

Abstract
Many important physiological roles of the urocortin (UCN) family of peptides as well as CRH involve the type 2 CRH receptor (CRH-R2) and downstream activation of multiple pathways. To characterize molecular determinants of CRH-R2 functional activity, we used HEK293 cells overexpressing recombinant CRH-R2β and investigated mechanisms involved in attenuation of CRH-R2 signaling activity and uncoupling from intracellular effectors. CRH-R2β-mediated adenylyl cyclase activation was sensitive to homologous desensitization induced by pretreatment with either UCN-II or the weaker agonist CRH. CRH-R2β activation induced transient β-arrestin1 and β-arrestin2, as well as clathrin, recruitment to the plasma membrane. β-Arrestin2 appeared to be the main β-arrestin subtype associated with the receptor. This was followed by CRH-R2β endocytosis in a mechanism that exhibited distinct agonist-dependent temporal characteristics. CRH-R2β also induced transient activation of the ERK1/2 and p38MAPK signaling cascades that peaked at 5 min and returned to basal within 20–30 min. Unlike p38MAPK, activated ERK1/2 was localized both in the cytoplasm and nucleus. Experiments employing inhibitors of receptor endocytosis showed that CRH-R2β-MAPK interaction does not require β-arrestin, clathrin, or receptor endocytosis. Site-directed mutagenesis studies on CRH-R2β C terminus showed that the amino acid cassette TAAV at the end of the C terminus is important for CRH-R2β signaling because loss of a potential phospho-acceptor site in mutant receptors containing deletion or Ala substitution of the cassette TAAV resulted in reduced ERK1/2 activation and accelerated receptor internalization. These findings provide new insights about the signaling mechanisms regulating CRH-R2β functional activity and determining its biological responses.
CRH, UROCORTIN (UCN)-I, UCN-II, and UCN-III are members of the CRH-like family of peptides that play diverse roles in mammalian development and physiology, especially the adaptation to stressful stimuli (1). Two classes of CRH receptors termed R1 and R2, mediate the distinct actions of CRH and UCNs in the brain, pituitary, and peripheral tissues (2).
Mammals express three known CRH-R2 variants: R2α and R2β, expressed both centrally and peripherally, and R2γ, which has so far been found only in the limbic regions of the human central nervous system (2). These variants have identical transmembrane and C-terminal domains and differ only in their N-terminal extracellular domains. Pharmacological characterization of the CRH-R2 has shown that this seven-heptahelical G-protein coupled receptor (GPCR) preferentially binds UCNs rather than CRH (3). CRH-R2 activation is central in mediating UCNs diverse physiological actions including anxiolytic and feeding reduction effects in the brain as well as cardioprotection, tissue angiogenesis, and gastrointestinal regulatory effects at the periphery (4, 5, 6, 7). Recent findings showing involvement of UCNs in the control of glucose utilization and insulin sensitivity in skeletal muscle as well as stimulation of insulin and glucagon secretion in pancreatic β-cells implicate CRH-R2 in the control of energy balance and homeostasis (8, 9).
The CRH-R2, similar to the CRH-R1, is primarily a Gsα/adenylyl cyclase/cAMP-coupled GPCR (10). In addition, two members of the family of MAPK, ERK1/2 and p38 MAPK, have also been implicated as key signaling intermediates downstream of CRH-R2 activation (11, 12, 13). Various studies (14, 15) investigating the mechanisms of CRH-R2-ERK1/2 interactions have shown involvement of multiple G proteins (Gq, Gi, and Go) and a number of signaling molecules including phosphatidylinositol-3 OH kinase, MAPK kinase 1, phospholipase C, Raf-1 kinase, tyrosine kinases, and intracellular Ca2+.
At present, there is little information about the intracellular mechanisms regulating CRH-R2 signaling efficiency. A characteristic feature of most GPCRs’ function is their ability to adjust their biological activity in response to prolonged agonist stimulation. This is achieved through a series of fine-tuned signaling events involving receptor phosphorylation by protein kinases, β-arrestin recruitment to the plasma membrane, and association with the receptor that leads to GPCR signaling desensitization and uncoupling from G proteins; this is followed by receptor endocytosis, sometimes via a clathrin/dynamin-mediated process (16). Interestingly, for many GPCRs, this mechanism also appears to be essential for activation of the ERK1/2 signaling pathway in a G protein-independent manner (17).
Previous studies addressing CRH-R1 signaling properties (18, 19, 20) demonstrated that the CRH-R1 is sensitive to β-arrestin-dependent signaling desensitization and internalization. Receptor endocytosis is also one of the mechanisms employed by CRH-R1 to induce ERK1/2 and p38 MAPK phosphorylation and activation (21). Accordingly, in this study, we sought to determine the internalization characteristics of CRH-R2 and explore the possible link between the CRH-R2 internalization and MAPK activation. We used recombinant human CRH-R2β stably expressed in HEK293 cells. We also analyzed some of the spatiotemporal characteristics of MAPK activation and investigated potential receptor structural domains that are involved in this process.
RESULTS
UCN-II-Induced cAMP Response in HEK293-R2β Cells and Regulation by CRH-R2β Desensitization
The functional coupling of CRH-R2 to the Gsα/adenylyl cyclase/cAMP pathway in response to different agonists was assessed in HEK293 cells stably expressing CRH-R2β receptor (293-R2β cells). Preliminary studies using RT-PCR and immunoblotting using a specific CRH-R1/2 antibody in untransfected HEK293 cells failed to identify endogenous CRH-R2 mRNA or protein expression (Fig. 1A). Successful expression of CRH-R2β in HEK293 cells was confirmed by Western blot analysis using a specific CRH-R1/2 antibody raised against the C terminus of the CRH-R, which showed a single immunoreactive protein with an apparent molecular mass of approximately 60 kDa, in cell membranes prepared from 293-R2β cells (Fig. 1A). No immunoreactive proteins were detected when a synthetic CRH-R2 blocking peptide (1 μm) was added, thus confirming the specificity of these data. In addition, indirect immunofluorescent confocal microscopy experiments demonstrated exclusive localization of CRH-R2β receptors on the cell surface (data not shown). Treatment of cells with various concentrations of CRH, UCN-I, or UCN-II (1–100 nm) for 15 min resulted in a dose-dependent increase of intracellular cAMP accumulation (Fig. 1B). In agreement with previous studies (3), both UCN-II and UCN-I were equipotent in inducing adenylyl cyclase activation and were 40% more potent than CRH.
Fig. 1.
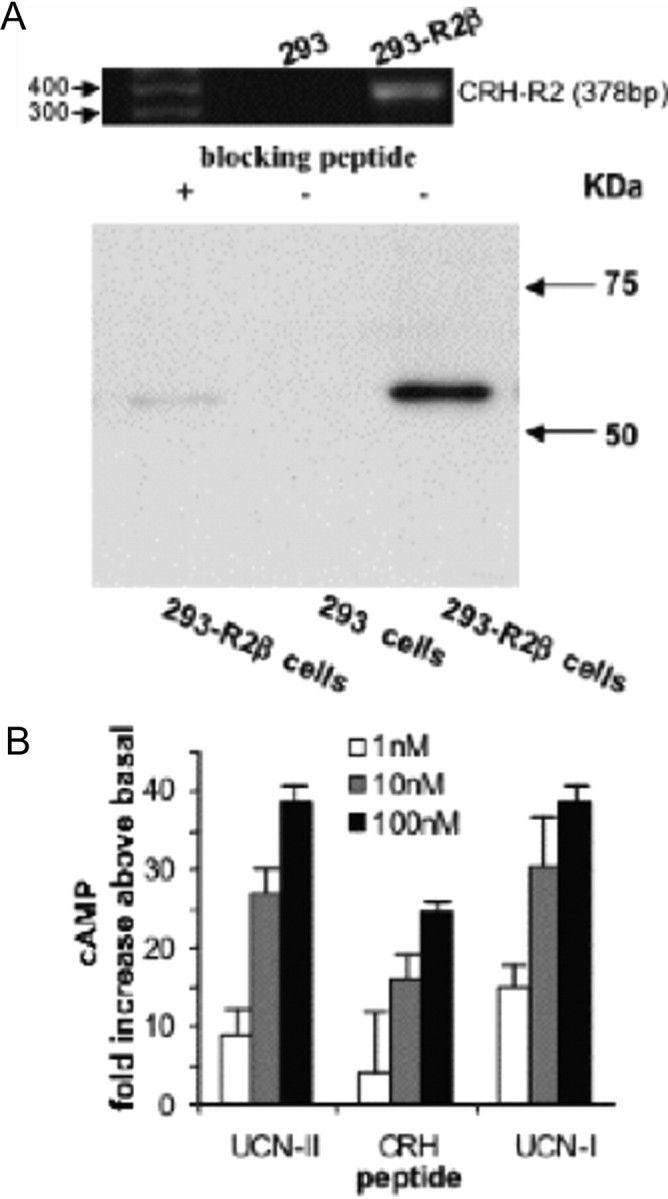
Receptor Protein and mRNA Expression and Agonist-Stimulated cAMP Production in 293-R2β Cells
A, Cell lysates from untransfected HEK293 or HEK293 cells stably expressing CRH-R2β receptors were fractionated on SDS-PAGE and subjected to immunoblotting with a specific CRH-R2 antibody in the presence or absence of blocking peptide as described in Materials and Methods. Identical results were obtained from three independent experiments. Inset, RT-PCR of CRH-R2 mRNA present in HEK293 cells using specific oligunucleotide primers. The size of the amplified DNA fragment was 378 bp. B, Cells were pretreated with various concentrations of CRH, UCN-I, or UCN-II for 15 min. cAMP production was determined by ELISA. Results are expressed as the mean ± sem of three estimations.
We next determined whether CRH-R2β functional coupling to the cAMP-signaling pathway is susceptible to agonist-induced (homologous) desensitization. UCN-II was used because of its specificity for CRH-R2 binding and activation. For that purpose, 293-R2β cells were pretreated with 100 nm UCN-II for various time intervals (0–30 min), and the maximal UCN-II-induced cAMP stimulation was determined. Results showed (Fig. 2A) that agonist pretreatment for only 15 min was sufficient to induce a 80% decrease in CRH-R2β responsiveness, and a 30-min preexposure to 100 nm UCN-II almost completely abolished subsequent UCN-II (100 nm)-induced cAMP response. Interestingly, when CRH (100 nm) was used to induce receptor desensitization, we observed a weaker effect on CRH-R2β responsiveness; CRH pretreatment for 15 min caused a 60% decrease only in subsequent UCN-II (100 nm)-induced cAMP response, whereas a 30-min preexposure to 100 nm CRH reduced the UCN-II effect on cAMP stimulation by 70–75%.
Fig. 2.

Desensitization and Internalization Characteristics of CRH-R2β Stably Expressed in HEK293 Cells
Cells were pretreated with CRH or UCN-II (100 nm) for various time intervals (2–45 min). A, After extensive washing, cAMP response to a second UCN-II stimulus (100 nm for 15 min) was determined. Data represent the mean ± sem of two estimations from three independent experiments. *, P < 0.05 compared with cells without pretreatment; +, P < 0.05 compared with each other’s agonist pretreatment. B, Alternatively, CRH-R2β distribution was monitored over the ensuing time period by indirect immunofluorescence using specific primary antibodies and Alexa Fluor 594 secondary antibodies for CRH-R2β (red). Identical results were obtained from four independent experiments. For quantification of cytoplasmic CRH-R2β, 20 individual cells in five random fields of view were examined. *, P < 0.05 compared with cells without pretreatment; +, P < 0.05 compared with each other’s agonist pretreatment. C, CRH-R2β loss from the cell surface after agonist stimulation (UCN-II or CRH, 100 nm) for 30 min was quantified by an On-cell Western assay with near-IR dyes in nonpermeabilized cells, using antibodies against the CRH-R2β N terminus and the Odyssey IR imaging system. In the right panel, the IR-specific signal intensity characteristics are presented. Data represent the mean ± sem of two estimations from three independent experiments. *, P < 0.05 compared with cells without pretreatment; +, P < 0.05 compared with each other’s agonist pretreatment. Inset, After agonist stimulation, cell membrane-rich fractions were immunoblotted with antibodies against CRH-R1/2 C terminus. Identical results were obtained from four independent experiments.
The fate of CRH-R2β receptors after activation by 100 nm UCN-II or CRH was also investigated by indirect confocal microscopy to monitor temporal distribution of the receptor. In the absence of agonist, CRH-R2β receptors were exclusively localized on the cell surface of 293-R2β cells (Fig. 2B). Within 15 min of UCN-II treatment, there was evidence of some receptor trafficking in the cytoplasm, and after 30 min of treatment, intense receptor fluorescent signal was observed in the cytoplasm indicative of substantial receptor internalization (Fig. 2B, right column). Use of CRH as the receptor internalization-inducing agonist revealed important agonist-specific temporal differences in CRH-R2β internalization kinetics (Fig. 2B, left column); substantial CRH-R2β internalization was evident only after 30–45 min of treatment with 100 nm CRH (Fig. 2B, left column). These observations were confirmed by using the Image J software and quantification of intracellular fluorescence spectra (4–18 μm) of 20 individual cells, which were randomly selected.
We also attempted to quantify the loss of CRH-R2β receptors from the cell surface after agonist stimulation, by using an On-cell Western assay with near-infrared (IR) dyes in nonpermeabilized cells. For that purpose, antibodies targeted against the CRH-R2β N terminus and the Odyssey IR imaging system was employed, as previously described (22). In preliminary experiments, the IR signal characteristics were established; signal intensity was typically 80% higher than signal obtained when secondary antimouse IRDye 800-conjugated IgG was used alone (data not shown). The calculated percent specific signal of the antibodies used in this assay was determined to be 45–55% of total signal. Results showed that UCN-II (100 nm) treatment for 30 min induced a 58–67% decrease in CRH-R2β immunoreactivity from the cell surface of 293-R2β cells compared with basal, whereas an equimolar concentration of CRH reduced CRH-R2β immunoreactivity by only 43–52% (Fig. 2C). Cell membrane-rich fractions were also prepared from cells treated with CRH or UCN-II (100 nm) for 30 min. CRH-R2 proteins were detected as a single immunoreactive protein with an apparent molecular mass of approximately 60 kDa, by Western blotting using a specific antibody raised against the CRH-R1/2 C terminus (Fig. 2C, inset). Densitometric analysis revealed UCN-II treatment decreased receptor expression in the cell membrane by 50–55% compared with untreated (control) cells, whereas CRH exerted a weaker effect (25–35% decrease), thus confirming the findings of the On-cell Western assay and the indirect fluorescence confocal microscopy. In the last set of experiments, the purity of the membrane-rich fractions and equal protein loading was confirmed by immunoblotting; results showed that all samples contained equal amounts of the plasma membrane protein cadherin, whereas glyceraldehyde-3-phosphate dehydrogenase (GAPDH), a cytosolic protein, was not detected in any of the samples (data not shown).
CRH-R2β Desensitization and Endocytosis and β-Arrestin
Immunostaining for β-arrestin (using an antibody that recognizes both β-arrestin1 and -2) demonstrated widespread distribution in 293-R2β cells, primarily in the cytoplasm. Treatment of cells with 100 nm UCN-II for 2–5 min induced a rapid redistribution of β-arrestin to the plasma membrane (Fig. 3, right column) that appear to colocalize with CRH-R2β receptors, as demonstrated by a significant increase in plasma membrane immunostaining of β-arrestin (green) signal and the appearance of yellow signals in the overlap images. Within 15 min of UCN-II treatment, there was evidence of some receptor trafficking in the cytoplasm, and after 30 min of treatment, intense receptor fluorescent signal was observed in the cytoplasm, indicative of substantial receptor internalization (Fig. 3, right column). Interestingly, receptor association with β-arrestin was transient and restricted on or near to the plasma membrane because CRH-R2β receptors internalized without any β-arrestin. Similar to UCN-II, CRH (100 nm) induced β-arrestin recruitment to the plasma membrane after 2 min after CRH treatment (Fig. 3, left column), although CRH-induced CRH-R2β receptor internalization was evident only after 30 min of treatment and did not colocalize with β-arrestin. These observations were confirmed by quantification of cell membrane green fluorescence (1–3 and 19–21 μm, respectively) and red fluorescence present in the intracellular space (4–18 μm) of 20 individual cells, which were randomly selected (Fig. 4).
Fig. 3.

CRH-R2β and β-Arrestin Subcellular Distribution after CRH or UCN-II Treatment: Visualization by Fluorescent Confocal Microscopy
HEK293 cells expressing CRH-R2β were stimulated with either CRH or UCN-II (100 nm) for 2–30 min. CRH-R2β and β-arrestin distribution was monitored over the ensuing time period by indirect double immunofluorescence using specific primary antibodies and Alexa Fluor 594 secondary antibodies for CRH-R2β (red) and Alexa Fluor 488 secondary antibody for β-arrestin (green). Identical results were obtained from four independent experiments.
Fig. 4.

Cytoplasmic CRH-R2β and Plasma Membrane β-Arrestin Distribution
Fluorescence intensity of cytoplasmic CRH-R2β and plasma membrane β-arrestin distribution from 20 individual cells in five random fields of view was examined, and the sum of fluorescence intensity of either cytoplasmic (distance 4–18 μm) or plasma membrane (1–3 and 19–21 μm) fluorescence was measured by the ImageJ software. Results are expressed as the mean ± sem of three estimations from 20 individual cells. *, P < 0.05 compared with basal.
Additional experiments were performed to characterize β-arrestin activation and membrane translocation in response to agonist-occupied receptor. Distribution of β-arrestin immunoreactivity was monitored in cell membrane fractions prepared from 293-R2β cells treated with either 100 nm CRH or UCN-II for various time intervals (0–30 min). In resting 293-R2β cells, small amounts of β-arrestin1 (49 kDa) and β-arrestin2 (47 kDa) were detected in the cell membrane fraction. UCN-II or CRH treatment for 2 min resulted in a 3- to 4-fold increase in the membrane fraction content of both β-arrestin1 and β-arrestin2 (Fig. 5A). This increase was transient, and after 10–15 min of UCN-II treatment, membrane levels of both β-arrestins were significantly reduced (by 70%). In contrast, after CRH (100 nm) treatment, substantial membrane levels of β-arrestin1 and β-arrestin2 were observed up to 15 min of treatment (2- and 3.5-fold above basal for β-arrestin1 and β-arrestin2, respectively) that were significantly decreased only after 30 min of CRH treatment. As described previously, the purity of cell membrane preparations was assessed by immunoblotting for the membrane protein cadherin, which appeared as a single band with an apparent molecular mass of approximately 140 kDa (Fig. 5A, bottom panel). In contrast, immunoreactive GAPDH was not detected in these preparations (data not shown).
Fig. 5.

Role of β-Arrestin in CRH-R2β Desensitization and Internalization in 293-R2β Cells
A, Agonist-dependent β-arrestin recruitment to the plasma membrane. Membrane fractions were prepared from 293-R2β cells stimulated with or without UCN-II or CRH (100 nm) for various time intervals, and proteins were resolved on SDS-PAGE gels, followed by immunoblotting with pan-arrestin antibodies, as described in Materials and Methods. Densitometry scanning was carried out to quantify agonist-induced β-arrestin translocation to the plasma membrane. Equal protein loading and membrane purity was confirmed by immunoblotting with pan-cadherin antibodies, as described in Materials and Methods. Top panel, Representative immunoblots; bottom panel, mean ± sem of three independent experiments. *, P < 0.05 compared with basal. B, Identification of β-arrestin and CRH-R2β complex formation by coimmunoprecipitation. 293-R2β cells were stimulated with UCN-II (100 nm) for 2 min before solubilization of membrane-rich fractions and immunoprecipitation with specific CRH-R1/2 antibodies. Proteins were resolved on SDS-PAGE gels, followed by immunoblotting with CRH-R1/2 (middle panel) or pan-arrestin (bottom panel) antibodies to identify potential complex formation. Total cell lysates were also prepared and immunoblotted with pan-arrestin antibodies (top panel). Representative immunoblots are presented. Identical results were obtained from four independent experiments. C and D, Effect of DN β-arrestin on UCN-II-induced CRH-R2β desensitization (C) and internalization (D) in 293-R2β cells. Cells were transfected with either 5 μg empty pcDNA3 vector (control) or β-arrestin (319–418). and the effect on CRH-R2β homologous desensitization (induced by pretreatment with 100 nm UCN-II for 30 min) was determined by measurement of UCN-II-induced cAMP production (C). Data represent the mean ± sem of two estimations from three independent experiments. *, P < 0.05 compared with cells without UCN-II pretreatment; +, P < 0.05 compared with control cells. Alternatively, internalization of UCN-II CRH-R2β was monitored by indirect confocal microscopy (B and C) using specific primary antibodies and Alexa Fluor 594 secondary antibody for CRH-R2β (red). Identical results were obtained from four independent experiments. At least 20 individual cells in five random fields of view were examined. Inset, Fluorescence intensity of cytoplasmic CRH-R2β distribution from 20 individual cells in five random fields of view was examined, and the sum of fluorescence intensity of cytoplasmic (distance 4–18 μm) fluorescence was measured by the ImageJ software. Results are expressed as the mean ± sem of three estimations from 20 individual cells. *, P < 0.05 compared with basal; +, P < 0.05 compared with control (empty vector) cells.
To investigate whether β-arrestin recruitment to the plasma membrane resulted in binary complex formation with the CRH-R2β receptor, membranes from 293-R2β cells, treated with or without UCN-II (100 nm) for 2 min, were solubilized, CRH-R2β receptors were immunoprecipitated by a specific CRH-R1/2 antibody, and β-arrestin copurified with CRH-R2β receptors was detected by immunoblotting using the pan-arrestin antibody. Immunodetection of β-arrestin (Fig. 5B, top panel) in total cell lysates showed equal amounts of β-arrestin present in UCN-II-treated or untreated cells. Furthermore, detection by immunoblotting of resolved CRH-R2β receptors in the immunoprecipitated membrane fractions showed a similar amount of immunoprecipitated receptors between UCN-II-treated or untreated cell membranes (Fig. 5B, middle panel). Under basal conditions, only small amounts of β-arrestin1 and -2 were found co-associated with the CRH-R2β receptors (Fig. 5B, bottom panel). In contrast, when the receptor was activated by UCN-II, there was a significant increase (4- to 5-fold above basal) in the amount of β-arrestin that could be immunoprecipitated with the receptor. β-Arrestin2 was the major β-arrestin subtype associated with the CRH-R2β receptor.
In addition, we used adenylyl cyclase activity assays and confocal microscopy to investigate whether the presence of β-arrestin is essential for CRH-R2β desensitization and internalization. For that purpose, we determined agonist-induced CRH-R2β desensitization and internalization in the presence of a dominant-negative (DN) β-arrestin, β-arrestin (319–418). In 293-R2β cells overexpressing DN β-arrestin, the ability of UCN-II pretreatment (100 nm for 30 min) to desensitize CRH-R2β responsiveness and cAMP activation was significantly impaired (by 50%) (Fig. 5C). Furthermore, UCN-II failed to induce significant CRH-R2β internalization (Fig. 5D), suggesting that CRH-R2β homologous internalization is β-arrestin dependent.
The relative contribution of β-arrestin1 and -2 in CRH-R2β desensitization and internalization was further assessed by RNA interference (RNAi) to deplete cells from either β-arrestin1 or -2 or both. Reduction of each β-arrestin subtype expression (in excess of 90%) in small interfering RNA (siRNA)-transfected cells was confirmed by immunoblotting (Fig. 6A). Adenylyl cyclase activity assays in siRNA-transfected cells demonstrated that the ability of UCN-II pretreatment (100 nm for 30 min) to desensitize CRH-R2β responsiveness and cAMP activation was significantly impaired in β-arrestin1 and β-arrestin2-depleted cells (by 30 and 65%, respectively) (Fig. 6B). Simultaneous depletion of both β-arrestin1 and -2 reversed UCN-II-induced desensitization almost completely. Moreover, confocal microscopy experiments demonstrated that UCN-II-induced CRH-R2β internalization (determined by quantification of cytoplasmic red fluorescent signal) was significantly impaired by 35–45 and 70–85% in β-arrestin1 and β-arrestin2-depleted cells, respectively (Fig. 6C). Depletion of both β-arrestin subtypes completely abolished agonist-induced internalization. Similar results were obtained when receptor expression was quantified by immunoblotting in solubilized cell membrane fractions of RNAi-treated cells (data not shown).
Fig. 6.
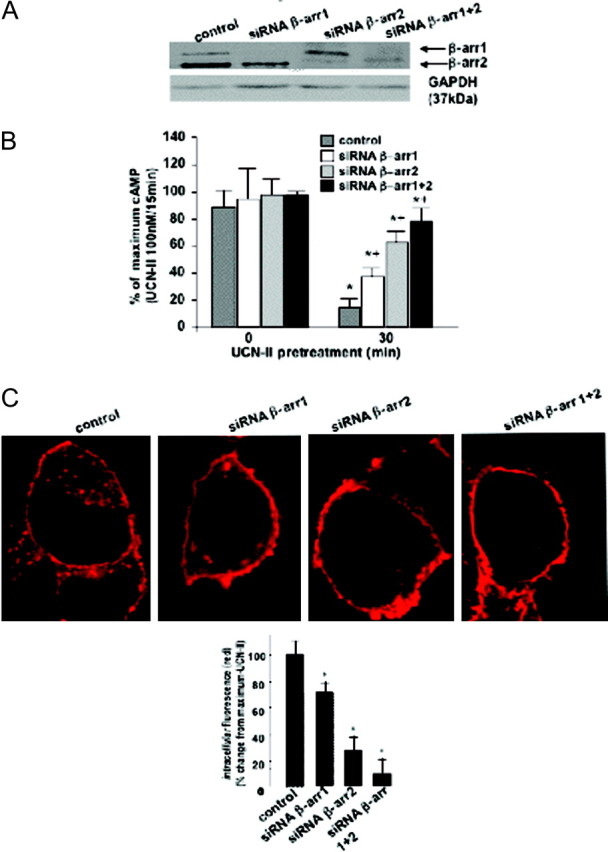
Effect of β-Arrestin RNAi on UCN-II-Induced CRH-R2β Desensitization and Internalization in 293-R2β Cells
A, Cells were transfected with 1 nmol siRNA oligonucleotides for β-arrestin1 or -2 or scrambled oligonucleotide, and the effect on β-arrestin1 or -2 protein expression was determined in cell lysates by immunoblotting with a pan-arrestin antibody. Level of expression of GAPDH was also determined by immunoblotting (bottom panel). B, CRH-R2β homologous desensitization (induced by pretreatment with 100 nm UCN-II for 30min) was determined by measurement of UCN-II-induced cAMP production. Data represent the mean ± sem of two estimations from three independent experiments. *, P < 0.05 compared with cells without UCN-II pretreatment; +, P < 0.05 compared with control cells. C, Alternatively, internalization of UCN-II CRH-R2β was monitored by indirect confocal microscopy using specific primary antibodies and Alexa Fluor 594 secondary antibody for CRH-R2β (red). Identical results were obtained from four independent experiments. At least 20 individual cells in five random fields of view were examined. Inset, Fluorescence intensity of cytoplasmic CRH-R2β distribution, from 20 individual cells in five random fields of view was examined, and the sum of fluorescence intensity of cytoplasmic (distance 4–18 μm) fluorescence was measured by the ImageJ software. Results are expressed as the mean ± sem of three estimations from 20 individual cells. *, P < 0.05 compared with control (scrambled sequence) cells.
CRH-R2β Desensitization and Endocytosis and Clathrin
Immunostaining for clathrin (heavy chain) demonstrated widespread distribution, primarily in the cytoplasm. Treatment of cells with 100 nm UCN-II for 2–5 min induced a rapid redistribution of clathrin to the plasma membrane (Fig. 7A). Similar results were obtained when CRH was used to stimulate 293-R2β cells (data not shown). Increased recruitment of clathrin to the plasma membrane (by 7-fold compared with basal) in response to UCN-II treatment for 2 min was also confirmed by detection of clathrin heavy chain (CHC) immunoreactivity in solubilized cell membrane fractions (Fig. 7A, inset).
Fig. 7.
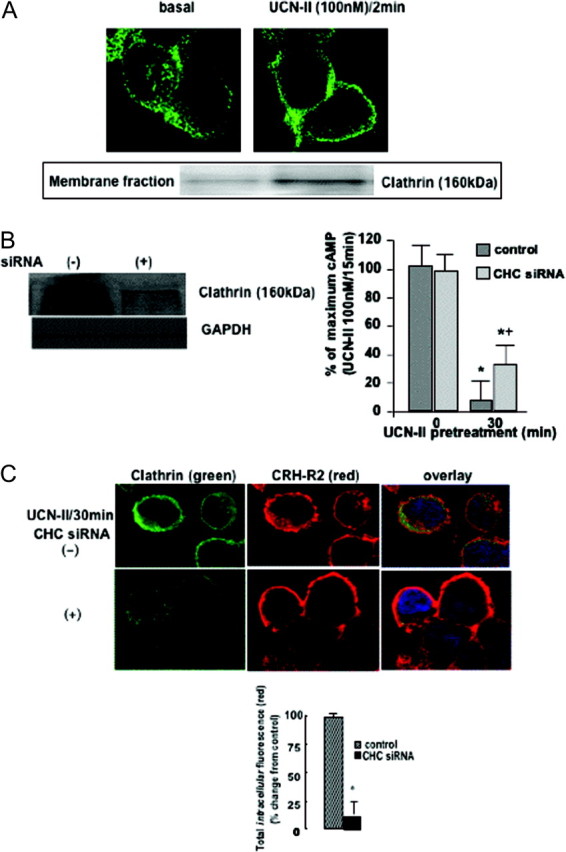
Role of Clathrin on UCN-II-Induced CRH-R2β Desensitization and Internalization in 293-R2β Cells
A, Clathrin membrane translocation after UCN-II treatment. HEK293 cells expressing CRH-R2β were stimulated with UCN-II (100 nm) for 2 min and CHC distribution was monitored over the ensuing time period by indirect immunofluorescence using specific primary antibodies and Alexa Fluor 488 secondary antibody (green). Identical results were obtained from four independent experiments. Also, CHC immunoreactivity was determined in membrane-rich fractions immunoblotted with antibodies against CHC (inset). Identical results were obtained from four independent experiments. B and C, Effect of CHC siRNA on UCN-II induced CRH-R2β desensitization (B) and internalization (C). 293-R2β Cells were transfected with 1 nmol CHC siRNA or scrambled oligonucleotide, and the effect on CHC protein expression was assessed by immunoblotting of total cell lysates with specific CHC or GAPDH antibodies. CRH-R2β homologous desensitization (induced by pretreatment with 100 nm UCN-II for 30 min) was also determined by measurement of UCN-II-induced cAMP production (B). Data represent the mean ± sem of two estimations from three independent experiments. *, P < 0.05 compared with cells without UCN-II pretreatment; +, P < 0.05 compared with control cells. Alternatively, internalization of UCN-II CRH-R2β was monitored by indirect confocal microscopy (C) using specific primary antibodies and Alexa Fluor 594 secondary antibody for CRH-R2β (red) and Alexa Fluor 488 secondary antibody for CHC (green). Identical results were obtained from four independent experiments. Scale bar, 20 μm. At least 20 individual cells in five random fields of view were examined. Inset, Fluorescence intensity of cytoplasmic CRH-R2β distribution from 20 individual cells in five random fields of view was examined, and the sum of fluorescence intensity of cytoplasmic (distance 4–18 μm) fluorescence was measured by the ImageJ software. Results are expressed as the mean ± sem of three estimations from 20 individual cells. *, P < 0.05 compared with control (scrambled sequence) cells.
The role of clathrin on CRH-R2β desensitization and internalization was also evaluated by RNAi to deplete cells from CHC and prevent assembly of functional clathrin-coated pits at the plasma membrane. Significant reduction (in excess of 70%) of CHC protein expression in siRNA-transfected cells was confirmed by immunoblotting of total cell lysates with antibodies directed against the CHC (Fig. 7B, left panel). In 293-R2β cells transfected with CHC siRNA, the ability of UCN-II pretreatment (100 nm for 30 min) to desensitize CRH-R2β responsiveness and cAMP activation was significantly impaired (by 25–35%) (Fig. 7B). Confocal microscopy experiments demonstrated that siRNA-treated cells exhibited a significant reduction in CHC immunofluorescent signal intensity, and this was associated with an almost complete absence of red fluorescent signal in the cytoplasm, indicative of inhibition of CRH-R2β internalization after UCN-II treatment (Fig. 7C). These observations were confirmed by using the Image J software and quantification of intracellular fluorescence spectra (4–18 μm) of 20 individual cells, which were randomly selected (Fig. 7C, inset).
CRH-R2β Endocytosis and Regulation of UCN-II-Induced MAPK Activation
Because GPCR internalization can potentially lead to activation of the MAPK signaling cascade, we further investigated the possibility that CRH-R2β endocytosis is involved in UCN-II-induced activation of ERK1/2 and p38MAPK signaling cascades. In preliminary experiments, the concentration dependency and spatiotemporal characteristics of ERK1/2 and p38MAPK activation by UCN-II were determined. In 293-R2β cells, UCN-II induced increases in both phospho-ERK1/2 and phospho-p38MAPK immunoreactivity in a concentration-dependent manner. The UCN-II effect on both ERK1/2 and p38MAPK activation was significant at concentrations greater than 1 nm and was maximal at concentrations of 100 nm; maximal ERK1/2 activation was 8- to 10-fold above basal levels, whereas maximal p38MAPK activation was found to be 2- to 2.5-fold above basal (data not shown). In addition, UCN-II induced ERK1/2 activation was transient, and maximal stimulation was observed after 5 min of treatment and returned to basal levels after 30 min of treatment (Fig. 8A). The UCN-II effect on p38MAPK phosphorylation showed a similar response with maximal stimulation after 5–10 min of treatment and return to basal levels after 30 min of treatment (Fig. 8B).
Fig. 8.
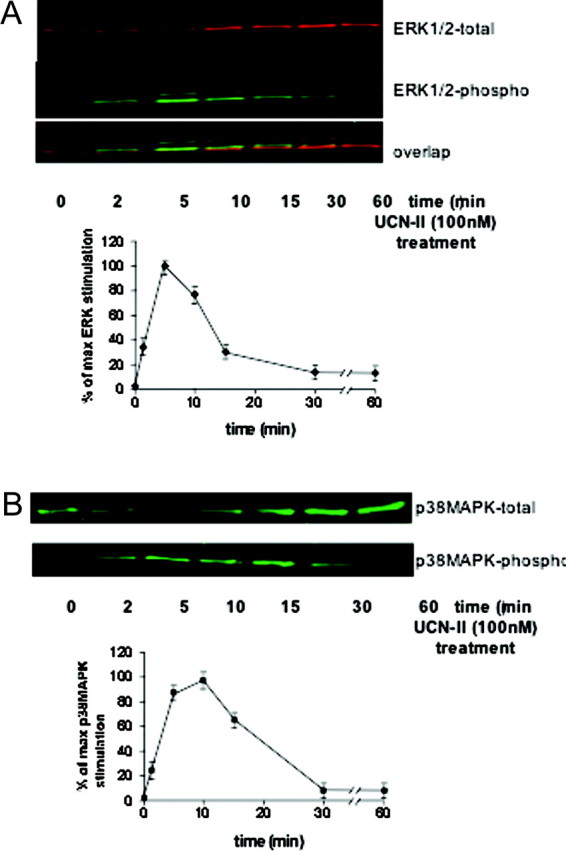
Time-Dependent ERK1/2 (A) and p38 MAPK (B) Activation by UCN-II in 293-R2β Cells
Top panels are representative Western blots of cells stimulated with UCN-II (100 nm) for various time points (2–60 min). After cell lysis and centrifugation, supernatants were subjected to SDS-PAGE and immunoblotted with antibodies for phospho-ERK1/2 and total ERK1/2 to determine the phosphorylated/activated ERK1/2 and secondary antibodies conjugated to IRDye800 and Alexa Fluor 680 (near-IR fluorophore dyes) as described in Materials and Methods. Alternatively, samples were immunoblotted with antibody for phospho-p38MAPK and total p38MAPK. Data represent the mean ± sem of three estimations from three independent experiments.
To examine the spatiotemporal characteristics of MAPK activation, indirect immunofluorescence confocal microscopy was employed with phospho-specific MAPK antibodies to monitor the relative subcellular distribution of activated MAPK and CRH-R2β after agonist stimulation (Fig. 9). In unstimulated 293-R2β cells, low levels of activated (phosphorylated) ERK1/2 and p38MAPK were found in the cytoplasm. As expected, CRH-R2β was primarily localized in the plasma membrane. UCN-II treatment for 5–10 min led to a rapid increase in the amount of fluorescent signal for both phospho-ERK1/2 and p38MAPK, indicating increased activity. Phospho-ERK1/2 immunoreactive signal was widespread throughout the intracellular space, suggesting cytoplasmic as well as nuclear localization of activated ERK1/2. In contrast, phospho-p38MAPK immunoreactivity was present only in the cytoplasm, and no phospho-p38 MAPK immunoreactivity was detected in the nucleus. In agreement with our immunoblotting experiments suggesting transient MAPK activation, prolonged treatment (15–20 min) with UCN-II (100 nm), sufficient to induce CRH-R2β internalization, caused only a small increase in the amount of phospho-ERK1/2 and p38MAPK immunofluorescent signal, reflecting deactivation of the signaling pathway. Moreover, no colocalization between internalized CRH-R2β and phospho-ERK1/2 or p38MAPK was found to be present (Fig. 9).
Fig. 9.

CRH-R2β and Phospho-ERK1/2 (A) or Phospho-p38MAPK (B) Subcellular Distribution Induced by UCN-II in 293-R2β Cells: Visualization by Confocal Microscopy
The 293-R2β cells were stimulated with or without UCN-II (100 nm) for various time intervals (5–20 min). CRH-R2β and phospho-ERK1/2 or phospho-p38MAPK distribution was monitored over the ensuing time period by indirect double immunofluorescence using specific primary antibodies for CRH-R2β and Alexa Fluor 594 secondary antibody (red) and Alexa Fluor 488 secondary antibody for phospho-ERK1/2 or p38MAPK (green) as described in Materials and Methods. Cell nuclei were stained with the DNA-specific dye DAPI (blue). Identical results were obtained from four independent experiments, and at least 20 cells were examined in each experiment. Scale bar, 20 μm.
We next investigated whether UCN-II-induced ERK1/2 and p38MAPK activation is sensitive to disruption of the CRH-R2β internalization pathways. 293-R2β cells were pretreated with concanavalin A, which blocks receptor clustering. Alternatively, cells were transfected with either DN β-arrestin (319–418) or siRNA duplexes targeting CHC. All compounds strongly inhibited agonist-stimulated CRH-R2β internalization (see Figs. 4D and 7C). However, these compounds exerted no inhibitory effects on ERK1/2 and p38MAPK activation (Fig. 10); in contrast, disruption of CRH-R2β internalization by β-arrestin (319–418) and CHC siRNA amplified by 70 and 30%, respectively, ERK1/2 phosphorylation stimulated by 100 nm UCN-II. Studies on UCN-II-induced p38MAPK activation showed similar effects. These results suggested that an intact receptor endocytosis pathway is not essential for UCN-II-induced MAPK activation.
Fig. 10.

Effect of Concanavalin, DN β-Arrestin, and CHC siRNA on UCN-II-Induced ERK1/2 and p38MAPK Activation in 293-R2β Cells
Cells were pretreated with or without concanavalin A (0.25 mg/ml for 40 min) or alternatively transfected with 5 μg of either empty pcDNA3 vector (control) or β-arrestin (319–418) or 1 nmol CHC siRNA or scrambled oligonucleotide,and the effect on UCN-II (100 nm for 5 in) stimulation on ERK1/2 and p38MAPK activation was determined, by measurement of ERK1/2 and p38MAPK phosphorylation as described in Materials and Methods. Data represent the mean ± sem of two estimations from three independent experiments. *, P < 0.05 compared with control cells.
The Role of the TAAV Amino Acid Cassette in the in CRH-R2β C Terminus in Receptor Signaling and Endocytosis
The CRH-R1 and -R2 receptors share considerable amino acid homology between their C termini. A notable diversity is found within the last four amino acid residues of the C terminus (TAAV instead of STAV). In contrast to the CRH-R2, the CRH-R1-MAPK interactions display different spatiotemporal characteristics involving β-arrestin-dependent pathways; thus, we explored the possibility that this amino acid cassette plays a role in determining CRH-R2β desensitization and internalization characteristics as well as MAPK activation. We used site-directed mutagenesis to create CRH-R2β receptors, in which the cassette TAAV was replaced by AAAA or STAV or deleted (Del) (Fig. 11, inset). Wild-type and mutant CRH-R2β receptors, CRH-R2β(STAV), CRH-R2β(4A), and CRH-R2β(Del), were transiently expressed in HEK293 cells. These mutant receptors were expressed normally at the cell membrane (Fig. 11A) and were able to bind human [125I-]UCN-II with binding affinity and maximal binding capacity that were similar to that of the wild-type CRH-R2β (data not shown). All mutant receptors were able to stimulate intracellular cAMP production in response to 100 nm UCN-II (Fig. 11B). Interestingly, although CRH-R2β(4A) and CRH-R2β(Del) potency in stimulating cAMP response was comparable to the wild-type receptor, the CRH-R2β(STAV) cAMP response was 50% greater than the wild type CRH-R2β. Furthermore, the sensitivity of both wild-type and mutant receptors to desensitization, induced by UCN-II (100 nm) pretreatment for 30 min, was comparable (Fig. 11B). Immunoprecipitation of membrane CRH-R2β-β-arrestin binary complexes from cell membrane-rich fractions by using a CRH-R1/2 antibody followed by immunoblotting using a pan-arrestin antibody showed that wild-type and mutant receptors exhibited comparable agonist-induced β-arrestin recruitment to the plasma membrane and interaction with the CRH-R2β (Fig. 11C). Surprisingly, confocal microscopy studies showed that the CRH-R2β(4A) and CRH-R2β(Del) mutant receptors exhibited distinct temporal internalization characteristics and increased rate of internalization compared with the wild-type receptor; significant receptor (red) fluorescent signal was observed in the cytoplasm after only 5 min of UCN-II (Fig. 12). This was confirmed by determination of CRH-R immunoreactivity in cell-membrane-rich fractions prepared from UCN-II-treated 293-R2β cells for various time points (data not shown).
Fig. 11.
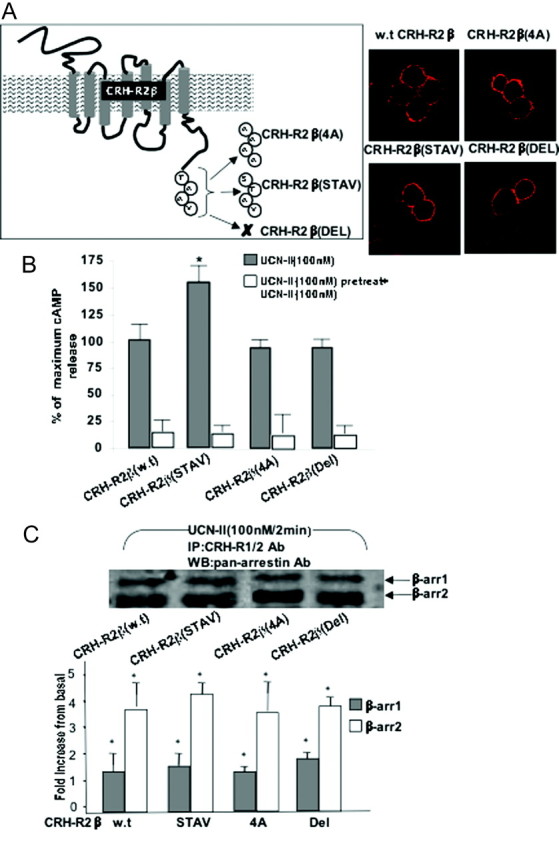
Role of CRH-R2β C-Terminal Amino Acid Cassette TAAV on Receptor Desensitization and β-Arrestin Recruitment
A, Receptor (wild-type or mutant) expression was monitored by indirect fluorescent confocal microscopy using specific primary antibodies and Alexa Fluor 594 secondary antibodies for CRH-R2β (red). Identical results were obtained from three independent transfections. B, Alternatively, cells were pretreated with UCN-II (100 nm) for 30 min to induce desensitization. After extensive washing, cAMP response to a second UCN-II stimulus (100 nm for 15 min) was determined. Data represent the mean ± sem of two estimations from three independent experiments. *, P < 0.05 compared with wild type CRH-R2 cAMP response. C, To identify mutant CRH-R2β and β-arrestin complex formation, 293-R2β cells transiently expressing wild-type or mutant CRH-R2β were stimulated with UCN-II (100 nm) for 2 min before solubilization of membrane-rich fractions and immunoprecipitation with specific CRH-R1/2 antibodies. Proteins were resolved on SDS-PAGE gels, followed by immunoblotting with pan-arrestin (bottom panel) antibodies to identify potential complex formation. Representative immunoblots are presented. Identical results were obtained from four independent experiments. Quantification was carried out by densitometry scanning. Data represent the mean ± sem of two estimations from three independent experiments. *, P < 0.05 compared with basal.
Fig. 12.
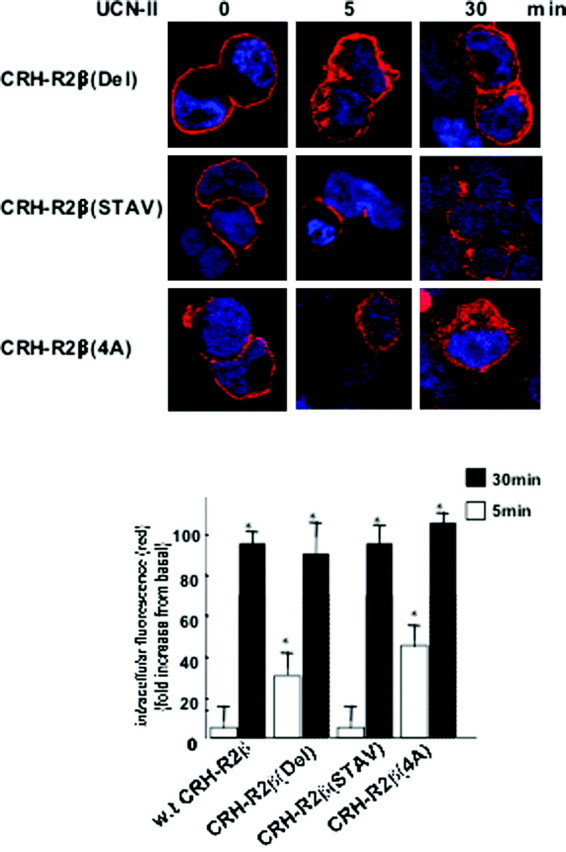
Role of CRH-R2β C-Terminal Amino Acid Cassette TAAV on Receptor Endocytosis
Cells were stimulated with 100 nm UCN-II for various time intervals, and CRH-R2β internalization was monitored by indirect confocal microscopy as described in Materials and Methods. Cell nuclei were stained with the DNA-specific dye DAPI (blue). Identical results were obtained from four independent experiments, and at least 20 cells were examined in each experiment. Inset, Fluorescence intensity of cytoplasmic CRH-R2β distribution. The sum of fluorescence intensity of cytoplasmic (distance 4–18 μm) fluorescence was measured by the ImageJ software. Results are expressed as the mean ± sem of three estimations from 20 individual cells. *, P < 0.05 compared with basal (unstimulated).
The temporal characteristics of ERK1/2 and p38MAPK activation of the CRH-R2β mutant receptors were also determined. The CRH-R2β(STAV) mutant receptor showed comparable potency to the wild-type receptor in activating both ERK1/2 (Fig. 13A) and p38MAPK (Fig. 13B) in response to UCN-II. In contrast, the ability of CRH-R2β(4A) and CRH-R2β(Del) to induce ERK1/2 and p38MAPK activation was significantly impaired by 35 and 20%, respectively. For all mutant receptors examined, no difference in the temporal characteristics of ERK1/2 p38MAPK phosphorylation was observed.
Fig. 13.

Time Course of UCN-II-Stimulated ERK1/2 (A) and p38MAPK (B) Phosphorylation in HEK293 Cells Transiently Expressing Wild-Type or Mutant CRH-R2β
HEK293 cells transiently expressing wild-type or mutant CRH-R2β receptors were treated with or without UCN-II (100 nm) for various time intervals to induce MAPK activation. After cell lysis and centrifugation, supernatants were subjected to SDS-PAGE and immunoblotted with antibodies for phospho-ERK1/2 and total-ERK1/2 to determine the phosphorylated/activated ERK1/2 and secondary antibodies conjugated to IRDye 800 and Alexa Fluor 680 (near-IR fluorophore dyes) as described in Materials and Methods. Alternatively, samples were immunoblotted with antibodies for phospho-p38MAPK and total p38MAPK. The data represent the mean ± sem of three estimations from three independent experiments.
DISCUSSION
The present study investigated the intracellular mechanisms regulating CRH-R2 responsiveness to agonist stimulation and its potential link to activation of distinct signaling cascades. Our studies provided novel evidence that the CRH-R2β functional activity is sensitive to homologous desensitization after UCN-II binding and receptor activation. CRH-R2β desensitization leads to significant down-regulation of cAMP production of the main intracellular mediators of CRH-R2 signaling in tissues (2). Desensitization of the CRH-R2β appears to be considerably rapid, and exposure to UCN-II for 15 min was sufficient to diminish receptor activity by 80–90%. This is remarkably different from the response of the CRH-R1α, which required agonist (CRH) treatment for 2–3 h to achieve a similar level of desensitization (18), and might reflect distinct requirements of the two CRH-Rs’ signal propagation in mammalian pathophysiology.
Investigations on the molecular mechanisms downstream of CRH-R2β desensitization showed that UCN-II induces a rapid and transient recruitment of both β-arrestin1 and -2 to the plasma membrane, a step also important for receptor endocytosis. Many other GPCRs that belong to the subfamily B1 (brain-gut neuropeptide receptors) exhibit high affinity for both β-arrestin1 and -2. Selective targeting of each β-arrestin subtype suggested that β-arrestin2 is the major subtype interacting with the CRH-R2β at the plasma membrane, similar to the CRH-R1α (24), although it appears that both β-arrestins are involved in CRH-R2β desensitization and endocytosis.
β-Arrestins, by binding to clathrin, promote GPCR endocytosis via clathrin-coated pits (25); the mechanism of CRH-R2β endocytosis also appears to require clathrin, which rapidly translocates to the plasma membrane after CRH-R2β activation, because depletion of clathrin significantly impairs CRH-R2β endocytosis. These findings suggest that CRH-R2β endocytosis might occur via clathrin-coated pits, similar to CRH-R1α (20). However, no receptor colocalization with β-arrestins inside the cells was observed, a characteristic of class A GPCRs. This is in contrast to the mixed class A/class B picture previously reported for the CRH-R1α-β-arrestin interactions (19). This might have important consequences on the fate of internalized receptor, because lack of interaction between β-arrestins and internalized class A GPCRs allows a more rapid recycling and resensitization by facilitating receptor dephosphorylation (26).
Our studies on CRH-R2β desensitization and endocytosis also revealed a number of interesting features. First, the efficiency and kinetics of this process appear to be dependent on CRH-R2β-agonist potency because CRH, which has a low affinity for the receptor, induced a weaker desensitization and delayed receptor endocytosis compared with UCN-II. The temporal characteristics of β-arrestin translocation to the plasma membrane also appeared to be differentially regulated by UCN-II and CRH, and although both peptides induced rapid translocation of β-arrestins and clathrin, CRH actions led to a prolonged association of β-arrestins to the membrane. At present, the precise functional consequences of these differences are not known; however, it is possible that these distinct characteristics are linked to differences in receptor active conformations in response to UCN-II or CRH that potentially lead to diverse signaling pathways in agreement with the hypothesis of agonist-directed trafficking (27). Second, the processes of CRH-R2β receptor desensitization and endocytosis appeared to be intrinsically linked, because molecular inhibitors of receptor internalization such as DN β-arrestin (319–418), which bind clathrin and prevent receptor from being targeted to clathrin-coated pits (28), are able to inhibit receptor desensitization. This effect on GPCR desensitization has been demonstrated for other receptors (29). Our data also suggest that receptor internalization might be important in receptor desensitization. It is possible that both agonist-bound and unbound (activated and resting) receptors become internalized as one single cohort of receptors. Thus, rather than affecting desensitization per se, inhibition of CRH-R2β internalization may increase the number of available resting receptors resulting in increased response to further agonist stimulation.
However, our studies suggest discrete differences on the receptor structural determinants involved in the mechanisms regulating CRH-R2β desensitization and internalization. Our mutant receptor studies showed that amino acid residues within the cassette -TAAV, present at the end of the CRH-R2β receptor cytoplasmic tail, appear to be important for determining the rate of receptor endocytosis, but not β-arrestin recruitment to plasma membrane, receptor association, and receptor desensitization as well as activation of the cAMP pathway. Interestingly, the C-tail of CRH-R1α, which exhibits a slower rate of desensitization and endocytosis, contains one extra Ser/Thr residue (-STAV). Our mutagenesis studies suggest that lack of potential phospho-acceptor residues in this amino acid cassette results in accelerated receptor endocytosis. The molecular determinants of this are unknown; it is possible that absence of Ser/Thr residues from the end of the C-tail might facilitate interaction and association with signaling molecules involved in receptor endocytosis. Future studies will test this hypothesis.
We also found that UCN-II binding to CRH-R2β leads to a robust activation of ERK1/2 and p38MAPK, important signaling cascades for mediating CRH-R2 physiological effects (11, 12, 13). In particular, in certain cellular models, p38MAPK but not ERK1/2, is capable of modulating CRH-R2β expression and signaling because it has the potential to down-regulate CRH-R2β mRNA levels (30). Furthermore, previous studies on recombinant CRH-R1α expressed in HEK293 cells, (21) directly linked CRH-R1α trafficking and endocytosis to activation of ERK1/2 and p38MAPK. However, our present studies do not support a direct role for receptor endocytosis-dependent pathways in CRH-R2β-mediated ERK1/2 and p38MAPK activation. However, the amount of receptors in the cell membrane accessible for activation appears to determine the overall magnitude of MAPK response, because inhibition of receptor endocytosis (by DN β-arrestin and depletion of clathrin) enhanced by 30–40% UCN-II MAPK activation, whereas faster receptor internalization rate, such as seen with mutant CRH-R2β receptors [CRH-R2β(4A), and CRH-R2β(Del)], results in decreased MAPK response to UCN-II. In the latter, the potential effect of mutating the CRH-R2β C-tail on G-protein coupling should also be considered as a contributing factor to the impaired MAPK response to UCN-II.
CRH-R2β-mediated ERK1/2 and p38MAPK activation was characterized by 1) a transient MAPK phosphorylation response, 2) a lack of association between internalized CRH-R2β receptors and phospho-ERK1/2 or p38MAPK, and 3) cytoplasmic as well as nuclear distribution. Current hypotheses on GPCR-ERK1/2 activation suggest that G protein-dependent pathways produce a transient activation of nuclear ERK1/2, whereas β-arrestin-dependent pathways lead to sustained activation of ERK1/2 that is localized to the cytosol and endosomes (31). Thus, the absence of receptor endocytosis-dependent pathways in CRH-R2β-mediated ERK1/2 and p38MAPK activation might explain the distinct spatiotemporal characteristics of CRH-R2β-mediated ERK1/2 phosphorylation. The previously reported CRH-R1α-MAPK interactions also seem to follow these principles; the CRH-R1α mediates a sustained ERK1/2 activation that is β-arrestin dependent and is restricted primarily to the cytoplasm in stable complex formation with internalized CRH-R1α (21).
In summary, we demonstrated that the CRH-R2β receptor desensitization involves β-arrestin recruitment to the plasma membrane and depends on the binding and signaling potency of the desensitizing agonist. Subsequently, the receptor internalizes via a β-arrestin- and clathrin-dependent mechanism. The kinetics and pattern of trafficking partners differ significantly from the corresponding mechanism regulating CRH-R1α functional activity, a finding that might have important implications for the elucidation of CRH and UCN signal propagation in mammalian pathophysiology. Furthermore, we showed that this mechanism is not involved in the activation of ERK1/2 and p38MAPK cascades, and this functional independence allows distinct spatiotemporal control of MAPK activity by UCNs and CRH-R2β .
MATERIALS AND METHODS
Chemicals
CRH, UCN-I, and UCN-II were purchased from Bachem UK Ltd. (Helens, Merseyside, UK). Radiodinated rat [tyr°]UCN-I was obtained from Amersham (GE Healthcare UK Ltd., Little Chalfont, Buckinghamshire, UK). U0126 (MEK inhibitor) and forskolin were from Calbiochem/Merck Biosciences (Nottingham, UK). CRH-R1/2, CRH-R2 antibody, and their blocking peptides were from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-ERK1/2 (Thr202/Tyr204), total ERK1/2, phospho-p38MAPK (Thr180/Tyr182), total p38MAPK, and phospho-β-arrestin1 (Ser 412) were from Cell Signaling (Chandlers Ford, Hampshire, UK). The N terminus CRH-R2 antibody, pan-arrestin, and total β-arrestin1 antibodies were from Abcam (Cambridge, UK). Secondary antibodies Alexa Fluor 405, Alexa Fluor 488, Alexa Fluor 594, Alexa Fluor 680, and Gold Slowfade mounting solution with 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Invitrogen/Molecular Probes (Paisley, UK), and IRDye 800-conjugated goat antirabbit IgG was from Rockland Immunochemicals (Gilbertsville, PA). Vectashield mounting medium without DAPI was from Vector Laboratories, Inc. (Orton Southgate, Peterborough, UK). Cell culture media, gentamicin (G-418), Lipofectamine 2000, pcDNA3.1(+), restriction enzymes, and Pfu polymarase were from GIBCO/Invitrogen (Paisley, UK). dNTPs and DNA ladder were purchased from Bioline Ltd. (London, UK). Primers were purchased from TANG (Gateshead, UK). All other chemicals were purchased from Sigma Aldrich Co. Ltd. (Gillingham, UK).
CRH-R2β RT-PCR and Site-Directed Mutagenesis
Total RNA from 293 or 293-R2β cells was extracted by using GeneElute Mammalian Total RNA Kit (Sigma), according to the manufacturer’s instructions. The purity and concentration of total RNA were determined spectroscopically, with 0.2–1 μg RNA (1–3 μl), 1.5 μl random hexamers (Promega, Madison, WI), and up to 16.5 μl of molecular biology-grade water mixed and heated at 70 C for 5 min and then allowed to return to room temperature. Then, the RNA was centrifuged and the following mixture was added in: 3 μl dNTP (10 mm), 6 μl 5× buffer, 0.5 μl avian myeloblastosis virus (25 U/μl), 1.5 μl RNasin, and 2.5 μl water. For cDNA synthesis, the mixture was heated to 37 C for 60 min, 95 C for 5 min, and 4 C for 5 min. The resulting cDNA was stored at −20 C. CRH-R2β PCR assays were performed as previously described (15).
Site-directed mutagenesis was used to create CRH-R2β receptors, in which the cassette TAAV at the end of C-tail was replaced by AAAA, STAV, or deleted (Del) [CRH-R2β(STAV), CRH-R2β(4A), and CRH-R2β(Del)]. Human CRH-R2β cDNA was subcloned into the mammalian cell expression vector pcDNA3.1(+) (Invitrogen Life Technologies) and was used as a template for mutagenesis using Pfu polymerase. The PCR mixture (20 μl) contained 5 ng cDNA, 5 ng/μl of each primer, 1 U Pfu polymerase, and 20 nm of each dNTP. cDNAs were amplified at 48 C in a total of 35 cycles. The forward primer contained a sequence for EcoRI restriction site adjusted to the 5′-end of the receptor sequence 5′-ATTCCGATGAGGGGTCCCTCAGGGCCCCCAGG-3′, whereas the reverse primer was specific for each mutant and contained the sequence for XhoI restriction site (CRH-R2β-STAV, 5′-CCGCTCGAGCGGGTCACACAGCGGTCGACTGCTTGATGCTGTGGAAGCT-3′; CRH-R2β-Ala, 5′-CC GCTCGAGCGGGTC-ACGC-AGCGGCCGCCTGCTTGATGCTGTGGAGCT-3′, CRH-R2β-Del, 5′-TCGAGCGGGTCACTGCTTGATGCTGTGGAAG-CT-3′). The resulting PCR products were run on 1.2% agarose gel, purified using QIAquick gel extraction kit (QIAGEN Ltd., Crawley, West Sussex, UK), digested with EcoRI and XhoI, purified again, and ligated into pcDNA3.1(+) using FastLiga System from Promega according to manufacturer’s instruction. Escherichia coli DH-5α was transformed with the resulting plasmids, and after selective overnight growth on LB plates containing 100 μg/ml ampicillin, five colonies were picked up for further analysis. The entire regions amplified by PCR were sequenced to ensure the fidelity of the mutant cDNAs and confirm presence of mutations. DNA sequence analysis was performed by the Core Facility of the Department of Biological Sciences, University of Warwick.
Transfection of CRH-R2β and DN β-Arrestin to HEK293 Cells
Human wild-type or mutant CRH-R2β or DN β-arrestin cDNAs subcloned in pcDNA3.1(+) were transfected in HEK293 cells using Lipofectamine 2000 reagent, according to the manufacturer’s protocol. Briefly, 5 μg DNA was mixed with 5μl Lipofectamine 2000 reagent and added to 50–70% confluent cells seeded in 25-cm2 flasks in 5 ml OptiMEM + GlutaMax (Invitrogen/GIBCO) for overnight transfection. The next morning, the transfection mixture was replaced with high-glucose DMEM with Glutamax containing 10% fetal calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Eight hours later, cells were transferred onto 12-well plates coated with 100 μg/ml poly-d-lysine in PBS or on coated glass coverslips (for confocal microscopy studies). All required experiments were carried out 48–96 h after transfection. For HEK293 stably expressing CRH-R2β (293-R2β), 10 μg cDNA was transfected into HEK293 cells grown in 75-cm2 vented flasks using Lipofectamine 2000, as described above. After 3 d of nonselective growth in normal growth media, followed by 15 d growth in media containing 500 μg/ml gentamicin (G418; GIBCO), clones were selected by serial dilution of surviving foci and maintained in 250 μg/ml gentamicin. The optimal concentration of gentamicin and the length of cell growth was determined by growing nontransfected HEK293 cells in media containing different concentrations of the antibiotic (50–1000 μg/ml) for 21 d and performing a cell viability tetrazolium salt 3,[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) (thioazolyl blue) assay. After 2 months, the expression of CRH-R2β was verified by RT-PCR, immunoblotting, confocal microscopy analysis, and functional assays (including cAMP production and ERK activation). At least two clones were selected and used for further studies. HEK293 cells stably expressing recombinant CRH-R2β (293-R2β) were maintained in high-glucose DMEM with Glutamax containing 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 250 μg/ml gentamicin.
Clathrin and β-Arrestin1 and -2 siRNA Transfection to 293-R2β
Three sets of CHC Stealth RNAi (3216–3241 oligo 1, 3543–3528 oligo 2, and 3928–3953 oligo 3) were obtained from Invitrogen. β-Arrestin1 and -2 siRNA (32) were purchased from QIAGEN (West Sussex, UK). AllStars-negative siRNA with a 3′-Alexa Fluor 488 modification (QIAGEN) was used as a control. One day before transfection, 293-R2β cells were plated in six-well plates in normal growth medium without any antibiotics. Lipofectamine 2000 was used for the delivery of 0.5 and 1 nmol siRNA into cells according to the manufacturer’s instructions. Subsequent experiments were performed 48 h after transfections.
CRH-R2 Radioreceptor Assay
Radioiodinated rat [tyr°]UCN binding characteristics in st.293-R2β or HEK293 cells transiently expressing wild-type or mutant CRH-R2β receptors were determined as previously described (19). The binding data were analyzed using the EBDA program (33) and LIGAND (34) (EBDA/LIGAND; Elsevier-Biosoft, Cambridge, UK)
Receptor Desensitization/cAMP Studies
Cells were grown in 12-well plates and when 70–80% confluency was reached , medium was aspirated and the cells washed with plain DMEM. Stimulation buffer (DMEM containing 10 mm MgCl2 and 0.1 mg/ml 3-isobutyl-1-methylxanthine) was added to the cells for 20 min at 37 C, and then cells were washed with plain DMEM and stimulated with 1–100 nm UCN-II, CRH, or 10 μm forskolin for 15 min. The reaction was stopped by adding 10 μl concentrated HCl for 15–20 min, and cells were transferred in 1.5-ml tubes. After a brief spin, the cells and media were stored at −20 C. The cAMP levels were determined by commercially available ELISA Direct cAMP Enzyme Immunoassay Kit (Assay Designs Inc., Ann Arbor, MI).
Receptor desensitization was assessed as previously described (18). Briefly, cells plated in 12-well plates were pretreated for various time intervals (0–30 min) with 100 nm UCN-II or CRH, and after a brief wash, cells were stimulated with 100 nm UCN-II for 15 min. Intracellular cAMP levels were measured as described above.
Receptor Desensitization/Endocytosis Studies and Western Blot Analysis
Cells reached 60–70% confluency and were serum starved overnight. The next morning, appropriate inhibitors were added (concanavalin A at 0.25 mg/ml for 40 min, U0126 at 10 μm for 1 h) followed by stimulation of cells with different concentrations (1–100 nm) of UCN-II or CRH for various time periods (2–60 min). At the end the stimulation, medium was removed, and cells were briefly washed with ice-cold PBS and then lysed by addition of 150 μl 2× SDS-PAGE sample buffer (Sigma). Solubilized material was collected, sonicated for 20 sec, and boiled for an additional 5min. After a brief centrifugation step, protein lysates were stored at −20 C until used. Before electrophoresis, protein extracts were centrifuged at 4000 rpm for 5 min to remove insoluble material, and 15 μl of the supernatant was loaded on 10% SDS-PAGE gels. For β-arrestin1 phosphorylation studies, cells were lysed in 100 μl RIPA buffer containing protease and phosphatase inhibitors cocktail (Santa Cruz). Protein concentration was determined with BCA Protein Assay Kit (Pierce, Rockford, IL), and 5 or 10 μg proteins were loaded on 10% SDS-PAGE gels that were run at 200 V for approximately 1 h. Proteins were electrophoretically transferred to polyvinylidene difluoride membrane at 100V for 1 h. After completion of transfer, the polyvinylidene difluoride membrane was incubated with 8–10 ml LI-COR blocking buffer (LI-COR Biosciences, Cambridge, UK) for 1 h at room temperature with gentle agitation. To determine ERK1/2 phosphorylation, the membrane was incubated simultaneously with the phospho-ERK1/2 (Thr202/Tyr204) antibody and total ERK2 antibody (dilutions 1:1500 and 1:2000, respectively) in 10 ml LI-COR blocking buffer with gentle agitation overnight at room temperature. For β-arrestin1 phosphorylation, membranes were incubated simultaneously with phospho-β-arrestin1 (Ser412) and total β-arrestin1 antibodies (dilutions 1:500 and 1:1000, respectively) in 10 ml LI-COR blocking buffer with gentle agitation overnight at 4 C. The next day, the antiserum was removed and the membrane was three times for 10 min each with 15 ml Tris-buffered saline (TBS) with 0.1% Tween (TBS-T) before addition of secondary antibody conjugated to a fluorescent entity: IRDye 800-conjugated goat antirabbit IgG and/or Alexa Fluor 680-conjugated goat antimouse IgG (dilution 1:6000) in 10 ml LI-COR blocking buffer with gentle agitation for 1 h at room temperature. At the end of the incubation period, membranes were washed twice with 15 ml TBS-T and once with 15 ml TBS. The membrane was dried, visualized, and analyzed on the Odyssey IR imaging system (LI-COR Biosciences). To monitor p38MAPK phosphorylation, membranes were incubated with the phospho-p38MAPK antibody (1:1000 in LI-COR buffer) with gentle agitation overnight at 4 C. After three washes with TBS-T, goat antimouse Alexa Fluor 680-conjugated IgG (dilution 1:6000 in 10 ml blocking buffer) was added for 1 h at room temperature before visualization and analysis by the Odyssey IR imaging system (LI-COR Biosciences). Membranes were then stripped with 25 mm glycine buffer (pH 2) containing 2% SDS and reprobed with total p38MAPK antibody (dilution 1:500 in LI-COR blocking buffer) with gentle agitation overnight at room temperature.
For plasma membrane fractionation, 293-R2β were grown in 10-cm petri dishes, and when 80% confluency was reached, cells were deprived of FCS for 1 h before treatment with 100 nm UCN-II or CRH for the indicated time intervals. Membrane proteins were prepared using ProteoExtract Native Membrane Protein Extraction Kit (Calbiochem/Merck Biosciences, Nottingham, UK). Protein concentration was determined using a BCA Protein Assay Kit (Pierce). Protein fractions (5 μg) were analyzed by Western blot analysis. After SDS-PAGE and electrophoretic transfer, membranes were incubated overnight at 4 C with pan-arrestin antibody (recognizes both arrestins, 49-kDa β-arrestin1 and 47-kDa β-arrestin2) (dilution 1:500), after a 1-h incubation step in TBS-T containing 5% BSA at room temperature. After incubation with the secondary horseradish peroxidase-conjugated antirabbit IgG (Dako UK Ltd., Ely, UK) (1:2000 dilution in TBS-T for 1 h at room temperature), proteins were visualized using ECL reagent from Amersham/Pharmacia Biotech (GE Healthcare).
Coimmunoprecipitation Studies
Plasma membrane-rich fractions were prepared from 293-R2β as described above. Treated plates were rinsed twice with ice-cold PBS, and lysates were then incubated overnight at 4 C with antibodies specific for CRH-R1/2 (Santa Cruz Biotechnology) and protein A/G agarose (Pierce). The immunoprecipitates were collected and washed four times in PBS before being resuspended in Laemmli sample buffer. After heating at 95 C for 5 min, the samples were centrifuged briefly and the supernatants analyzed by SDS-PAGE on 10% gels. After electrophoretic transfer, membranes were incubated overnight at 4 C with pan-arrestin antibody (recognizes both arrestins, 49-kDa β-arrestin1 and 47-kDa β-arrestin2) (dilution 1:500) or pan-cadherin (dilution 1:500 in TBS-T) or GAPDH (1:60,000 in TBS-T) after a 1-h incubation step in TBS-T containing 5% BSA at room temperature. After incubation with the secondary horseradish peroxidase-conjugated antirabbit IgG (Dako) (1:2000 dilution in TBS-T for 1 h at room temperature), proteins were visualized using ECL reagent from Amersham/Pharmacia Biotech (GE Healthcare).
Immunofluorescent Confocal Microscopy and Internalization Studies
Cells were seeded on coverslips coated with 100 μg/ml poly-d-lysine in PBS. When 70–80% confluency was reached, the cells were treated with various concentrations of agonists and fixed with 4% paraformaldehyde. Nonspecific binding was blocked with 1% BSA in PBS-Triton X-100 (0.01%) for 1 h at room temperature. After a 5-min wash with PBS, a PBS solution containing 1:100 monoclonal mouse antibody against CHC and 1:75 polyclonal rabbit pan-arrestin antibody were added for overnight incubation at 4 C. The next morning, the antibody mixture was removed, slides were washed with PBS three times for 5 min each, and polyclonal goat antibody against the C terminus of CRH-R1/2 (1:100 in PBS) was added for 2 h at room temperature. After three PBS washes for 5 min each, donkey antirabbit Alexa Fluor 488 and donkey anti-goat Alexa Fluor 594 were added in 1:400 dilution each for 1 h at room temperature. After three 5-min washes with PBS, solution containing 1:400 goat anti-mouse Alexa Fluor 405 was added for 1 h at room temperature. After three final washes with PBS, slides were mounted with Vectashield mounting medium without DAPI after three final washes with PBS. When double staining was carried out, both primary antibodies, 1:100 CRH-R1/2 and 1:75 pan-arrestin or 1:100 CHC or β-arrestin1 or phospho-p38MAPK, were placed on slides for overnight incubation at 4 C, whereas incubation with 1:100 CRH-R1/2 and 1:100 phospho-ERK1/2 was overnight at room temperature. After three washes with PBS, slides were incubated with donkey antirabbit (or mouse) Alexa Fluor 488 and donkey antigoat Alexa Fluor 594 (1:400), before mounting with Gold Slowfade mounting solution with DAPI (Molecular Probes). The slides were examined under an oil immersion objective (×63) using a Leica model DMRE laser scanning confocal microscope (Leica Microsystems UK, Milton Keynes, Buckinghamshire, UK) with TCS SP2 scan head. Laser 543 nm at 50% of power and emission filter set at 555–620 nm was used to examine Alexa Fluor 594 staining, and Laser 488 nm at 30% of power and emission filter set at 500–535 nm was used to examine Alexa Fluor 488 staining. DAPI or Alexa Fluor 405 staining was examined with Laser 405 nm at 10% of power and emission filter set at 410–450 nm. The scan speed was set at 400 Hz, and the format was 1024 × 1024 pixels. Optical sections (0.5 μm) were taken, and representative sections corresponding to the middle of the cells are presented. After indirect immunofluorescent staining, no specific fluorescence was observed in cells treated with secondary antibody only or when blocking peptide was incubated with the primary antibody before incubation on cells. For each treatment, between 20 and 30 individual cells in five random fields of view were randomly selected and examined. Fluorescence intensity profiles were generated along multiple line axes, analyzed, and quantified using ImageJ software developed at the National Institutes of Health (http://rsb.info.nih.gov/ij/). Relative quantification of intracellular (internalized) CRH-R2 was carried out by measuring the amount of total fluorescence along the longitudinal axis corresponding to the intracellular space (average 4–18 μm).
On-Cell Western
Cells were cultured on 24-well plates. When 80% confluent, cells were treated with 100 nm UCN-II or CRH for 15–45 min. Cells were fixed with 4% paraformaldehyde for 20 min, and after three brief washes with PBS, nonspecific binding was blocked with LI-COR blocking buffer. A rabbit antibody raised against the N terminus of CRH-R2 was used at a dilution of 1:2000 (Abcam) in LI-COR blocking buffer at 4 C overnight. After three washes with PBS, cells were incubated with the secondary antirabbit IRDye-800 antibody (dilution 1:800) for 1 h at room temperature. The signal was detected and analyzed using the Odyssey IR imaging system (LI-COR Biosciences).
Statistics
The results obtained are presented as the mean ± sem of each measurement. Data were tested for homogeneity, and comparison between group means was performed by one- or two-way ANOVA. Probability values of P < 0.05 are considered to be significant.
Footnotes
This work was supported by a Wellcome Trust University Award (to D.K.G.).
Disclosure Statement: The authors have nothing to disclose.
First Published Online November 29, 2007
Abbreviations: CHC, Clathrin heavy chain; DAPI, 4′,6-diamidino-2-phenylindole; DN, dominant-negative; FCS, fetal calf serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GPCR, G protein-coupled receptor; IR, infrared; RNAi, RNA interference; siRNA, small interfering RNA; TBS, Tris-buffered saline; TBS-T, TBS with 0.1% Tween; UCN, urocortin.
References
- 1.Bale TL, Vale WW 2004. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 244:525–557 [DOI] [PubMed] [Google Scholar]
- 2.Hillhouse EW, Grammatopoulos DK 2006. The molecular mechanisms underlying the regulation of the biological activity of corticotropin-releasing hormone receptors: implications for physiology and pathophysiology. Endocr Rev 27:260–286 [DOI] [PubMed] [Google Scholar]
- 3.Lewis K, Li C, Perrin MH, Blount A, Kunitake K, Donaldson C, Vaughan J, Reyes TM, Gulyas J, Fischer W, Bilezikjian L, Rivier J, Sawchenko PE, Vale WW 2001. Identification of urocortin III, an additional member of the corticotropin-releasing factor (CRF) family with high affinity for the CRF2 receptor. Proc Natl Acad Sci USA 98:7570–7575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bale TL, Contarino A, Smith GW, Chan R, Gold LH, Sawchenko PE, Koob GF, Vale WW, Lee KF 2000. Mice deficient for corticotropin-releasing hormone receptor-2 display anxiety-like behaviour and are hypersensitive to stress. Nat Genet 24:410–414 [DOI] [PubMed] [Google Scholar]
- 5.Lawrence KM, Latchman DS 2006. The urocortins: mechanisms of cardioprotection and therapeutic potential. Mini Rev Med Chem 6:1119–1126 [DOI] [PubMed] [Google Scholar]
- 6.Bale TL, Giordano FJ, Hickey RP, Huang Y, Nath AK, Peterson KL, Vale WW, Lee KF 2002. Corticotropin-releasing factor receptor 2 is a tonic suppressor of vascularization. Proc Natl Acad Sci USA 99:7734–7739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carlin KM, Vale WW, Bale TL 2006. Vital functions of corticotropin-releasing factor (CRF) pathways in maintenance and regulation of energy homeostasis. Proc Natl Acad Sci USA 103:3462–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen A, Brar B, Choi CS, Rousso D, Vaughan J, Kuperman Y, Kim SN, Donaldson C, Smith SM, Jamieson P, Li C, Nagy TR, Shulman GI, Lee KF, Vale W 2006. Urocortin 2 modulates glucose utilization and insulin sensitivity in skeletal muscle. Proc Natl Acad Sci USA 103:16580–16585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C, Chen P, Vaughan J, Blount A, Chen A, Jamieson PM, Rivier J, Smith MS, Vale W 2003. Urocortin III is expressed in pancreatic β-cells and stimulates insulin and glucagon secretion. Endocrinology 144:3216–3224 [DOI] [PubMed] [Google Scholar]
- 10.Hoare SR, Sullivan SK, Fan J, Khongsaly K, Grigoriadis DE 2005. Peptide ligand binding properties of the corticotropin-releasing factor (CRF) type 2 receptor: pharmacology of endogenously expressed receptors, G-protein-coupling sensitivity and determinants of CRF2 receptor selectivity. Peptides 26:457–470 [DOI] [PubMed] [Google Scholar]
- 11.Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J 2003. Mitogen-activated protein kinase signaling in the hippocampus and its modulation by corticotropin-releasing factor receptor 2: a possible link between stress and fear memory. J Neurosci 23:11436–11443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karteris E, Hillhouse EW, Grammatopoulos D 2004. Urocortin II is expressed in human pregnant myometrial cells and regulates myosin light chain phosphorylation: potential role of the type-2 corticotropin-releasing hormone receptor in the control of myometrial contractility. Endocrinology 145:890–900 [DOI] [PubMed] [Google Scholar]
- 13.Dermitzaki E, Tsatsanis C, Gravanis A, Margioris AN 2002. Corticotropin-releasing hormone (CRH) induces Fas ligand production and apoptosis in PC12 cells via activation of p38 MAPK. J Biol Chem 277:12280–12287 [DOI] [PubMed] [Google Scholar]
- 14.Grammatopoulos D, Randeva H, Levine MA, Katsanou E, Hillhouse EW 2000. Urocortin but not corticotropin-releasing hormone (CRH) activates the MAP kinase signal transduction pathway in human pregnant myometrium: an effect mediated via R1α and R2β CRH receptor subtypes and stimulation of Gq-proteins. Mol Endocrinology 14:2076–2091 [DOI] [PubMed] [Google Scholar]
- 15.Brar BK, Chen A, Perrin MH, Vale W 2004. Specificity and regulation of extracellularly regulated kinase 1/2 phosphorylation through corticotropin-releasing factor (CRF) receptors 1 and 2β by the CRF/urocortin family of peptides. Endocrinology 145:1718–1729 [DOI] [PubMed] [Google Scholar]
- 16.Claing A, Laporte SA, Caron MG, Lefkowitz RJ 2002. Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and β-arrestin proteins. Prog Neurobiol 66:61–79 [DOI] [PubMed] [Google Scholar]
- 17.Lefkowitz RJ, Shenoy SK 2005. Transduction of receptor signals by β-arrestins. Science 308:512–517 [DOI] [PubMed] [Google Scholar]
- 18.Teli T, Markovic D, Levine MA, Hillhouse EW, Grammatopoulos D 2005. Regulation of corticotrophin releasing hormone (CRH) receptor type 1α signaling: structural determinants for G protein-coupled receptor kinase-mediated phosphorylation and agonist-mediated desensitization. Mol Endocrinol 19:474–490 [DOI] [PubMed] [Google Scholar]
- 19.Markovic D, Papadopoulou N, Teli T, Levine MA, Hillhouse EW, Grammatopoulos DK 2006. Differential responses of CRH receptor type 1 variants to PKC phosphorylation. J Pharmacol Exp Ther 19:1032–1042 [DOI] [PubMed] [Google Scholar]
- 20.Perry SJ, Junger S, Kohout TA, Hoare SR, Struthers RS, Grigoriadis DE, Maki RA 2005. Distinct conformations of the corticotropin releasing factor type 1 receptor adopted following agonist and antagonist binding are differentially regulated. J Biol Chem 280:11560–11568 [DOI] [PubMed] [Google Scholar]
- 21.Punn A, Levine MA, Grammatopoulos DK 2006. Identification of signaling molecules mediating CRH-R1α-MAPK interactions: the critical role of PI3-K in regulating ERK1/2 but not p38 MAPK activation. Mol Endocrinol 20:3179–3195 [DOI] [PubMed] [Google Scholar]
- 22.Miller JW 2004. Tracking G protein-coupled receptor trafficking using Odyssey Imaging. LI-COR Biosciences Application notes, pp 1–4 (http://www.licor.com/bio/PDF/Miller_GPCR.pdf)
- 23.Lin FT, Miller WE, Luttrell LM, Lefkowitz RJ 1999. Feedback regulation of β-arrestin1 function by extracellular signal-regulated kinases. J Biol Chem 274:15971–15974 [DOI] [PubMed] [Google Scholar]
- 24.Holmes KD, Babwah AV, Dale LB, Poulter MO, Ferguson SS 2006. Differential regulation of corticotropin releasing factor 1α receptor endocytosis and trafficking by β-arrestins and Rab GTPases. J Neurochem 96:934–949 [DOI] [PubMed] [Google Scholar]
- 25.Shenoy SK, Lefkowitz RJ 2003. Multifaceted roles of β-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem J 375:503–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG 1999. Association of β-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem 274:32248–32257 [DOI] [PubMed] [Google Scholar]
- 27.Kenakin T 1997. Agonist-specific receptor conformations. Trends Pharmacol Sci 18:232–238 [DOI] [PubMed] [Google Scholar]
- 28.Krupnick JG, Santini F, Gagnon AW, Keen JH, Benovic JL 1997. Modulation of the arrestin-clathrin interaction in cells. Characterization of β-arrestin dominant-negative mutants. J Biol Chem 272:32507–32512 [DOI] [PubMed] [Google Scholar]
- 29.Smith MP, Ayad VJ, Mundell SJ, McArdle CA, Kelly E, Lopez Bernal A 2005. Internalization and desensitization of the oxytocin receptor is inhibited by dynamin and clathrin mutants in human embryonic kidney 293 cells. Mol Endocrinol 20:379–388 [DOI] [PubMed] [Google Scholar]
- 30.Kageyama K, Hanada K, Suda T 2005. Regulation of corticotropin-releasing factor receptor type 2β mRNA by mitogen-activated protein kinases in aortic smooth muscle cells. Regul Pept 126:223–231 [DOI] [PubMed] [Google Scholar]
- 31.Luttrell DK, Luttrell LM 2003. Signaling in time and space: G protein-coupled receptors and mitogen-activated protein kinases. Assay Drug Dev Technol 1:327–338 [DOI] [PubMed] [Google Scholar]
- 32.Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, Lefkowitz RJ 2006. Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem 281:10856–10864 [DOI] [PubMed] [Google Scholar]
- 33.McPherson G 1983. A practical computer based approach to the analysis of radioligand binding experiments. Prog Biomed 17:107–114 [DOI] [PubMed] [Google Scholar]
- 34.Munson P, Rodgbard D 1980. LIGAND: a versatile computerized approach for characterization of ligand binding systems. Anal Biochem 107:220–239 [DOI] [PubMed] [Google Scholar]