

Abstract
The nuclear receptor steroidogenic factor-1 (SF1) is critical for development and function of steroidogenic tissues. Posttranslational modifications are known to influence the transcriptional capacity of SF1, and it was previously demonstrated that serine 203 is phosphorylated. In this paper we report that serine 203 is phosphorylated by a cyclin-dependent kinase 7 (CDK7)-mediated process. As part of the CDK-activating kinase complex, CDK7 is a component of the basal transcription factor TFIIH, and phosphorylation of SF1 as well as SF1-dependent transcription was clearly reduced in cells carrying a mutation that renders the CDK-activating kinase complex unable to interact with the TFIIH core. Coimmunoprecipitation analyses revealed that SF1 and CDK7 reside in the same complex, and kinase assays demonstrated that immunoprecipitated CDK7 and purified TFIIH phosphorylate SF1 in vitro. The CDK inhibitor roscovitine blocked phosphorylation of SF1, and an inactive form of CDK7 repressed the phosphorylation level and the transactivation capacity of SF1. Structural studies have identified phosphoinositides as potential ligands for SF1. Interestingly, we found that mutations designed to block phospholipid binding dramatically decreased the level of SF1 phosphorylation. Together our results suggest a connection between ligand occupation and phosphorylation and association with the basic transcriptional machinery, indicating an intricate regulation of SF1 transactivation.
THE TRANSCRIPTION FACTOR (TF) steroidogenic factor 1 (SF1, also known as Ad4BP; systematic name NR5A1) is a member of the nuclear receptor superfamily and a key regulator of endocrine functions within the hypothalamus-pituitary gonadal and adrenal axes. SF1 induces the expression of essentially all factors involved in steroid hormone biosynthesis and is fundamental for adrenal and gonadal organogenesis (1). SF1 has been classified as a constitutively active orphan nuclear receptor, and although the nature of a bona fide ligand in eukaryotic cells remains unknown, structural analyses revealed the presence of phospholipids in the ligand-binding pocket (LBP) of bacterially expressed SF1 (2, 3, 4). Eukaryotic phospholipids, including phosphoinositides, stabilize SF1 in an active conformation and increase the interaction with coactivators with subsequent effects on transcription (2, 3, 4). Recently, another class of lipids, sphingolipids (i.e. sphingosine and lyso-sphingomyelin), was identified in the LBP of SF1 isolated from mammalian cells (5). In contrast to the positive effect of phosphoinositides, sphingolipids were found to inhibit SF1 activity (5), indicating that the relative levels of phosphatidylinositol phosphates and sphingolipids might regulate the transcriptional capacity of SF1. The identification of lipids in the LBP encourages further studies on the mechanisms whereby SF1 activates transcription. For instance, it is yet to be established how ligand occupation affects posttranslational modifications of SF1, and vice versa.
Steroid hormone biosynthesis is regulated by pituitary-derived peptide hormones (i.e. FSH, LH, and ACTH). ACTH induces multiple signaling cascades in adrenocortical cells, such as cAMP and Ca2+-dependent pathways, as well as protein kinase C (PKC) and MAPK pathways (6). Because most of the genes that are regulated by SF1 are hormone responsive, substantial effort has been directed to dissect the molecular mechanisms whereby SF1 integrates hormone-induced signaling and gene expression. Hormonal stimulation does not affect SF1 mRNA levels (7, 8, 9, 10) but induces posttranslational modifications such as phosphorylation (11) and acetylation (12) with subsequent effects on stability and transcriptional capacity. Immunoprecipitation of SF1 from radioactively labeled cells demonstrates that SF1 is a phosphoprotein (13, 14, 15). The only identified phosphorylation site in SF1 until now is serine 203 (S203), and the MAPK ERK2 has been proposed to phosphorylate this residue (14). Phosphorylation of S203 leads to alterations in the recruitment of the coactivator glucocorticoid receptor-interacting protein-1 (GRIP-1) (16), and mutation of S203 to alanine (S203A) causes decreased transcription from some SF1-dependent reporter genes (14, 17). Several reports support that activation of MAPK cascades stimulates SF1 transactivation and steroid hormone biosynthesis. These studies demonstrate that inhibition of ERK1/2 reduces transcription from SF1-dependent promoters (17, 18), reduces steroid hormone production in adrenocortical cells (19), and affects the intramolecular stabilization of SF1 (16, 17). Notably however, ERK1/2 inhibition appears to have minimal effects on the phosphorylation status of SF1 when monitored with an S203-phospho-specific antibody (16, 17), suggesting that other kinases might target this residue. It is well documented that cAMP-dependent kinase [protein kinase A (PKA)] is essential for optimal steroidogenesis. Although PKA clearly affects SF1-dependent transcription (7, 11, 20, 21), a putative phosphorylation site for PKA has not yet been identified. In fact, results from metabolically labeled cells indicate that cAMP does not induce phosphorylation of SF1 (13, 14, 15).
Similar to other nuclear receptors, SF1 contains two activation function (AF) domains, AF1 and AF2, that work together for maximal transactivity (14, 22, 23). AF2 is found at the C terminus of the ligand-binding domain (LBD), but in contrast to most nuclear receptor, the AF1-domain is located between the DNA-binding domain (DBD) and the hinge region and not in the N-terminal region. Proline-dependent kinases, such as MAPKs and cyclin-dependent kinases (CDKs) are known to target the AF1 domain of several nuclear receptors (24, 25), and notably, S203 is located in the AF1 domain of SF1. In line with this, the AF1 domain of the progesterone receptor (26, 27), the estrogen receptors (ER)α (28, 29) and -β (30), the androgen receptor (AR) (31), the retinoic acid receptor (RAR)γ (32), and the peroxisome proliferator-activated receptors (PPARs) α and γ (33, 34), are phosphorylated by ERK1/2 or p38 MAPKs. In the case of ERα (35) and PPARα/γ (36), the very same residues that are targeted by MAPK are also phosphorylated by the cyclin-dependent kinase 7 (CDK7), an event that is paralleled with increased transcriptional response. CDK7 is part of the trimeric CDK-activating kinase (CAK) complex, which includes the two other subunits, cyclin H (cycH) and ménage à trois 1 (MAT1). CAK, as part of the basic transcription factor TFIIH, phosphorylates the Pol II C-terminal domain (CTD) and is associated with transcriptional elongation (37). CAK also phosphorylates other transcriptional regulators in addition to nuclear receptors [e.g. p53 (38), Oct-factors (39), and E2F-1 (40)] and is also involved in cell cycle progression through phosphorylation of other CDKs (for a review see Ref.37).
SF1 is evidently a phosphoprotein and integrates kinase activities and gene expression in steroidogenic cells. Our current knowledge of which kinases that modify SF1 activity, as well as how peptide hormone-induced signaling control these modifications, is limited. To gain further information of how the transcriptional competence of SF1 is modified by phosphorylation, we attempted to identify kinases that target SF1. We found that SF1 interacts with a CDK7-containing complex and that SF1 is phosphorylated in a TFIIH-dependent fashion on S203. Furthermore, we show that phosphorylation of S203 is coupled to the integrity of the LBP and transcriptional activation, indicating a correlation between ligand binding, phosphorylation status, and functionality.
RESULTS
SF1 Is Recognized by a Phospho-(Ser) CDKs Substrate (P-S-sub-CDKs) Antibody
To search for kinases that phosphorylate SF1, we asked whether antibodies that recognize phosphorylated serine and/or threonine residues within peptide motifs specific for CDKs, PKA, Akt, and PKC would interact with SF1. Endogenous SF1 was isolated from human adrenocortical nuclei (H295R cells) by DNA pull down (DPD) using a biotinylated oligonucleotide containing SF1-responsive elements (SFREs) (41). When the DPD material was visualized by Coomassie staining, several bands were apparent (Fig. 1A, left panel). The identity of the band corresponding to the molecular weight of SF1 was validated as SF1 by mass spectrometry and Western blot analyses (data not shown and Fig. 1A, right panel). The other proteins that were visible on the Coomassie-stained gel were identified by mass spectrometry to be acetyl-coenzyme A carboxylase, poly (ADP-ribose) polymerase 1, heat shock protein 70, Y-box binding protein 1, and histone H1. None of these proteins has been described to interact with SF1 or SFRE and may represent nonspecific binding to DNA. In this study, it was not investigated further whether these proteins, in fact, interact with SF1 or SFRE. The DPD-protein mixture was subjected to Western blotting using phospho-(Ser/Thr) kinase substrate antibodies for CDKs, PKA, Akt, and PKCs as shown in Fig. 1B. SF1 was only reactive toward the P-S-sub-CDKs antibody, thus indicating a potential phosphorylation by CDKs on a serine residue. The P-S-sub-CDKs antibody also detected phosphorylated SF1 from murine or bovine origin, consistent with phosphorylation on a conserved site (c.f. Fig. 3A, and data not shown). Under the same conditions, we did not detect any interaction between the P-S-sub-PKA antibody and SF1, suggesting that PKA does not phosphorylate SF1, at least not on a typical PKA recognition site. A corresponding antibody specifically interacting with phosphorylated serine residues in MAPK target sites was not available.
Fig. 1.

SF1 Is Phosphorylated by CDKs
A, Endogenous SF1 was isolated from H295R nuclear extract (10 mg) by DPD using a biotinylated double-stranded oligonucleotide containing SFREs and streptavidin-conjugated agarose beads. Eluates (DPD) were run on 10% SDS-PAGE and stained with Coomassie blue (left panel) or subjected to Western blotting with an SF1 antibody (right panel). The proteins present in the bands (in the left panel were identified by mass spectrometry (please see main text for details). MW, Molecular weight standard. B, H295R cells were treated with okadaic acid (100 nm) for 1 h, and SF1 was isolated by DPD. Eluates were subjected to Western blotting with the anti-phospho-(Ser and/or Thr) kinase substrate antibodies for CDKs, PKA, Akt, and PKC. WB, Western blot.
Fig. 3.
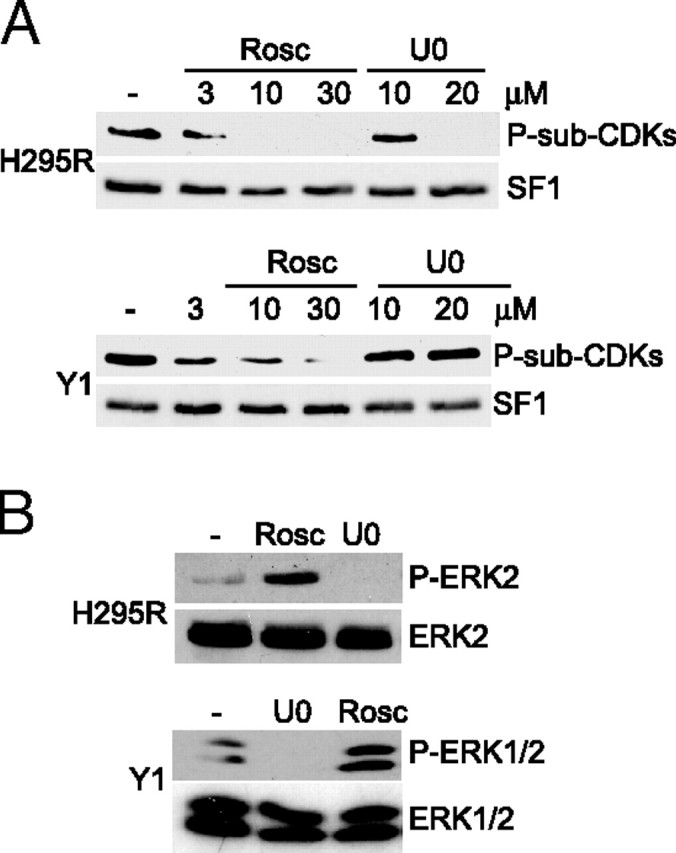
Inhibition of CDKs Blocks Phosphorylation of SF1 in Adrenocortical Cells
A, H295R and Y1 cells were incubated with DMSO (−), increasing concentrations of roscovitine (Rosc; 3, 10, or 30 μm) or U0186 (U0; 10 or 20 μm) for 1 h. SF1 was isolated by DPD, and Western blot analyses were performed using the anti-P-S-sub-CDKs antibody followed by the SF1 antibody. B, H295R and Y1 cells were incubated with DMSO (−), U0186 (U0; 10 μm), or roscovitine (Rosc; 30 μm) for 45 min. Cell extracts were run on SDS-PAGE and blotted with the phospho-ERK1/2 antibody (upper panel) followed by the ERK1/2 antibody (lower panel).
CDKs Phosphorylate SF1 on S203
There is no consensus site for CDKs in SF1. Deletion constructs were therefore generated to narrow down the region of P-S-sub-CDKs antibody recognition. Flag-tagged murine SF1 and deletion mutants spanning amino acids (aa) 1–110 [SF1(1–110)] or 1–279 [SF1(1–279)] (Fig. 2A) were expressed in COS-1 cells and isolated by immunoprecipitation. Both full-length SF1 and SF1(1–279), but not SF1(1–110), were recognized by the P-S-sub-CDKs antibody (Fig. 2B, left panel), demonstrating that SF1 is phosphorylated on a residue located between aa 110 and 279, spanning the AF1 domain and part of the LBD. Further examination of this region in the human, murine, and bovine protein sequences (Fig. 2C), pointed to S203 as the likely phosphorylation site because it is followed by a proline, CDKs being proline-dependent kinases. This was confirmed by site-directed mutagenesis of this residue to glutamic acid (S203E) or alanine (S203A). As shown in Fig. 2D, the P-sub-CDKs antibody did not detect these mutants. As a control, we also mutated another conserved serine residue (S219) to alanine (S219A). This modification did not reduce phosphorylation of SF1 (Fig. 2D). These results thus indicate that the P-S-sub-CDKs antibody recognizes phosphorylated SF1 at a unique site, S203, which was previously suggested to be targeted by another proline-dependent kinase, ERK2 (14). To investigate the functional significance of the S203 phosphorylation, SF1(WT), SF1(S203A), and SF1(S203E) were transfected into COS-1 cells together with a luciferase construct containing a natural SF1-responsive promoter from the human gene encoding cytochrome P450 17α-steroid hydroxylase (CYP17; CYP17/Luc). SF1(WT) efficiently induced transcription from CYP17/Luc (Fig. 2E). Mutation of S203 to alanine led to a small decrease in luciferase activity, whereas the activity of SF1(S203E) was equal to that of SF1(WT) (Fig. 2E).
Fig. 2.
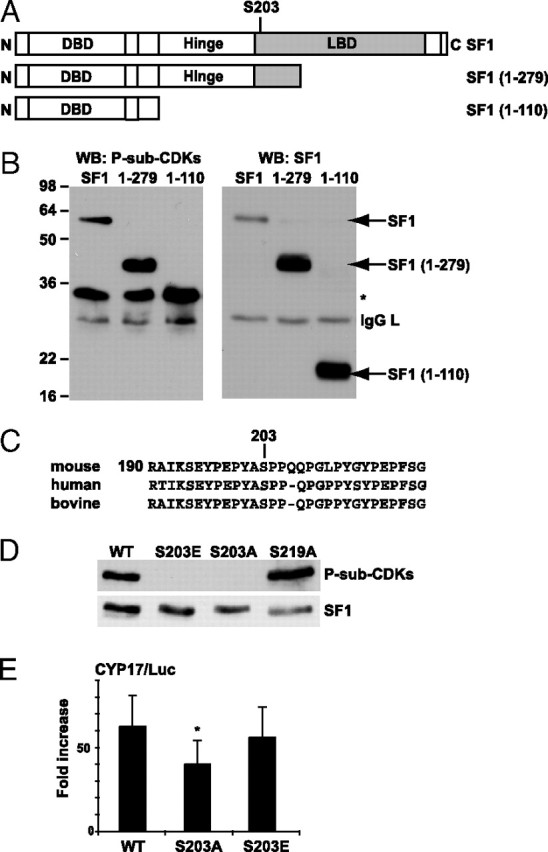
SF1 Is Phosphorylated by CDKs on S203
A, Schematic representation of full-length SF1 and the deletion constructs of SF1 [SF1(1–279), SF1(1–110)] used in this study. The numbering corresponds to the aa sequence. B, COS-1 cells were transfected with Flag-pCMV2/SF1 (SF1), Flag-pCMV2/SF1(1–110) (1–110), and Flag-pCMV2/SF1(1–279) (1–279), and SF1 was immunoprecipitated with the SF1 antibody and subjected to Western blot analyses using the anti-P-S-sub-CDKs antibody and subsequently the SF1 antibody. *, Unknown reactive protein. C, Sequence alignment of human, mouse, and bovine SF1. D, COS-1 cells were transfected with Flag-pCMV2/SF1 (WT) and mutants Flag-pCMV2/SF1(S203A) (S203A), Flag-pCMV2/SF1(S203E) (S203E), or Flag-pCMV2/SF1(S219A) (S219A). SF1 was immunoprecipitated with Flag antibodies, and Western blot analyses were performed using the anti-P-sub-CDKs antibody (upper panel) followed by incubation with SF1 antibodies (lower panel). E, COS-1 cells were transfected with CYP17/Luc (1 μg) together with Flag-pCMV2/SF1 WT, S203A, or S203E (100 ng). The luciferase activity is shown as fold increase and relative to β-gal activity (n = 7–10; *, P < 0.05 as determined by ANOVA, Bonferoni test). DBD, DNA-binding domain; WB, Western blot.
Inhibition of CDKs Abolishes SF1 Phosphorylation
Even though the P-sub-CDKs antibody recognized phosphorylated S203, the results presented until now did not provide direct information as to which proline-dependent kinase targets SF1. To investigate the putative role of CDKs, we used the inhibitor roscovitine, which specifically inhibits CDK1, CDK2, CDK5, CDK7, and CDK9 (42, 43). H295R cells and mouse adrenocortical tumor Y1 cells were incubated with either dimethylsulfoxide (DMSO) or increasing amounts of roscovitine, and endogenous SF1 was isolated by DPD. As shown in Fig. 3A, phosphorylation of SF1 was prevented by roscovitine in both cell lines. However, the sensitivity toward this compound differed, because phosphorylation was completely abolished by 10 μm in H295R cells whereas 30 μm roscovitine was required to inhibit phosphorylation in Y1 cells. An inhibitor (U0126) of the MAPK kinases MEK-1/-2, acting upstream of ERK1/2 (44, 45), was also included in the experiment to differentiate the roles of CDKs and ERK1/2. Interestingly, U0126 did not affect SF1 phosphorylation at a concentration (10 μm) that totally blocked ERK1/2 activity, as monitored by an antibody detecting phosphorylated ERK1/2 [active ERK1/2 is phosphorylated on tyrosine 204 (46)] (Fig. 3B). However, treatment with U0126 did prevent phosphorylation of SF1 in H295R cells at a higher concentration (20 μm) (Fig. 3A), presumably because of nonspecific effects, because the lower dose (that completely inhibited ERK1/2 phosphorylation) was without effect. In contrast, U0126 had no effect in Y1 cells, and it is unknown why the phosphorylation of SF1 is affected differently by these compounds in Y1 and H295R cells. The results in Fig. 3B also demonstrated that whereas U0126 completely inhibited ERK1/2 phosphorylation, roscovitine did not in either cell line, arguing against a nonspecific effect of roscovitine on ERK1/2. Taken together, the results presented in Fig. 3 further indicated the involvement of CDKs in phosphorylation of S203.
SF1 Interacts with CDK7 and Is Phosphorylated by TFIIH
Because the AF1 domain of several nuclear receptors (i.e. RARγ, RΑRα, PPARα/γ, and ERα) is phosphorylated by CDK7 on serine residues (Table 1) (35, 36, 47), we investigated whether SF1 is a substrate for CDK7. First, we determined whether SF1 interacted with CDK7 in cells. Flag-SF1(WT and S203A) were overexpressed in COS-1 cells and immunoprecipitated with anti-Flag antibodies, followed by Western blot analyses using anti-CDK7 antibodies. A faint, but consistent, band corresponding to the size of CDK7 was detected in the immunoprecipitated material from COS-1 cells expressing Flag-SF1(WT) but not in extract from COS-1 cells expressing β-Gal (Fig. 4A, upper panel). Interestingly, SF1(S203A) interacted equally well with CDK7 as SF1(WT) did, indicating that lack of phosphorylation and transcriptional enhancement of this mutant by CAK subunits (see below) were not due to a lack of interaction (Fig. 4A). Reprobing the blot with antibodies against SF1 verified the presence of SF1 in the precipitated material (Fig. 4A, lower panel). To determine whether SF1 could act as a substrate for CDK7, an in vitro kinase assay was performed. Because CDK2 is also involved in nuclear receptor phosphorylation (48), this kinase was included in the experiment. CDK2 and CDK7 were immunoprecipitated from H295R cells and incubated with bacterially expressed glutathione-S-transferase (GST)-SF1 fusion proteins containing the hinge and LBD domains [GST-SF1(179–431) and GST-SF1(179–431/S203A)], or histone H1 in the presence of radioactive ATP. As evident from Fig. 4B, CDK7 phosphorylated SF1(WT) but did not induce phosphorylation of the S203A mutant. Although CDK2 was active and phosphorylated its substrate histone H1, it did not act on SF1 (Fig. 4B, upper panel). The levels of immunoprecipitated kinases were confirmed to be equal by Western blotting with anti-CDK7 or anti-CDK2 (Fig. 4B, middle and lower panel). Because we could not exclude that the immunoprecipitated material contained additional kinases, we also performed kinase assays with the same GST-SF1 proteins and TFIIH isolated from HeLa nuclear extracts. This experiment demonstrated that TFIIH phosphorylated SF1(WT), whereas the S203A mutant was not phosphorylated above background levels (Fig. 4C). Because CDK7 is the only kinase known to be associated with the TFIIH complex (49), these results strongly suggest that SF1 is indeed a target for CDK7, and that the phosphorylated residue is S203. To obtain support for these findings in an in vivo system, H295R cells were transfected with expression plasmids encoding either Myc-tagged wild-type (WT) CDK7 or a catalytically inactive form of CDK7 (CDK7-K41R), followed by DPD of SF1. The CDK7-K41R mutant, which is frequently used to validate the involvement of CDK7 in cellular processes, participates in the formation of a CAK complex, but renders it inactive (50). As shown in Fig. 4D, overexpression of WT CDK7 led to an increase in the phosphorylation of SF1 compared with control cells transfected with an unrelated plasmid. In contrast, phosphorylation of SF1 was reduced by approximately 50% after overexpression of CDK7-K41R, as determined by densitometric analyses (Fig. 4D, upper panel). These results thus further support that SF1 is a target for CDK7. Western analyses verified the expression of WT CDK7 and CDK7-K41R (Fig. 4D, lower panel).
Table 1.
CDK7 Phosphorylation Motifs in Nuclear Receptors (NRs) and Other Transcription Factors
| NR (Phospho Site) | CDK7 Phospho Sequence | Ref. Nos. |
|---|---|---|
| SF1 (S203) | PEPYASPPQP | This report |
| PPARγ1 (S84) | VEPASPPYY | 36 |
| PPARγ2 (S112) | ||
| PPARα (S12/S21) | CPLSPLEAGDLESPL | 36 |
| RARα (S77) | PSPPPSPPPL | 51 |
| RARγ (S77–S79) | MVPSSPSPPPP | 47 ) |
| ERα (S118) | PPPQLSPFLQ | 35 |
| E2F1 (S403/T433) | EFISLSPPHE | 40 |
| DFGDLTPLDF | ||
| RNA pol II CTD | YSPTSPS | 49 |
Underlined residues are phosphorylated. RNA pol II CTD, RNA polymerase II CTD.
Fig. 4.
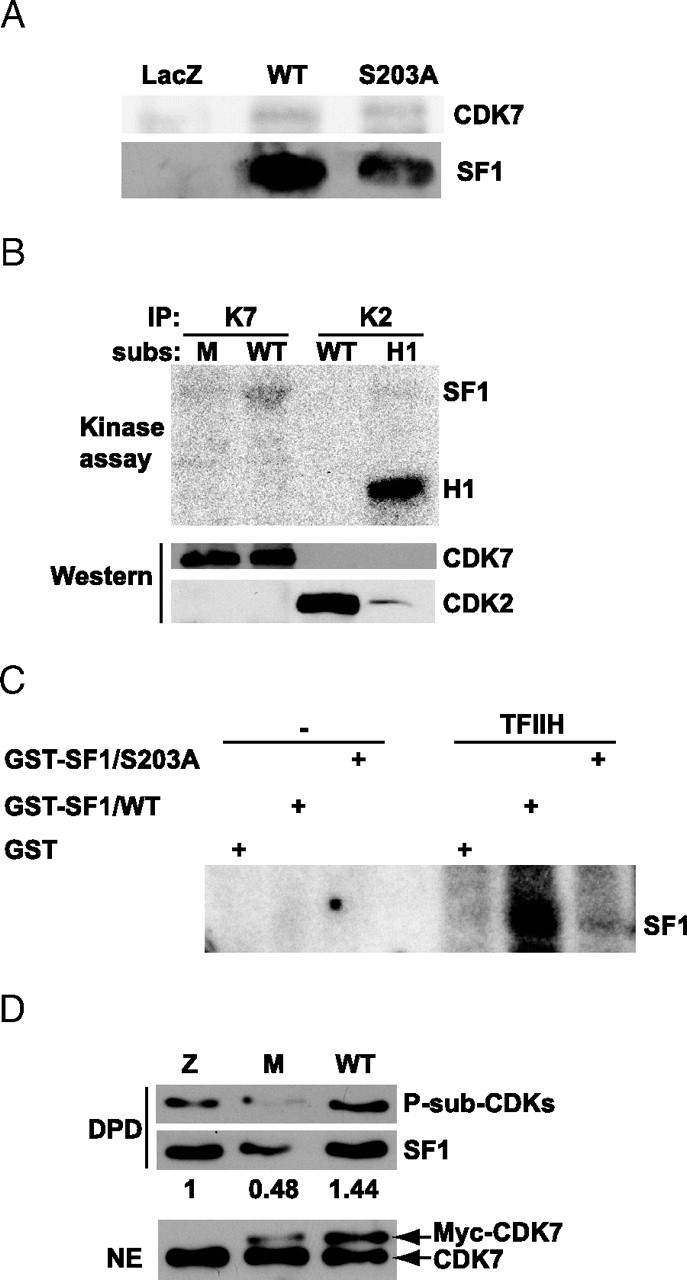
SF1 Coimmunoprecipitates with CDK7 and Is Phosphorylated by CDK7 and TFIIH
A, COS-1 cells were transfected with Flag-pCMV2/SF1 WT or S203A, or the control vector pCMV5/LacZ and immunoprecipitated with the Flag antibody. Eluates were run on a 10% SDS-PAGE and subjected to Western blot analyses using CDK7 (upper panel) and SF1 (lower panel) antibodies. B, CDK7 and CDK2 were immunoprecipitated from H295R cells and incubated with different substrates (subs), GST-SF1(179–431) (WT), GST-SF1(179–431/S203A) (M), or histone H1 (H1) for kinase assays. Membranes were subjected to autoradiography (O/N exposure) followed by Western blot analyses with anti-CDK7 and anti-CDK2. C, GST, GST-SF1(179–431), and GST-SF1(179–431/S203A) were incubated in the absence or presence of TFIIH isolated from HeLa cells. Gels were subjected to autoradiography (1–2 h exposure). D, H295R cells were transfected with control vector pCMV5/LacZ, Myc-CDK7 WT, or the Myc-CDK7-K41R (5 μg). SF1 was then isolated by DPD, and Western blotting was performed with anti-P-Sub-CDK (upper panel) and SF1 (middle panel) antibodies. Nuclear extracts (20 μg) were also subjected to Western blotting with anti-CDK7 (lower panel). Densitometric analyses were performed and are shown as ratios between phosphorylated SF1 and total SF1. The value from control cells was set to 1. IP, Immunoprecipitation; NE, nuclear extract.
CDK7, cycH, and MAT1 Enhance SF1-Dependent Transcription
Because the activity of CDK7 is dependent on its association with cycH and MAT1 to form the ternary CAK complex, we investigated the functional relevance of all three factors with regard to SF1 transactivation. All three subunits of the CAK complex stimulated transcription from a reporter construct carrying four copies of an optimized SF1 response element (SFRE/Luc) when overexpressed in H295R cells (Fig. 5A). A similar increase in luciferase activity was observed from CYP17/Luc after overexpression of CDK7 and cycH, whereas MAT1 did not stimulate transcription (Fig. 5B). In contrast, this reporter construct was stimulated by MAT1 in nonsteroidogenic COS-1 cells (Fig. 5C), and the difference between H295R cells and COS-1 cells with regard to MAT1 stimulation could be explained by the absence of background activity from this promoter in COS-1 cells. Although the stimulatory effect of CDK7, cycH, and MAT1 is restricted (fold induction of 1.2–1.6), it is in the same range as previously observed for ERα and RARα (51, 52). Concurrent overexpression of all three factors did not increase transcription further (data not shown). To further assess the functional importance of S203 as the target for CDK7, we transfected COS-1 cells with SF1(S203A) together with CYP17/Luc and expression plasmids encoding CDK7, cycH, or MAT1 (Fig. 5D). Importantly, the members of the CAK complex did not activate reporter gene activity in the presence of this mutant, demonstrating the requirement for a phosphorylatable residue at position S203. As expected, based on the effects of overexpressed CDK7-K41R on SF1 phosphorylation (Fig. 4D), this mutated form of CDK7 led to a decrease in luciferase activity (Fig. 6A, left panel). In contrast, an inactive form of CDK2 (53) did not reduce transcription from the CYP17 promoter (Fig. 6B), arguing, together with the results in Fig. 4B, that SF1 is specifically targeted by CDK7. CDK7-K41R did not have a general repressive effect on transcription, because transcription from LacZ/Luc (measured as βGal activity) was not affected (Fig. 6A, right panel).
Fig. 5.
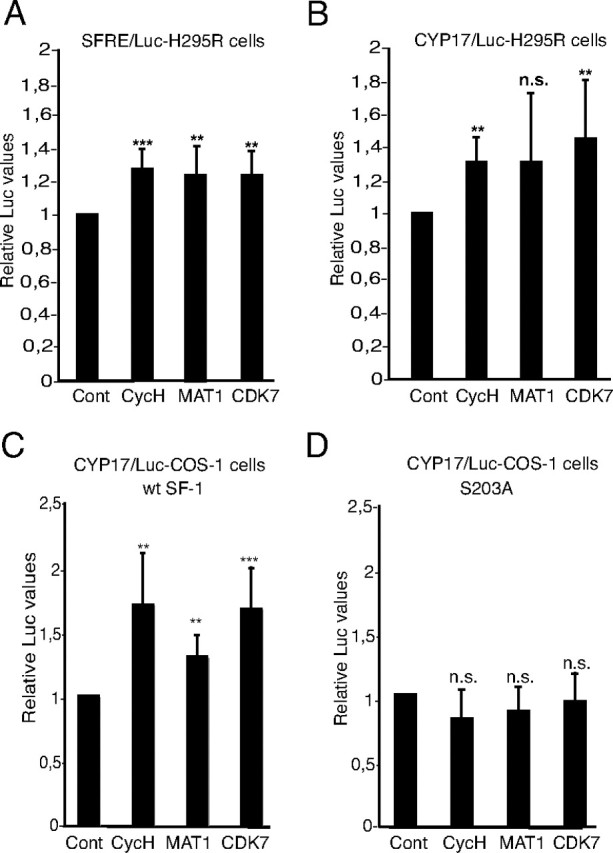
Subunits of the CAK Complex Enhance SF1-Dependent Transcription
A, H295R cells were transfected with SFRE/Luc (1 μg) together with expression plasmids encoding cycH (25 ng), MAT1 (10 ng), or CDK7 (10 ng). B, H295R cells were transfected as described in panel A, but with CYP17/Luc instead of SFRE/Luc. C, COS-1 cells were transfected with the CYP17/Luc, and an expression plasmid encoding SF1 (50 ng) together with cycH (50 ng), MAT1 (10 ng), or CDK7 (25 ng) expression plasmids as indicated in the figure. D, COS-1 cells were transfected as described in panel C, but with SF1(S203A) instead of SF1(WT). For all experiments, the luciferase activities in extracts from cells without exogenous expression of cycH, MAT1, or CDK7 were set to 1. Luciferase activities were calculated relative to β-gal activity (from pCMV5/βgal; 100 ng). All transfections were performed in triplicate repeated at least three times, shown as means ± sds. *, P < 0.05, as determined by Student’s t test. Cont, Control; n.s., not significant [P = 0.12 in panel B; P = 0.91 (cycH), 0.83 (MAT1), and 0.97 (CDK7) in panel D].
Fig. 6.

Mutated CDK7 Inhibits SF1-Dependent Transactivation
COS-1 cells were transfected with CYP17/Luc (1 μg) together with expression plasmids encoding SF1 and a mutated form of CDK7 (CDK7-K41R) (mCDK7; 50–250 ng) (panel A) or CDK2 (mCDK2; 50–250 ng) (panel B). The luciferase activities are given as fold induction relative to the luciferase activities in cells not transfected with Flag-pCMV2/SF1. Luciferase activities were calculated relative to β-gal activity (from pCMV5/βgal; 100 ng). The experiments were repeated at least three times (n ≥11), shown as means ± sds. *, P < 0.05; **, P < 0.01, as determined by one-way ANOVA Bonferoni posttest. The effect of mCDK7 on the pCMV5/βGAL promoter activity is shown in A, right panel.
Defective TFIIH Prevents SF1 Phosphorylation and Decreases SF1-Dependent Transcription
To further confirm a role for CDK7 in SF1-dependent transactivation, we used HD2 cells that result from the fusion of HeLa cells and fibroblasts harboring the point mutation R683W in the xeroderma pigmentosum group D (XPD) gene (54). This mutation weakens the interaction between the subunits XPD and the p44, causing a destabilization of the anchoring of CAK to the TFIIH core and rendering the CAK complex inactive (55). HD2 and HeLa cells were transfected with three SF1-inducible reporter gene constructs, SFRE/Luc and constructs containing promoter fragments from the genes encoding the steroidogenic acute regulatory protein (StAR; StAR/Luc), and the ACTH receptor (MC2R; MC2R/Luc) together with an SF1 expression vector. As demonstrated in Fig. 7A, SF1-dependent transactivation was decreased from all three constructs in HD2 cells compared with HeLa cells. For clarification, the results are presented as fold increase in luciferase activity in response to overexpression of SF1 (Fig. 7A, left panels). To visualize the overall effects on transcription in HD2 cells, the absolute luciferase activities are shown in the middle panels. The transcriptional activities were reduced from the SFRE/Luc reporter construct in the absence of SF1 compared with HeLa cells. However, transcription from StAR/Luc and MC2R/Luc, as well as the activities from a control plasmid that was included in all transfections (pCMV5/LacZ, right panels), was not affected in HD2 cells. These results therefore argue against a general repression of transcription in these cells and indicate that SF1-dependent transcription is specifically affected by mutations in XPD. The slight increase in luciferase activity observed for SFRE/Luc and MC2R/Luc in HD2 cells in response to SF1 suggests that SF1 retains transactivation ability also when not phosphorylated by CDK7. The cell lysates were subsequently subjected to Western blotting to ensure equal expression of SF1 in both cell lines (Fig. 7B). Importantly, assessment of the phosphorylation status of SF1 demonstrated low levels of phosphorylation in HD2 cells compared with HeLa cells (Fig. 7C, upper panel), demonstrating that the phosphorylation status of SF1 correlated with its capacity for transactivation, and furthermore that a functional TFIIH complex is required for phosphorylation of SF1. Western blot analyzes confirmed that SF1 was equally isolated from both cell lines (Fig. 7C, lower panel).
Fig. 7.
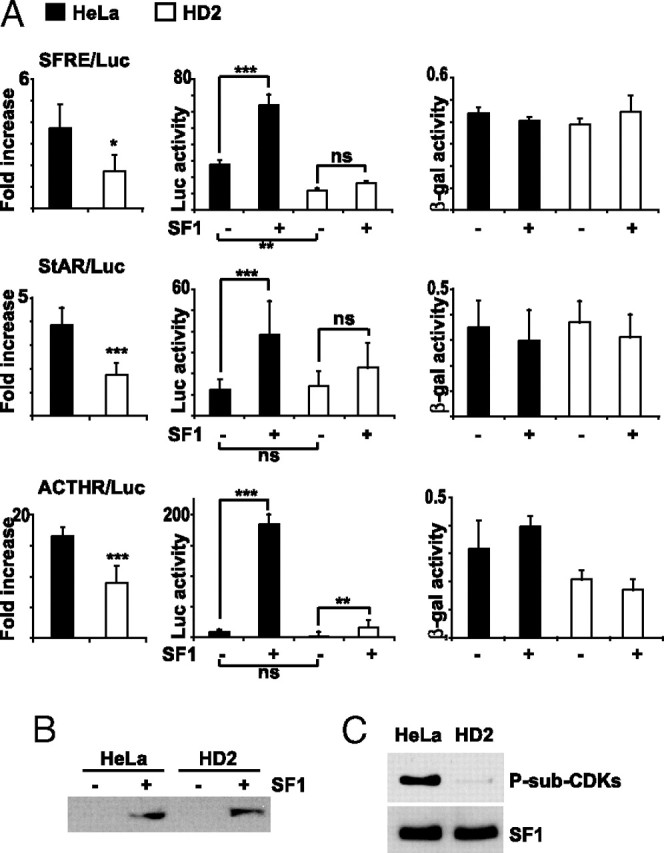
Mutation in XPD of TFIIH Decreases SF1-Dependent Transcription and Phosphorylation
A, Hela and HD2 cells were transfected with SFRE/Luc, StAR/Luc, or MC2R/Luc (0.5 μg) and pCMV5/LacZ, in the absence or presence of Flag-pCMV2/SF1 (50 ng). The luciferase values are shown as fold increase in the presence of SF1 and relative to the β-gal activity (left panels). The luciferase and the β-gal activities are also shown separately in the middle and right panels, respectively. The experiments were repeated two or three times, shown as means ± sds (SFRE/Luc, n = 5; StAR/Luc, n =12; MC2R/Luc, n = 6). *, P < 0.05; **, P < 0.01; ***, P < 0.001; as determined by unpaired t test (folds) or by ANOVA, Bonferonni (luciferase and β-gal activities). B, Lysates from panel A were incubated with 0.1% SDS and subjected to Western blot analyses with a SF1 antibody to verify equal SF1 expression. C, HeLa and HD2 cells were transfected with Flag-pCMV2/SF1. SF1 was isolated by DPD and Western blotted with anti-P-Sub-CDK (upper panel) and SF1 (lower panel) antibodies. ACTHR, ACTH receptor.
Mutations in the LBP that Inhibit Phospholipid Binding Reduce SF1 Phosphorylation
Mutations in the LBP of SF1 that disrupt binding of phosphoinositides decrease its ability to interact with the coactivator GRIP-1 (4). Because binding to GRIP-1 is also influenced by the phosphorylation status of S203 (14), we hypothesized that the inability to bind ligands might cause abnormal interaction with the basal transcription machinery and reduced phosphorylation of SF1 by CDK7. The transcriptional abilities of the LBP mutants were previously tested on the optimized SF1-responsive promoter p65-luc [containing five copies of the 21-hydroxylase −65 SF1 binding site (56)] (4). We therefore initially investigated the effect of these mutants on natural SF1-dependent promoters. CYP17/Luc and MC2R/Luc were transfected into COS-1 cells together with expression plasmids encoding the different LBP mutations. One mutant, SF1(V349W), had reduced capability to activate transcription from both promoters (Fig. 8, A and B), and transcription from CYP17/Luc was also affected by the SF1(A434W) mutant (Fig. 8A). Both these mutations completely abolish phospholipid binding to SF1 (4). When the different LBP mutations were isolated by DPD and assessed for their phosphorylative status, it became evident that SF1(V349W) was not phosphorylated (Fig. 8C, upper panel). The phosphorylation level was also reduced when L345 was mutated, and although we did not detect any significant decrease in transcription from CYP17/Luc and MC2R/Luc (Fig. 8A), this mutation has reduced ability to activate transcription from p65-luc (4). Mutations of A270 and L348 to tryptophan did not significantly affect transactivation or phosphorylation under the conditions used here (Fig. 8). Western blotting verified that SF1(WT) and the different SF1 LBP mutants were equally isolated (Fig. 8C, lower panel). These results therefore indicate a connection between ligand binding and phosphorylation by CDK7 and provide a mechanism by which the AF1 and the LBD of SF1 might interact.
Fig. 8.
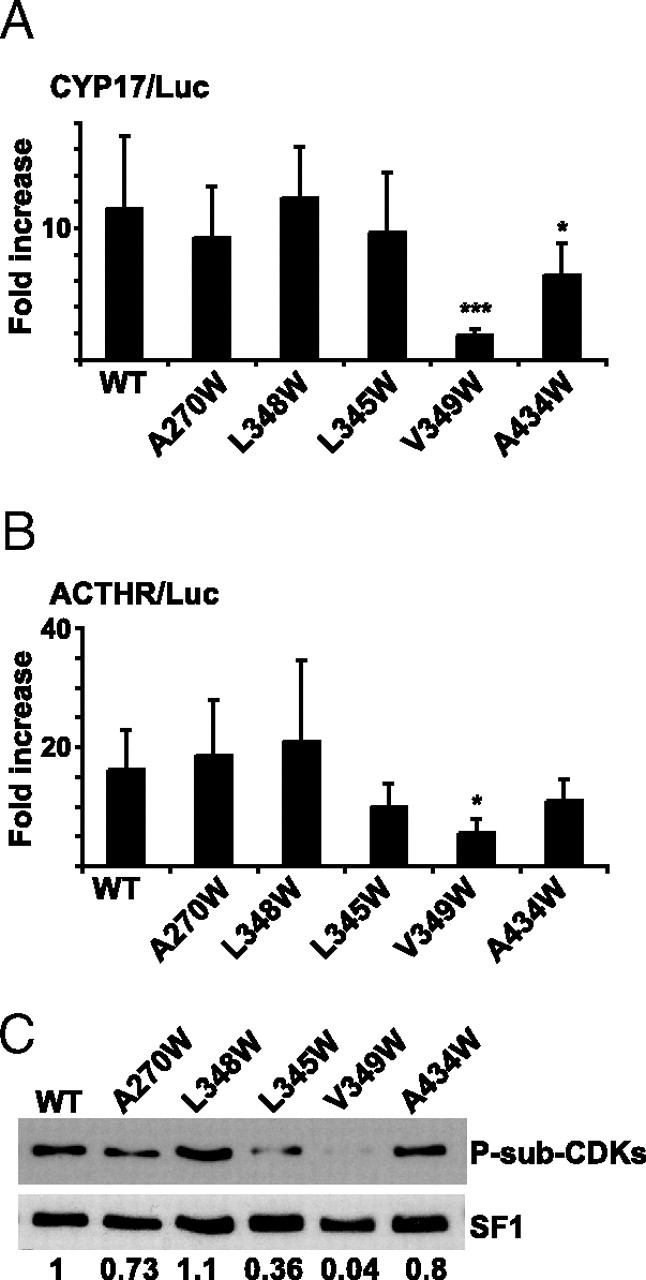
Mutations in the LBP that Reduce Transcriptional Activity Is Less Phosphorylated by CDKs
COS-1 cells were transfected with pCDNA3.1+/SF1 (WT; 50 ng) or LBP mutants (pCDNA3.1Zeo+/SF1-A270W, L348W, L345W, V349W, and A434W; 50 ng) together with the reporter gene construct CYP17/Luc (0.5 μg) (panel A) or MC2R/Luc (panel B). The luciferase activities are presented as fold induction compared with the activity in the absence of SF1 and relatively to the activities from pCMV5/βgal. The experiments were repeated three times, shown as means ± sds (CYP17/Luc, n = 9; *, P < 0.05; ***, P < 0.001; as determined by one-way ANOVA, Bonferoni posttest; MC2R/Luc, n = 6; *, P < 0.05, as determined by nonparametric ANOVA, Dunn posttest). C, COS-1 cells were transfected with SF1 (WT) or LBP mutants (5 μg), and SF1 was isolated by DPD. The isolated proteins were subjected to Western blotting analyses using anti-P-Sub-CDK (upper panel) and SF1 antibodies (lower panel). Densitometric analyses were performed and are shown as ratios between phosphorylated SF1 and total SF1. ACTHR, ACTH receptor.
DISCUSSION
In addition to being ligand-inducible transcription factors, nuclear receptors are subjected to rapid posttranslational modifications, such as phosphorylation. These processes can occur in response to ligand binding or activation of other signaling pathways, allowing nuclear receptors to integrate the responses from different signals into a transcriptional output (for a review see Ref.25). We demonstrate that SF1 is phosphorylated on S203 by immunoprecipitated CDK7 and by purified TFIIH, and that this modification correlates with increased transcriptional activity. Moreover, our results indicate that ligand binding might affect phosphorylation of S203, because two mutations that render SF1 unable to bind phospholipids are hypophosphorylated. To our knowledge, this is the first report that potentially links SF1-ligand binding to a posttranslational event, although further studies are required to formally prove this. For instance, we cannot exclude that the lack of phosphorylation is caused by conformational changes induced by the mutations in the LBP. However, it is interesting to note that other nuclear receptors are phosphorylated by CDK7 in a ligand-dependent fashion, indicating a general mode of activation for this class of transcription factors.
Typically, nuclear receptors that are targets for CDK7 do not contain consensus CDK recognition sequences (S/T)PX(K/R) and diverge from the well-characterized CDK7 phosphorylation site in the RNA pol II CTD (see Table 1). The peptide sequence in SF1, PYASPP (underlined S corresponds to S203) shows highest similarity to the site identified in PPARγ when compared with the hitherto recognized CDK7 target motifs in nuclear receptors (see Table 1). In fact, the S203 phosphorylation site shows more similarity to the MAPK consensus site PXn(S/T)P, which is also a proline-dependent S/T kinase, and, in line with this, it was previously demonstrated that SF1 is phosphorylated on this residue by recombinant ERK2 (14). As shown in Fig. 3A, we found that phosphorylation of SF1 was more sensitive to inhibitors of CDKs than of MEK1. Consistently, treatment of pituitary αT3-1 cells with U0126 and overexpression of the dual phosphatase MKP-1 in Y1 cells only slightly reduce the phosphorylation of SF1 on S203 (16, 17). Despite these findings, we will not dispute that MAPK-dependent phosphorylation might be of significance for SF1 phosphorylation and activity. Rather, the possibility of two seemingly independent pathways converging on the same residue is interesting, and PPARα and -γ (34, 36, 57) and ERα (28, 52) are also modified by CDK7 and MAPK on the same serine residue. Consequently, whereas nuclear receptors might be affected by mitogenic signals under certain conditions, signals directed toward CDK7 might induce the formation of a transactivation complex bringing the nuclear receptor in close proximity to TFIIH. In this regard it is interesting to note that CDK7-induced phosphorylation of ERα (on S118) is ligand inducible, whereas MAPK mediates phosphorylation of the same residue in a ligand-independent manner (35). Hence, estradiol and the partial agonist, 4-hydroxytamoxifen, induce CDK7-dependent phosphorylation whereas the complete antagonist ICI 182,780 does not, suggesting that conformational changes induced by agonists enable phosphorylation of S118 (35).
Is CDK7-induced phosphorylation of SF1 also ligand dependent? The results presented in Fig. 8 indicated that this might be the case. We found that mutation of either L345 or V349 to tryptophan (W), which completely block phospholipid binding (4), leads to significantly less phosphorylation of SF1 (Fig. 8C). In contrast, mutation of A270 (located in the back of the LBP) or L348 (located distal from the ligand) that still allow binding of short-chain phospholipids (4) did not affect phosphorylation (Fig. 8C). When monitored from p65-luc, Li and collaborators (4) reported that all four mutations reduce coactivator recruitment and transcriptional activation, but that the transactivation potential of A270W and L348W is rescued by addition of small phospholipids, demonstrating a connection between LBP occupation and activity. As shown in Fig. 8, A and B, we found reporter gene expression driven by natural SF1-dependent promoters to be less sensitive to alterations in the LBP because only two of the mutations caused reduced transcription from CYP17/Luc (V349W and A434W) and MC2R/Luc (V349W). It is somewhat puzzling that SF1(A434W) is phosphorylated equally well as SF1(WT) (Fig. 8C) because this mutant affects transcription from both CYP17/Luc and p65-luc (Fig. 8A and Ref.4). At present we cannot explain this inconsistency, but it might be related to the fact that L345 and V349 are located in helix 7 and packed closely toward the bound ligand, whereas A434 is in helix 10 and located at the bottom of the LBP (4). Thus, despite the lack of a perfect correlation between LBP mutations, phosphorylation, and transactivation, our results, together with published data, suggest an interesting mechanism whereby ligand binding followed by a posttranslational modification might connect SF1 to the basic transcriptional machinery. This would subsequently lead to the activation of SF1 target genes, possibly by increased cofactor recruitment or by increased promoter clearance due to cyclic phosphorylation and dephosphorylation of SF1 as described to occur on the Mc2r promoter (11). Furthermore, these findings suggest a mechanism of cooperation between the AF1 and LBD of SF1. In recent years, several reports have demonstrated cooperativity between the AF1 and AF2 domains after ligand binding and phosphorylation by CDK7 (35, 58), and, along this line, phosphorylation of S203 was demonstrated to enhance the stability of SF1 by increased intramolecular association between AF1 and the LBD (16). Hence, a general mechanism for nuclear receptors emerges in which, as a result of ligand binding, CDK7-dependent phosphorylation induces intramolecular cooperativity of transactivation domains. Intriguingly, disruption of the integrity of the LBP by point mutations (V349W and L345W) abolishes or greatly reduces phosphorylation of SF1 on S203 (Fig. 8C), whereas the deletion mutant SF1 (1–279), in which most of the LBD is removed, is still phosphorylated (Fig. 2). As mentioned above, the region encompassing S203 is believed to be tightly associated with the LBD. Perhaps, the lack of ligand binding affects the conformational structure and/or stability, thereby causing a hindrance for the phosphorylation event, whereas the truncated SF1 may be more easily accessed by the kinase when the whole LBD is removed.
Transcription from the CYP17 promoter was only slightly reduced when SF1(WT) was substituted by SF1(S203A) in transfection experiments in COS-1 cells (Fig. 2E). Furthermore, the S203E mutant activated transcription similarly to SF1(WT). These results are somewhat puzzling but are most likely explained by the fact that SF1 maintains transactivation properties in the absence of phosphorylation. It is known that SF1(S203A) retains approximately 50% binding capacity toward the coactivator GRIP-1 and that it does not differ from SF1(WT) with regard to protein stability, cellular localization, and DNA binding (14). Thus, reduced transactivation capacity in response to mutation of S203 might be difficult to detect in overexpression experiments but could still be of physiological significance in vivo. In line with this, the existing literature is conflicting with regard to the effects of the S203A mutation (see Table 2). For instance, Sewer and Waterman (59) find this mutant to be without any effect on transcription mediated by the CYP17 promoter in H295R cells. Furthermore, no effect is observed for the promoter from SR-B1 (gene encoding the high-density lipoprotein receptor) in HTB9 cells (human bladder carcinoma cell line) (60) or for the cyp1B1 promoter (gene encoding cytochrome P4501B1) in Y1 cells (61). In contrast, decreased transcriptional activity as a consequence of the S203A mutation has been reported for the α glycoprotein subunit (αGSU) promoter in αT3-1 cells (17), and for reporter constructs carrying several copies of SFREs from the Müllerian-inhibiting substance (Mis) promoter and cyp21 promoter in JEG-3 cells (human choriocarcinoma cell line) (14). Furthermore, this mutation exhibits decreased capability to activate transcription from the StAR promoter using HTB9 cells (60). Thus, the transactivation capacity of SF1(S203A) might depend on experimental conditions and on the cell line and promoter studied.
Table 2.
Transcriptional Effects of the S203A Mutation
| Promoter | Cell Line | Endogenous SF1 Expression | Effect on Transcription | Ref. Nos. |
|---|---|---|---|---|
| hCYP17 | COS-1 | − | Small decrease | This study |
| StAR | HTB9 | − | Small decrease | 60 |
| αGSU | αT3–1 | + | Major decrease | 7 |
| MIS | JEG3 | − | Major decrease | 4 |
| CYP11A | JEG3 | − | Major decrease | 4 |
| hCYP17 | H295R | + | No effect | 59 |
| HDL-R | HTB9 | − | No effect | 60 |
| CYP1B1 | Y1 | + | No effect | 61 |
See text for details.
HD2 cells harbor a mutation in the XPD subunit (R683W) that destabilizes the binding of the CAK complex. These cells were previously employed to demonstrate CDK7-dependent phosphorylation of both PPARγ (36) and RARα (62). The finding that SF1 is hypophosphorylated in these cells (Fig. 7) is therefore consistent with a role for CDK7/TFIIH in phosphorylation of S203. Because XPD is part of the general transcription factor TFIIH, it could be speculated that the observed effect on SF1-dependent promoters simply reflects a general defect in basal transcription. This does not appear to be the case however, because the transcription of several genes is not affected in these cells (36, 62). Moreover, it is not believed that CDK7 has a general effect on transcription in mammalian cells, although this remains to be formally proven (37). Mutations in the XPD gene result in the genotype- and phenotype-interrelated hereditary diseases xeroderma pigmentosum (XP) and trichothiodystrophy (TTD) (63). The complex phenotypic picture observed in the affected patients is explained by functional defects in both the nucleotide-excision repair (NER) activity and transcriptional roles of XPD (64). The TTD-mouse model, which carries a mutation originally identified in patients (R722W/R722W), exhibits the characteristic features of the human TTD phenotype (i.e. brittle hair and nails and carcinogenesis caused by photosensitivity) and also show other abnormalities linked to this syndrome such as hypoplasia of adipose tissue and immature sexual development (65, 66, 67). Interestingly, some patients suffer from hypogonadism and undescended testes (66), and although these characteristics can be caused by various factors, they are also observed in the SF1 pituitary-specific knockout model (68). Moreover, it is interesting to note that SF1(S203A) has significantly reduced ability to transactivate the αGSU-promoter in pituitary cells compared with SF1(WT) (17), and it is tempting to speculate that a link exists between the gonadal/genitalial phenotype, the disorganization of the CDK7/TFIIH complex, and phosphorylation of SF1.
In conclusion, our results indicate that CDK7, as part of the CAK complex and TFIIH, phosphorylates SF1 at S203 followed by increased transcriptional activity of SF1. Furthermore, we show that the integrity of the LBP is important for full SF1 phosphorylation, implying that ligand binding may be critical for recruiting the kinase.
MATERIALS AND METHODS
Materials
Roscovitine and U0126 were from Sigma Chemical Co. (St. Louis, MO). The anti-flag (M2) antibody was from Sigma (F3165), the anti-SF1 antibody (07–618) was from Upstate Biotechnology, Inc. (Lake Placid, NY), and the anti-phospho-S/T substrate-kinase antibodies (CDKs: no. 2324, PKA: no. 9621, PKC: no. 2261, Akt: no. 9611) were from Cell Signaling Technology (Beverly, MA). Antibodies against CDK2 (sc-6248), CDK7 (sc-7344), ERK1 (sc-94), ERK2 (sc-153), and phosho-Tyr 204-ERK1/2 (sc-7383), as well as antimouse and antirabbit conjugated to horseradish peroxidase (HRP), were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The protein G-agarose beads were from Invitrogen (Carlsbad, CA) and the streptavidin-agarose beads were from Pierce Chemical Co. (Rockford, IL).
Plasmids
The cDNAs encoding human SF1 (in pcDNA3.1/Zeo+) and mouse SF1 (in pBS) were obtained from Dr. K. L. Parker (University of Texas Southwestern Medical Center, Dallas, TX). The murine SF1 cDNA was cloned into pcDNA3.1+ (Invitrogen) at the EcoRI site or into the EcoRI site of the expression plasmid vector Flag-pCMV2 (Sigma) to create pcDNA3.1+/SF1 and Flag-pCMV2/SF1. pCMV5/SF1(1–279) and pCMV5/SF1 (1–110) (69), expressing truncated murine SF1, were obtained from Dr. B.-C. Chung (Institute of Molecular Biology, Academia Sinica, Taipei, Taiwan) and subcloned into the EcoRI/Bpu and EcoRI/SalI sites, respectively, of the Flag-pCMV2 expression vector to generate Flag-pCMV2/SF1(1–110) and Flag-pCMV2/SF1(1–279). The Flag-pCMV2/SF1S203A, Flag-pCMV2/SF1S203E, and Flag-pCMV2/SF1S219A mutants were generated by site-directed mutagenesis (QuikChange Site Directed Mutagenesis kit, Stratagene) using the respective primers, 5′-CCAGAGCCCTATGCCGCACCCCCACAACAGCCAG, 5′-CCAGAGCCCTATGCCGAGCCCCCACAACAGCCAG and 5′-CAGAGCCCTTCGCAGGAGGGCCCA. GST-SF1(179–431) was a gift from Dr. G. Hammer (University of Michigan Medical School, Ann Arbor, MI) and the GST-SF1(179–431/S203A) mutant was generated by site-directed mutagenesis (QuikChange Site-Directed Mutagenesis kit) using the primer 5′-CCA GAG CCC TAT GCC GCA CCC CCA CAA CAG CCA G. The SFRE/pGL3 reporter construct contains four optimized SF1-binding sites (5′-TCAAGGTCAGGCGCGCCTCAAGGTCAGGCGCGCCTCAAGGTCAGGCGCGCCTCAAGGTCA) upstream of a minimal thymidine kinase promoter in pGL3basic (Invitrogen). All constructs were verified by sequencing. The luciferase reporter constructs, pGL3/CYP17-1114 bp (CYP17/Luc), pGL3/MC2R-1000 bp (MC2R/Luc), and pGL2/StAR-150 bp (StAR/Luc), were provided by Drs. W. E. Rainey (Medical College of Georgia, Augusta, GA), M. Reincke (Ludwig-Maximilians-Universitaet, München, Germany), and B. Clark (University of Louisville, Louisville, KY) respectively. The SF1 ligand-binding pocket mutants, A270W, A434W, L345W, L348W, and V349W, cloned into pcDNA3.1/Zeo+, were from Dr. S.A. Kliewer (University of Texas Southwestern Medical School) (4). The expression plasmids pRc-CMV/Myc-CDK7, the kinase-deficient mutant pRc-CMV/Myc-CDK7-K41R, and pGEX-KG/MAT1 were from Dr. E. Nigg (Max-Planck Institute for Biochemistry, Martinsried, Germany). The MAT1 cDNA was excised from pGEX-KG and subcloned into the EcoRI and EcoRV sites of pCDNA3. The plasmid encoding dominant-negative CDK2 (pCMV/dnCDK2) was obtained from Dr. D. Ucker (University of Illinois, Chicago, IL). pcDLSRa/hcyclin H was from Dr. D. O. Morgan (University of California, San Francisco, CA).
Cell Culture
Y1, HeLa, and HD2 cells were cultured in DMEM (high glucose) supplemented with 10% fetal calf serum, penicillin (100 U/ml), and streptomycin (100 μg/ml). COS-1 cells were maintained in DMEM (low glucose) supplemented with 10% fetal calf serum, penicillin (100 U/ml), streptomycin (100 μg/ml), and l-glutamine. H295R cells were cultured in a 1:1 mixture of DMEM:Hams F12 supplemented with ITS+ (Collaborative Research, Bedford, MA), 2% Nu-Serum (Collaborative Research), penicillin (100 U/ml), and streptomycin (100 μg/ml).
Transfection of Cells
Cells were plated at a density of 2.5 × 105 cells per well onto six-well plates (5 × 104 cells for 12-well plates) and transiently transfected the following day. H295R cells were transfected with Effectene (QIAGEN, Chatsworth, CA), Hela and HD2 cells were transfected with Lipofectamine 2000 (Invitrogen), and COS-1 cells were transfected using Superfect (QIAGEN). Cells were transfected with reporter plasmid (1 μg or 0.5 μg for six- or 12-well plates, respectively) and expression plasmids as indicated in the figures. The total amount of plasmid was kept constant by compensating with empty expression vectors. To control for transfection efficiency, an expression plasmid encoding β-gal (pCMV5/LacZ, 100 or 50 ng) was included. The cells were washed twice with PBS 24 h after transfection and lysed in luciferase buffer [10 mm Tris-HCl (pH 8), 4 mm EDTA, 150 mm NaCl, 0.65% Nonidet P-40 (NP-40)]. Cell extracts were then assayed for luciferase and β-gal activity on a LUCY-1 luminometer (Anthos, Eugendorf, Austria) using the Luciferase Assay Kit from BIO Thema AB (Dalarö, Sweden).
Western Blotting
Samples separated by SDS-PAGE were blotted to nitrocellulose membranes and incubated with 7.5% milk in PBS-Tween (PBS-T) for 1 h at room temperature. Membranes were rinsed with PBS-T and incubated overnight in primary antibodies and subsequently with antirabbit-HRP or antimouse-HRP (1:10,000 dilution; Santa Cruz Biotechnology, Inc). Detection was performed by SuperSignal West Pico Chemiluminescent Substrate (Pierce). When needed, membranes were stripped after a first probing by incubation with Restore Western Blot Stripping Solution (Pierce) for 30 min at room temperature, extensively washed in PBS-T, blocked, and subsequently reprobed with anti-SF1 to confirm even expression levels.
DNA Pull Down
DPD was carried out as described by Mukai et al. (41) with some modifications. Cells were grown in 10-cm dishes, rinsed twice with PBS, and scraped in buffer A (400 μl) [10 mm HEPES, pH 7.9; 1.5 mm MgCl2, 10 mm KCl; 0.5 mm dithiothreitol (DTT); 0.05% NP-40; and 1× complete-EDTA-free protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN)] and then incubated on ice for 10 min. Cell pellets were resuspended in buffer B (400 μl) (20 mm HEPES, pH 7.9; 1.5 mm MgCl2; 0.5 m NaCl; 0.5 mm DTT; 25% glycerol; 0.05% NP-40; and 1× complete-EDTA-free protease inhibitor cocktail), incubated for 30 min on ice and centrifuged for 30 min at 4 C. Supernatants (nuclear extracts) were diluted with the same volume of buffer C (25 mm HEPES, pH 7.9; 2.5 mm EDTA; 0.1% NP-40; and 1× complete-EDTA-free protease inhibitor cocktail). Nuclear extracts (300–400 μg) were incubated with 20 μg polydeoxyinosinic-deoxycytidylic acid (polydI-dC), 400 μg BSA, and 3 μg biotin-labeled double-stranded oligonucleotide containing SF1 DNA binding consensus sequence (5′-ACTAGCTCTTTGATCTCAAGGTCAGAACTT) overnight at 4 C on a rotator. A slurry of streptavidin agarose-conjugated beads (30 μl) was added for 1 h at 4 C. Samples were boiled and then subjected to SDS-PAGE and Western blotting.
Proteomic Analysis
The identifications of the proteins in Fig. 1A were performed by the proteomic unit PROBE (FUGE, Norwegian Research Council, University of Bergen) using MALDI-ToF acquisition.
Immunoprecipitation
COS-1 cells were transfected with expression plasmids (5 μg for 10-cm dishes, Fig. 2B; 12 μg for 15-cm dishes, Fig. 4A) using Superfect. Cells were rinsed twice with PBS 48 h after transfection, lysed with ice-cold immunoprecipitation buffer (500 μl) [50 mm Tris-HCl (pH 7.5), 100 mm NaCl, 0.5% sodium dodecyl sulfate (SDS), 1% Triton X-100, freshly added 1 mm phenylmethylsulfonylfluoride (PMSF), 1 μg aprotinin and leupeptin, 1 mm Na3VO4, and 1 mm NaF], as described by Børud et al. (70). After sonication, samples were centrifuged to remove insoluble debris. Cell extract (400 μg) (diluted 5× in 50 mm Tris-HCl, pH 7.5; 100 mm NaCl) were incubated with 2 μg anti-flag (M2) antibodies (15 μg for coimmunoprecipitation) overnight and subsequently with protein G agarose-conjugated beads (30 μl slurry) for 1 h at 4 C on a rotator. After three washes with 50 mm Tris-HCl (pH 7.5), 100 mm NaCl, 0.1% SDS, 0.2% Triton X-100, 1 mm Na3VO4, and 1 mm NaF, the precipitates were boiled for 5 min in reducing buffer and then subjected to SDS-PAGE and Western blotting.
Expression of GST-SF1 in Bacteria
GST-SF1 (179–431) and GST-SF1 (179–431/S203A) were expressed from the pGEX-4T1 vector in BL21-CodonPlus(DE3)-RIPL bacteria at 28 C and induced with 0.4 mm isopropyl-β-d-thiogalactopyranoside for 6 h at an optical density of 0.5 at 600 nm. Bacterial pellets were resuspended with 50 mm Tris (pH 7.5), 2 mm EDTA (and 1 mm DTT, 1 mm PMSF, 10 μg/ml aprotinin and leupeptin) and lysed by sonication, and debris was removed by centrifugation. GST-tagged proteins were purified with glutathione-agarose 4B beads, and analyzed by SDS-PAGE for purity.
CDK7 and TFIIH in Vitro Kinase Assays
For CDK kinase assays, H295R cells plated on 10-cm plates were washed twice with PBS, lysed in RIPA buffer (400 μl; 50 mm Tris-HCl, pH 8; 150 mm NaCl; 0.5% deoxycholic acid; 1% NP-40; 0.1% SDS; containing freshly added protease inhibitors, as above) and centrifuged to remove insoluble debris. Cell extracts (600 μg) were incubated with anti-CDK7 or CDK2 antibodies (3 μg) for 1 h or O/N at 4 C, and with a 50% slurry r-protein G-agarose (30 μl) for 1 h at 4 C. Immune complexes were washed three times in RIPA buffer, two times in kinase buffer (50 mm HEPES, pH 7.5; 10 mm MgCl2; 1 mm dithiothreitol), and resuspended in kinase buffer (40 μl) containing 5 μCi [γ32P]ATP (3000 Ci/mmol), 10–50 μm cold ATP, and 5 μg GST-SF1 (179–431) or GST-SF1(179–431/S203A). After incubation for 30 min at 30 C, the samples were boiled for 5 min with reducing buffer, separated by electrophoresis on a 10% gel, and transferred to nitrocellulose membrane. Membranes were subjected to autoradiography and Western blotting with anti-CDK7 and CDK2antibodies to verify the levels of immunoprecipitated materials. For TFIIH kinase assays, GST-SF1(179–431) or GST-SF1(179–431/S203A) (2 μg) bound to Sepharose beads was incubated with human TFIIH complex (150 ng; affinity purified with M2-agarose from nuclear extract from a cell line expressing Flag-p89), in buffer A [50 mm Tris-HCl, pH 7.5; 100 mm KCl; 10% glycerol; 0.1 mg/ml BSA; 1 mm DTT; 1 mm PMSF; 1× phosphatase inhibitor cocktail 2 (Sigma)] containing 5 μCi [γ32P]-ATP (3000 Ci/mmol) and cold ATP (100 μm) at 30 C for 30 min. Beads were washed two times with buffer A (500 μl), and eluted proteins were resolved by SDS-PAGE and analyzed by autoradiography.
Acknowledgments
We thank T. Ellingsen for excellent technical assistance, and Drs. J.-M. Egly and P. Drané for discussions and experimental support.
NURSA Molecule Pages:
Coregulators: CDK7 | MAT1;
Nuclear Receptors: SF-1.
Footnotes
This work was supported by The National Program for Research in Functional Genomics in Norway (FUGE) in the Research Council of Norway, The Norwegian Cancer Society, and Helse Vest.
Disclosure Statement: The authors have nothing to declare.
First Published Online September 27, 2007
Abbreviations: aa, Amino acid; AF, activation function; CAK, CDK-activating kinase; CDK, cyclin-dependent kinase; CTD, C-terminal domain; cycH, cyclin H; DMSO, dimethylsulfoxide; DPD, DNA pull down; DTT, dithiothreitol; ER, estrogen receptor; GRIP, glucocorticoid receptor-interacting protein; GST, glutathione-S-transferase; HRP, horseradish peroxidase; LBD, ligand-binding domain; LBP, ligand-binding pocket; MAT1, ménage à trois 1; NP-40, Nonidet P-40; PKA, protein kinase A; PKC, protein kinase C; PMSF, phenylmethylsulfonylfluoride; PPAR, peroxisome proliferator-activated receptor; P-S-sub-CDK, phospho-(Ser) CDKs substrate; RAR, retinoic acid receptor; S203, serine 203; SDS, sodium dodecyl sulfate; SF1, steroidogenic factor 1; SFRE, SF1-responsive element; TF, transcription factor; TTD, trichothiodystrophy; WT, wild type; XPD, xeroderma pigmentosum group D.
References
- 1.Parker KL, Rice DA, Lala DS, Ikeda Y, Luo X, Wong M, Bakke M, Zhao L, Frigeri C, Hanley NA, Stallings N, Schimmer BP 2002. Steroidogenic factor 1: an essential mediator of endocrine development. Recent Prog Horm Res 57:19–36 [DOI] [PubMed] [Google Scholar]
- 2.Wang W, Zhang C, Marimuthu A, Krupka HI, Tabrizizad M, Shelloe R, Mehra U, Eng K, Nguyen H, Settachatgul C, Powell B, Milburn MV, West BL 2005. The crystal structures of human steroidogenic factor-1 and liver receptor homologue-1. Proc Natl Acad Sci USA 102:7505–7510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, Lebedeva L, Suzawa M, Williams JD, Williams SP, Guy RK, Thornton JW, Fletterick RJ, Willson TM, Ingraham HA 2005. Structural analyses reveal phosphatidyl inositols as ligands for the NR5 orphan receptors SF-1 and LRH-1. Cell 120:343–355 [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Choi M, Cavey G, Daugherty J, Suino K, Kovach A, Bingham NC, Kliewer SA, Xu HE 2005. Crystallographic identification and functional characterization of phospholipids as ligands for the orphan nuclear receptor steroidogenic factor-1. Mol Cell 17:491–502 [DOI] [PubMed] [Google Scholar]
- 5.Urs AN, Dammer E, Sewer MB 2006. Sphingosine regulates the transcription of CYP17 by binding to steroidogenic factor-1. Endocrinology 147:5249–5258 [DOI] [PubMed] [Google Scholar]
- 6.Gallo-Payet N, Payet MD 2003. Mechanism of action of ACTH: beyond cAMP. Microsc Res Tech 61:275–287 [DOI] [PubMed] [Google Scholar]
- 7.Zhang P, Mellon SH 1996. The orphan nuclear receptor steroidogenic factor-1 regulates the cyclic adenosine 3′,5′-monophosphate-mediated transcriptional activation of rat cytochrome P450c17 (17α-hydroxylase/c17–20 lyase). Mol Endocrinol 10:147–158 [DOI] [PubMed] [Google Scholar]
- 8.Chau YM, Crawford PA, Woodson KG, Polish JA, Olson LM, Sadovsky Y 1997. Role of steroidogenic-factor 1 in basal and 3′,5′-cyclic adenosine monophosphate-mediated regulation of cytochrome P450 side-chain cleavage enzyme in the mouse. Biol Reprod 57:765–771 [DOI] [PubMed] [Google Scholar]
- 9.Mamluk R, Greber Y, Meidan R 1999. Hormonal regulation of messenger ribonucleic acid expression for steroidogenic factor-1, steroidogenic acute regulatory protein, and cytochrome P450 side-chain cleavage in bovine luteal cells. Biol Reprod 60:628–634 [DOI] [PubMed] [Google Scholar]
- 10.Aesoy R, Mellgren G, Morohashi K, Lund J 2002. Activation of cAMP-dependent protein kinase increases the protein level of steroidogenic factor-1. Endocrinology 143:295–303 [DOI] [PubMed] [Google Scholar]
- 11.Winnay JN, Hammer GD 2006. Adrenocorticotropic hormone-mediated signaling cascades coordinate a cyclic pattern of steroidogenic factor 1-dependent transcriptional activation. Mol Endocrinol 20:147–166 [DOI] [PubMed] [Google Scholar]
- 12.Chen WY, Juan LJ, Chung BC 2005. SF-1 (nuclear receptor 5A1) activity is activated by cyclic AMP via p300-mediated recruitment to active foci, acetylation, and increased DNA binding. Mol Cell Biol 25:10442–10453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lund J, Bakke M, Mellgren G, Morohashi K, Doskeland SO 1997. Transcriptional regulation of the bovine CYP17 gene by cAMP. Steroids 62:43–45 [DOI] [PubMed] [Google Scholar]
- 14.Hammer GD, Krylova I, Zhang Y, Darimont BD, Simpson K, Weigel NL, Ingraham HA 1999. Phosphorylation of the nuclear receptor SF-1 modulates cofactor recruitment: integration of hormone signaling in reproduction and stress. Mol Cell 3:521–526 [DOI] [PubMed] [Google Scholar]
- 15.Sewer MB, Waterman MR 2002. Adrenocorticotropin/cyclic adenosine 3′,5′-monophosphate-mediated transcription of the human CYP17 gene in the adrenal cortex is dependent on phosphatase activity. Endocrinology 143:1769–1777 [DOI] [PubMed] [Google Scholar]
- 16.Desclozeaux M, Krylova IN, Horn F, Fletterick RJ, Ingraham HA 2002. Phosphorylation and intramolecular stabilization of the ligand binding domain in the nuclear receptor steroidogenic factor 1. Mol Cell Biol 22:7193–7203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fowkes RC, Desclozeaux M, Patel MV, Aylwin SJ, King P, Ingraham HA, Burrin JM 2003. Steroidogenic factor-1 and the gonadotrope-specific element enhance basal and pituitary adenylate cyclase-activating polypeptide-stimulated transcription of the human glycoprotein hormone α-subunit gene in gonadotropes. Mol Endocrinol 17:2177–2188 [DOI] [PubMed] [Google Scholar]
- 18.Weck J, Mayo KE 2006. Switching of NR5A proteins associated with the inhibin α-subunit gene promoter after activation of the gene in granulosa cells. Mol Endocrinol 20:1090–1103 [DOI] [PubMed] [Google Scholar]
- 19.Gyles SL, Burns CJ, Whitehouse BJ, Sugden D, Marsh PJ, Persaud SJ, Jones PM 2001. ERKs regulate cyclic AMP-induced steroid synthesis through transcription of the steroidogenic acute regulatory (StAR) gene. J Biol Chem 276:34888–34895 [DOI] [PubMed] [Google Scholar]
- 20.Carlone DL, Richards JS 1997. Functional interactions, phosphorylation, and levels of 3′,5′-cyclic adenosine monophosphate-regulatory element binding protein and steroidogenic factor-1 mediate hormone-regulated and constitutive expression of aromatase in gonadal cells. Mol Endocrinol 11:292–304 [DOI] [PubMed] [Google Scholar]
- 21.de Santa Barbara P, Mejean C, Moniot B, Malcles MH, Berta P, Boizet-Bonhoure B 2001. Steroidogenic factor-1 contributes to the cyclic-adenosine monophosphate down-regulation of human SRY gene expression. Biol Reprod 64:775–783 [DOI] [PubMed] [Google Scholar]
- 22.Crawford PA, Polish JA, Ganpule G, Sadovsky Y 1997. The activation function-2 hexamer of steroidogenic factor-1 is required, but not sufficient for potentiation by SRC-1. Mol Endocrinol 11:1626–1635 [DOI] [PubMed] [Google Scholar]
- 23.Jacob AL, Lund J 1998. Mutations in the activation function-2 core domain of steroidogenic factor-1 dominantly suppresses PKA-dependent transactivation of the bovine CYP17 gene. J Biol Chem 273:13391–13394 [DOI] [PubMed] [Google Scholar]
- 24.Weigel NL 1996. Steroid hormone receptors and their regulation by phosphorylation. Biochem J 319:657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rochette-Egly C 2003. Nuclear receptors: integration of multiple signalling pathways through phosphorylation. Cell Signal 15:355–366 [DOI] [PubMed] [Google Scholar]
- 26.Lange CA, Shen T, Horwitz KB 2000. Phosphorylation of human progesterone receptors at serine-294 by mitogen-activated protein kinase signals their degradation by the 26S proteasome. Proc Natl Acad Sci USA 97:1032–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen T, Horwitz KB, Lange CA 2001. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol Cell Biol 21:6122–6131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P 1995. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 270:1491–1494 [DOI] [PubMed] [Google Scholar]
- 29.Bunone G, Briand PA, Miksicek RJ, Picard D 1996. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J 15:2174–2183 [PMC free article] [PubMed] [Google Scholar]
- 30.Tremblay A, Tremblay GB, Labrie F, Giguere V 1999. Ligand-independent recruitment of SRC-1 to estrogen receptor β through phosphorylation of activation function AF-1. Mol Cell 3:513–519 [DOI] [PubMed] [Google Scholar]
- 31.Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C 1999. From HER2/Neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci USA 96:5458–5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gianni M, Bauer A, Garattini E, Chambon P, Rochette-Egly C 2002. Phosphorylation by p38MAPK and recruitment of SUG-1 are required for RA-induced RAR γ degradation and transactivation. EMBO J 21:3760–3769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barger PM, Browning AC, Garner AN, Kelly DP 2001. p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor α: a potential role in the cardiac metabolic stress response. J Biol Chem 276:44495–44501 [DOI] [PubMed] [Google Scholar]
- 34.Hu E, Kim JB, Sarraf P, Spiegelman BM 1996. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science 274:2100–2103 [DOI] [PubMed] [Google Scholar]
- 35.Chen D, Washbrook E, Sarwar N, Bates GJ, Pace PE, Thirunuvakkarasu V, Taylor J, Epstein RJ, Fuller-Pace FV, Egly JM, Coombes RC, Ali S 2002. Phosphorylation of human estrogen receptor α at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene 21:4921–4931 [DOI] [PubMed] [Google Scholar]
- 36.Compe E, Drane P, Laurent C, Diderich K, Braun C, Hoeijmakers JH, Egly JM 2005. Dysregulation of the peroxisome proliferator-activated receptor target genes by XPD mutations. Mol Cell Biol 25:6065–6076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fisher RP 2005. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci 118:5171–5180 [DOI] [PubMed] [Google Scholar]
- 38.Ko LJ, Shieh SY, Chen X, Jayaraman L, Tamai K, Taya Y, Prives C, Pan ZQ 1997. p53 is phosphorylated by CDK7-cyclin H in a p36MAT1-dependent manner. Mol Cell Biol 17:7220–7229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inamoto S, Segil N, Pan ZQ, Kimura M, Roeder RG 1997. The cyclin-dependent kinase-activating kinase (CAK) assembly factor, MAT1, targets and enhances CAK activity on the POU domains of octamer transcription factors. J Biol Chem 272:29852–29858 [DOI] [PubMed] [Google Scholar]
- 40.Vandel L, Kouzarides T 1999. Residues phosphorylated by TFIIH are required for E2F-1 degradation during S-phase. EMBO J 18:4280–4291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukai T, Kusaka M, Kawabe K, Goto K, Nawata H, Fujieda K, Morohashi K 2002. Sexually dimorphic expression of Dax-1 in the adrenal cortex. Genes Cells 7:717–729 [DOI] [PubMed] [Google Scholar]
- 42.Bain J, McLauchlan H, Elliott M, Cohen P 2003. The specificities of protein kinase inhibitors: an update. Biochem J 371:199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bach S, Knockaert M, Reinhardt J, Lozach O, Schmitt S, Baratte B, Koken M, Coburn SP, Tang L, Jiang T, Liang DC, Galons H, Dierick JF, Pinna LA, Meggio F, Totzke F, Schachtele C, Lerman AS, Carnero A, Wan Y, Gray N, Meijer L 2005. Roscovitine targets, protein kinases and pyridoxal kinase. J Biol Chem 280:31208–31219 [DOI] [PubMed] [Google Scholar]
- 44.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM 1998. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 273:18623–18632 [DOI] [PubMed] [Google Scholar]
- 45.Davies SP, Reddy H, Caivano M, Cohen P 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351:95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Payne DM, Rossomando AJ, Martino P, Erickson AK, Her JH, Shabanowitz J, Hunt DF, Weber MJ, Sturgill TW 1991. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J 10:885–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bastien J, Adam-Stitah S, Riedl T, Egly JM, Chambon P, Rochette-Egly C 2000. TFIIH interacts with the retinoic acid receptor γ and phosphorylates its AF-1-activating domain through cdk7. J Biol Chem 275:21896–21904 [DOI] [PubMed] [Google Scholar]
- 48.Narayanan R, Adigun AA, Edwards DP, Weigel NL 2005. Cyclin-dependent kinase activity is required for progesterone receptor function: novel role for cyclin A/Cdk2 as a progesterone receptor coactivator. Mol Cell Biol 25:264–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, Morgan DO, Reinberg D 1995. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature 374:283–287 [DOI] [PubMed] [Google Scholar]
- 50.Tassan JP, Jaquenoud M, Fry AM, Frutiger S, Hughes GJ, Nigg EA 1995. In vitro assembly of a functional human CDK7-cyclin H complex requires MAT1, a novel 36 kDa RING finger protein. EMBO J 14:5608–5617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rochette-Egly C, Adam S, Rossignol M, Egly JM, Chambon P 1997. Stimulation of RARα activation function AF-1 through binding to the general transcription factor TFIIH and phosphorylation by CDK7. Cell 90:97–107 [DOI] [PubMed] [Google Scholar]
- 52.Chen D, Riedl T, Washbrook E, Pace PE, Coombes RC, Egly JM, Ali S 2000. Activation of estrogen receptor α by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol Cell 6:127–137 [PubMed] [Google Scholar]
- 53.Harvey KJ, Lukovic D, Ucker DS 2000. Caspase-dependent Cdk activity is a requisite effector of apoptotic death events. J Cell Biol 148:59–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson RT, Squires S, Elliott GC, Koch GL, Rainbow AJ 1985. Xeroderma pigmentosum D-HeLa hybrids with low and high ultraviolet sensitivity associated with normal and diminished DNA repair ability, respectively. J Cell Sci 76:115–133 [DOI] [PubMed] [Google Scholar]
- 55.Coin F, Marinoni JC, Rodolfo C, Fribourg S, Pedrini AM, Egly JM 1998. Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat Genet 20:184–188 [DOI] [PubMed] [Google Scholar]
- 56.Wilson TE, Fahrner TJ, Milbrandt J 1993. The orphan receptors NGFI-B and steroidogenic factor 1 establish monomer binding as a third paradigm of nuclear receptor-DNA interaction. Mol Cell Biol 13:5794–5804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Juge-Aubry CE, Hammar E, Siegrist-Kaiser C, Pernin A, Takeshita A, Chin WW, Burger AG, Meier CA 1999. Regulation of the transcriptional activity of the peroxisome proliferator-activated receptor α by phosphorylation of a ligand-independent trans-activating domain. J Biol Chem 274:10505–10510 [DOI] [PubMed] [Google Scholar]
- 58.Bour G, Gaillard E, Bruck N, Lalevee S, Plassat JL, Busso D, Samama JP, Rochette-Egly C 2005. Cyclin H binding to the RARα activation function (AF)-2 domain directs phosphorylation of the AF-1 domain by cyclin-dependent kinase 7. Proc Natl Acad Sci USA 102:16608–16613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sewer MB, Waterman MR 2003. CAMP-dependent protein kinase enhances CYP17 transcription via MKP-1 activation in H295R human adrenocortical cells. J Biol Chem 278:8106–8111 [DOI] [PubMed] [Google Scholar]
- 60.Lopez D, Nackley AC, Shea-Eaton W, Xue J, Schimmer BP, McLean MP 2001. Effects of mutating different steroidogenic factor-1 protein regions on gene regulation. Endocrine 14:353–362 [DOI] [PubMed] [Google Scholar]
- 61.Zheng W, Jefcoate CR 2005. Steroidogenic factor-1 interacts with cAMP response element-binding protein to mediate cAMP stimulation of CYP1B1 via a far upstream enhancer. Mol Pharmacol 67:499–512 [DOI] [PubMed] [Google Scholar]
- 62.Keriel A, Stary A, Sarasin A, Rochette-Egly C, Egly JM 2002. XPD mutations prevent TFIIH-dependent transactivation by nuclear receptors and phosphorylation of RARα. Cell 109:125–135 [DOI] [PubMed] [Google Scholar]
- 63.Lehmann AR 2001. The xeroderma pigmentosum group D (XPD) gene: one gene, two functions, three diseases. Genes Dev 15:15–23 [DOI] [PubMed] [Google Scholar]
- 64.Dubaele S, Proietti De Santis L, Bienstock RJ, Keriel A, Stefanini M, Van Houten B, Egly JM 2003. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell 11:1635–1646 [DOI] [PubMed] [Google Scholar]
- 65.Chu G, Mayne L 1996. Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy: do the genes explain the diseases? Trends Genet 12:187–192 [DOI] [PubMed] [Google Scholar]
- 66.de Boer J, de Wit J, van Steeg H, Berg RJ, Morreau H, Visser P, Lehmann AR, Duran M, Hoeijmakers JH, Weeda G 1998. A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol Cell 1:981–990 [DOI] [PubMed] [Google Scholar]
- 67.Bergmann E, Egly JM 2001. Trichothiodystrophy, a transcription syndrome. Trends Genet 17:279–286 [DOI] [PubMed] [Google Scholar]
- 68.Zhao L, Bakke M, Krimkevich Y, Cushman LJ, Parlow AF, Camper SA, Parker KL 2001. Steroidogenic factor 1 (SF1) is essential for pituitary gonadotrope function. Development 128:147–154 [DOI] [PubMed] [Google Scholar]
- 69.Li LA, Chiang EF, Chen JC, Hsu NC, Chen YJ, Chung BC 1999. Function of steroidogenic factor 1 domains in nuclear localization, transactivation, and interaction with transcription factor TFIIB and c-Jun. Mol Endocrinol 13:1588–1598 [DOI] [PubMed] [Google Scholar]
- 70.Børud B, Hoang T, Bakke M, Jacob AL, Lund J, Mellgren G 2002. The nuclear receptor coactivators p300/CBP/cointegrator-associated protein (p/CIP) and transcription intermediary factor 2 (TIF2) differentially regulate PKA-stimulated transcriptional activity of steroidogenic factor 1. Mol Endocrinol 16:757–773 [DOI] [PubMed] [Google Scholar]