (Fig. 7A), Ca2+ influx (Fig. 7C), and glutamate release (Fig. 7D) were inhibited by the MAPK pathway inhibitor U0126. Taking these results together, DEX might influence the action of BDNF via reduction of MAPK activation. In the present study, the influence on the exocytotic efficiency of individual synaptic sites was not significant (Fig. 4). Jovanovic et al. (50) reported that acute application of BDNF enhances glutamate release from synaptosomes obtained from cerebral cortex and suggested the involvement of synapsin I phosphorylation through the MAPK pathway. The importance of the MAPK pathway in phosphorylation of synapsin I and exocytosis (triggered by glucose) was also suggested in pancreatic β-calls (51). In their experiment, cell responses through the MAPK pathway after stimulation are acute actions. In our experiment, neuronal function at DIV7 (5 d after BDNF addition) was examined; thus, the difference in experimental conditions including the timescale, maturity, or cell types might contribute to these differences.
The MAPK pathway is also involved in dendritic formation (28). We also reported the importance of the MAPK pathway for dendrite outgrowth of developing cortical neurons (52). Therefore, inhibition of neurite outgrowth with DEX treatment in our study may also be due to reduction in the activity of MAPK signaling. The inhibitory effect of DEX on MAPK activation has recently been reported in the analysis of another growth factor. Platelet-derived neurotrophic factor induced the activation of MAPK/ERK, and pretreatment of DEX inhibited it (53). As expected, DEX inhibited the platelet-derived neurotrophic factor-dependent outgrowth of the neuronal progenitor cell line HIB5. The glucocorticoid-induced leucine zipper protein GILZ is reported to inhibit the phosphorylation of ERK in the T-cell cell line 3DO (54). Furthermore, glucocorticoid induces the expression of MAPK phosphatase-1 (MKP-1), resulting in a decrease in the phosphorylation of MAPK/ERK in the mast cell line RBL-2H3 (55). Therefore, up-regulation of the phosphatase or a similar functional molecule may be involved in our neuronal system. On the other hand, GR function is also suggested to be reduced by MAPK activation (56); thus, it is interesting to study the interaction between the glucocorticoid/GR function and BDNF/MAPK signaling.
In the present study, we focused on the cross-talk between the BDNF function and the effect of glucocorticoid. In particular, the essential role of BDNF involved in synaptic protein expression and function was down-regulated after glucocorticoid exposure. We recently reported that chronic treatment with antidepressant potentiated the BDNF-induced glutamate release via enhancing the activation of BDNF-stimulated intracellular signaling (23). Our current results may provide critical information regarding the interaction of BDNF signaling and stress hormone.
MATERIALS AND METHODS
Chemicals
DEX was dissolved in dimethylsulfoxide (DMSO; Wako Pure Chemical Industries, Ltd.). Thus, the effect of DMSO (vehicle) was checked, and we confirmed that DMSO alone did not have any effects compared with no treatment (data not shown). RU486 was obtained from LKT Laboratories (St. Paul, MN). With regard to intracellular signaling inhibitors, U0126, a MAPK pathway inhibitor, was purchased from Promega (Madison, WI) and used at a final concentration of 10 μm. Other reagents were obtained from Sigma Chemical Co. (St. Louis, MO). Regeneron Pharmaceutical Co., Takeda Chemical Industries (Osaka, Japan), and Sumitomo Co. Ltd. (Osaka, Japan) donated the BDNF. BDNF was applied to neurons at a final concentration of 100 ng/ml.
Cell Culture
Dissociated hippocampal neurons prepared from postnatal 2-d-old rats (SLC, Shizuoka, Japan) were maintained as reported previously (27). All animals were treated according to the institutional guideline for care and use of animals. Hippocampal cells were plated on polyethyleneimine-coated culture dishes (Corning, Corning, NY), plates (Corning), or coverglasses (Matsunami, Osaka, Japan) attached to flexiPERM (Vivascience, Gottingen, Germany). The cell density was 5 × 105/cm2 for Western blot analysis and glutamate release detection, 2 × 105/cm2 for Ca2+ imaging, or 5 × 104/cm2 for immunocytochemistry and exocytosis imaging. The culture medium consisted of 5% fetal bovine serum, 5% heat-inactivated horse serum, and 90% of a 1:1 mixture of DMEM and Ham’s F-12 medium (Invitrogen, Carlsbad, CA). Four hours after cell plating, the medium was exchanged to Neurobasal-A medium (Invitrogen) containing 2% B27 supplement (Invitrogen) and 0.5 mm l-glutamine (Invitrogen).
Immunocytochemistry
Two days after cell plating (DIV2), BDNF was applied with or without pretreatment with 10 μm DEX for 24 h. Forty-eight hours after BDNF application, cultured cells were fixed with 4% paraformaldehyde for 30 min and then rinsed three times with PBS. Cells were then permeabilized with 0.1% Triton X-100 and 5% heat-inactivated horse serum in PBS for 20 min at room temperature. Afterward, the fixed cells were incubated with first antibodies overnight at 4 C. We used anti-MAP2 (1:1000; Sigma), anti-GAD (1:2000; Sigma), anti-glutamate (1/8000; Sigma), and anti-synapsin I (1:2000; Chemicon, Temecula, CA) antibodies. Alexa fluor 488- or Alexa fluor 594-conjugated antimouse IgG (1:1000; Invitrogen Molecular Probes, Tokyo, Japan) or antirabbit IgG (1:1000; Invitrogen Molecular Probes) were used as secondary antibodies. Immunoreactivity was observed with a fluorescence microscope (Axiovert 200; Zeiss, Tokyo, Japan), and the obtained images were analyzed with the Slide Book 3.0 software (Intelligent Imaging Innovations Inc., Denver, CO). To estimate the neurite outgrowth, the number of primary dendrites per cell after immunostaining was examined (52). MAP2 and GAD (or glutamate) double-positive dendritic neurites was examined. The number of primary dendrites (from the cell body) that were two times the length of the cell diameter or longer was counted. The number of presynaptic sites was estimated by counting the synapsin I-positive dots on dendrite shafts per 50-μm length.
3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyl Tetrazolium Bromide (MTT) Assay
MTT assay (mitochondrial-dependent conversion of tetrazolium salts) was performed as reported previously (57). In brief, after treatment with 10 μm DEX (DIV1) and/or BDNF (DIV2), cultured hippocampal neurons were incubated with MTT solution at DIV4. Two hours later, cultures were lysed, and the metabolic activity of the mitochondria was estimated to determine the cell viability.
Western Blotting
Cells were lysed in SDS lysis buffer containing 1% SDS, 20 mm Tris-HCl (pH 7.4), 5 mm EDTA (pH 8.0), 10 mm NaF, 2 mm Na3VO4, 0.5 mm phenylarsine oxide, and 1 mm phenylmethylsulfonyl fluoride. After boiling for 5 min, lysates were centrifuged at 15,000 rpm for 60 min at 4 C, and the supernatants were collected for analysis. The protein concentration of the supernatants was quantified by BCA Protein Assay kit (Pierce, Rockford, IL), and the same amount of total protein was assayed for each Western blot. For primary antibodies, anti-NR2A (1:500; Sigma), anti-NR2B (1:500; Sigma), anti-GluR1 (1:1000; Chemicon), anti-synapsin I (1:2000; Chemicon), anti-synaptotagmin (1:3000; Transduction Laboratories, Lexington, KY), anti-SNAP25 (1:1000; Synaptic Systems, Gottingen, Germany), anti-TUJ1 (1:5000; Berkeley Antibody Co., Berkeley, CA), anti-pERK (1:1000; Cell Signaling Technology, Beverly, MA), anti-ERK (1:1000; Cell Signaling), anti-Akt (1:1000; Cell Signaling), anti-pAkt (1:1000; Cell Signaling), anti-GR (1:500; Santa Cruz Biotechnology Inc., Santa Cruz, CA), anti-MR (1:500; Santa Cruz), and anti-BDNF (1:200; Santa Cruz) antibodies were used. The changes in protein expression are indicated as a ratio that was normalized to sole BDNF application in each experiment. The n indicates the number of experiments performed with separate cultures.
Imaging of Intracellular Ca2+
Ca2+ imaging analysis was carried out as reported previously (27). Briefly, cultured cells maintained on polyethyleneimine-coated coverglasses attached to flexiPERM were incubated for 1 h at 37 C with 10 μm Fluo-3 AM (Molecular Probes, Eugene, OR) diluted in HEPES-buffered Krebs Ringer assay buffer (KRH; 130 mm NaCl, 5 mm KCl, 1.2 mm NaH2PO4, 1.8 mm CaCl2, 10 mm glucose, 1% BSA, and 25 mm HEPES, pH 7.4). The dye intensity was monitored using a fluorescent microscope (Axiovert 200 controlled by Slide Book 3.0). The emitted fluorescence was guided through a ×20 objective. Image data were obtained every 2 sec. High KCl (50 mm, final concentration) solution or glutamate (1 μm, final concentration) was applied by bath application to trigger cell depolarization. Data were stored and analyzed with the Slide Book 3.0. The experiment was performed at least three times with separate cultures, and the reproducibility was confirmed. Representative data from neurons in a sister culture are shown in the figures.
Exocytosis Imaging
The efficiency of exocytosis in synaptic sites was measured with FM-43 fluorescent imaging as reported previously (58). Briefly, after washing three times with KRH buffer, FM-43 dye (2 μm in KRH) was loaded for 30 min at 37 C. After washing cells three times with KRH, the fluorescence was monitored using a fluorescent microscope. The emitted fluorescence was guided through a ×40 objective. Image data were obtained every 2 sec. The exocytosis was evoked by 50 mm (final concentration) KCl. The efficiency of exocytosis was estimated as the quenching ratio of fluorescence before and after the stimulation. The fluorescence elimination was expressed by the value of (F0 − F)/F0, where F0 is basal fluorescence 4 sec before KCl stimulation and F is fluorescence 20 sec after the stimulation.
Detection of Amino Acid Neurotransmitters
The amounts of amino acids released from cultured hippocampal neurons were measured as described previously (59). Briefly, amino acids released into KRH assay buffer were measured by HPLC (Shimadzu Co., Kyoto, Japan). Initially, KRH assay buffer was collected without stimulation (1 min); that is, the amount of glutamate in the sample was considered a basal release. Next, 50 mm (final concentration) KCl was added to cultures for 1 min, and the samples were collected as KCl-stimulated samples. The amount of released glutamate is indicated as a relative release amount vs. basal release in control (without DEX and BDNF). Representative data from a sister culture are shown in the figures. The n indicates the number of wells for each experimental condition on a plate. The reproducibility was confirmed.
siRNA
SiRNA transfection was performed as reported (60). We used 21-nucleotide siRNA duplexes with two 3′ overhanging nucleotides of the rat GR mRNA coding region (1801–1819, 5′-TGACCACACTCAACATGTT-3′, NM_012576). Sense (5′-UGACCACACUCAACAUGUUTT-3′) and antisense (5′-AACAUGUUGAGUGUGGUCATT-3′) strands were chemically synthesized by Nippon EGT Co., Ltd. (Toyama, Japan). The siRNA (GCGCGCUUUGUAGGAUUCG) named ScrambleII from Dharmacon Research Inc. (Lafayette, CO) was used as a control. Transfection of both siRNAs (final 100 nm) was performed using Lipofectamine 2000 reagent (Invitrogen, Tokyo, Japan). We carried out the siRNA transfer 24 h before DEX exposure. After the addition of 10 μm DEX (at DIV2) and/or BDNF (at DIV3), the DIV5 hippocampal neurons were lysed for Western blotting.
Statistical Analysis
Data shown in the present study are expressed as mean ± sd. For statistical analysis comparing two groups, Student’s t test was used. For multiple groups, analysis using nonparametric Kruskal-Wallis test and Mann-Whitney U test were carried out. To analyze the large sample number (n > 30), ANOVA was also performed. All analyses were conducted using the Statistical Package for Social Science (SPSS) version 11.0 (SPSS Japan, Tokyo, Japan). P values < 5% were considered significant.
NURSA Molecule Pages:
Ligands: Dexamethasone | RU486;
Nuclear Receptors: GR | MR.
Footnotes
This work was supported by The Ichiro Kanehara Foundation (T.N.), the Japan Health Sciences Foundation (Research on Health Sciences focusing on Drug innovation) (H.K.), Health and Labor Sciences Research Grants (Research on Psychiatric and Neurological Diseases and Mental Health) (H.K.), the Mitsubishi Pharma Research Foundation (H.K.), a Grant from the Japan Foundation for Neuroscience and Mental Health (H.K.), the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO) (H.K.), and a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) (T.N.).
Disclosure Statement: The authors have nothing to disclose.
First Published Online December 20, 2007
Abbreviations: BDNF, Brain-derived neurotrophic factor; CNS, central nervous system; DEX, dexamethasone; DIV1, 1 d in vitro; DMSO, dimethylsulfoxide; GABA, γ-aminobutyric acid; GAD, glutamic acid decarboxylase; GluR1, glutamate receptor 1; GR, glucocorticoid receptor; KRH, HEPES-buffered Krebs Ringer assay buffer; MAP2, microtubule-associated protein 2; MR, mineralocorticoid receptor; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; NMDA, N-methyl-d-aspartate; NR2A, NMDA receptor 2A; P2, postnatal d 2; pERK1/2, phosphorylated ERK1/2; siRNA, small interfering RNA; SNAP25, synaptosome-associated protein 25; TUJ1, class III β-tubulin.
References
- 1.de Kloet ER, Joels M, Holsboer F 2005. Stress and the brain: from adaptation to disease. Nat Rev Neurosci 6:463–475 [DOI] [PubMed] [Google Scholar]
- 2.Smoak KA, Cidlowski JA 2004. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev 125:697–706 [DOI] [PubMed] [Google Scholar]
- 3.Holsboer F 2000. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23:477–501 [DOI] [PubMed] [Google Scholar]
- 4.Kunugi H, Ida I, Owashi T, Kimura M, Inoue Y, Nakagawa S, Yabana T, Urushibara T, Kanai R, Aihara M, Yuuki N, Otsubo T, Oshima A, Kudo K, Inoue T, Kitaichi Y, Shirakawa O, Isogawa K, Nagayama H, Kamijima K, Nanko S, Kanba S, Higuchi T, Mikuni M 2006. Assessment of the dexamethasone/CRH test as a state-dependent marker for hypothalamic-pituitary-adrenal (HPA) axis abnormalities in major depressive episode: a multicenter study. Neuropsychopharmacology 31:212–220 [DOI] [PubMed] [Google Scholar]
- 5.Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP, Thakur M, McEwen BS, Hauger RL, Meaney MJ 1998. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci 1:69–73 [DOI] [PubMed] [Google Scholar]
- 6.Kim JJ, Diamond DM 2002. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci 3:453–462 [DOI] [PubMed] [Google Scholar]
- 7.Perlis RH, Brown E, Baker RW, Nierenberg AA 2006. Clinical features of bipolar depression versus major depressive disorder in large multicenter trials. Am J Psychiatry 163:225–231 [DOI] [PubMed] [Google Scholar]
- 8.de Quervain DJ, Roozendaal B, McGaugh JL 1998. Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature 394:787–790 [DOI] [PubMed] [Google Scholar]
- 9.Sousa N, Lukoyanov NV, Madeira MD, Almeida OF, Paula-Barbosa MM 2000. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience 97:253–266 [DOI] [PubMed] [Google Scholar]
- 10.Liu HH, Payne HR, Wang B, Brady ST 2006. Gender differences in response of hippocampus to chronic glucocorticoid stress: role of glutamate receptors. J Neurosci Res 83:775–786 [DOI] [PubMed] [Google Scholar]
- 11.Magariños AM, McEwen BS, Flügge G, Fuchs E 1996. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci 16:3534–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe Y, Gould E, McEwen BS 1992. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res 588:341–345 [DOI] [PubMed] [Google Scholar]
- 13.Woolley CS, Gould E, McEwen BS 1990. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res 531:225–231 [DOI] [PubMed] [Google Scholar]
- 14.Brown SM, Henning S, Wellman CL 2005. Mild, short-term stress alters dendritic morphology in rat medial prefrontal cortex. Cereb Cortex 15:1714–1722 [DOI] [PubMed] [Google Scholar]
- 15.Huot RL, Plotsky PM, Lenox RH, McNamara RK 2002. Neonatal maternal separation reduces hippocampal mossy fiber density in adult Long Evans rats. Brain Res 950:52–63 [DOI] [PubMed] [Google Scholar]
- 16.Takahashi T, Kimoto T, Tanabe N, Hattori TA, Yasumatsu N, Kawato S 2002. Corticosterone acutely prolonged N-methyl-d-aspartate receptor-mediated Ca2+ elevation in cultured rat hippocampal neurons. J Neurochem 83:1441–1451 [DOI] [PubMed] [Google Scholar]
- 17.Cho K, Little HJ 1999. Effects of corticosterone on excitatory amino acid responses in dopamine-sensitive neurons in the ventral tegmental area. Neuroscience 88:837–845 [DOI] [PubMed] [Google Scholar]
- 18.Karege F, Vaudan G, Schwald M, Perroud N, La Harpe R 2005. Neurotrophin levels in postmortem brains of suicide victims and the effects of antemortem diagnosis and psychotropic drugs. Brain Res Mol Brain Res 136:29–37 [DOI] [PubMed] [Google Scholar]
- 19.Gervasoni N, Aubry JM, Bondolfi G, Osiek C, Schwald M, Bertschy G, Karege F 2005. Partial normalization of serum brain-derived neurotrophic factor in remitted patients after a major depressive episode. Neuropsychobiology 51:234–238 [DOI] [PubMed] [Google Scholar]
- 20.Smith MA, Makino S, Kvetnansky R, Post RM 1995. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci 15:1768–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansson AC, Sommer W, Rimondini R, Andbjer B, Stromberg I, Fuxe K 2003. c-fos reduces corticosterone-mediated effects on neurotrophic factor expression in the rat hippocampal CA1 region. J Neurosci 23:6013–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nibuya M, Morinobu S, Duman RS 1995. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15:7539–7547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yagasaki Y, Numakawa T, Kumamaru E, Hayashi T, Su TP, Kunugi H 2006. Chronic antidepressants potentiate via sigma-1 receptors the brain-derived neurotrophic factor-induced signaling for glutamate release. J Biol Chem 281:12941–12949 [DOI] [PubMed] [Google Scholar]
- 24.Bibel M, Barde YA 2000. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev 14:2919–2937 [DOI] [PubMed] [Google Scholar]
- 25.Gartner A, Polnau DG, Staiger V, Sciarretta C, Minichiello L, Thoenen H, Bonhoeffer T, Korte M 2006. Hippocampal long-term potentiation is supported by presynaptic and postsynaptic tyrosine receptor kinase B-mediated phospholipase Cγ signaling. J Neurosci 26:3496–3504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsumoto T, Numakawa T, Yokomaku D, Adachi N, Yamagishi S, Numakawa Y, Kunugi H, Taguchi T 2006. Brain-derived neurotrophic factor-induced potentiation of glutamate and GABA release: different dependency on signaling pathways and neuronal activity. Mol Cell Neurosci 31:70–84 [DOI] [PubMed] [Google Scholar]
- 27.Numakawa T, Yamagishi S, Adachi N, Matsumoto T, Yokomaku D, Yamada M, Hatanaka H 2002. Brain-derived neurotrophic factor-induced potentiation of Ca2+ oscillations in developing cortical neurons. J Biol Chem 277:6520–6529 [DOI] [PubMed] [Google Scholar]
- 28.Miller FD, Kaplan DR 2003. Signaling mechanisms underlying dendrite formation. Curr Opin Neurobiol 13:391–398 [DOI] [PubMed] [Google Scholar]
- 29.Tartaglia N, Du J, Tyler WJ, Neale E, Pozzo-Miller L, Lu B 2001. Protein synthesis-dependent and -independent regulation of hippocampal synapses by brain-derived neurotrophic factor. J Biol Chem 276:37585–37593 [DOI] [PubMed] [Google Scholar]
- 30.Gross C, Hen R 2004. The developmental origins of anxiety. Nat Rev Neurosci 5:545–552 [DOI] [PubMed] [Google Scholar]
- 31.Kamphuis PJGH, Gardoni F, Kamal A, Croiset G, Bakker JM, Cattabeni F, Gispen WH, van Bel F, Luca MD, Wiegant VM 2003. Long-lasting effects of neonatal dexamethasone treatment on spatial learning and hippocampal synaptic plasticity. Involvement of the NMDA receptor complex. FASEB J 17:911–913 [DOI] [PubMed] [Google Scholar]
- 32.Roceri M, Hendriks W, Racagni G, Ellenbroek BA, Riva MA 2002. Early maternal deprivation reduces the expression of BDNF and NMDA receptor subunits in rat hippocampus. Mol Psychiatry 7:609–616 [DOI] [PubMed] [Google Scholar]
- 33.Castren E 2005. Is mood chemistry? Nat Rev Neurosci 6:241–246 [DOI] [PubMed] [Google Scholar]
- 34.de Kloet ER, Vreugdenhil E, Oitzl MS, Joels M 1998. Brain corticosteroid receptor balance in health and disease. Endocr Rev 19:269–301 [DOI] [PubMed] [Google Scholar]
- 35.Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA 2007. Human glucocorticoid receptor β binds RU-486 and is transcriptionally active. Mol Cell Biol 27:2266–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, DeFranco DB 2005. Alternative effects of the ubiquitin-proteasome pathway on glucocorticoid receptor down-regulation and transactivation are mediated by CHIP, an E3 ligase. Mol Endcrinol 19:1474–1482 [DOI] [PubMed] [Google Scholar]
- 37.Vaynman SS, Ying Z, Yin D, Gomez-Pinilla F 2006. Exercise differentially regulates synaptic proteins associated to the function of BDNF. Brain Res 1070:124–130 [DOI] [PubMed] [Google Scholar]
- 38.Sudhof TC 1995. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 375:645–653 [DOI] [PubMed] [Google Scholar]
- 39.Silhol M, Bonnichon V, Rage F, Tapia-Arancibia L 2005. Age-related changes in brain-derived neurotrophic factor and tyrosine kinase receptor isoforms in the hippocampus and hypothalamus in male rats. Neuroscience 132:613–624 [DOI] [PubMed] [Google Scholar]
- 40.Yamada MK, Nakanishi K, Ohba S, Nakamura T, Ikegawa Y, Nishiyama N, Matsuki N 2002. Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J Neurosci 22:7580–7585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez-Otano I, Ehlers M 2005. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci 28:229–238 [DOI] [PubMed] [Google Scholar]
- 42.Schinder AF, Poo M 2000. The neurotrophin hypothesis for synaptic plasticity. Trends Neurosci 23:639–645 [DOI] [PubMed] [Google Scholar]
- 43.Hartmann M, Heumann R, Lessmann V 2001. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20:5887–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balkowiec A, Katz DM 2002. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci 22:10399–10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patapoutian A, Reichardt LF 2001. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11:272–280 [DOI] [PubMed] [Google Scholar]
- 46.Rodgers EE, Theibert AB 2002. Functions of PI3-kinase in development of the nervous system. Int J Dev Neurosci 20:187–197 [DOI] [PubMed] [Google Scholar]
- 47.Zheng WH, Quirion R 2004. Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J Neurochem 89:844–852 [DOI] [PubMed] [Google Scholar]
- 48.Numakawa Y, Matsumoto T, Yokomaku D, Taguchi T, Niki E, Hatanaka H, Kunugi H, Numakawa T 2007. 17β-Estradiol protects cortical neurons against oxidative stress-induced cell death through reduction in the activity of mitogen-activated protein kinase and in the accumulation of intracellular calcium. Endocrinology 148:627–637 [DOI] [PubMed] [Google Scholar]
- 49.Luo Y, DeFranco DB 2006. Opposing roles for ERK1/2 in neuronal oxidative toxicity: distinct mechanisms of ERK1/2 action at early versus late phases of oxidative stress. J Biol Chem 281:16436–16442 [DOI] [PubMed] [Google Scholar]
- 50.Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS 2000. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci 3:323–329 [DOI] [PubMed] [Google Scholar]
- 51.Longuet C, Broca C, Costes S, Hani EH, Bataille D, Dalle S 2005. Extracellularly regulated kinases 1/2 (p44/42 mitogen-activated protein kinases) phosphorylate synapsin I and regulate insulin secretion in the MIN6 β-cell line and islets of Langerhans. Endocrinology 146:643–654 [DOI] [PubMed] [Google Scholar]
- 52.Numakawa T, Ishimoto T, Suzuki S, Numakawa Y, Adachi N, Matsumoto T, Yokomaku D, Koshimizu H, Fujimori KE, Hashimoto R, Taguchi T, Kunugi H 2004. Neuronal roles of the integrin-associated protein (IAP/CD47) in developing cortical neurons. J Biol Chem 279:43245–43253 [DOI] [PubMed] [Google Scholar]
- 53.Obradovic D, Gronemeyer H, Lutz B, Rein T 2006. Cross-talk of vitamin D and glucocorticoids in hippocampal cells. J Neurochem 96:500–509 [DOI] [PubMed] [Google Scholar]
- 54.Ayroldi E, Zollo O, Macchiarulo A, Di Marco B, Marchetti C, Riccardi C 2002. Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol 22:7929–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC 2001. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J 20:7108–7116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szatmary Z, Garabedian MJ, Vilcek J 2004. Inhibition of glucocorticoid receptor-mediated transcriptional activation by p38 mitogen-activated protein (MAP) kinase. J Biol Chem 279:43708–43715 [DOI] [PubMed] [Google Scholar]
- 57.Numakawa Y, Numakawa T, Matsumoto T, Yagasaki Y, Kumamaru E, Kunugi H, Taguchi T, Niki E 2006. Vitamin E protected cultured cortical neurons from oxidative stress-induced cell death through the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. J Neurochem 97:1191–1202 [DOI] [PubMed] [Google Scholar]
- 58.Numakawa T, Yokomaku D, Kiyosue K, Adachi N, Matsumoto T, Numakawa Y, Taguchi T, Hatanaka H, Yamada M 2002. Basic fibroblast growth factor evokes a rapid glutamate release through activation of the MAPK pathway in cultured cortical neurons. J Biol Chem 277:28861–28869 [DOI] [PubMed] [Google Scholar]
- 59.Numakawa T, Takei N, Yamagishi S, Sakai N, Hatanaka H 1999. Neurotrophin-elicited short-term glutamate release from cultured cerebellar granule neurons. Brain Res 842:431–438 [DOI] [PubMed] [Google Scholar]
- 60.Numakawa T, Nakayama H, Suzuki S, Kubo T, Nara F, Numakawa Y, Yokomaku D, Araki T, Ishimoto T, Ogura A, Taguchi T 2003. Nerve growth factor-induced glutamate release is via p75 receptor, ceramide, and Ca2+ from ryanodine receptor in developing cerebellar neurons. J Biol Chem 278:41259–41269 [DOI] [PubMed] [Google Scholar]