

Abstract
Ginsenoside Re (Re), a compound derived from Panax ginseng, shows an antidiabetic effect. However, the molecular basis of its action remains unknown. We investigated insulin signaling and the antiinflammatory effect by Re in 3T3-L1 adipocytes and in high-fat diet (HFD) rats to dissect its anti-hyperglycemic mechanism. Glucose uptake was measured in 3T3-L1 cells and glucose infusion rate determined by clamp in HFD rats. The insulin signaling cascade, including insulin receptor (IR) β-subunit, IR substrate-1, phosphatidylinositol 3-kinase, Akt and Akt substrate of 160 kDa, and glucose transporter-4 translocation are examined. Furthermore, c-Jun NH2-terminal kinase (JNK), MAPK, and nuclear factor (NF)-κB signaling cascades were also assessed. The results show Re increases glucose uptake in 3T3-L1 cells and glucose infusion rate in HFD rats. The activation of insulin signaling by Re is initiated at IR substrate-1 and further passes on through phosphatidylinositol 3-kinase and downstream signaling cascades. Moreover, Re demonstrates an impressive suppression of JNK and NF-κB activation and inhibitor of NF-κBα degradation. In conclusion, Re reduces insulin resistance in 3T3-L1 adipocytes and HFD rats through inhibition of JNK and NF-κB activation.
THE PREVALENCE OF diabetes is dramatically increased in the world as well as in China. It is known that type 2 diabetes is caused by insulin resistance and loss of β-cell compensation for insulin resistance (1). Improvement of insulin resistance by antidiabetic drugs definitely plays a major role in treatment of diabetes and reduction of the related complications.
Diabetes has been known as Xiao Ke Zheng for 2000 yr in China and is attributable to Qi deficiency resulting from prolonged Yin and Yang deficiency. Ginseng has been used as a tonic to correct Qi deficiency and been prescribed for diabetic patients for hundreds of years (2). Ginseng means cure-all in Latin. Most pharmacological actions of ginseng are attributed to ginsenosides, which are major components extracted from different species of ginseng. There are two major classes of ginsenosides, namely, the derivatives of protopanaxatriol (Rg1, Rg2, Rg3, Re, and Rf) and protopanaxadiol (Rb1, Rb2, Rc, and Rd), which were mostly studied as triterpene saponins (3).
In recent years, accumulating evidence in vitro and in vivo has shown that ginseng and its extracts possess anti-hyperglycemic activities (4, 5, 6, 7, 8). Ginsenoside Re (Re), as an active compound demonstrates a significant anti-hyperglycemic effect as well as reduction of serum insulin levels in either fed or fasting ob/ob mice, which indicates insulin resistance improvement in peripheral tissues (8, 9). Re also reduces serum lipid levels and exerts protective actions against the occurrence of oxidative stress in eyes and kidneys of diabetic rats (10). Re even can reduce serum C-responsive protein (CRP) levels in streptozotocin-induced diabetic rats (11).
However, the molecular mechanism of Re improving insulin resistance in diabetic animals is still unknown. Insulin plays an important role in glucose homeostasis. Insulin-stimulated glucose uptake in muscle and adipose tissues is associated with the translocation of insulin-regulated glucose transporters (GLUTs), such as GLUT4, from intracellular vesicles to plasma membrane (PM). Insulin triggers its signal transduction by binding to insulin receptor (IR). The stimulated IR phosphorylates itself and IR substrate (IRS) family members at tyrosine residues, subsequently recruits phosphatidylinositol 3-kinase (PI3K) to the membrane, leading to activation of downstream Akt and protein kinase C (PKC)-ζ/λ, and results in GLUT4 translocation, which transports glucose into the cells (12). It is known that chronic low-grade inflammation is associated with insulin resistance (13, 14). The inhibition of signaling downstream of the IR is a primary mechanism through which inflammatory signaling leads to insulin resistance. Several serine/threonine kinases are activated by inflammatory or stressful stimuli and contribute to inhibition of insulin signaling, including c-Jun NH2-terminal kinase (JNK) and inhibitor of nuclear factor (NF)-κB kinase (IKK). The JNK group of serine/threonine kinases includes JNK-1, -2 and -3, which belong to MAPK family. JNK has recently emerged as a central metabolic regulator participating in the development of insulin resistance through phosphorylation of IRS-1 on Ser 307 (15). Exposure of cells to TNF-α stimulates this phosphorylation and reduces both tyrosine phosphorylation of IRS-1 in response to insulin and the ability of IRS-1 to associate with the IR and thereby inhibits downstream signaling and insulin action (16). Ginseng is reported to have beneficial effects on immune function and is widely used to improve health. In the present study, we investigated the insulin signaling pathway and JNK and NF-κB signaling cascade in 3T3-L1 adipocytes and high-fat diet (HFD) rats to dissect the molecular mechanism of Re in improving insulin resistance.
RESULTS
Re Increases Basal and Insulin-Stimulated Glucose Uptake in 3T3-L1 Adipocytes
The effect of Re on glucose transport in 3T3-L1 adipocytes was evaluated using 2-deoxyglucose (2-DOG) uptake. Treatment of 3T3-L1 adipocytes for 2 h with different concentrations of Re resulted in progressive increases of basal glucose uptake (Fig. 1A). The time course showed glucose uptake was maximally increased at 2 h of incubation (Fig. 1B). During coincubation with insulin for another 30 min, Re further increased glucose uptake by 27% as compared with insulin alone (Fig. 1C).
Fig. 1.
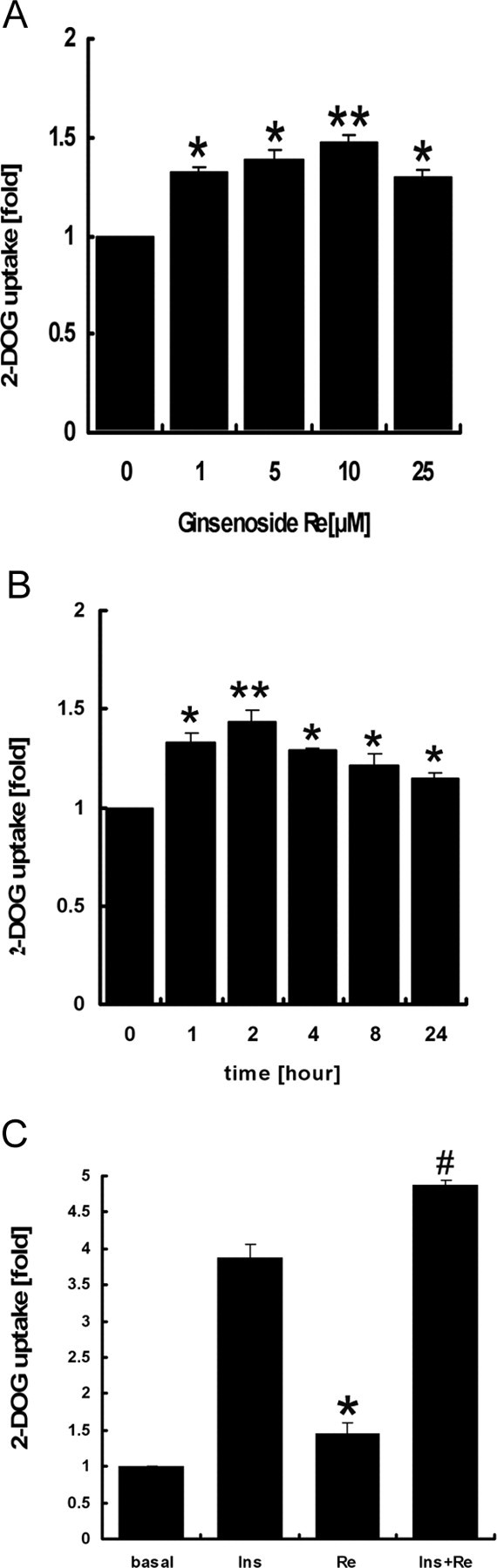
Re Enhances Glucose Uptake in 3T3-L1 Adipocytes
2-DOG uptake change is represented as fold of the control (no treatment). A, Dose-dependent change of 2-DOG uptake with Re incubation of 2 h; B, time course of glucose uptake with or without 10 μm Re; C, 2-DOG uptake after an incubation of Re (10 μm) for 2 h in the absence or presence of insulin (10 nm) for 30 min. The data are means ± sd of three independent experiments. *, P < 0.05 compared with the control; #, P < 0.05 compared with insulin. **, P < 0.01 compared with control (0 μM in A and 0 h in B).
Re Promotes GLUT4 Translocation
To determine the GLUT4 translocation, PM, and low-density membrane (LDM) fractions were separately made. The immunoblotting showed that GLUT4 translocated from cytoplasm to plasma membrane in Re-treated 3T3-L1 cells (Fig. 2A). Relative quantity of GLUT4 in PM fractionation was increased by Re as compared with LDM fractionation (Fig. 2B).
Fig. 2.

Immunoblotting of GLUT4 in PM and Intracellular Membranes (LDM) of 3T3-L1 Cells
A, GLUT4 translocation in LDM and PM fractions after an incubation of Re at 10 μm for 2 h and insulin at 10 nm for 30 min; B, relative quantities of GLUT4 in LDM and PM fractions are indicated as percentage of total LDM and PM. The data are shown as mean ± sd of three independent experiments.
Re Increases Tyrosine Phosphorylation of IRS-1 and PI3K Recruitment
We next explored the insulin signaling pathways through which Re might induce GLUT4 translocation. IR β-subunit (IRβ) and IRS-1 were immunoprecipitated, respectively, and then resolved by SDS-PAGE. Immunoblotting with anti-phosphotyrosine antibody (Py20) showed that phosphorylation of IRβ was not increased (Fig. 3A), whereas phosphorylation of IRS-1 was slightly increased by Re (Fig. 3B). We further identified that tyrosine phosphorylation of IRS-1 on 612 was increased by Re (Fig. 3C). The p85 subunit of PI3K was recruited to IRS-1 as a complex by Re treatment (Fig. 3B). These results indicated that Re could activate IRS-1 independent of IR activation.
Fig. 3.

Immunoprecipitation of IRβ and IRS-1 in 3T3-L1 Cells
A, Immunoprecipitation of IRβ after an incubation of Re at 10 μm for 2 h and insulin at 10 nm for 30 min, with anti-IRβ antibody used for pull-down and anti-phosphotyrosine antibody (PY20) for immunoblotting; B, coimmunoprecipitation of IRS-1 and p85 subunit of PI3K after the same treatment as in A, with anti-IRS-1 antibody for pull-down and anti-PY20 and anti-p85/PI3K for immunoblotting; C, immunoblotting for phosphorylation of IRS on tyrosine 612 in adipocytes after the same treatment as above. The representative immunoblots are shown from at least three independent experiments.
Re Activates PI3K, Akt/PKCζ/λ, and Akt Substrate of 160 kDa (AS160)
PI3K activation is crucial in insulin signaling. We are uncertain whether the slight tyrosine phosphorylation of IRS-1 in response to Re is responsible for PI3K activation. We measured PI3K activities for the complex obtained from immunoprecipitation with anti-p85 subunit, anti-IRS-1, and anti-phosphorylated tyrosine antibodies using 32P incorporation of phosphorylated inositol and found the PI3K activities were increased in 3T3-L1 adipocytes by Re (Fig. 4A). Meanwhile, Re-mediated glucose uptake was abrogated by wortmannin, a PI3K inhibitor (Fig. 4B). The downstream signaling cascade of PI3K was further activated by Re as an increase of phosphorylation of Akt/PKB on Ser473 and Thr308, phosphorylation of PKC-ζ/λ, p70 ribosomal S6 protein kinase (p70S6K), and glycogen synthase kinase-3β (GSK-3β) (Fig. 4C). Re also increased AS160 phosphorylation (Fig. 4D).
Fig. 4.

PI3K and Downstream Signaling Cascade
Cells were treated with Re at 10 μm for 2 h and insulin at 10 nm for 30 min respectively. A, PI3K activity assay. PI3K proteins were pulled down by anti-p85 subunit (top), anti-IRS-1 (middle), and anti-PY20 antibodies (bottom), and the enzymatic reaction was carried out by adding PI and [γ-32P]ATP. Phosphorylated inositol (PIP) is migrated on a thin-layer chromatograph (TLC) and visualized by autoradiography. B, 2-DOG uptake is abrogated by wortmannin (100 nm) in 3T3-L1 cells treated with either Re or insulin. Data represent mean ± sd of three experiments. **, P < 0.01 compared with Re alone; ##, P < 0.01 compared with insulin alone (Ins). C, Immunoblotting of phospho-Akt (Thr308 and Ser473), phospho-PKCζ/λ (Thr410/403), phospho-GSK-3β (Ser9) and phospho-p70S6K (Thr389). D, Immunoblotting of phosphorylation of AS160. The representative immunoblots are shown from at least three independent experiments.
Re Inhibits JNK MAPK and Serine 307 Phosphorylation of IRS-1 in 3T3-L1 Adipocytes and HFD Rats
Because it is known that ginseng has beneficial effects on low-grade inflammation, we adopted TNF-α-treated 3T3-L1 adipocytes and the adipose tissues from HFD rats to explore the antiinflammatory machinery of Re and its association with improvement of insulin signaling cascade. Phosphorylation of JNK MAPK was dramatically increased by TNF-α (Fig. 5A) and HFD (see Fig. 8D). Re impressively reduced phosphorylated JNK MAPK in vitro and vivo (Figs. 5A and 8D). It is known that JNK activation leads to Ser 307 phosphorylation of IRS-1. We further revealed that Ser 307 phosphorylation of IRS-1 was dramatically increased by TNF-α but greatly reduced by Re (Fig. 5B).
Fig. 5.
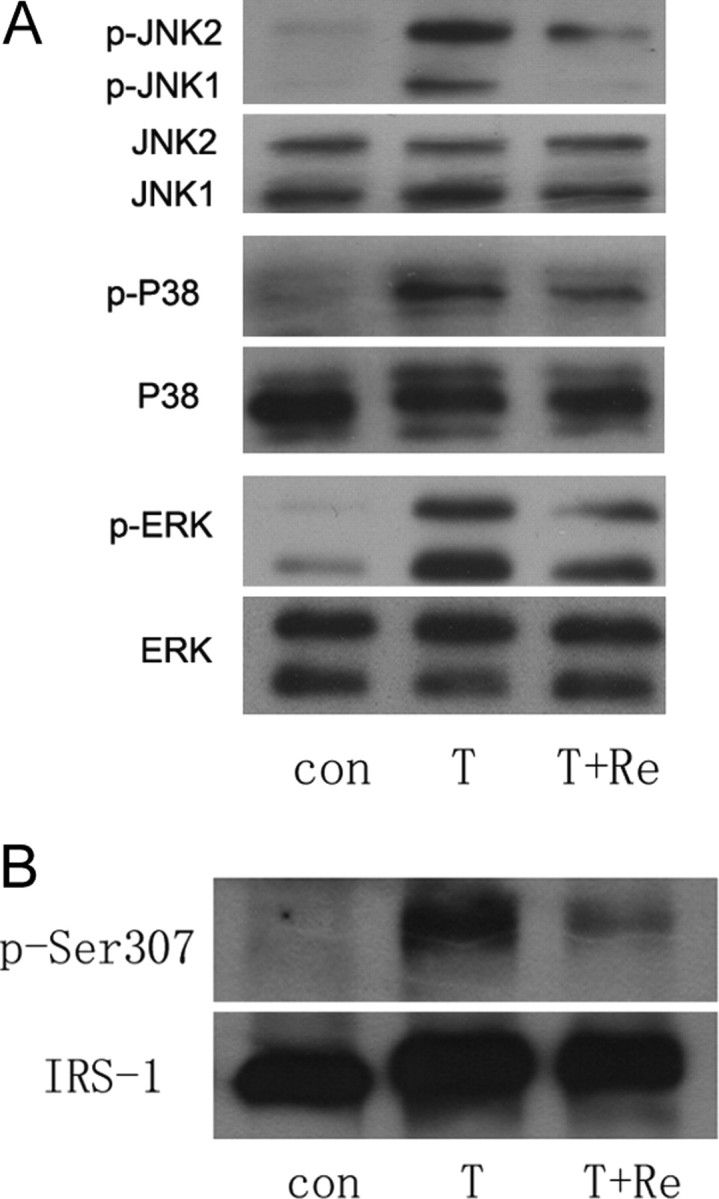
JNK and NF-κB Activation Suppressed by Re in 3T3-L1 Adipocytes
Cells were treated with Re at 10 μm for 24 h and then with TNF-α at 1 nm for 30 min. A, Immunoblotting shows phospho-JNK, phospho-p38 and phospho-ERK1/2 MAPKs are up-regulated by TNF-α and phospho-JNK particularly reduced by Re; B, immunoblotting shows serine 307 phosphorylation of IRS-1 is increased by TNF-α and reduced by Re. Representative blots are shown from at least three independent experiments.
Fig. 8.

HEC and Body Fat Change in HFD Rats
A, GIR was significantly reduced in HFD rats compared with normal chow rats. GIR was significantly increased by Re in HFD rats. B and C, The visceral fats represented by epididymal fats and sc fats were significantly reduced with Re treatment, which is shown as ratio of visceral fat to body weight and sc fat to body weight, respectively. The data are shown as means ± se of five normal chow, nine HFD, and five HFD plus Re rats. *, P < 0.05; **, P < 0.01 vs. HFD rats. D, Western blots of phospho-JNK, IκBα, phospho-AMPK, and phospho-AS160 for adipose tissues from HFD rats with 2 wk Re treatment.
Re Inhibits NF-κB in 3T3-L1 Adipocytes and HFD Rats
We also found that Re inhibited TNF-α-induced IKK activation and inhibitor of NF-κBα (IκBα) degradation in 3T3-L1 cells and in adipose tissues from HFD rats (Figs. 6A and 8D). NF-κB is a proinflammatory master switch in response to TNF-α to up-regulate some important inflammation factors, such as IL-6 and suppressor of cytokine signaling proteins-3 (SOCS-3). We found IL-6 and SOCS-3 expression was markedly reduced by Re in 3T3-L1 adipocytes treated with TNF-α (Fig. 6, B and C).
Fig. 6.
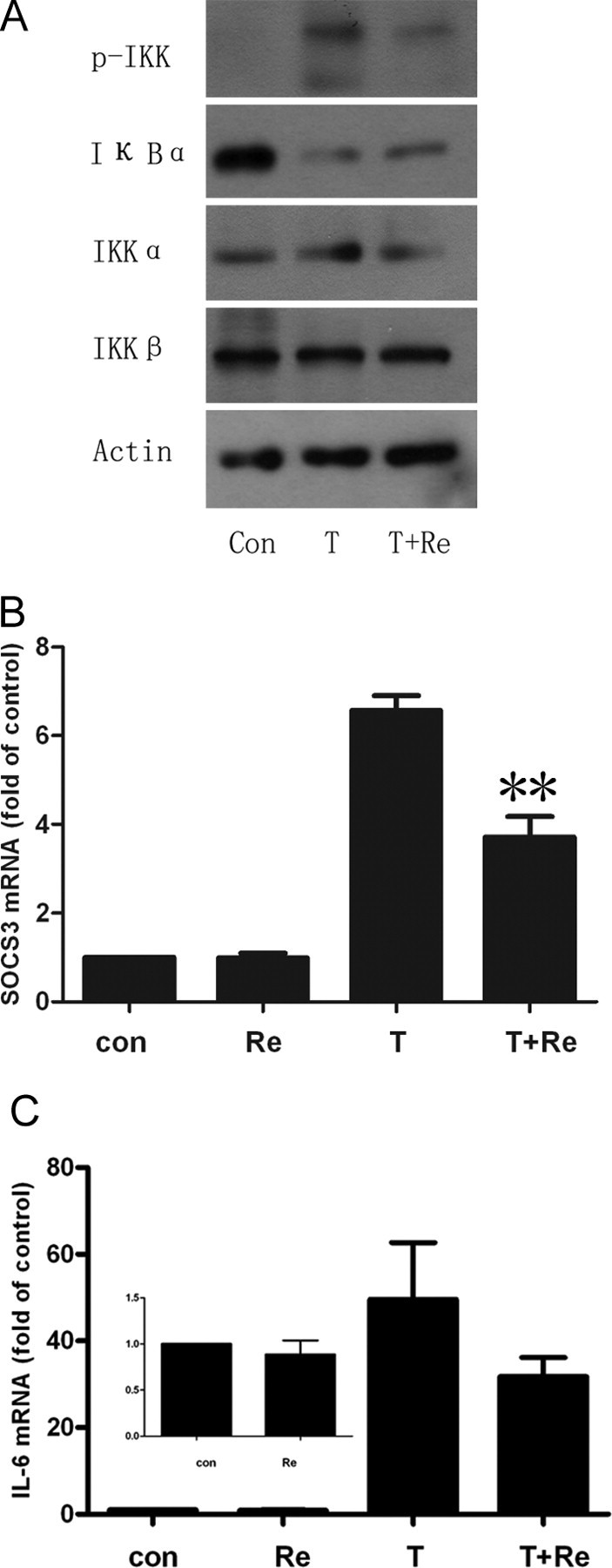
Inhibition of IKK/NF-κB Signaling Pathway by Re
A, Cells were treated with Re at 10 μm for 24 h and then with TNF-α at 1 nm for 30 min. The cell lysates were resolved by SDS-PAGE and analyzed using antibodies against total and phosphorylated IKKα/β and IκBα. Representative blots are shown from three independent experiments. B and C, Real-time PCR for SOCS-3 and IL-6 expression as described in Materials and Methods. 3T3-L1 adipocytes were incubated with 10 μm Re and with or without 10 ng/ml TNF-α for 24 h. SOCS-3 is significantly reduced and IL-6 remarkably decreased. **, P < 0.01 compared with TNF-α-treated group.
Re Protects Insulin Signaling Impairment by TNF-α
Re suppressed TNF-α-induced JNK activation and Ser 307 phosphorylation of IRS-1. This represents at least part of the mechanism by which Re enhances insulin sensitivity. Basal and insulin-stimulated glucose uptake was suppressed by 23 and 47%, respectively, in 3T3-L1 adipocytes treated with TNF-α. However, this suppression could be restored by Re at 10 μmol/liter for 24 h (Fig. 7A). This improvement was much greater in insulin-treated cells (Fig. 7A). It indicates that Re could antagonize TNF-α-mediated insulin resistance in 3T3-L1 adipocytes. In addition, the suppression of phosphorylation of Akt/PKB by TNF-α could be also restored by Re, which parallels glucose uptake improvement (Fig. 6B).
Fig. 7.
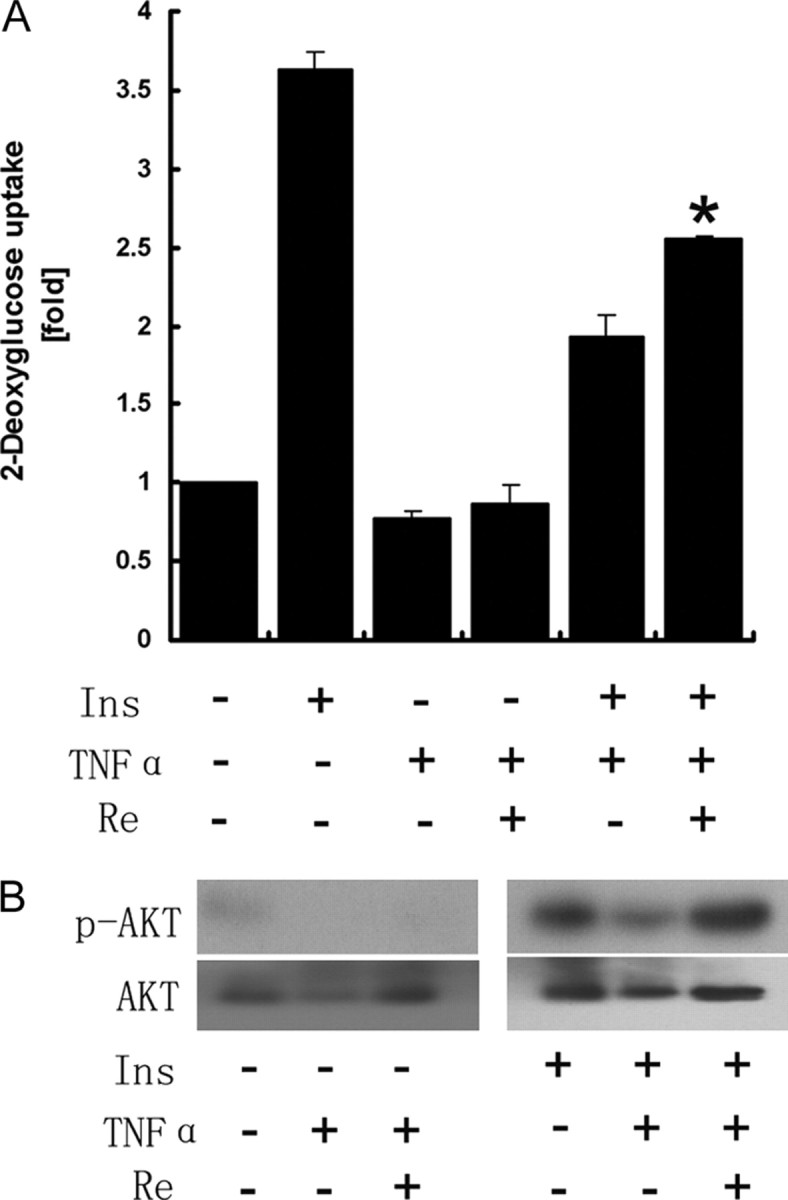
2-DOG Uptake Increased by Re in TNF-α-Treated Cells
Cells were treated with TNF-α at 10 ng/ml for 72 h and with Re at 10 μm for 24 h followed by insulin treatment at 10 nm for 30 min. A, 2-DOG uptake was decreased by TNF-α and significantly increased by Re. The data are shown as mean ± sd of at least three independent experiments. *, P < 0.05 compared with TNF-α plus insulin. B, Phosphorylation of Akt (Ser 473) is up-regulated by Re, consistent with glucose uptake as shown in A. Representative blots from three independent experiments are shown.
Re Increases Glucose Infusion Rate (GIR) in HFD Rats
We further investigated Re for the anti-hyperglycemic effect in HFD rats. During the 2-h hyperinsulinemic-euglycemic clamp (HEC) test, blood glucose levels were maintained between 4.7 and 5.3 mmol/liter in all groups by varying GIR. The GIR was significantly lower in HFD rats compared with the normal chow-fed rats (19.91 vs. 30.88, P < 0.0001), which indicates a severe whole-body insulin resistance in HFD rats (Fig. 8A). The GIR was significantly promoted by Re in HFD rats (25.60 vs. 19.91, P = 0.011), which demonstrates that Re can improve insulin resistance in obese rats (Fig. 8A). However, fasting plasma glucose levels were not different in normal chow-fed, HFD, and HFD/Re rats (data not shown).
Re led to reduction of fat storage of both visceral and sc fats, which is estimated by the ratio of epididymal fat over body weight and the ratio of sc fats over body weight, respectively (0.0065 vs. 0.0070, P = 0.039; 0.016 vs. 0.022, P = 0.005) (Fig. 8, B and C). However, the body weights were not significantly changed in HFD rats treated with Re (data not shown). In addition, we found that phosphorylation of AMP-activated protein kinase (AMPK) was pronouncedly increased in adipose tissues by Re and 3T3-L1 adipocytes (Fig. 8D and supplemental Fig. 1, published as supplemental data on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org), which implies lipid oxidation enhancement by Re treatment.
DISCUSSION
It has been reported that ginseng has an antidiabetic effect as a traditional Chinese medicine (2, 6). Recently, several studies have showed Re, extracted from ginseng, can reduce blood glucose in ob/ob diabetic mice (8, 9). In the present study, we demonstrated that Re promoted glucose uptake in 3T3-L1 adipocytes and improved insulin resistance by increasing the GIR in HFD rats. HEC was used to evaluate the state of insulin resistance as a gold standard. The GIR obtained from HEC depends on glucose uptake by peripheral tissues and therefore a measure of peripheral tissue sensitivity to insulin.
Our results demonstrate that Re initiates its effects at IRS-1 and through the IRS-1/PI3K/Akt-PKC-ζ/λ signaling cascade. IRS-1 is a major substrate of IR. However, IRS-1 can also be phosphorylated by some other receptor tyrosine kinases or protein tyrosine kinases even though we cannot tell which tyrosine kinase participate in IRS-1 phosphorylation by Re in the present study. Some studies showed dehydroepiandrosterone binds to a G protein-coupled receptor, increases IRS-1 phosphorylation independent of IR, and stimulates glucose uptake in 3T3-L1 cells (17). The ginsenoside also has a steroid skeleton in chemical structure, possibly interacts with some membrane receptor, and leads to changes in activity of steroid receptors and membrane fluidity (18). The interaction of Re and receptor might directly or indirectly result in IRS-1 phosphorylation. We have shown that tyrosine 612 phosphorylation of IRS-1 is increased by Re (Fig. 3C) and PI3K activated as shown in Fig. 4A. PI3K plays a pivotal role in insulin signaling and is important for insulin-mediated GLUT4 translocation (19). AS160 (20, 21), a key protein required for GLUT4 trafficking, was activated by Re. Re administration also increases AS160 phosphorylation in adipose tissue from HFD rats (Fig. 8D). Our results demonstrate that Re promotes glucose disposal by activation of the insulin signaling pathway.
Insulin-stimulated glucose disposal is markedly reduced in patients with type 2 diabetes due to multiple defects in the insulin signaling pathway. In the past decade, a number of studies have witnessed a close association of inflammation with insulin resistance (22). Elevation of inflammatory factors and potential mediators in diabetic patients indicates a preceding low-grade inflammation in the development of type 2 diabetes (23, 24). It has been known that TNF-α, as a major proinflammatory factor, mediates insulin resistance through multiple targets, including activation of JNK and NF-κB (25, 26, 27). JNK has been shown to promote insulin resistance through the phosphorylation of serine residues in IRS-1 (28, 29, 30). Suppression of JNK activation could be a therapeutic target for insulin resistance (31). In particular, JNK1 appears to be the predominant isoform present in muscle, liver, and adipose tissues. JNK activity is strikingly elevated in the above tissues in obese patients (32). JNK can phosphorylate IRS-1 on Ser 307 and inhibits insulin-stimulated tyrosine phosphorylation of IR (15, 31). In our present study, we revealed that Re suppressed JNK activities and further reduced Ser 307 phosphorylation of IRS-1 in 3T3-L1 cells.
IKK/NF-κB activation is also critical in the development of insulin resistance and metabolic dysfunction. Inactivated NF-κB stays in cytosol in a complex of the inhibitory subunit IκBα. IκBα undergoes proteasome-dependent degradation immediately by IKK phosphorylation (33). Our results also showed Re had a weak inhibition for TNF-α-induced IκBα degradation. It has been reported that aspirin can reduce serine 307 phosphorylation of IRS-1 through inhibition of JNK and IKK activity (34). Our study suggests that Re can block Ser307 phosphorylation of IRS-1 and attenuate the alteration in IκBα protein abundance in a similar way with aspirin although the exact role of IKK in serine phosphorylation of IRS-1 has not yet been well established under physiological conditions (13, 14).
NF-κB is a master switch in response to extracellular stimuli and intracellular stress to up-regulate inflammatory mediators participating in insulin resistance and the development of type 2 diabetes. We found that Re dramatically suppresses NF-κB initiated IL-6 and SOCS-3 expression. Our results indicate that Re has a potential antiinflammatory effect. However, we did not observe a significant increase of adiponectin expression (supplemental Fig. 2A), although pioglitazone can increase adiponectin production by enhancement of signal transducer and activator of transcription-3 activation via reduction of SOCS-3 expression (35).
Recent studies have shown that ginseng has potential beneficial effects on key players in the inflammation cascade. BST204, a fermented ginseng extract, could inhibit inducible nitric oxide synthase expression and subsequent nitric oxide production from lipopolysaccharide-treated RAW264.7, a murine macrophage cell line (36). Ginsenoside Rg3, a major ginsenoside derived from the heat-processed ginseng and belonging to the protopanaxatriol can inhibit phorbol ester-induced COX-2 production and NF-κB activation in mouse skin (37). Re can even reduce serum CRP levels in diabetic rats (11). Our study has provided the evidence that Re may play a role in antiinflammation through inhibition of JNK and NF-κB.
We have also observed that Re reduces visceral and sc fats in HFD rats. AMPK is known as an energy sensor and metabolic switch that targets key proteins and enzymes involved in lipid metabolism. Phosphorylation of AMPK and acetyl coenzyme A carboxylase are increased in 3T3-L1 adipocytes (supplemental Fig. 1) and adipose tissues of HFD rats treated with Re, which may explain the fat mass reduction by Re.
In the present study, we did not identify the primary target(s) of Re. However. we assume Re could have multiple targets because it has a steroid backbone structure (18). On one hand, we thought Re might interact with some membrane receptor and directly or indirectly result in IRS-1 phosphorylation and inhibition of JNK and IKK signaling as well. On the other hand, Re may also enter the cells to play some role, such as activation of AMPK. So we think Re could have multiple targets in the promotion of glucose uptake in 3T3-L1 adipocyte and improvement of insulin resistance in peripheral tissues. At any rate, we think the antiinflammatory effect of Re could be a major one, even though we did not identify the primary effect of increased insulin action.
In conclusion, it is certain that Re has an antiinflammatory effect, which is associated with an anti-hyperglycemic effect in the state of insulin resistance. Re may be promising to be developed as an antidiabetic drug by improving insulin resistance through inhibition of JNK and NF-κB activation.
MATERIALS AND METHODS
Materials
Re of HPLC grade and purity of more than 98% was obtained from Jilin University School of Medicine (Jilin, China). DMEM and fetal bovine serum were purchased from PAA Laboratories (Pasching, Austria). Isobutylmethylxanthine, dexamethasone, wortmannin, phosphatidylinositol, cytochalasin B, 2-deoxy-d-[1-3H]glucose were purchased from Sigma Chemical Co. (St. Louis, MO). [γ-32P]ATP was obtained from Amersham Pharmacia (Little Chalfont, UK) and Humulin R from Eli Lilly (Indianapolis, IN). Polyclonal anti-Akt antibody, anti-phospho-Akt (Thr308 and Ser473) antibody, anti-phospho-PKC-ζ/λ (Thr410/403) antibody, anti-phospho-GSK-3β (Ser9) antibody, anti-phospho-p70S6K (Thr389) antibody, phospho (Ser/Thr)-Akt substrate antibody, polyclonal anti-p38 antibody, anti-phospho-p38 (Thr180/Tyr182) antibody, anti-p44/42 MAPK antibody (ERK) antibody, anti-phospho-ERK1/2 (Thr202/Tyr204) antibody, anti-JNK antibody, and anti-phospho-SAPK/JNK (Thr183/Tyr185) antibody were all purchased from Cell Signaling Technology Inc. (Beverly, MA). GLUT4 antibody, IRβ antibody, and IRS-1 antibody were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-phospho-IRS-1 Ser307 antibody was obtained from Upstate Biotechnology (Lake Placid, NY). Aluminum-backed silica gel thin layer chromatographic plates were from Merck (Darmstadt, Germany).
Cell Culture
3T3-L1 fibroblasts were obtained from American Type Culture Collection (Rockville, MD) and cultured in DMEM supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum in an atmosphere of 7% CO2 at 37 C. Two days later, when the fibroblasts had reached confluence, differentiation was induced by treating the cells with medium containing 0.5 mmol/liter 3-isobutyl-1-methylxanthine, 1 μm dexamethasone, and 10 μg/ml insulin for 48 h. Cells were maintained in complete DMEM every other day for the next 4–6 d until more than 90% of cells were demonstrating adipocyte phenotype. The 3T3-L1 fibroblasts were used up to passage 10. Re was dissolved in dimethylsulfoxide. In chronic TNF-α treatment assay, 3T3-L1 adipocytes were treated with TNF-α for 72 h and Re for the last 24 h, unless otherwise indicated. Fresh TNF-α was added every 24 h. Re application is described in the figure legends.
2-DOG Uptake Assay
Glucose transport was determined by measuring the uptake of [3H]2-DOG. Cells were cultured in 24-well plates. Transport assay was initiated by washing the cells twice in a transport solution, Krebs-Ringer’s phosphate buffer (20 mm HEPES, 137 mm NaCl, 4.7 mm KCl, 1.2 mm MgSO4, 1.2 mm KH2PO4, 2.5 mm CaCl2, and 2 mm pyruvate, pH 7.4). Cells were then incubated in the transport solution for 15 min, which contained 0.1 mm 2-DOG and 0.5 μCi [3H]2-DOG (10 mCi/mmol). The cells were quickly washed three times with ice-cold PBS containing 10 mm glucose. The cells were lysed in 0.1 n NaOH and subsequently solubilized in scintillation fluids (Triton X-100/methylbenzene, 1:2.5) overnight. [3H]2-DOG was measured by liquid scintillation counter. The nonspecific glucose uptake was measured by subtracting values for [3H]2-DOG in the presence of 10 μm cytochalasin B.
Subcellular Fractionation
To obtain PM and LDM, cells grown on 10-cm dishes were treated with Re as described above. Then subcellular fractionation was carried out with a modification of the method provided by Shisheva et al. (38), Briefly, after being washed in ice-cold PBS, cells were scraped in HES buffer [20 mm Tris/HCl (pH 7.4), 255 mm sucrose, 1 mm EDTA, and 1× protease inhibitor mixture) on ice and homogenized. The homogenate was centrifuged at 16,000 × g for 20 min. The supernatant was further centrifuged at 36,000 × g for 5 min for HDM fraction and at 200,000 × g for 24 min for the LDM fraction. The PM fraction was collected from the interface of a 1.12 m sucrose cushion after centrifugation of the 16,000 × g pellet at 70,000 × g for 10 min. PM were then resuspended in homogenization buffer and pelleted at 200,000 × g for 4 min. The obtained fractions were then resuspended in HES buffer to a mixture about 1–2 mg/ml protein, quantitated by bicinchoninic protein assay kit (Pierce, Rockford, IL), and subjected to Western blotting analysis.
Immunoprecipitation
The cell lysates were extracted with lysis buffer (RIPA, 1× PBS, 1% Nonidet P-40, 5 mm EDTA, 0.1% SDS, 1 mm sodium orthovanadate, 1% phenylmethylsulfonyl fluoride, complete protease inhibitor cocktail, and complete phosphatase inhibitors). The whole-cell lysates were centrifuged at 12,000 × g for 20 min at 4 C to remove the insoluble materials. Protein concentrations were determined by Lowry method using DC protein assay reagent (Bio-Rad Laboratories, Hercules, CA). For immunoprecipitation, the supernatants were incubated with the indicated antibody for 16 h at 4 C, followed by incubation with protein A-agarose for 2 h. The beads were washed with the lysis buffer three times. The cell lysates or immunoprecipitates were boiled with Laemmli sample buffer for 5 min and then were resolved by SDS-PAGE.
Western Blotting
Cells were treated and cell lysates extracted as described above. The cell lysates were loaded onto SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA). The membranes were blocked and incubated with different antibodies, followed by incubation with horseradish peroxidase-conjugated secondary antibodies. The proteins were visualized with enhanced chemiluminescence reagents according to the manufacturer’s protocol (Amersham Pharmacia). In some experiments, the intensities of blots were quantitated using a scan densitometer.
PI3K Assay
The treated 3T3-L1 adipocytes were solubilized in a buffer containing 20 mmol/liter Tris, 100 mmol/liter NaCl, 0.8 mmol/liter MgCl2, 0.8 mmol/liter CaCl2, 0.1 mmol/liter Na3VO4, 1% Nonidet P-40, 10% glycerol, 1 mmol/liter phenylmethylsulfonyl fluoride, and 100 mg/ml aprotinin. The cell lysates were centrifuged at 10,000 ×g for 20 min at 4 C to remove the insoluble materials. The supernatant was incubated with anti-p85 subunit, anti-IRS-1, and anti-PY20 antibodies, respectively, overnight at 4 C, and then the immune complexes were collected by incubation with protein A-agarose for 2 h at 4 C. The beads were washed and suspended in 20 ml solution containing 0.5 mg/ml PI, 50 mmol/liter HEPES, 1 mmol/liter NaH2PO4, and 1 mmol/liter EGTA. The phosphorylation reaction was carried out by adding 10 μl reaction buffer [50 mm MgCl2, 100 mm HEPES (pH 7.6), 250 μm ATP containing 5 μCi [γ-32P]ATP] for 5 min at 25 C. The reaction was terminated by the addition of 15 μl 4 mol/liter HCl. The products were extracted by adding 130 μl chloroform/methanol (1:1) and centrifugation. The lower organic phase was removed and spotted on a silica gel 60 plate. The plates were developed in CHCl3/CH3OH/H2O/NH4OH (60:47:11.3:2) and dried. The phosphorylated inositol was visualized by autoradiography.
Real-Time PCR
Total RNA was isolated from mature 3T3-L1 adipocytes using Trizol reagent (Sigma). An aliquot of 1 μg total RNA from each sample was reverse transcribed to cDNA according to the protocol of the reverse transcription system (a3500; Promega, Madison, WI) with anchored oligo(dT) primer. Gene expression was analyzed by relative quantitation with the 2−ΔΔCt method using real-time PCR with an ABI Prism 7300 instrument (Applied Biosystems, Foster City, CA). Real-time PCR was performed in 96-well plates using a SYBR Premix Ex Taq (Takara, Shiga, Japan) according to the supplier’s instructions. All samples were normalized to values of β-actin and results expressed as fold changes of threshold cycle (Ct) value relative to controls. Cycling parameters were 95 C for 10 sec and then 40 cycles of 95 C for 5 sec and 60 C for 31 sec. Quantifications were performed in triplicate, and the experiments were repeated independently three times. Primers sequences were as follows: Socs3 sense, 5′-AGAGCGGATTCTACTGGAGC-3′, and antisense 5′-TGGATGCGTAGGTTCTTGGTC-3′; IL-6 sense, 5′-TTCCATCCAGTTGCCTTCTT-3′, and antisense, 5′-CAGAATTGCCATTGCACAAC-3′; and actin sense, 5′-GGCTGTATTCCCCTCCATCG-3′, and antisense, 5′-CCAGTTGGTAACAATGCCATGT-3′.
HEC in HFD Rats
Male Wistar rats (250 g) were purchased from Shanghai Slac Laboratory Animal Co. Ltd. (Shanghai, China), habituated to the new environment at 22 ± 0.5 C, and fed with a standard chow diet ad libitum for 1 wk. After this acclimatization, they were randomly allocated into three groups as chow, HFD, and HFD treated with Re (HFD/Re). They were fed with standard chow and HFD, respectively, for 2 wk. The nutrient compositions of HFD included 60% fat, 20% carbohydrate, and 20% protein, Thereafter, the rats were assigned to receive vehicle (50% propylene glycol) in chow and HFD groups and Re (40 mg/kg·d) in the HFD/Re group. Re was administrated by ip injection twice a day between 1300 and 1500 for 2 wk. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Ruijin Hospital.
One week before the HEC test, left and right jugular veins were cannulated under general anesthesia (2.5% pentobarbitone) (39). After the surgery, the rats were single housed and handled daily to minimize stress. Only those rats that regained body weight after surgery were used for the HEC test.
HEC tests were performed on conscious and postabsorptive (∼5 h fasted) rats that were allowed to move freely. After a period of settling (∼40–50 min), two basal blood samples were collected for measurements of plasma glucose and other parameters. Rats were then infused with insulin (Humulin R; Eli Lilly) via the jugular cannula at 0.5 U/kg·h, together with a variable rate of 25% glucose to maintain euglycemia based on regular blood glucose measurements. The GIR was measured to calculate whole-body insulin sensitivity.
Calculations and Statistical Analysis
Data are presented as means ± sd or means ± se. Comparisons between two groups were made using Student’s t test or ANOVA as appropriate. A P value of <0.05 was considered significant.
Acknowledgments
We are grateful to Dr. Marc Prentki from the Department of Nutrition, University of Montreal, Quebec, Canada, for critical reading of the manuscript.
Footnotes
This work was supported by 973 Project (No. 2006CB503904), Shanghai Commission for Science and Technology (No. 04DZ19502) and E-Institute of Shanghai Universities, Shanghai Education Commission (No. E03007).
Disclosure Summary: The authors have nothing to disclose.
First Published Online September 20, 2007
Abbreviations: AMPK, AMP-activated protein kinase; AS160, Akt substrate of 160 kDa; CRP, C-responsive protein; 2-DOG, 2-deoxyglucose; GIR, glucose infusion rate; GSK-3β, glycogen synthase kinase-3β; HEC, hyperinsulinemic-euglycemic clamp; HFD, high-fat diet; IκBα, inhibitor of NF-κBα; IKK, nuclear factor-κB kinase; IR, insulin receptor; IRβ, IR β-subunit; IRS, IR substrate; JNK, c-Jun NH2-terminal kinase; LDM, low-density membrane; NF, nuclear factor; PI3K, phosphatidylinositol 3-kinase; PM, plasma membrane; P70S6K, p70 ribosomal S6 protein kinase; PKC, protein kinase C; Re, ginsenoside Re; SOCS-3, suppressor of cytokine signaling proteins-3.
References
- 1.Saltiel AR 2001. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 104:517–529 [DOI] [PubMed] [Google Scholar]
- 2.Li WL, Zheng HC, Bukuru J, De Kimpe N 2004. Natural medicines used in the traditional Chinese medical system for therapy of diabetes mellitus. J Ethnopharmacol 92:1–21 [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization 1999. WHO monographs on selected medicinal plants. Geneva: World Health Organization; 168–183
- 4.Han KL, Jung MH, Sohn JH, Hwang JK 2006. Ginsenoside 20(S)-protopanaxatriol (PPT) activates peroxisome proliferator-activated receptor γ (PPARγ) in 3T3-L1 adipocytes. Biol Pharm Bull 29:110–113 [DOI] [PubMed] [Google Scholar]
- 5.Dey L, Xie JT, Wang A, Wu J, Maleckar SA, Yuan CS 2003. Antihyperglycemic effects of ginseng: comparison between root and berry. Phytomedicine 10:600–605 [DOI] [PubMed] [Google Scholar]
- 6.Sotaniemi EA, Haapakoski E, Rautio A 1995. Ginseng therapy in noninsulin-dependent diabetic patients. Diabetes Care 18:1373–1375 [DOI] [PubMed] [Google Scholar]
- 7.Yeh GY, Eisenberg DM, Kaptchuk TJ, Phillips RS 2003. Systematic review of herbs and dietary supplements for glycemic control in diabetes. Diabetes Care 26:1277–1294 [DOI] [PubMed] [Google Scholar]
- 8.Attele AS, Zhou YP, Xie JT, Wu JA, Zhang L, Dey L, Pugh, W, Rue PA, Polonsky KS, Yuan CS 2002. Antidiabetic effects of Panax ginseng berry extract and the identification of an effective component. Diabetes 51:1851–1858 [DOI] [PubMed] [Google Scholar]
- 9.Xie JT, Mehendale SR, Li X, Quigg R, Wang X, Wang CZ, Wu JA, Aung HH, A Rue P, Bell GI, Yuan CS 2005. Anti-diabetic effect of ginsenoside Re in ob/ob mice. Biochim Biophys Acta 1740:319–325 [DOI] [PubMed] [Google Scholar]
- 10.Cho WC, Chung WS, Lee SK, Leung AW, Cheng CH, Yue KK 2006. Ginsenoside Re of Panax ginseng possesses significant antioxidant and antihyperlipidemic efficacies in streptozotocin-induced diabetic rats. Eur J Pharmacol 550:173–179 [DOI] [PubMed] [Google Scholar]
- 11.Cho WC, Yip TT, Chung WS, Lee SK, Leung AW, Cheng CH, Yue KK 2006. Altered expression of serum protein in ginsenoside Re-treated diabetic rats detected by SELDITOF MS. J Ethnopharmacol 108:272–279 [DOI] [PubMed] [Google Scholar]
- 12.Watson RT, Kanzaki M, Pessin JE 2004. Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endocr Rev 25:177–204 [DOI] [PubMed] [Google Scholar]
- 13.Shoelson SE, Lee J, Goldfine AB 2006. Inflammation and insulin resistance. J Clin Invest 116:1793–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotamisligil GS 2006. Inflammation and metabolic disorders. Nature 444:860–867 [DOI] [PubMed] [Google Scholar]
- 15.Aguirre V, Uchida T, Yenush L, Davis R, White MF 2000. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J Biol Chem 275:9047–9054 [DOI] [PubMed] [Google Scholar]
- 16.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF 2002. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem 277:1531–1537 [DOI] [PubMed] [Google Scholar]
- 17.Perrini S, Natalicchio A, Laviola L, Belsanti G, Montrone C, Cignarelli A, Minielli V, Grano M, De Pergola G, Giorgino R, Giorgino F 2004. Dehydroepiandrosterone stimulates glucose uptake in human and murine adipocytes by inducing GLUT1 and GLUT4 translocation to the plasma membrane. Diabetes 53:41–52 [DOI] [PubMed] [Google Scholar]
- 18.Attele AS, Wu JA, Yuan CS 1999. Ginseng pharmacology: multiple constituents and multiple actions. Biochem Pharmacol 58:1685–1693 [DOI] [PubMed] [Google Scholar]
- 19.Lewis C. Cantley 2002. The phosphoinositide 3-kinase pathway. Science 296:1655–1657 [DOI] [PubMed] [Google Scholar]
- 20.Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE 2003. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 278:14599–14602 [DOI] [PubMed] [Google Scholar]
- 21.Zeigerer A, McBrayer MK, McGraw TE 2004. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell 15:4406–4415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wellen KE, Hotamisligil GS 2005. Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barzilay JI, Freedland ES 2003. Inflammation and its relationship to insulin resistance, type 2 diabetes mellitus, and endothelial dysfunction. Metab Syndrome 1:55–67 [DOI] [PubMed] [Google Scholar]
- 24.Festa A, D’Agostino Jr R, Tracy RP, Haffner SM 2002. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes 51:1131–1137 [DOI] [PubMed] [Google Scholar]
- 25.Hotamisligil GS, Shargill NS, Spiegelman BM 1993. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science 259:87–91 [DOI] [PubMed] [Google Scholar]
- 26.Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A 1993. Tumor necrosis factor-α suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J Biol Chem 268:26055–26058 [PubMed] [Google Scholar]
- 27.Hotamisligil GS, Spiegelman BM 1994. Tumor necrosis factor α: a key component of the obesity-diabetes link. Diabetes 143:1271–1278 [DOI] [PubMed] [Google Scholar]
- 28.Paz K, Hemi R, LeRoith D, Karasik A, Elhanany E, Kanety H, Zick YA 1997. Molecular basis for insulin resistance: elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem 272:29911–29918 [DOI] [PubMed] [Google Scholar]
- 29.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM 1996. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 271:665–668 [DOI] [PubMed] [Google Scholar]
- 30.Zick Y 2005. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci STKE 268:pe4 [DOI] [PubMed]
- 31.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS 2002. A central role for JNK in obesity and insulin resistance. Nature 420:333–336 [DOI] [PubMed] [Google Scholar]
- 32.Bennett BL, Satoh Y, Lewis AJ 2003. JNK: a new therapeutic target for diabetes. Curr Opin Pharmacol 3:420–425 [DOI] [PubMed] [Google Scholar]
- 33.Qi C, Pekala PH 2000. Tumour necrosis factor-α-induced insulin resistance in adipocytes. Proc Soc Exp Biol Med 223:128–135 [DOI] [PubMed] [Google Scholar]
- 34.Gao Z, Zuberi A, Quon MJ, Dong Z, Ye J 2003. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases. J Biol Chem 278:24944–24950 [DOI] [PubMed] [Google Scholar]
- 35.Kanatani Y, Usui I, Ishizuka K, Bukhari A, Fujisaka S, Urakaze M, Haruta T, Kishimoto T, Naka T, Kobayashi M 2007. Effects of pioglitazone on suppressor of cytokine signaling 3 expression. Diabetes 56:795–803 [DOI] [PubMed] [Google Scholar]
- 36.Seo JY, Lee JH, Kim NW, Kim YJ, Chang SH, Ko NY, Her E, Yoo YH, Kim JW, Lee BY, Lee HY, Kim YM, Choi WS 2005. Inhibitory effects of a fermented ginseng extract, BST204, on the expression of inducible nitric oxide synthase and nitric oxide production in lipopolysaccharide-activated murine macrophages. J Pharm Pharmacol 57:911–918 [DOI] [PubMed] [Google Scholar]
- 37.Keum YS, Han SS, Chun KS, Park KK, Park JH, Lee SK, Surh YJ 2003. Inhibitory effects of the ginsenoside Rg3 on phorbol ester-induced cyclooxygenase-2 expression, NF-κB activation and tumor promotion. Mutat Res 523–524:75–85 [DOI] [PubMed]
- 38.Shisheva A, Buxton J, Czech MP 1994. Differential intracellular localizations of GDP dissociation inhibitor isoforms. J Biol Chem 269:23865–23868 [PubMed] [Google Scholar]
- 39.Cleasby ME, Dzamko N, Hegarty BD, Cooney GJ, Kraegen EW, Ye JM 2004. Metformin prevents the development of acute lipid-induced insulin resistance in the rat through altered hepatic signaling mechanisms. Diabetes 53:3258–3266 [DOI] [PubMed] [Google Scholar]