

Abstract
The biological effects of dioxins are mediated by the aryl hydrocarbon receptor (AhR) and its dimerization partner, the AhR nuclear translocator (ARNT), and include interference with hormonal signaling pathways like the response to estrogens. The effects of estrogens are mediated by two estrogen receptor (ER) isoforms, ERα and ERβ, which belong to the family of nuclear receptors. We have previously shown that ARNT can act as coactivator of the ERs. In this study, we show that recruitment of ARNT to AhR or hypoxia-inducible factor-1α signaling pathways as well as small interfering RNA-mediated down-regulation of ARNT levels lead to a reduction in ER transcriptional activity. Using chromatin immunoprecipitation assays, we demonstrate that this decrease coincides with reduced recruitment of ARNT to estradiol-regulated promoters. We show further that coactivation by ARNT as well as inhibition by dioxin acts stronger on ERβ than on ERα activity. Additionally, we demonstrate that the effects of ARNT are dependent on the A/B domain of the ERs with the A/B domain of ERβ being considerably stronger in mediating the coactivating effects of ARNT. Taken together, our studies show that recruitment of ARNT to the AhR after dioxin treatment can account for the antiestrogenic effect of dioxins. Moreover, we show for the first time that the inhibitory effects of dioxin are more pronounced on ERβ than on ERα.
17β-ESTRADIOL (E2) is a steroid hormone and regulates several key biological processes such as bone development, reproduction, and brain development (1). The cellular response to estrogenic compounds is mediated by the two estrogen receptor (ER) isoforms, ERα (NR3A1) and ERβ (NR3A2). These receptors belong to the nuclear receptor (NR) superfamily, which includes, e.g. the receptors for glucocorticoids, progestins, and thyroid hormone. The members of the NR family share a conserved structural arrangement with a centrally located DNA-binding domain flanked by the N-terminal A/B domain that includes the transcriptional activation function AF-1, and the C-terminal ligand-binding domain (LBD), harboring both the ligand-binding pocket and a second transcriptional function known as the AF-2 domain (2).
Upon binding of agonists, ERα and ERβ undergo a complex activation process to acquire full transcriptional activity. Ligand binding induces a conformational change, which in turn promotes release of inhibitory repressive factors such as silencing mediator of retinoid and thyroid receptors or nuclear receptor corepressor. Subsequently, positive regulatory factors are recruited to the receptor, and this complex binds to specific DNA elements located in the regulatory regions of target genes (3). Extensive studies have identified numerous proteins that can interact with and coactivate the transcriptional activity of the NRs. These factors include the classical coactivators of the p160 family, like steroid receptor coactivator and transcriptional intermediary factor 2, comodulators such as p300/cAMP response element binding protein-binding protein (CBP)-associated factor, and factors with histone acetyl transferase activity like CBP and p300 (4).
In addition to their endogenous ligands, the activity of ERα and ERβ can be modulated by a wide array of exogenous compounds, such as dietary derived substances like isoflavonoids and chemical pollutants like polyaromatic hydrocarbons such as dioxins (3). Modulation of receptor activity by chemical contaminants is commonly referred to as endocrine disruption. The negative effect of dioxins and related compounds on estrogen signaling is a well-studied phenomenon at the organism level (5, 6, 7). However, the molecular mechanisms behind the antiestrogenic effect of dioxins are poorly understood but have been suggested to involve increased metabolism of E2, increased ER turnover, and promoter interference (8).
Dioxin (also known as TCDD, or 2,3,7,8-tetrachlorodibenzo-p-dioxin) is an environmental pollutant formed through incomplete combustion of waste material or as a side product in certain industrial processes. The biological responses to dioxin are mediated by the aryl hydrocarbon receptor (AhR) (5). The AhR is a member of the basic helix-loop-helix (bHLH)-Per-ARNT-Sim (PAS) family of proteins that includes transcription factors like the hypoxia-inducible factors hypoxia-inducible factor-1α (HIF-1α) and endothelial PAS domain protein-1 or the circadian regulatory proteins Clock and Per. To become transcriptionally active, bHLH-PAS proteins form heterodimers with their respective partner proteins. These partner factors are members of the bHLH-PAS subfamily AhR nuclear translocator (ARNT) (6). Three different ARNT factors have been identified, ARNT-1, ARNT-2, and ARNT-3, the last also known as bMAL. ARNT-1 and -2 display a high degree of sequence similarity (9, 10) and function as general dimerization partners for the AhR as well as the hypoxia-inducible factors HIF-1α and endothelial PAS domain protein-1. However, ARNT-1 has been described to be more important for the signal transduction of AhR (11). ARNT-3 is selectively recruited by the circadian regulator Clock and does not support HIF-1α or AhR function (12, 13).
We have previously shown that ARNT-1 and ARNT-2 can interact with and coactivate ERα and ERβ transcriptional activities, with the effects being more potent on the ERβ subtype (14). ARNT-3 had no effect on ER transcriptional activity. The C-terminal domain of ARNT was required for its coactivating capacity on the ERs. On the other hand, the key domains of ARNT in terms of AhR and HIF-1α function, the bHLH and PAS domains, were not needed to support E2 signaling, suggesting that the mechanism by which ARNT acts to stimulate ER function is distinct from the documented mechanism (15) of AhR or HIF-1α activation. Furthermore, both the LBD and the A/B domains of ERα and ERβ were necessary for functional interaction with ARNT, suggesting a role of ARNT in AF-1 and AF-2 synergism (14).
The identification of ARNT as a coactivator of the ERs (14) is interesting in several regards. It suggests a novel mechanism for how the antiestrogenic effects of dioxins are mediated, whereby the AhR and ER compete for its common partner ARNT. It can also implicate that activation of other signaling pathways using ARNT as cofactor, e.g. activation of HIF-1α, can decrease ER activity. Furthermore, the preference of ARNT for ERβ could indicate a more potent antiestrogenic effect of dioxins on this receptor subtype, which could have interesting implications when evaluating dioxins as endocrine disruptors. Thus, detailed analysis of the interplay between ARNT and the ERs is essential for understanding the effects of dioxin on the ER system.
In this study, we set out to investigate mechanistic details of the coactivating function of ARNT on the ERs and test our hypothesis that competition for ARNT between AhR and ER can account at least partly for the antiestrogenic effect of dioxin. Because ARNT-1 has been reported to be more important for AhR signaling than ARNT-2 (11), we focused our study on the cross-talk between the ERs and ARNT-1. We report here that reducing the available pool of ARNT by either activating the AhR or HIF-1α signaling pathway or by decreasing ARNT levels using small interfering RNA (siRNA), inhibits ER function, with a more drastic effect on ERβ. We show further that ARNT coactivation and, importantly, TCDD inhibition is more pronounced on ERβ than on ERα and is dependent on the A/B domains of the ERs.
RESULTS
Activation of Alternative ARNT-Dependent Pathways Impairs ER Transcriptional Activity
The mechanism(s) behind the inhibitory cross-talk between the AhR and the ERs are not fully understood, in particular at the molecular level. We have previously demonstrated that the AhR partner protein ARNT functions as an ER coactivator (14). It is therefore possible that sequestering of ARNT to alternative signaling pathways, e.g. the AhR pathway, would interfere with ER transcriptional activity and therefore explain the inhibitory effect of dioxin on ER signaling.
To test this hypothesis, we coexposed cells to E2 and either dioxin or a hypoxic environment and studied the effects on ER signaling. Hypoxia induces the formation of the HIF-1α/ARNT complex that regulates expression of genes involved in the adaptive response to low oxygen tension in the cell (16). Recruitment of ARNT to the hypoxia signaling network is therefore also expected to reduce the intracellular levels of ARNT available for the ERs. To activate AhR, we decided to use TCDD because this compound is highly resistant to cellular metabolism, in contrast to other AhR ligands such as 3-methylcholantrene (3-MC). HC11 cells stably transfected with a 3xERE-TATA-luciferase construct (H-ERE cells) were exposed to 10 nm TCDD and/or hypoxia (1% O2) alone or in combination with 10 nm E2 or the ER antagonist 4-hydroxytamoxifen (4-OHT) for 24 h. After incubation, luciferase activity was determined and normalized to protein concentrations. As expected, the H-ERE cells responded to E2 with a robust increase of reporter gene expression (Fig. 1A). Coexposure with TCDD or hypoxia, however, resulted in a significant down-regulation of reporter gene activity, showing that activation of either the AhR or HIF-1α pathway impairs the ER transcriptional capacity. In addition, the inhibition of E2-induced response in cells coexposed to TCDD and hypoxia resulted only in a slight further reduction in transcriptional activity, suggesting that the effects of hypoxia and dioxin are not additive but rather decrease ER activity by a similar mechanism.
Fig. 1.
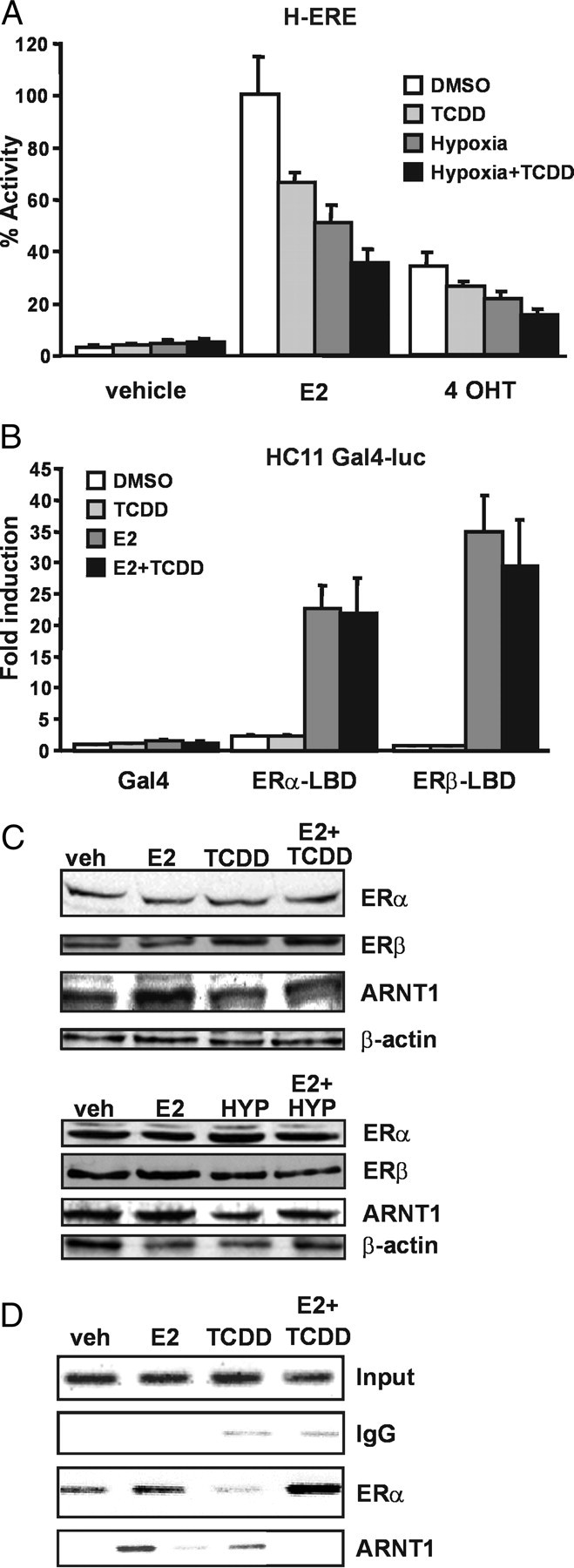
Recruitment of ARNT to AhR or HIF-1α Decreases ER Function
A, Activation of dioxin and/or hypoxia signaling inhibits ER transcriptional activity. H-ERE cells were treated with 10 nm E2 or 1 μm 4-OHT alone or combination with 10 nm TCDD, in hypoxic (<1% O2) or normoxic conditions, as indicated. After 24 h, luciferase activity was determined and adjusted to protein concentration in each sample. Treatment with E2 alone was set to 100%. Shown are means and sd for three independent experiments. B, Dioxin does not inhibit transcription by the ER-LBD. HC11 cells were transiently cotransfected with Gal4-ERα LBD or Gal4-ERβ LBD together with a Gal4-regulated luciferase reporter gene construct. The cells were treated with 10 nm E2 alone or in combination with 10 nm TCDD. After 48 h, reporter gene activity was determined. Luciferase activity without treatment with Gal4-vector only was set to 1. Shown are means and se for three independent experiments. C, Exposure to TCDD or hypoxia does not affect ERα and ERβ protein levels. H-ERE cells were incubated for 12 h with 10 nm E2 and/or 10 nm TCDD, under normal or hypoxic conditions, as indicated. Representative Western blots on whole-cell extracts with antibodies recognizing ERα, ERβ, ARNT, and actin are shown. D, ARNT c-Fos promoter occupancy is decreased by TCDD. HC11 cells were incubated for 45 min with 10 nm E2 alone or in combination with 10 nm TCDD and subsequently subjected to ChIP with the indicated antibodies. Shown here are representative agarose gels with PCR products for an ER binding site on the c-fos promoter.
Exposure to TCDD or hypoxia in HC11 cells treated with 1 μm 4-OHT resulted in a reduced inhibitory effect on 4-OHT-induced transcriptional activation compared with E2-induced activation (81% compared with 69%, Fig. 1A). 4-OHT is a so-called mixed ER agonist/antagonist known to activate mainly the transcriptional activity exerted by the N-terminal AF-1 domain of ERα (17). Our finding could suggest that TCDD or hypoxia mainly targets the AF-2 situated in the LBD and/or the cooperativity between the AF-1/AF-2 transcriptional domains of ERα. Alternatively, TCDD and hypoxic exposure may target mainly ERβ transcriptional activity that is not induced by 4-OHT.
Inhibition of ER by TCDD Is Not Mediated by the ER LBD Alone
To test whether the LBDs of ERα and ERβ are the main structural targets for the inhibitory actions of TCDD, we used constructs expressing the isolated LBD of the ERs fused to the Gal4 DNA-binding domain. HC11 cells were cotransfected with a Gal4-luciferase reporter construct and ER-LBD plasmids. After transfection, the cells were treated with indicated ER ligands, alone or in combination with TCDD, for 48 h (Fig. 1B). Interestingly, we note that the negative effects by TCDD on E2-induced activity previously observed in H-ERE cells did not occur using the Gal4 fusion of either ERα or ERβ. These results therefore suggest that TCDD does not target the individual transcriptional activation domains of the ER isoforms.
Exposure to TCDD and Hypoxia Has No Effect on ER Protein Levels in HC11 Cells
Previous studies have suggested that exposure to E2 and TCDD can lead to increased degradation of the ERα protein (18, 19). To investigate whether alterations in protein levels could account for the observed effects of TCDD and hypoxia, we performed Western blot analysis of extracts from cells treated with E2, TCDD, or hypoxia, alone or in combination (Fig. 1C). Compared with β-actin loading control, we observed no or minimal effects on the protein levels of ERα and ARNT by the different treatments, and the ERβ levels were even higher after hypoxic treatment. These results were confirmed by density measurement of the bands and normalization to β-actin levels (data not shown). The findings suggest that the decrease in ER-dependent transcriptional activity in H-ERE cells cannot be attributed to protein degradation. Somewhat surprisingly, ERα levels were not down-regulated by E2 treatment. ERα down-regulation after hormone treatment is a well-documented phenomenon that we observe in other cell lines (data not shown) but not in HC11 cells. In conclusion, these data show that activation of both AhR and HIF-1α pathways leads to reduced ER transcriptional activity, possibly by sequestering of ARNT.
Recruitment of ARNT to E2-Dependent Promoters Is Decreased upon TCDD Exposure
We have previously shown that ARNT is corecruited together with ER on the pS2 promoter in an E2-dependent fashion (14). To investigate whether TCDD treatment results in a reduced ARNT recruitment to an E2-regulated promoter, we performed chromatin immunoprecipitation (ChIP) experiments on the c-Fos promoter in HC11 cells. The pS2 gene is not induced by E2 in these cells, therefore the c-fos promoter was chosen. The cells were treated with E2 alone or in combination with 10 nm TCDD for 45 min. After this treatment, the cells were cross-linked and harvested, and ChIP assays were performed as described in Materials and Methods. In control experiments, ERα was recruited to the c-Fos promoter after treatment with E2 (Fig. 1D). No association of ERα with the c-Fos promoter was observed in the presence of TCDD alone. ARNT was also found on the c-Fos promoter in the presence of E2 but not in the presence of TCDD alone. Interestingly, the levels of ARNT recruited to the c-Fos promoter decreased when the cells were simultaneously treated with E2 and TCDD. These results strongly suggest that dioxin-activated AhR can sequester ARNT away from E2-regulated promoters, thereby reducing ER transcriptional activity.
Reduction of Intracellular Amounts of ARNT Inhibits the Transcriptional Activity of the ERs
The results presented above clearly indicate that recruitment of ARNT to alternative cellular signaling pathways affects the transcriptional activity of the ERs. To verify this notion directly, we performed siRNA experiments to lower the intracellular levels of ARNT in HeLa cells and test the consequence of this reduction on transcriptional activity of the ERs. Using Western blotting, we observed that treatment with siRNA against ARNT led to a substantial reduction of ARNT protein levels (Fig. 2A) but did not affect the levels of an unrelated protein, heat-shock protein 90 (hsp90). In contrast, a scrambled siRNA sequence did not affect ARNT or hsp90 levels (Fig. 2A). We next assessed the effects of reduced ARNT intracellular levels on ERα- or ERβ-regulated transcription. We cotransfected siRNA against ARNT or the scrambled sequence together with the 3xERE-TATA-luciferase reporter construct and expression plasmids for either ERα or ERβ. The cells were subsequently incubated with E2 for 24 h, and the transcriptional response was assessed. As shown in Fig. 2B, siRNA reduction of the intracellular levels of ARNT resulted in a clear reduction in ERα and ERβ transcriptional activity. Interestingly, the ERβ isoform was considerably more affected by low ARNT levels compared with ERα. In fact, whereas the transcriptional activity of ERα was reduced by 40% (Fig. 2B), the effect of reduced intracellular ARNT levels on ERβ was substantial with an almost 80% drop in transcriptional activity (Fig. 2B).
Fig. 2.
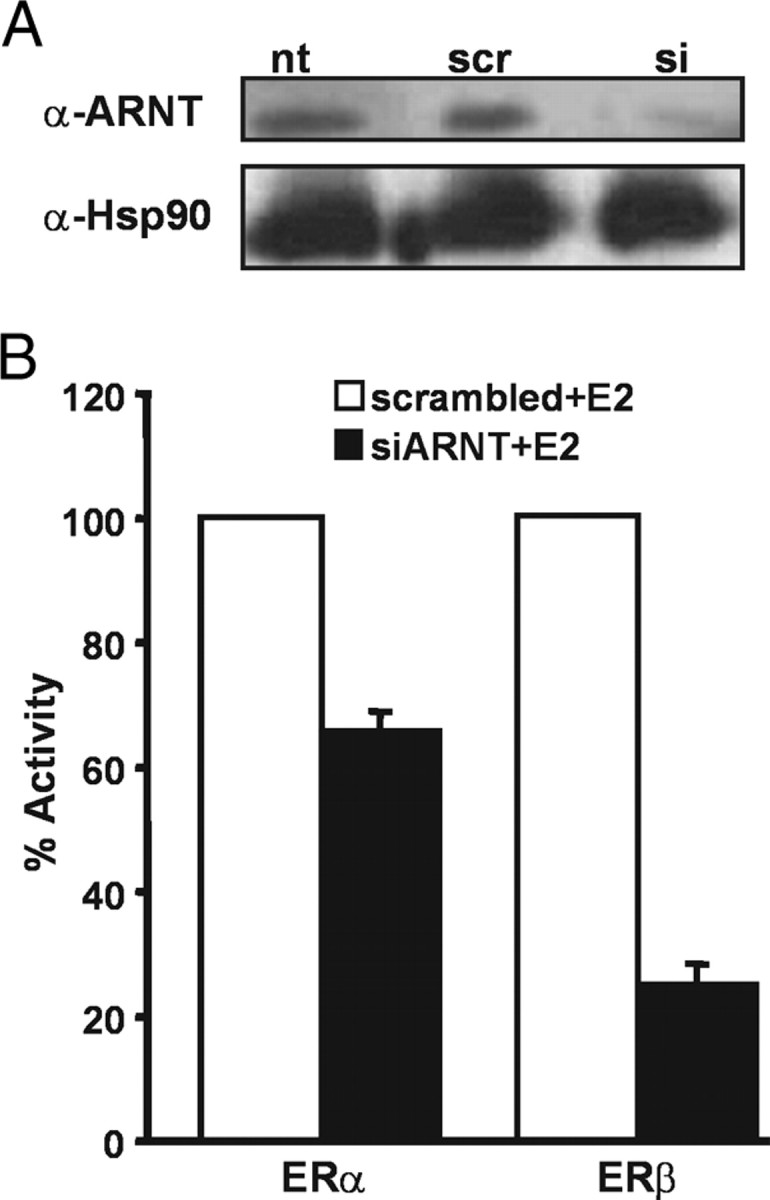
Reduction of Intracellular ARNT Levels Decreases ER-Dependent Transcription
A, siRNA against ARNT reduces ARNT protein levels. HeLa cells were transfected with siRNA oligonucleotides targeting ARNT (si) and as control, a scramble siRNA (scr), together with 3xERE-TATA-Luc reporter and expression plasmids for ERα and ERβ gene constructs and treated with 10 nm E2 for 48 h. Cells were lysed, and resulting whole-cell extracts were used for Western blots and luciferase assays shown in B. Shown are representative Western blots using antibodies against ARNT and hsp90 as control comparing nontransfected cells with scr- or si-transfected cells. B, siRNA against ARNT inhibits ER activity. Luciferase activities were measured in the same cell lysates and normalized to β-galactosidase activity. Shown are means and sd of three independent experiments; 100% reflects activity of ERα or ERβ, in the presence of E2 and scrambled RNA.
This experiment demonstrates that reduced availability of ARNT in a cell impairs ER function. Interestingly, we observe an ER isoform-specific sensitivity to reduced ARNT levels, which suggests that ARNT is more critical as a coactivator for ERβ than for ERα.
ARNT Coactivates ERα-ERβ Heterodimers to a Similar Extent As ERα
The preference of ARNT toward ERβ is consistent with our previous findings of a stronger coactivating effect of ARNT on ERβ-mediated transcription (14). In addition, this observation correlates well with our results that in cells treated with 4-OHT, TCDD only minimally affects ER-mediated transcription. It is known that in cells where both receptor isoforms are present, ERα and ERβ are able to form heterodimers (20, 21). Recent evidence suggests that ERα-ERβ heterodimers can activate a separate set of genes from the homodimers of each isoform (22). We therefore decided to investigate the effect of ARNT on ER heterodimers. To this end, single-chain ER homo- and heterodimers were used because they provide an efficient system to assay ERα/β heterodimeric transcriptional regulation (23). Single-chain ERs are transcribed by joined cDNAs of ERα and/or ERβ to simulate ER homo- and heterodimers. The fusion ERs have been shown to interact with estrogen response element (ERE) and E2 in a manner similar to that observed with the ER dimers (23). The single-chain receptors were transfected into HeLa cells together with a 3xERE-luciferase reporter and ARNT or the respective empty vector. The cells were treated with E2 for 48 h, and luciferase activity was measured. As described, the constructs were expressed and activated by E2 in a similar manner (data not shown). Luciferase activity in the presence of E2 without ARNT was set to 100%. As shown in Fig. 3, ARNT was much more potent in coactivating the ERβ than the ERα homodimer. Interestingly, the effect of ARNT on the ERα/ERβ heterodimers was comparable with that observed on ERα homodimers.
Fig. 3.

ARNT Coactivates ERα-ERβ Heterodimers to a Similar Extent as ERα Homodimers
Hela cells were transiently transfected with single-chain ER constructs together with a 3xERE-TATA-Luc reporter and an ARNT expression vector or empty plasmid. Cells were treated with 10 nm E2 for 48 h, and reporter gene activity was determined and normalized to β-galactosidase activity. Luciferase activity in the presence of E2 and empty vector was set to 100%. Shown are means and se of three independent experiments.
The A/B Domain Is Responsible for the Differential Effect of ARNT on ERα and ERβ
We have shown previously that, in contrast to the wild-type receptors, ERα and ERβ mutants lacking the N terminus are coactivated by ARNT to a similar extent. Additionally, the coactivating effect of ARNT on these mutants lacking the A/B domains was considerably weaker. We concluded that the A/B domain of the ERs might be important for the differential effect of ARNT on ERα and ERβ (14). Our observations presented here provide additional support for an important role of the A/B domain of ERβ. To directly test the involvement of the N-terminal domain of ERβ, we conducted transactivation assays in HeLa cells using ER swap mutants that consist of the A/B domain of either ERα or ERβ, combined with ERβ (ERβABα) or ERα (ERαABβ), respectively (Fig. 4A). Cells were transfected with the 3xERE-TATA-luciferase reporter and wild-type receptors or the swap mutants as well as increasing amounts of ARNT. After transfection, the cells were stimulated with E2 for 48 h, and luciferase activity was measured. The receptor constructs were all expressed to a similar extent (data not shown). The reporter activity of treated cells in the absence of ARNT was set to 100%. As shown in Fig. 4B, ARNT coactivated ERβ more potently than ERα. Interestingly, ARNT was not able to coactivate ERβABα at all, whereas the ERαABβ construct was similarly coactivated as ERβ. This confirms that the A/B domain of ERβ is essential for the differential effect of ARNT on ERα and ERβ. Furthermore, the combination of AF-1 of ERβ and AF-2 of ERα seems to be sufficient for ARNT to coactivate the receptor, whereas ARNT is ineffective on the opposite combination. The results suggest that the N-terminal region of ERβ includes feature(s) necessary for the coactivating function of ARNT. We decided to narrow down the region necessary for the effect of ARNT. To this end, we introduced N-terminal mutants of ERβ and performed transient transfection experiments (Fig. 4A, described in Ref. 24). Luciferase assays were carried out as described above. As shown in Fig. 4C, both full-length ERβ and the short form of ERβ, ERβ485 lacking the first 45 amino acids, were coactivated by ARNT in the presence of E2. Strikingly, however, deletion of the additional 31 amino acids from the ERβ530 N-terminus generating the Δ76 mutant resulted in a complete abolishment of ARNT coactivation. This could imply that this region of ERβ comprises a binding site for a factor that is involved in the mechanism of ARNT coactivation.
Fig. 4.
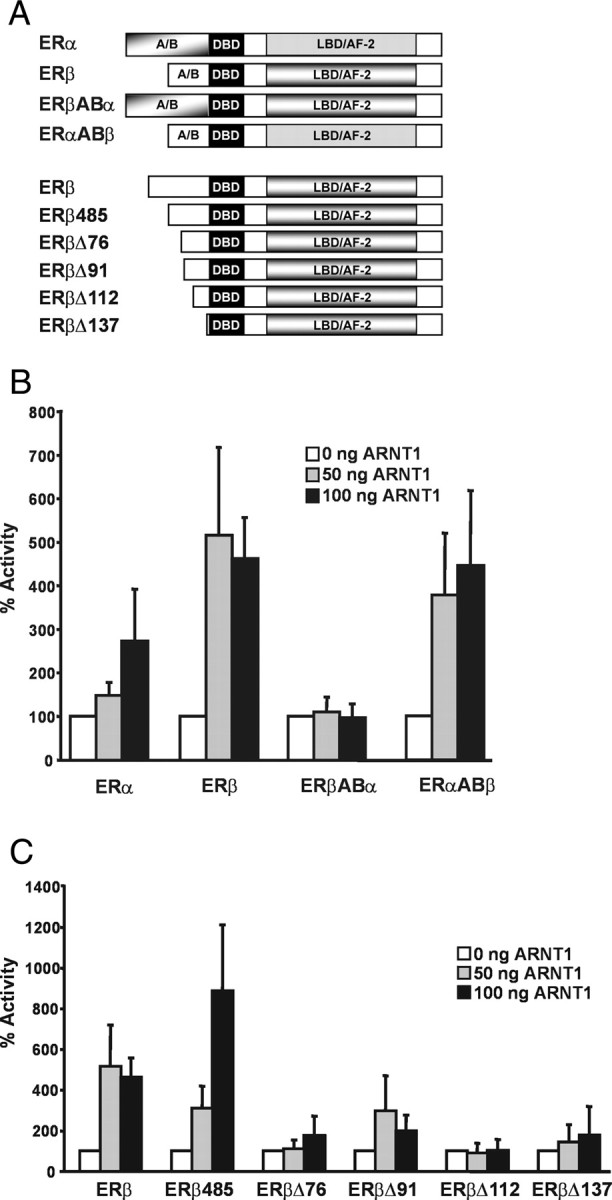
Analysis of the Effect of ARNT on N-Terminal ER Mutants
A, Schematic representation of the mutants used for the experiments in B and C. ERβ 530 serves as a reference for the numbering of the deleted amino acids. A/B, A/B domain; DBD, DNA-binding domain. B, The A/B domain of the ERs is important for ARNT coactivation. Hela cells were transiently transfected with ERα, ERβ, ERβABα, or ERαABβ together with a 3xERE-TATA-Luc reporter and increasing amounts of ARNT expression vector. Cells were treated with 10 nm E2 for 48 h, and reporter gene activity was determined and normalized to β-galactosidase activity. Luciferase activity in the presence of E2 and empty vector was set to 100%. Shown are means and sd of three independent experiments. C, Deletion of the very N terminus of ERβ abolishes its responsiveness to ARNT coactivation. Experiments were carried out as in B but with the ERβ constructs shown in the figure. Luciferase activity in the presence of E2 and empty vector was set to 100%. Shown are means and sd of three independent experiments.
ARNT Recruitment to E2-Responsive Promoters Is More Pronounced in the Presence of ERβ than of ERα
In Fig. 1D, we showed that ARNT is E2-dependently recruited to the c-fos promoter in HC-11 cells and that this recruitment is diminished by TCDD exposure. HC-11 cells express both ER isoforms. Thus, this experiment does not reveal whether ARNT recruitment is also isoform specific. To distinguish between recruitment after ERα and after ERβ stimulation, we used two different MCF-7 cell lines, a wild-type MCF-7 cell line that predominantly expresses ERα and a cell line with a stably integrated flag-tagged ERβ construct under the control of an inducible tet-off system (MCF-7ERβ). ChIP assays were carried out after removal of tet (i.e. allowing expression of ERβ) for 48 h and treatment of wild-type and MCF7-ERβ cells with E2 and diarylpropionitrile (DPN, an ERβ-specific agonist) (25) for 45 min, respectively. In MCF-7 cells, GREB-1 and pS2 are prominently regulated by E2 (26); thus, we investigated transcription factor recruitment to these promoters. Recruitment of the ERs and ARNT on the GREB-1 promoter is shown in Fig. 5A. Interestingly, we observe low levels of ARNT recruitment in wild-type cells (i.e. after ERα activation) compared with that in MCF-7ERβ cells. Similar results were observed on the pS2 promoter (data not shown). To quantify these data, we carried out real-time PCR on the ChIP samples. As shown in Fig. 5B, ARNT is recruited after activation of ERβ, whereas no ARNT recruitment is detectable after stimulation of ERα. Upon TCDD treatment, ARNT recruitment is significantly diminished in ERβ-expressing cells. These experiments corroborate the findings that ARNT is an ERβ-specific coactivator and is primarily recruited by ERβ to E2-induced genes.
Fig. 5.

Exposure to TCDD Results in Reduced Recruitment of ARNT to E2-Responsive Promoters in ERβ-Expressing Cells
A, MCF-7 wild type or ERβ cells were incubated for 45 min with 10 nm E2 or DPN, respectively, alone or in combination with 10 nm TCDD. Subsequently, cells were subjected to ChIP analysis with the indicated antibodies. Shown here are representative agarose gels with PCR products for an ER binding site on the GREB-1 promoter. B, Quantification of the ChIP experiments in A. Real-time PCR was performed on precipitated DNA of at least three independent experiments. The results were normalized to input DNA, and vehicle treatment was set to 1.
ARNT Interacts Directly with ERβ on E2-Regulated Promoters of Activated Genes
Using immunoprecipitation, we have previously shown that ARNT directly interacts with the ERs (14). To test whether ERβ and ARNT are found in the same complex on E2-regulated promoters, re-ChIP experiments were performed. MCF-7 ERβ cells were treated as for the ChIP experiments, and then two immunoprecipitations were carried out. In the first immunoprecipitation, we used antibodies against ARNT; in the second precipitation, we used flag antibodies against the tagged ERβ isoform. In control experiments, antimouse IgG was used instead of antiflag antibody. Upon E2 treatment, ARNT-ERβ complexes were found on the GREB-1 promoter (Fig. 6) and the pS2 promoter (data not shown). No promoter enrichment was found under control conditions. As expected, TCDD treatment reduced the ARNT-ERβ recruitment to the promoter.
Fig. 6.

ARNT Forms Complexes with ERβ and RNA Polymerase II on E2-Responsive Promoters after E2 Treatment
MCF-7 ERβ cells were incubated for 45 min with 10 nm DPN alone or in combination with 10 nm TCDD. Subsequently, re-ChIP analysis was performed with the indicated antibodies as described in Materials and Methods. Shown here are representative agarose gels with PCR products for an ER binding site on the GREB-1 promoter.
To verify that ARNT indeed was present on transcriptionally active promoters, re-ChIP analysis with ARNT and RNA polymerase II were carried out. As shown in Fig. 6, ARNT and RNA polymerase II bind in the same complex to the GREB-1 promoter after E2 treatment. This suggests that ARNT is part of the receptor complex on promoters of E2-activated genes.
TCDD Affects ERβ Activity More than that of ERα, and This Is Dependent on the A/B Domain of the Receptors
According to our findings, competition for ARNT between AhR and the ERs contributes to the inhibitory effect of dioxin on ER signaling. The fact that ARNT is more potent on ERβ than on ERα transcriptional activity and that it is recruited to E2-regulated promoters almost exclusively in the presence of ERβ suggests that dioxins could also impair the ERs in an isoform-specific manner. To test this hypothesis, we compared the effect of TCDD on ERα and ERβ transcriptional activity separately. We decided to use the H-ERE cells again, treating them with E2 and additionally with the ERα-specific agonist propyl pyrazole triol (PPT) (27) and with the ERβ-specific agonist DPN, alone or in combination with TCDD. As shown in Fig. 7A, treatment of the cells with TCDD reduced ER activity in the presence of E2 by approximately 40%. A similar reduction was observed with PPT. Interestingly, treatment with the ERβ-specific agonist DPN resulted in a much more drastic decrease of ER activity by approximately 80%.
Fig. 7.
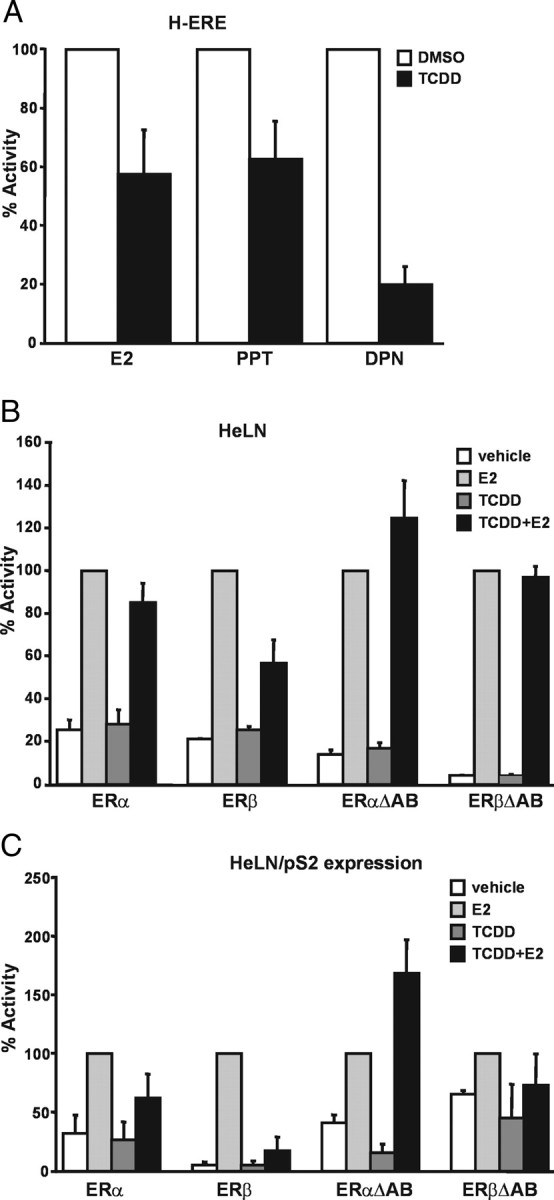
TCDD Shows Selectivity for the ERβ Isoform and Acts on the A/B Domain on the ERs
A, Differential effects of TCDD on ERα and ERβ transcriptional activity in HC11 cells. H-ERE cells were treated with 10 nm E2, 10 nm PPT, or 10 nm DPN alone or combination with 10 nm TCDD. After 24 h, luciferase activity was determined and adjusted to protein concentration in each sample. Treatment with the respective agonist alone was set to 100%. Shown are means and sd of three independent experiments. B, Effect of TCDD in HELN cell lines measured by the stably integrated ERE-β glob-luciferase activity. HELN cells were treated with 10 nm E2 alone or combination with 10 nm TCDD. After 24 h, luciferase activity was determined and adjusted to protein concentration in each sample. Treatment with E2 alone was set to 100%. Shown are means and sd of three independent experiments. C, Effect of TCDD in HELN cell lines measured by induction of the endogenous pS2 gene. HELN cells were treated with 10 nm E2 alone or combination with 10 nm TCDD. After 6 h, cells were harvested, total RNA extracted, and mRNA transcribed into cDNA. Amounts of pS2 cDNA was measured by real-time PCR and normalized to amount of 18S RNA. Treatment with E2 alone was set to 100%. Shown are means and sd of four independent experiments.
These findings were verified in HeLa cells stably transfected with the ERE-β-glob-luciferase reporter construct together with either ERα or ERβ (HELN ERα and HELN ERβ). Moreover, we investigated cell lines with mutant receptors lacking the N-terminal A/B domain (ERαΔAB and ERβΔAB). These cells were treated with E2 and TCDD, and luciferase activity was measured and normalized to total amount of protein. As expected, E2 induced luciferase expression, and TCDD had no effect on basal ER activity (Fig. 7B). However, TCDD reduced the E2-induced ER activity, again with a more pronounced effect on ERβ than on ERα. Interestingly, the negative effect of TCDD was completely abolished in cells expressing the ER mutants.
To confirm these results on an endogenous promoter, we performed real-time PCR to measure the induction of the ER-regulated pS2 gene in the stable HeLa cells. After treatment with E2, pS2 transcription was induced in all four cell lines (Fig. 7C). TCDD alone had no effect on pS2 induction, whereas treatment with TCDD decreased E2-induced pS2 transcription for ERα by about 40%, and particularly for ERβ with a robust reduction of 80%. ERβΔAB activity was only marginally affected by TCDD treatment, whereas activity of ERαΔAB was somewhat increased in the presence of TCDD.
In conclusion, TCDD decreased ERα and ERβ activity in different cell lines and on different promoters. In addition, our experiments reveal a remarkably stronger sensitivity of the ERβ isoform to the inhibitory actions of TCDD. Moreover, our results suggest that the inhibitory effect of TCDD requires the ER A/B domain because the ΔAB mutants were only marginally or not at all affected upon exposure to dioxin.
DISCUSSION
Competition between AhR and ER for ARNT Can Account for the Antiestrogenic Effect of TCDD
The ability of industrial chemicals and environmental pollutants to interfere with hormonal signaling pathways, a phenomenon known as endocrine disruption, has caused considerable attention and concern for decades. The potency of dioxins as endocrine disruptors is well documented, in particular with regard to their negative effects on E2 signaling pathways (28). The biological effects of dioxins are mediated by the AhR, a ligand-dependent transcription factor. The activation process of the AhR has been thoroughly studied and involves the recruitment of its dimerization partner ARNT (6). We have previously shown that ARNT can act as coactivator of the ERs (14). The ability of ARNT to coregulate E2 signaling pathways prompted us to suggest that competition for the intracellular pool of ARNT can be an important regulatory component that may explain, at least in part, the disruptive effects of dioxins such as TCDD on estrogen signaling. In this study, our experiments show that activation of alternative ARNT-dependent signaling pathways such as exposure to TCDD or hypoxic conditions, which leads to the formation of AhR/ARNT or HIF-1α/ARNT complexes, respectively, results in a decrease in ER transcriptional activity (Fig. 8). In addition, exposure of cells to TCDD and the subsequent formation of the AhR/ARNT complex resulted in reduced occupation of ARNT on ER-regulated promoters in ChIP assays. ARNT recruitment was observed on the c-fos promoter in HC11 cells and the GREB-1 and pS2 promoters in MCF-7 cells; all of them are part of E2-regulated genes. This suggests that ARNT could be a general coactivator for ERβ and not only recruited to specific genes. However, this hypothesis has to be studied more closely, for example by using ChIP-on-chip technology. Reducing the cellular levels of ARNT by siRNA significantly attenuated the transcriptional response of ERα and in particular ERβ, supporting our notion and demonstrating an important role of ARNT in ER-mediated transcription. The siRNA was directed against a sequence that is homologous between ARNT-1 and ARNT-2. We therefore cannot conclude at that point whether it is sufficient to knock down one form or whether both have to be down-regulated to see an effect on ER transcription. Obviously, an even more conclusive experiment would be to investigate ER activity in ARNT knockout animals. However, unconditional knockout of ARNT-1 as well as ARNT-2 is lethal in mice (29). Knockout mice have been used where ARNT-1 is disrupted in liver (30), in T cells (31), in endothelial cells (32), and in mammary epithelium (33). Using the last, the authors reported that disruption of ARNT has no implication for mammary gland development, which would suggest that ARNT is dispensable for ER function (33). This observation is in line with the results presented in this study, because the main ER isoform in the breast is ERα. Yet our experiments suggest that ERβ rather than ERα is affected by ARNT-1 down-regulation. In addition, it is possible that ARNT-2 still interacts and coactivates ER in the breast-specific ARNT-1 knockout mouse. Only ARNT1-ARNT2 double-knockout mice can give information about the necessity of ARNT in ER signaling in vivo.
Fig. 8.

Model for the Proposed Mechanism of the Antiestrogenic Effects of TCDD and Hypoxia
Transcriptional activity of the ERs, particularly of ERβ, is enhanced by the presence of ARNT. Activation of the AhR or HIF-1α signaling pathway leads to reduction of ARNT available for ER and thus to decreased ER function.
We show here that ARNT is a much more potent coactivator for ERβ than for ERα. Interestingly, ERβ activity is also affected to a much greater extent by TCDD than that of ERα. Furthermore, we demonstrate that both coactivation by ARNT and inhibition by TCDD are dependent on the presence of the N-terminal A/B domain of both ERα and ERβ. This parallel behavior between coactivation and inhibition by ARNT and TCDD, respectively, substantially corroborates our hypothesis that recruitment of ARNT to AhR is at least partly responsible for the antiestrogenic effect of dioxins.
A recent publication showed that in the presence of the AhR agonist 3-MC, the AhR/ARNT complex activated the ERs in the absence of E2, suggesting a proestrogenic function of the activated AhR/ARNT complex (34). This study is inconsistent with numerous epidemiological studies and experimental data demonstrating an antiestrogenic effect of AhR ligands (7, 35, 36, 37, 38). We speculate that this inconsistency can be explained by the choice of AhR ligand. Although TCDD, the most potent dioxin in the environment, remains biochemically stable, metabolism of polycyclic aromatic hydrocarbons such as 3-MC potentially leads to the formation of metabolites with the ability to activate alternative signaling pathways. This has been shown to occur for instance with benzo-(a)-pyrene, a compound related to 3-MC. For this study, we chose to use TCDD and could not observe a proestrogenic effect in any of our experimental settings.
Our results suggest that TCDD inhibits ER activity and that this antiestrogenic action involves competition for limited intracellular levels of ARNT (Fig. 8). This makes ARNT an interesting factor in the context of endocrine disruption and puts forward the importance of studying the mechanistic details of the interplay between ARNT and the ERs.
The N-Terminal Region of ER Plays an Important Role for ARNT and TCDD Action on the Receptor
We have shown previously that, in contrast to the wild-type receptors, ARNT coactivated deletion mutants of ERα and ERβ lacking the N terminus to a similar extent. Additionally, the coactivating effect of ARNT on these mutants was weaker. We concluded that the A/B domain of the ERs might be responsible for the differential effect of ARNT on ERα and ERβ (14). Here, by swapping the A/B domains of the two isoforms, we show that the A/B domain of ERβ is responsible for the preference of ARNT toward ERβ. Additionally, the combination between ERα AF-1 and ERβ AF-2 was not responsive to ARNT coactivation at all. These findings were corroborated by the fact that TCDD was less potent in inhibiting the function of A/B domain deletion mutants of the receptors (and constructs exhibiting only the ER LBD). In addition, we investigated the effect of ARNT on ER heterodimers using single-chain ER constructs. We show that the effect of ARNT coactivation on heterodimers is comparable with that on ERα. We can only speculate about the reason for this finding. It is possible, for instance, that the presence of one ERα receptor is sufficient for the dimer to adopt a conformation that resembles an ERα homodimer. Previous reports have shown that ER heterodimers emulate ERα properties with respect to their potency on different E2 response elements and DNA-independent pathways (23) as well as the activation of AF-1 by 4-OHT and their recruitment of coactivators to AF-2 and AF-1 (23, 39). It is therefore possible that recruitment of additional coactivators besides ARNT is involved in the activation process of the ERα/β heterodimer function.
The role of ERβ AF-1 remains to be fully solved. Several publications suggest that the N terminus of ERβ has an inhibitory effect on ERβ AF-2 (24, 40, 41). The mechanism behind this finding is unclear; it has been suggested, however, that steric hindrance could account for less efficient recruitment of coactivators to AF-2 (41). It is possible that binding of ARNT to ERβ AF-1 overcomes the interference between the two activation functions and hence leads to enhanced recruitment of coactivators to the AF-2. Alternatively, ARNT could enhance recruitment of other coactivators to ERβ AF-1. Several coactivators are reported to interact not only with AF-2 but also with AF-1 of the ERs, e.g. p300/CBP (42). Interestingly, p300 has been shown to bind to amino acids 62–72 of ERβ (42). We found here that this region is crucial for the coactivating effect of ARNT-1 on ERβ. P300 has also been shown to interact with the C-terminal domain of ARNT. This coincides with our previous finding that the C terminus of ARNT is essential for its coactivating function on the ERs (14). In addition, CBP/p300 has been demonstrated to mediate the interaction between ARNT and HIF-1α (43). Thus, our results together with previous findings could put forward a role of p300 in mediating the coactivating effect of ARNT on ERβ.
TCDD Is Considerably More Potent in Disrupting Activity of ERβ than of ERα
In accordance with our previous findings, we show here that ARNT exhibits a remarkable preference for ERβ compared with ERα. Coactivation by ARNT overexpression as well as inhibition by ARNT down-regulation using siRNA was more pronounced for ERβ. This prompted us to investigate whether TCDD shows a similar preference for ERβ. Indeed, we could show in two different cell lines and on stably transfected reporters as well as on an endogenous E2-regulated gene that the inhibitory effect of TCDD is much more pronounced on the activity of ERβ than of ERα.
ERα and ERβ are known to be expressed in different tissues and to have distinct functions and target genes. In cells expressing both receptors, stimulation of ERα induces proliferation and cell growth, whereas activation of ERβ inhibits cell growth and leads to increased apoptosis (44). This is of particular interest because the literature about the endocrine-disruptive effects of TCDD is controversial and focused on ERα-containing cells and tissues. For example, its antiestrogenic effect has led to the development of related drugs to treat estrogen-dependent breast cancer (45). On the other hand, studies in nonhuman primates have shown that TCDD is associated with an increased prevalence and severity of endometriosis (46). Furthermore, follow-up studies of the TCDD-exposed population in Seveso, Italy, indicate that both all-cancer and lung cancer incidences tended to be higher than expected (47). Thus, TCDD seems to be both carcinogenic and antiproliferative, depending on the tissue type and the receptors expressed. Studies investigating the effect of TCDD on the estrogen system mostly refer to classical ERα-expressing tissues like breast and uterus and rarely investigated organs predominantly expressing ERβ. Our findings may be critical with regard to the sensitivity of different tissues to the biological effects of dioxin-like pollutants. Future studies should evaluate the impact of dioxins on tissues predominantly expressing ERβ.
Taken together, we present evidence that the recruitment of the ER coactivator ARNT to the AhR signaling pathway after dioxin exposure can account for the antiestrogenic action of dioxins. The effects of both ARNT and dioxin are considerably more pronounced on ERβ than on ERα. More studies have to be carried out to unravel the exact mechanism behind the ER-ARNT cross-talk and to evaluate the physiological importance of the selectivity of TCDD for ERβ.
MATERIALS AND METHODS
Plasmids and Reagents
TCDD, 3-MC, and benzo(a)pyrene were purchased from AccuStandard (New Haven, CT); E2 and 4-OHT from Sigma Chemical Co. (St. Louis, MO); ICI 182,780 from AstraZeneca (Södertälje, Sweden); and DPN and PPT from Tocris Bioscience (Bristol, UK).
The plasmids encoding for single-chain ERs were a kind gift from Dr. Mesut Muyan (23). The plasmids pSG5-hERα, pSG5-hERβ, 3xERE-TATA-Luc, pCMV-ARNT1, Gal4-Luc, pCMV-βGal, Gal4-ERα-LBD, and Gal4-ERβ-LBD have been described elsewhere. ERβ deletion mutants have been described previously (24). In the original publication, deletions were made on ERβ 485. Here, we refer to ERβ 530 as full-length receptor; the nomenclature is therefore different, e.g. Δ31 becomes Δ76. Additional details can be obtained from the authors upon request.
Antibodies
Monoclonal anti-ERα Ab10 was from NeoMarkers (Fremont, CA) and ERβ GTX14021 from GeneTex (San Antonio, TX), and monoclonal anti-flag M2 (Sigma), polyclonal anti-ERα H-184, polyclonal anti-ARNT1 H-172, polyclonal anti-polII, monoclonal anti-hsp90 F-8, and actin antibody sc-8432 were all from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Cell Culture and Transient Transfection Assays
HC11 cells and stably transfected 3xERE HC11 cells (H-ERE) (previously described in Ref. 48) were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 8% fetal calf serum (FCS; Invitrogen), l-glutamine (GIBCO/BRL, Carlsbad, CA), 50 μg/ml gentamycin (GIBCO/BRL), 10 ng/ml epidermal growth factor (Sigma), and 5 μg/ml insulin (Sigma). HeLa and MCF-7 cells were routinely maintained in DMEM supplemented with 10% FCS and penicillin (100 U/ml) and streptomycin (100 μg/ml). HELN ERα and HELN ERβ cells have been already described (49) and were kept in DMEM supplemented with 5% dextran-coated charcoal (DCC)-treated FCS (Hyclone, Logan, UT), penicillin (100 U/ml), streptomycin (100 μg/ml), 1 mg/ml G418, and 0.5 μg/ml puromycin. MCF7-ERβ cells were kindly provided by Dr. Anders Ström and were generated as follows: MCF-7 cells were first transfected with pTet-tTAk (GIBCO/BRL) modified to contain puromycin resistance by using Lipofectin according to the manufacturer’s instructions (GIBCO/BRL). Selection was performed with 0.5 μg/ml puromycin in the presence of 1 μg/ml tetracycline. A clone showing high levels of induction upon tetracycline withdrawal and low basal activity was selected by using the pUHC13-3 control plasmid (GIBCO/BRL). The short form of ERβ encoding 485 amino acids (ERβ 485) was fused to the flag tag and cloned into PBI-EGFP (Clontech, Palo Alto, CA). This construct was then transfected into the above-described inducible clone together with a neomycin resistance plasmid, and selection was performed with 700 μg/ml G418 (Calbiochem, La Jolla, CA). The cells were maintained in medium containing 0.5 μg/ml puromycin and 2 mg/ml tetracycline.
Cells were transfected as described earlier (14). Typically, cells were seeded in 12- or 24-well plates in phenol red-free medium 24 h before transfection. Cells were transfected using Lipofectamine reagent (Invitrogen) according to the manufacturer’s recommendations. We used 100 ng of the appropriate reporter plasmid (3xERE-TATA-Luc or Gal4-Luc) together with 20 ng pCMV5-βGal as internal transfection control and 5 ng receptor plasmids. After transfection, the medium was exchanged with phenol red-free medium supplemented with 5% DCC-treated FCS, and the cells were allowed to grow for an additional 24–48 h. At this point, cells were harvested, and luciferase and β-galactosidase activities were determined. Each graph represents the mean of at least three independent transfections performed in duplicate or triplicate. Stably transfected cells (H-ERE) were grown, treated, and lysed similarly before luciferase activity was determined and protein concentrations measured by the Bradford method. The luciferase values are correlated to the protein concentration of each sample.
Western Blot Analysis
HC11 cells were grown on 10-cm plates in phenol red-free medium for 24 h before they were treated with 10 nm E2, 10 nm TCDD, 10 μm 3-MC, and/or 10 nm TCDD for another 6 h. For the hypoxia treatment, cells were placed in a hypoxic chamber (O2 < 1%). Whole-cell extracts were prepared by lysing the cells on ice in RIPA buffer [20 mm Tris (pH 7.5), 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mm EDTA, 0.1% SDS] supplemented with protease inhibitor cocktail (Complete Protease Inhibitor; Roche Diagnostics, Basel, Switzerland). Protein concentrations were measured by the Bradford method. The protein extracts were separated on a 7.5% SDS-PAGE gel, and the proteins were detected with the respective antibodies. Western blots measuring siRNA efficiency were carried out directly on the whole-cell extracts used for luciferase assays.
ChIP and Re-ChIP Assays
ChIP assays for HC11 cells were performed as described (50). Assays with MCF-7 cells were performed as described previously (51) with slight modifications. Briefly, MCF-7 cells were grown on 15-cm plates to 80–90% confluency in phenol red-free DMEM supplemented with 5% DCC-stripped FCS for 48 h. After treating with 10 nm E2/DPN and/or 10 nm TCDD for 45 min, cells were washed with PBS and chromatin was cross-linked for 15 min with 1.5% formaldehyde. Cells were harvested and nuclear extracts produced. Chromatin was then sonicated 3 times for 10 sec with a Branston sonifier 250, duty cycle 90%, output 2.5, and a fraction of the soluble chromatin was put aside as input material. ChIP was performed overnight with various antibodies and protein Sepharose A or G (Pharmacia, Uppsala, Sweden). After washing, the Sepharose beads were eluted three times with 50 μl elution buffer (0.1 m NaHCO3/1% SDS), and bound chromatin was reverse cross-linked overnight at 65 C. For re-ChIP assays, beads were washed and eluted with 10 mm dithiothreitol after the first immunoprecipitation. Eluates were diluted, and the second antibody was added overnight. Then the samples were processed as for normal ChIP assays. Eluted DNA fragments were isolated and purified using QIAquick Spin Kit (QIAGEN, Valencia, CA), and the PCR-amplified fragments were analyzed on 2% agarose gels and by real-time PCR as described below. Specific primers used for the 3′-flanking region of the mouse c-Fos gene were 5′-GGCAGTTGTAAACTAGC-3′ and 5′-GGAACTTGGAGAAACC-3′; the 5′-flanking region of GREB-1, 5′-AGCAGTGAAAAAAAGTGTGGCAACTGGG-3′ and 5′-CGACCCACAGAAATGAAAAGGCAGCAAACT-3′; and the pS2 5′-flanking region, 5′-CCGGCCATCTCTCACTATGAA-3′ and 5′-CCTCCCGCCAGGGTAAATAC-3′.
siRNA-Mediated Down-Regulation of ARNT Levels
HeLa or MCF-7ERβ cells were seeded in six-well plates 24 h before transfection. Cells were transfected with 500 pmol siRNA (5′-CCAUCUUACGCAUGGCAGUTT-3′) (published sequence in Ref. 22), 200 ng 3xERE-TATA-Luc, 40 ng β-gal, and 30 ng pSG5-ERα or pSG5-ERβ per well, using Lipofectamine (Invitrogen) according to the manufacturer’s recommendations. After transfection, cells were treated with vehicle or 10 nm E2 or DPN for 48 h. Luciferase activity was measured as described above.
Detection of pS2 Transcription by Real-Time PCR
HELN cells were seeded out into six-well plates and grown in phenol red-free medium with 5% DCC-treated FCS for 48 h. After treatment with E2 and/or TCDD for 6 h, RNA was isolated using Trizol (Invitrogen) according to the manufacturer’s recommendations. One microgram of total RNA was treated with DNase I and reverse transcribed using random hexamer primers (Invitrogen). One microliter of the resulting cDNA was then used for real-time PCR with SYBR green (Invitrogen). The pS2 primer sequences were 5′-CCTCCCAGTGTGCAAATAAGG-3′ and 5′-TGGAGGGACGTCGATGGTAT-3′. Gene transcripts were normalized to the 18S rRNA content and to the untreated samples.
Acknowledgments
We thank Dr. Malin Hedengran-Faulds and Dr. Anders Ström for kindly providing the H-ERE and MCF-7ERβ cell lines, respectively. We also thank Mesut Muyan for kindly making available the single-chain ER constructs.
NURSA Molecule Pages:
Ligands: 17β-Estradiol | 4-Hydroxytamoxifen;
Nuclear Receptors: ERα | ERβ.
Footnotes
This work was supported by the German Research Foundation (DFG), the Swiss National Science Foundation (SNF), the European Union (EU)-funded CASCADE Network of Excellence, the EU-funded CRESCENDO project, the Swedish Cancer Foundation, and the Swedish Research Council.
Disclosure Statement: The authors have nothing to disclose.
First Published Online November 8, 2007
Abbreviations: AF, Activation function; AhR, aryl hydrocarbon receptor; ARNT, AhR nuclear translocator; bHLH basic helix-loop-helix; ChIP, chromatin immunoprecipitation; CBP, cAMP response element binding protein-binding protein; DCC, dextran-coated charcoal; DPN, diarylpropionitrile; E2, 17β-estradiol; ER, estrogen receptor; ERE, estrogen response element; FCS, fetal calf serum; HIF-1α, hypoxia-inducible factor-1α; hsp90, heat-shock protein 90; LBD, ligand binding domain; 3-MC, 3-methylcholantrene; NR, nuclear receptor; 4-OHT, 4-hydroxy-tamoxifen; PAS, Per-ARNT-Sim; PPT, propyl pyrazole triol; siRNA, small interfering RNA; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
References
- 1.Nilsson S, Gustafsson JA 2002. Biological role of estrogen and estrogen receptors. Crit Rev Biochem Mol Biol 37:1–28 [DOI] [PubMed] [Google Scholar]
- 2.Gronemeyer H, Gustafsson JA, Laudet V 2004. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov 3:950–964 [DOI] [PubMed] [Google Scholar]
- 3.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA 2001. Mechanisms of estrogen action. Physiol Rev 81:1535–1565 [DOI] [PubMed] [Google Scholar]
- 4.Pettersson K, Gustafsson JA 2001. Role of estrogen receptor β in estrogen action. Annu Rev Physiol 63:165–192 [DOI] [PubMed] [Google Scholar]
- 5.Poland A, Knutson JC 1982. 2,3,7,8-Tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol 22:517–554 [DOI] [PubMed] [Google Scholar]
- 6.Gu YZ, Hogenesch JB, Bradfield CA 2000. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol 40:519–561 [DOI] [PubMed] [Google Scholar]
- 7.Safe S 2001. Molecular biology of the Ah receptor and its role in carcinogenesis. Toxicol Lett 120:1–7 [DOI] [PubMed] [Google Scholar]
- 8.Wormke M, Stoner M, Saville B, Safe S 2000. Crosstalk between estrogen receptor α and the aryl hydrocarbon receptor in breast cancer cells involves unidirectional activation of proteasomes. FEBS Lett 478:109–112 [DOI] [PubMed] [Google Scholar]
- 9.Drutel G, Kathmann M, Heron A, Schwartz JC, Arrang JM 1996. Cloning and selective expression in brain and kidney of ARNT2 homologous to the Ah receptor nuclear translocator (ARNT). Biochem Biophys Res Commun 225:333–339 [DOI] [PubMed] [Google Scholar]
- 10.Hirose K, Morita M, Ema M, Mimura J, Hamada H, Fujii H, Saijo Y, Gotoh O, Sogawa K, Fujii-Kuriyama Y 1996. cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt). Mol Cell Biol 16:1706–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sekine H, Mimura J, Yamamoto M, Fujii-Kuriyama Y 2006. Unique and overlapping transcriptional roles of arylhydrocarbon receptor nuclear translocator (Arnt) and Arnt2 in xenobiotic and hypoxic responses. J Biol Chem 281:37507–37516 [DOI] [PubMed] [Google Scholar]
- 12.Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ 1998. Role of the CLOCK protein in the mammalian circadian mechanism. Science 280:1564–1569 [DOI] [PubMed] [Google Scholar]
- 13.Takahata S, Sogawa K, Kobayashi A, Ema M, Mimura J, Ozaki N, Fujii-Kuriyama Y 1998. Transcriptionally active heterodimer formation of an Arnt-like PAS protein, Arnt3, with HIF-1a, HLF, and clock. Biochem Biophys Res Commun 248:789–794 [DOI] [PubMed] [Google Scholar]
- 14.Brunnberg S, Pettersson K, Rydin E, Matthews J, Hanberg A, Pongratz I 2003. The basic helix-loop-helix-PAS protein ARNT functions as a potent coactivator of estrogen receptor-dependent transcription. Proc Natl Acad Sci USA 100:6517–6522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reisz-Porszasz S, Probst MR, Fukunaga BN, Hankinson O 1994. Identification of functional domains of the aryl hydrocarbon receptor nuclear translocator protein (ARNT). Mol Cell Biol 14:6075–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang BH, Rue E, Wang GL, Roe R, Semenza GL 1996. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem 271:17771–17778 [DOI] [PubMed] [Google Scholar]
- 17.Metzger D, Berry M, Ali S, Chambon P 1995. Effect of antagonists on DNA binding properties of the human estrogen receptor in vitro and in vivo Mol Endocrinol 9:579–591 [DOI] [PubMed] [Google Scholar]
- 18.Wormke M, Castro-Rivera E, Chen I, Safe S 2000. Estrogen and aryl hydrocarbon receptor expression and crosstalk in human Ishikawa endometrial cancer cells. J Steroid Biochem Mol Biol 72:197–207 [DOI] [PubMed] [Google Scholar]
- 19.Wormke M, Stoner M, Saville B, Walker K, Abdelrahim M, Burghardt R, Safe S 2003. The aryl hydrocarbon receptor mediates degradation of estrogen receptor α through activation of proteasomes. Mol Cell Biol 23:1843–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cowley SM, Hoare S, Mosselman S, Parker MG 1997. Estrogen receptors α and β form heterodimers on DNA. J Biol Chem 272:19858–19862 [DOI] [PubMed] [Google Scholar]
- 21.Pettersson K, Grandien K, Kuiper GG, Gustafsson JA 1997. Mouse estrogen receptor β forms estrogen response element-binding heterodimers with estrogen receptor α. Mol Endocrinol 11:1486–1496 [DOI] [PubMed] [Google Scholar]
- 22.Monroe DG, Secreto FJ, Subramaniam M, Getz BJ, Khosla S, Spelsberg TC 2005. Estrogen receptor α and β heterodimers exert unique effects on estrogen- and tamoxifen-dependent gene expression in human U2OS osteosarcoma cells. Mol Endocrinol 19:1555–1568 [DOI] [PubMed] [Google Scholar]
- 23.Li X, Huang J, Yi P, Bambara RA, Hilf R, Muyan M 2004. Single-chain estrogen receptors (ERs) reveal that the ERα/β heterodimer emulates functions of the ERα dimer in genomic estrogen signaling pathways. Mol Cell Biol 24:7681–7694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delaunay F, Pettersson K, Tujague M, Gustafsson JA 2000. Functional differences between the amino-terminal domains of estrogen receptors α and β. Mol Pharmacol 58:584–590 [DOI] [PubMed] [Google Scholar]
- 25.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA 2001. Estrogen receptor-β potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem 44:4230–4251 [DOI] [PubMed] [Google Scholar]
- 26.Lin CY, Strom A, Vega VB, Kong SL, Yeo AL, Thomsen JS, Chan WC, Doray B, Bangarusamy DK, Ramasamy A, Vergara LA, Tang S, Chong A, Bajic VB, Miller LD, Gustafsson JA, Liu ET 2004. Discovery of estrogen receptor α target genes and response elements in breast tumor cells. Genome Biol 5:R66 [DOI] [PMC free article] [PubMed]
- 27.Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA 2000. Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-α-selective agonists. J Med Chem 43:4934–4947 [DOI] [PubMed] [Google Scholar]
- 28.Safe SH 1995. Modulation of gene expression and endocrine response pathways by 2,3,7,8-tetrachlorodibenzo-p-dioxin and related compounds. Pharmacol Ther 67:247–281 [DOI] [PubMed] [Google Scholar]
- 29.Kozak KR, Abbott B, Hankinson O 1997. ARNT-deficient mice and placental differentiation. Dev Biol 191:297–305 [DOI] [PubMed] [Google Scholar]
- 30.Tomita S, Sinal CJ, Yim SH, Gonzalez FJ 2000. Conditional disruption of the aryl hydrocarbon receptor nuclear translocator (Arnt) gene leads to loss of target gene induction by the aryl hydrocarbon receptor and hypoxia-inducible factor 1α. Mol Endocrinol 14:1674–1681 [DOI] [PubMed] [Google Scholar]
- 31.Tomita S, Jiang HB, Ueno T, Takagi S, Tohi K, Maekawa S, Miyatake A, Furukawa A, Gonzalez FJ, Takeda J, Ichikawa Y, Takahama Y 2003. T cell-specific disruption of arylhydrocarbon receptor nuclear translocator (Arnt) gene causes resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced thymic involution. J Immunol 171:4113–4120 [DOI] [PubMed] [Google Scholar]
- 32.Yim SH, Shah Y, Tomita S, Morris HD, Gavrilova O, Lambert G, Ward JM, Gonzalez FJ 2006. Disruption of the Arnt gene in endothelial cells causes hepatic vascular defects and partial embryonic lethality in mice. Hepatology 44:550–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Provost F, Riedlinger G, Hee Yim S, Benedict J, Gonzalez FJ, Flaws J, Hennighausen L 2002. The aryl hydrocarbon receptor (AhR) and its nuclear translocator (Arnt) are dispensable for normal mammary gland development but are required for fertility. Genesis 32:231–239 [DOI] [PubMed] [Google Scholar]
- 34.Ohtake F, Takeyama K, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, Yanagisawa J, Fujii-Kuriyama Y, Kato S 2003. Modulation of oestrogen receptor signalling by association with the activated dioxin receptor. Nature 423:545–550 [DOI] [PubMed] [Google Scholar]
- 35.Zacharewski TR, Bondy KL, McDonell P, Wu ZF 1994. Antiestrogenic effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on 17β-estradiol-induced pS2 expression. Cancer Res 54:2707–2713 [PubMed] [Google Scholar]
- 36.Kharat I, Saatcioglu F 1996. Antiestrogenic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin are mediated by direct transcriptional interference with the liganded estrogen receptor. Cross-talk between aryl hydrocarbon- and estrogen-mediated signaling. J Biol Chem 271:10533–10537 [DOI] [PubMed] [Google Scholar]
- 37.Safe S, Wormke M, Samudio I 2000. Mechanisms of inhibitory aryl hydrocarbon receptor-estrogen receptor crosstalk in human breast cancer cells. J Mammary Gland Biol Neoplasia 5:295–306 [DOI] [PubMed] [Google Scholar]
- 38.Chen I, Hsieh T, Thomas T, Safe S 2001. Identification of estrogen-induced genes downregulated by AhR agonists in MCF-7 breast cancer cells using suppression subtractive hybridization. Gene 262:207–214 [DOI] [PubMed] [Google Scholar]
- 39.Tremblay GB, Tremblay A, Labrie F, Giguere V 1998. Ligand-independent activation of the estrogen receptors α and β by mutations of a conserved tyrosine can be abolished by antiestrogens. Cancer Res 58:877–881 [PubMed] [Google Scholar]
- 40.Hall JM, McDonnell DP 1999. The estrogen receptor β-isoform (ERβ) of the human estrogen receptor modulates ERα transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140:5566–5578 [DOI] [PubMed] [Google Scholar]
- 41.Huang J, Li X, Maguire CA, Hilf R, Bambara RA, Muyan M 2005. Binding of estrogen receptor β to estrogen response element in situ is independent of estradiol and impaired by its amino terminus. Mol Endocrinol 19:2696–2712 [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi Y, Kitamoto T, Masuhiro Y, Watanabe M, Kase T, Metzger D, Yanagisawa J, Kato S 2000. p300 mediates functional synergism between AF-1 and AF-2 of estrogen receptor α and β by interacting directly with the N-terminal A/B domains. J Biol Chem 275:15645–15651 [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi A, Numayama-Tsuruta K, Sogawa K, Fujii-Kuriyama Y 1997. CBP/p300 functions as a possible transcriptional coactivator of Ah receptor nuclear translocator (Arnt). J Biochem (Tokyo) 122:703–710 [DOI] [PubMed] [Google Scholar]
- 44.Helguero LA, Faulds MH, Gustafsson JA, Haldosen LA 2005. Estrogen receptors α (ERα) and β (ERβ) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene 24:6605–6616 [DOI] [PubMed] [Google Scholar]
- 45.Chen I, McDougal A, Wang F, Safe S 1998. Aryl hydrocarbon receptor-mediated antiestrogenic and antitumorigenic activity of diindolylmethane. Carcinogenesis 19:1631–1639 [DOI] [PubMed] [Google Scholar]
- 46.Rier S, Foster WG 2003. Environmental dioxins and endometriosis. Semin Reprod Med 21:145–154 [DOI] [PubMed] [Google Scholar]
- 47.Mandal PK 2005. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol [B] 175:221–230 [DOI] [PubMed] [Google Scholar]
- 48.Faulds MH, Olsen H, Helguero LA, Gustafsson JA, Haldosen LA 2004. Estrogen receptor functional activity changes during differentiation of mammary epithelial cells. Mol Endocrinol 18:412–421 [DOI] [PubMed] [Google Scholar]
- 49.Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavailles V, Balaguer P 2006. Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor α or β. Biochem Pharmacol 71:1459–1469 [DOI] [PubMed] [Google Scholar]
- 50.Burakov D, Crofts LA, Chang CP, Freedman LP 2002. Reciprocal recruitment of DRIP/mediator and p160 coactivator complexes in vivo by estrogen receptor. J Biol Chem 277:14359–14362 [DOI] [PubMed] [Google Scholar]
- 51.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M 2000. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103:843–852 [DOI] [PubMed] [Google Scholar]