

Key Points
Anfibatide potently inhibits platelet agglutination under static and arterial shear conditions.
Anfibatide is efficacious in treating spontaneous or shigatoxin-induced murine models of thrombotic thrombocytopenic purpura.
Abstract
Thrombotic thrombocytopenic purpura (TTP), a potentially fatal blood clot disorder, is primarily caused by severe deficiency of plasma ADAMTS13 activity resulting from acquired autoantibodies. Plasma exchange is the only effective initial therapy. However, the high mortality rate and the complications associated with plasma exchange therapy remain a major concern. To address unmet clinical needs, therapeutic efficacies of anfibatide, a snake venom-derived platelet glycoprotein Ib antagonist, in murine models of spontaneous thrombocytopenia and shigatoxin-induced TTP were determined. A light scattering platelet aggregometry, microfluidic shear-based assay, and murine models of TTP were used in the study. We showed that purified anfibatide inhibits ristocetin- or botrocetin-induced human or murine platelet agglutination in the presence of von Willebrand factor in a concentration-dependent manner. Anfibatide could also dramatically inhibit the adhesion and aggregation of murine and human platelets on a collagen surface under arterial shear stress, in the presence or absence of plasma ADAMTS13 activity. Most importantly, we demonstrated that an intraperitoneal administration of anfibatide at the dose of 60 ng/g body weight twice daily mitigated spontaneous thrombocytopenia and prevented shigatoxin-induced TTP in Adamts13−/− and disease-susceptible mice (CAST/Ei strain). Thus, we conclude that anfibatide, when administered at the optimal dosage, route, and interval, is efficacious in treating spontaneous and bacterial shigatoxin-induced TTP in the murine models. Our findings may provide the basis for further development of anfibatide for the treatment of acute TTP in humans.
Visual Abstract
Introduction
Thrombotic thrombocytopenic purpura (TTP) is characterized by severe thrombocytopenia and microangiopathic hemolytic anemia with or without signs and symptoms of organ dysfunctions.1,2 Acquired adult TTP is primarily caused by autoantibodies against a plasma metalloprotease ADAMTS13.3,4 Rarely, TTP is caused by germ line mutations of the ADAMTS13 gene that result in hereditary deficiency of plasma ADAMTS13 activity.5 ADAMTS13 cleaves a large adhesion protein, von Willebrand factor (VWF). The specific cleavage site is the Tyr1605-Met1606 bond located in the central A2 domain.6 This proteolytic cleavage is essential for normal hemostasis that creates a VWF-free endothelial surface.7 In addition, plasma ADAMTS13 can further degrade the released circulating ultra-large VWF (ULVWF) multimers once being exposed to high shear. Shear is necessary to unfold the VWF-A2 domain for cleavage, which reduces the VWF multimer sizes in circulation.4 Deficiency of plasma ADAMTS13 activity results in accumulation of ULVWF polymers on the endothelium8 and in blood,9,10 leading to heightened platelet adhesion and agglutination on injured vessel walls and formation of disseminated thrombosis in small arterioles and capillaries, the pathognomonic feature of TTP.
Without prompt treatment, patients with acute TTP may die within 24 to 48 hours.11 Plasma exchange is the only initial effective therapy available to date, resulting in the significant reduction of in-hospital mortality to <20%.1,11 However, ∼30% of patients who survive the acute episode may experience 1 to multiple episodes of exacerbation and/or relapses.12 This often requires installation of another round of intensive plasma exchange therapy in addition to other adjunctive therapies including cyclophosphamide, vincristine, cyclosporine, and rituximab.13 Plasma exchange is thought to remove the immunoglobulin G (IgG) autoantibodies against ADAMTS13 and the circulating ULVWF multimers, and perhaps inflammatory cytokines while replenishing ADAMTS13 to overcome the inhibition by autoantibodies. The clinical efficacy may be determined by the titer of anti-ADAMTS13 IgG as more ADAMTS13 may be required for overcoming the inhibition based on an in vitro mixing study.14,15 Other adjunctive immunotherapies are used to eliminate anti-ADAMTS13 autoantibody production13,16 and to suppress inflammatory responses.17,18 More recently, therapies that disrupt the interactions between VWF and platelet glycoprotein Ib (GPIb) such as anti-VWF RNA aptamer19,20 or VWF nanobody21-23 appear to be highly efficacious in the prevention and treatment of acquired TTP in preclinical models and in patients.
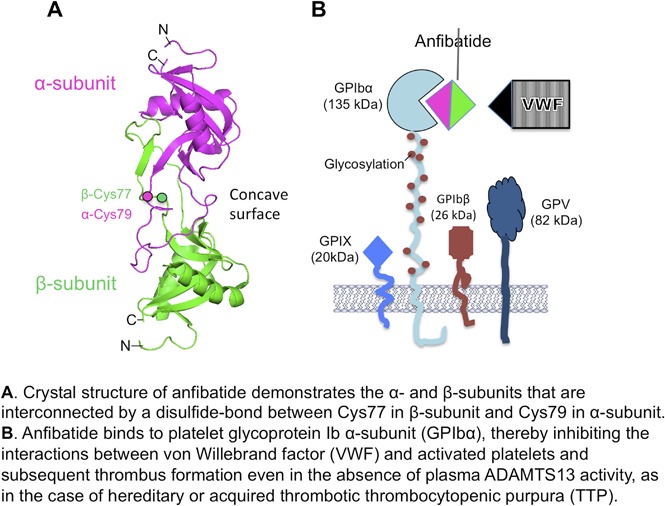
Anfibatide (also named agkicetin), a protein derived from the venom of Agkistrodon acutus,24 is another potent platelet GPIb antagonist.25 Anfibatide is a ∼30-kDa protein comprising 2 mature subunits (∼15 and ∼14 kDa) that are linked by a disulfide bond.26,27 The crystal structure of anfibatide at 1.9 Å resolution demonstrates how each subunit of anfibatide interacts with platelet GPIb.28 Anfibatide has been shown to inhibit thrombus growth in the flow chamber under high shear conditions, it markedly inhibits thrombosis in laser-injured cremaster vessels, and it prevented vessel occlusion in FeCl3-injured mesenteric vessels.25 Our results demonstrate, for the first time, that anfibatide, given at the optimal dosage, route, and interval, appears to be efficacious in preventing shigatoxin-induced TTP or in treating spontaneous thrombocytopenia resulting from severe Adamts13 deficiency.
Methods
Materials
HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid], bovine serum albumin, botrocetin, and apyrase were purchased from Sigma-Aldrich. Prostaglandin E1 was obtained from Biomol Research Laboratories. Murine recombinant VWF (mVWF) was expressed in HEK293 cells and purified according to the protocol described elsewhere.29 Human VWF was purified from normal human plasma as described previously.30
Purification of snake venom anfibatide
Anfibatide was purified from venom of the A acutus snake by anion exchange and monoclonal antibody affinity chromatography, followed by a Sephacryl S-100 column as previously described.27,31 Purified anfibatide exhibited a molecular weight of ∼30 kDa on a sodium dodecyl sulfate–polyacrylamide gel under denaturing but nonreducing conditions.27
TTP patient samples
The University of Alabama at Birmingham Institutional Review Board has reviewed and approved the protocols using human blood samples for the study. Whole blood (5 mL) was collected from a 41-year-old patient with acute TTP prior to the initiation of plasma exchange therapy. The blood sample was anticoagulated with sodium citrate (0.32%). The patient had a platelet count of 16.6 × 109/L, hematocrit of 29 g/dL, lactate dehydrogenase of 781 U/mL, and creatinine of 1.2 mg/dL. Plasma ADAMTS13 activity was <5% (normal, ≥67%) and inhibitor was <0.4 U/mL (normal, <0.4) but anti-ADAMTS13 IgG was 46 U/mL (normal, <18).
Ristocetin- or botrocetin-induced platelet agglutination
The University of Alabama at Birmingham Institutional Animal Care and Use Committee has reviewed and approved all studies involving animals. Murine platelets were isolated from anticoagulated (sodium citrate) whole blood obtained from Adamts13−/− mice by cardiac puncture after anesthesia with a ketamine (100 mg/kg) and xylazine (15 mg/kg) cocktail. Washed murine platelets (2 × 108/mL) in a modified Tyrode buffer (25 mM HEPES, pH 7.4 containing 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 10 mM glucose) were supplemented with mVWF (10 µg/mL) and the agglutination between platelets and mVWF was induced by addition of botrocetin (final concentration, 1.0 µg/mL). To assess the effect on platelet agglutination, purified anfibatide, at various concentrations, was added to the tube containing the washed murine platelets (2 × 108/mL) and mVWF (10 µg/mL) for 10 minutes, prior to initiation of platelet agglutination with botrocetin (1 µg/mL). The rate of platelet agglutination was determined by changes in the light transmission using the PAP-4 Platelet Aggregation Profiler (Bio/Data Corporation).
Microfluidic shear-based assay
BioFlux microfluidic channels (Fluxion Bioscience, Inc) were coated with fibrillar collagen (10 µg/mL) in 0.01 N hydrochloric acid under shear 500 s−1 for 10 minutes. The surface was blocked with 1% bovine serum albumin in phosphate-buffered saline (PBS). The platelets in the whole blood from the healthy donor (sodium citrate anticoagulated), the TTP patient’s whole blood (sodium citrate anticoagulated), and the Adamts13−/− mouse whole blood (PPACK [D-phenylalanyl-L-prolyl-L-arginine chloromethyl ketone] anticoagulated) were labeled with fluorescein isothiocyanate anti-human or anti-murine CD41 antibody and incubated with or without anfibatide, at various concentrations, for 15 minutes. The whole blood samples were then flowed under 50 dyne/cm2 over the collagen-coated surfaces for 3 minutes. The time-lapse digital images were collected every 3 seconds for a total of 3 minutes. The relative fluorescent intensity or platelet coverage was determined offline using Montage software and data were analyzed using GraphPad Prism 7 software.
Murine models of TTP
Complete blood counts were determined in all Adamts13−/− mice with and without Stx-2 challenge or treatment with anfibatide. Approximately 60 μL of whole blood was collected via retro-orbital puncture for analysis with the Hemavet 950 and Sysmex 200 hematology analyzer in EDTA or citrated anticoagulated whole blood, respectively. To trigger the TTP phenotype, Adamts13−/− mice (CAST/Ei strain) were given a single IV injection of Stx-2 (250 pg/g body weight) via the tail vein. Mice were then treated with PBS (control) or anfibatide (0, 30, 60, and 90 ng/g body weight) via intraperitoneal injection twice daily for 3 days immediately following Stx-2 challenge. The twice-daily schedule was based on the half-life of anfibatide determined in rats, which was 5 to 7 hours (data not shown).
Histology analysis
Major organ tissues (brain, heart, lung, kidney, and spleen) were obtained 4 to 6 hours post mortem after natural death or sacrifice at 3 days after Stx-2 injection, fixed in 4% paraformaldehyde in PBS, paraffin-embedded, sectioned, and stained with hematoxylin and eosin.
Statistical analysis
Statistical analysis was performed using the Student t test or analysis of variance using Prism 6 software. P values <.05 and <.01 were considered to be statistically significant and highly significant, respectively.
Results
Anfibatide inhibits botrocetin-induced platelet agglutination under stirring conditions
Although ristocetin has been used in vitro to induce agglutination of human platelets,32 botrocetin is the agent of choice for inducing conformational changes of murine VWF, which allows the murine VWF A1 domain to bind platelet GPIb with higher affinity.33,34 When purified anfibatide (0-12 μg/mL) is added to the washed murine platelets in the presence of mVWF (10 µg/mL) and botrocetin (1 µg/mL), the anfibatide inhibited platelet agglutination in a concentration-dependent manner. At the final concentration of 6 µg/mL and 9 µg/mL, anfibatide nearly abolished the platelet agglutination with mVWF (Figure 1A). As expected, anfibatide at its highest concentration (9 µg/mL) exhibited little to no inhibitory activity on PAR4 agonist (AYPGKH)-induced aggregation of washed murine platelets (Figure 1B). These results suggest that purified anfibatide can actively inhibit the interactions between murine VWF and platelets under stirring conditions.
Figure 1.

Effect of anfibatide on botrocetin-induced agglutination of murine platelets. Washed murine platelets (2 × 108/mL) were incubated with various concentrations (0, 3, 6, 9, and 12 μg/mL or 0 and 9 μg/mL) of purified anfibatide in the presence of mVWF (10 μg/mL). (A-B) The botrocetin (1 μg/mL) and PAR-4 agonist (AYPGKF) (500 μM) induced platelet agglutination, respectively. The change in light transmission (%) was recorded continuously for 3 minutes and plotted against the time. Each experiment was repeated 3 times. Only the representative images and tracing are shown.
Anfibatide inhibits platelet adhesion and aggregation on a collagen surface under shear
To assess the efficacy of anfibatide on VWF-platelet interaction under more physiological conditions, we performed microfluidic shear-based assays. Whole blood was obtained from Adamts13−/− mice, incubated with PBS (control) or anfibatide (1.5 µg/mL) in vitro, and flowed over a collagen-coated surface under arterial shear. The adhesion and aggregation of fluorescein-labeled platelets in anfibatide-treated blood samples were dramatically reduced (Figure 2A-B; supplemental Video 1). Also, when administered into Adamts13−/− mice via peritoneal cavity, anfibatide (60 ng/g body weight) at 6 hours postadministration (the maximal expected plasma concentration of anfibatide would be 0.2 µg/mL) appeared to have a more dramatic inhibitory activity on platelet adhesion and aggregation on the collagen surface under arterial flow (Figure 2C-D; supplemental Video 2). Similar inhibitory effects of anfibatide were observed when added to normal human whole blood and TTP patient whole blood (anticoagulated with sodium citrate). As shown, the surface coverage of fluorescein-labeled platelets from healthy individuals (Figure 3A-B) or a TTP patient (Figure 3C-D) under arterial shear in the presence of 1.5 μg/mL (or 0.75 μg/mL) anfibatide was dramatically reduced when compared side by side with the buffer controls. Together, these results demonstrated that anfibatide is a much more potent antithrombotic agent under physiological shear.
Figure 2.

Effect of anfibatide on murine platelet adhesion and aggregation on a collagen surface under flow. (A-B) Representative images of platelet coverage at the end of 3 minutes and the rate of platelet accumulation on a collagen surface over time (means/standard error of the mean [SEM], n = 4), respectively, after perfusion (50 dyne/cm2) of anticoagulated murine (Adamts13−/−) whole blood in the presence of PBS (buffer) or anfibatide (1.5 μg/mL). (C-D) Representative images of platelet coverage at the end of 3 minutes and the rates of platelet accumulation on a collagen surface over time (mean/SEM, n = 3), respectively, after perfusion of anticoagulated whole blood obtained from Adamts13−/− mice 6 hours after an intraperitoneal administration of anfibatide 60 ng/g body weight. The photomicrographs shown are 44% of the original size.
Figure 3.

Effect of anfibatide on human platelet adhesion and aggregation on a collagen surface under flow. (A-B) Representative images of platelet coverage and the rate of platelet accumulation on a collagen surface over time, respectively, after perfusion (50 dyne/cm2) of anticoagulated normal whole blood (n = 4) in the absence or presence of anfibatide (1.5 µg/mL). (C-D) Representative images of platelet coverage at the end of 3 minutes and the rate of platelet accumulation on a collagen surface, respectively, after perfusion (50 dyne/cm2) of an anticoagulated TTP patient’s whole blood, in the absence (buffer) or presence of 2 different concentrations (0.75 and 1.5 µg/mL) of anfibatide. The photomicrographs shown are 44% of the original size.
Anfibatide is efficacious in mitigating spontaneous thrombocytopenia in Adamts13−/− mice
Although Adamts13−/− mice (CAST/Ei) in a clean facility rarely developed thrombocytopenia, ∼30% of the same mice housed in our current animal facility developed mild to moderate spontaneous thrombocytopenia with occasional findings of fragmentation of red blood cells, but normal creatinine. This provided us an opportunity to determine whether anfibatide had a therapeutic effect on spontaneous thrombocytopenia associated with severe ADAMTS13 deficiency. Adamts13−/− mice were given a fixed dose (60 ng/g body weight) via intraperitoneal cavity twice daily for 3 days. As shown, mice with baseline platelet counts <400 000/µL exhibited significantly increased platelet counts 3 days after therapy and remained constant 4 days after cessation of therapy (Figure 4A). However, anfibatide at the same dosage had little or no effect in Adamts13−/− mice with baseline platelet count ≥400 000/µL (Figure 4B). Overall, 76% of mice showed an increase in platelet count from the baseline; 46% of mice increased by 30% and 24% of mice increased by 50% (Figure 4C) after anfibatide treatment despite ongoing severe deficiency of plasma ADAMTS13 activity. There appeared to be a nonlinear inversed correlation between the increment of platelet counts and the baseline platelet counts: the lower initial platelet counts, the greater the increase of platelet counts after therapy (Figure 4D). These results indicate the ongoing VWF-platelet interaction and low-grade platelet consumption in Adamts13−/− mice in which anfibatide may be effective.
Figure 4.

Effect of anfibatide in mitigating spontaneous thrombocytopenia in Adamts13−/−mice. (A-B) Platelet counts before (day 0 [D0]), during (day 3 [D3]), and after (day 7 [D7]) anfibatide therapy in mice with initial platelet counts <400 000/μL and ≥400 000/μL, respectively. (C) The percentage of mice with any increase, 30%, and 50% increase in platelet counts after anfibatide therapy. (D) The nonlinear correlation between platelet count increment on day 3 (D3) and day 7 (D7) after anfibatide therapy compared with the initial (day 0 [D0]) platelet counts. * and ** indicate that P values are <.05 and <.01, respectively. ns, No statistical significance (P >.05).
Anfibatide is efficacious in treating shigatoxin-induced TTP
As in humans, severe deficiency of plasma ADAMTS13 activity in most mice is not sufficient to cause an acute TTP episode.35 Stx2,35,36 or its B-subunit,37 has been used to stimulate the release of VWF from endothelial cells, thereby triggering an acute onset of TTP. Adamts13−/− mice (CAST/Ei strain) were challenged with Stx2 (250 pg/g body weight). The mice were then injected intraperitoneally with various doses (0, 30, 60, and 90 ng/g body weight) of purified anfibatide twice daily, for 3 days. As shown, 100% of Adamts13−/− mice that received the PBS control developed severe thrombocytopenia within 3 days (Figure 5A). Adamts13−/− mice that received the anfibatide treatment at a dose of 30 ng/g body weight showed little to no effect in preventing thrombocytopenia (Figure 5B). However, those that received anfibatide at the doses of 60 ng/g body weight (Figure 5C) and 90 ng/g body weight (Figure 5D) showed dramatically reduced rates of thrombocytopenia, defined by a 30% drop of platelet counts from their baseline. The Kaplan-Meier survival analysis showed the dose-dependent improvement of thrombocytopenia-free survival with the 60 ng/g anfibatide being the optimal dosage under these conditions (Figure 5E), despite a similar 3-day mortality rate (not shown). These results demonstrated that anfibatide, when administered at its optimal dosage, route, and interval, is efficacious, protecting Adamts13−/− mice from developing TTP-like phenotypes after a bacterial toxin challenge.
Figure 5.

Effect of anfibatide on shigatoxin-induced TTP in Adamts13−/−mice. Platelet counts in Adamts13−/− (CAST/Ei) mice treated with (A) PBS, (B) 30 ng/g body weight, (C) 60 ng/g body weight, and (D) 90 ng/g body weight, respectively, after being challenged with Stx-2 (250 pg/g body weight). (E) The thrombocytopenia-free survival over time. Here, thrombocytopenia was defined by 30% drop in platelet counts from the baseline. Statistical analysis was determined using analysis of variance (ANOVA) by Prism 7. **, ***, and **** indicate that P values are <.05, <.01, and <.001, respectively, when comparing with the platelet counts on day 0 (D0). ns, No statistical significance (P >.05).
Histological analysis revealed that Adamts13−/− mice without anfibatide treatment died of Stx-2–induced TTP; hyaline microvascular thrombi were found in multiple organ tissues including the brain, heart, lung, kidneys, and liver (Figure 6A-F). In the same mutant mice treated with anfibatide at 60 ng/g body weight, fewer occlusive thrombi in small arterioles were detected in all major organ tissues (Figure 6G-L). Together, these results demonstrated, for the first time, that anfibatide alone may be efficacious in preventing and treating murine models of TTP either triggered by bacterial toxin or occurring spontaneously.
Figure 6.

Histological analysis of mouse major organ tissues. Adamts13−/− mice (CAST/Ei) were treated with PBS or anfibatide (60 ng/g body weight) as indicated twice daily for 3 days after a single administration of Stx-2 (250 pg/g body weight). Major organ tissues from brain (A, G), heart (B, H), lung (C, I), liver (D, J), and kidney (E, K), as well as spleen (F, L) were collected from 3 mice in each group (either naturally died or sacrificed 3 days of the initial Stx-2 challenge) and processed for hematoxylin and eosin staining. Panels A-F are the control groups; panels G-L are the anfibatide-treated group here. Arrows indicate the presence of microvascular thrombosis. The images are representatives of all major organ tissues from 3 mice in each group. Original magnification ×200 for all panels.
Discussion
The present study demonstrates potent inhibitory activity of anfibatide purified from snake venom on ristocetin- or botrocetin-induced platelet agglutination under stirring conditions and on platelet adhesion and aggregation on a VWF/collagen surface under arterial flow. Most importantly, our results demonstrate the therapeutic efficacy of anfibatide in mitigating spontaneous thrombocytopenia and preventing shigatoxin-induced TTP in a murine model.
Unlike other platelet GPIb antagonists (eg, echicetin and flavocetin) which cause platelet agglutination rather than inhibition,38,39 anfibatide binds to platelet GPIb with high affinity (KD = 38 nM) but does not cross-link these receptors at the concentrations tested, thereby acting as a competitive inhibitor for the VWF-platelet GPIb binding.24 The inhibitory potency of anfibatide on shear-induced platelet agglutination in blood with severe ADAMTS13 deficiency is much greater (fivefold to 10-fold) than in blood with normal ADAMTS13 activity and on ristocetin- or botrocetin-induced platelet agglutination under stirring conditions. Approximately 0.75 μg/mL anfibatide may be sufficient to completely inhibit the adhesion and aggregation of both murine and human ADAMTS13-deficient platelets on a collagen surface under arterial flow. These results demonstrate the specific and potent inhibitory activity of purified anfibatide on platelet-VWF interaction under physiological shear.
Approximately 30% of our Adamts13−/− mice (CAST/Ei) housed in our current facility develop mild to moderate thrombocytopenia with occasional detection of fragmentation of red blood cells, consistent with ongoing spontaneous TTP. Administration of anfibatide at the dose of 60 ng/g body weight for 3 days results in an increase of platelet counts in 76% of mice, regardless of their initial platelet counts. The lower the initial platelet counts, the greater the platelet counts after anfibatide therapy. Such a therapeutic benefit appears to extend beyond the treatment period, suggesting that once the initial insult is over, TTP becomes quiescent. This phenomenon is also observed clinically in patients. Patients can stay in remission after initial therapy40 or remain asymptomatic for a long period of time41,42 despite ongoing severe deficiency of ADAMTS13 activity.
Most patients with severe deficiency of plasma ADAMTS13 do not develop acute TTP without additional triggers. Infection or flare of a systemic inflammatory disease often precedes the acute episode of TTP. To mimic the human disease, Adamts13−/− mice that had normal baseline platelet counts were challenged with a single dose of IV Stx-2. Nearly all Adamts13−/− mice developed severe thrombocytopenia the next day and worsened over 3 days, resulting in the formation of peripheral blood schistocytes, microvascular thrombosis in many major organ tissues, consistent with the development of acute TTP in these mice.35,36 Anfibatide exhibits a dose-dependent protective effect in this model with the optimal dose of 60 ng/g body weight at the twice-daily interval. Only 1 mouse in this treatment group failed to show a protective effect, likely due to the poor absorption of the drug from the intraperitoneal cavity. Also, it may be attributed by the toxicity of contaminated lipopolysaccharides in the commercial source of Stx-2. A 10% to 30% mortality rate has been observed with a single injection of Stx-2 in wild-type mice (normal ADAMTS13 activity).35 It is not clear why the effective dose range is narrow. We did not observe any intracranial or intra-abdominal bleeding complications in the few mice that died after treatment with anfibatide at the dose of 90 ng/g. The twice-daily schedule of anfibatide is critical for therapeutic efficacy because of its short half-life (t1/2 = 5-7 hours) determined in rats (data not shown) with a potential caveat of a minor difference in pharmacokinetics of anfibatide between rats and mice. Also, anfibatide, like anti-VWF nanobody caplacizumab,23 may be best used to treat acute TTP in conjunction with other therapies including plasma exchange, recombinant ADAMTS13 (if available), and immunosuppressives due to the fact that anfibatide does not correct underlying etiology which is the formation of autoantibodies against the ADAMTS13 protease.
We conclude that anfibatide can inhibit ristocetin or botrocetin-induced agglutination of purified human and murine platelets in the presence of VWF. Anfibatide can also efficiently inhibit shear-induced adhesion and aggregation of human and murine platelets on a collagen surface under arterial flow. When administered at the optimal dosage, route, and interval, anfibatide appears to be highly efficacious in treating murine models of TTP. Thus, anfibatide may be further developed as a novel therapy for acute TTP in humans, either alone or in conjunction with therapeutic plasma exchange and other therapeutic modalities.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank David Ginsburg from University of Michigan, Ann Arbor, Michigan for providing Adamts13−/− mice for the study.
This work was partially supported by Lee’s Pharmaceutical Holdings Limited, the Answering T.T.P. Foundation, and National Institutes of Health National Heart, Lung, and Blood Institute R01HL115187.
Authorship
Contribution: L.Z., Y.M., and X.L.Z. designed and performed research, interpreted data, and wrote the manuscript; M.S.A. and N.K.K. performed flow-based experiments, helped with mice, analyzed the data, and revised the manuscript; and M.L., X.D., and B.L. provided critical reagents and revised the manuscript.
Conflict-of-interest disclosure: X.L.Z. is a consultant for Alexion and Ablynx, receives research funding from Lee’s Pharmaceutical Holdings Limited, and provides services in the speaker’s bureau for Alexion. M.L. and B.L. are employees of Lee’s Pharmaceutical Holdings Limited and X.D. is an employee of Zhaoke Pharmaceutical. The remaining authors declare no competing financial interests.
Correspondence: X. Long Zheng, Division of Laboratory Medicine, Department of Pathology, The University of Alabama at Birmingham, WP P230K, 619 19th St South, Birmingham, AL 35233; e-mail: xzheng@uabmc.edu.
References
- 1.Rock GA, Shumak KH, Buskard NA, et al. ; Canadian Apheresis Study Group. Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med. 1991;325(6):393-397. [DOI] [PubMed] [Google Scholar]
- 2.Zheng XL, Sadler JE. Pathogenesis of thrombotic microangiopathies. Annu Rev Pathol. 2008;3:249-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai HM, Lian EC. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med. 1998;339(22):1585-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng XL. ADAMTS13 and von Willebrand factor in thrombotic thrombocytopenic purpura. Annu Rev Med. 2015;66:211-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy GG, Nichols WC, Lian EC, et al. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001;413(6855):488-494. [DOI] [PubMed] [Google Scholar]
- 6.Zheng X, Majerus EM, Sadler JE. ADAMTS13 and TTP. Curr Opin Hematol. 2002;9(5):389-394. [DOI] [PubMed] [Google Scholar]
- 7.Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100(12):4033-4039. [DOI] [PubMed] [Google Scholar]
- 8.Dong JF, Moake JL, Bernardo A, et al. ADAMTS-13 metalloprotease interacts with the endothelial cell-derived ultra-large von Willebrand factor. J Biol Chem. 2003;278(32):29633-29639. [DOI] [PubMed] [Google Scholar]
- 9.Moake JL, Rudy CK, Troll JH, et al. Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. N Engl J Med. 1982;307(23):1432-1435. [DOI] [PubMed] [Google Scholar]
- 10.Niiya M, Endo M, Shang D, et al. Correction of ADAMTS13 deficiency by in utero gene transfer of lentiviral vector encoding ADAMTS13 genes. Mol Ther. 2009;17(1):34-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell WR, Braine HG, Ness PM, Kickler TS. Improved survival in thrombotic thrombocytopenic purpura-hemolytic uremic syndrome. Clinical experience in 108 patients. N Engl J Med. 1991;325(6):398-403. [DOI] [PubMed] [Google Scholar]
- 12.Kremer Hovinga JA, Vesely SK, Terrell DR, Lämmle B, George JN. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115(8):1500-1511. [DOI] [PubMed] [Google Scholar]
- 13.Zheng X, Pallera AM, Goodnough LT, Sadler JE, Blinder MA. Remission of chronic thrombotic thrombocytopenic purpura after treatment with cyclophosphamide and rituximab. Ann Intern Med. 2003;138(2):105-108. [DOI] [PubMed] [Google Scholar]
- 14.Plaimauer B, Schiviz A, Kaufmann S, Höllriegl W, Rottensteiner H, Scheiflinger F. Neutralization of inhibitory antibodies and restoration of therapeutic ADAMTS-13 activity levels in inhibitor-treated rats by the use of defined doses of recombinant ADAMTS-13. J Thromb Haemost. 2015;13(11):2053-2062. [DOI] [PubMed] [Google Scholar]
- 15.Tersteeg C, Schiviz A, De Meyer SF, et al. Potential for recombinant ADAMTS13 as an effective therapy for acquired thrombotic thrombocytopenic purpura. Arterioscler Thromb Vasc Biol. 2015;35(11):2336-2342. [DOI] [PubMed] [Google Scholar]
- 16.Cataland SR, Wu HM. Acquired thrombotic thrombocytopenic purpura: new therapeutic options and their optimal use. J Thromb Haemost. 2015;13(suppl 1):S223-S229. [DOI] [PubMed] [Google Scholar]
- 17.Katler E, Weissmann G. Steroids, aspirin, and inflammation. Inflammation. 1977;2(4):295-307. [DOI] [PubMed] [Google Scholar]
- 18.Chapman KE, Odermatt A. Steroids: modulators of inflammation and immunity. J Steroid Biochem Mol Biol. 2010;120(2-3):67-68. [DOI] [PubMed] [Google Scholar]
- 19.Cataland SR, Peyvandi F, Mannucci PM, et al. Initial experience from a double-blind, placebo-controlled, clinical outcome study of ARC1779 in patients with thrombotic thrombocytopenic purpura. Am J Hematol. 2012;87(4):430-432. [DOI] [PubMed] [Google Scholar]
- 20.Jilma-Stohlawetz P, Gorczyca ME, Jilma B, Siller-Matula J, Gilbert JC, Knöbl P. Inhibition of von Willebrand factor by ARC1779 in patients with acute thrombotic thrombocytopenic purpura. Thromb Haemost. 2011;105(3):545-552. [DOI] [PubMed] [Google Scholar]
- 21.Callewaert F, Roodt J, Ulrichts H, et al. Evaluation of efficacy and safety of the anti-VWF nanobody ALX-0681 in a preclinical baboon model of acquired thrombotic thrombocytopenic purpura. Blood. 2012;120(17):3603-3610. [DOI] [PubMed] [Google Scholar]
- 22.Holz JB. The TITAN trial--assessing the efficacy and safety of an anti-von Willebrand factor nanobody in patients with acquired thrombotic thrombocytopenic purpura. Transfus Apheresis Sci. 2012;46(3):343-346. [DOI] [PubMed] [Google Scholar]
- 23.Peyvandi F, Scully M, Kremer Hovinga JA, et al. ; TITAN Investigators. Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med. 2016;374(6):511-522. [DOI] [PubMed] [Google Scholar]
- 24.Chen YL, Tsai IH. Functional and sequence characterization of agkicetin, a new glycoprotein Ib antagonist isolated from Agkistrodon acutus venom. offf2p4. Biochem Biophys Res Commun. 1995;210(2):472-477. [DOI] [PubMed] [Google Scholar]
- 25.Lei X, Reheman A, Hou Y, et al. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thromb Haemost. 2014;111(2):279-289. [DOI] [PubMed] [Google Scholar]
- 26.Li TT, Fan ML, Hou SX, et al. A novel snake venom-derived GPIb antagonist, anfibatide, protects mice from acute experimental ischaemic stroke and reperfusion injury. Br J Pharmacol. 2015;172(15):3904-3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng X, Qian Y, Liu Q, Li BX, Zhang M, Liu J. Purification, characterization, and cDNA cloning of a new fibrinogenlytic venom protein, Agkisacutacin, from Agkistrodon acutus venom. Biochem Biophys Res Commun. 1999;265(2):530-535. [DOI] [PubMed] [Google Scholar]
- 28.Gao Y, Ge H, Chen H, et al. Crystal structure of agkisacucetin, a Gpib-binding snake C-type lectin that inhibits platelet adhesion and aggregation. Proteins. 2012;80(6):1707-1711. [DOI] [PubMed] [Google Scholar]
- 29.Pickens B, Mao Y, Li D, et al. Platelet-delivered ADAMTS13 inhibits arterial thrombosis and prevents thrombotic thrombocytopenic purpura in murine models. Blood. 2015;125(21):3326-3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skipwith CG, Cao W, Zheng XL. Factor VIII and platelets synergistically accelerate cleavage of von Willebrand factor by ADAMTS13 under fluid shear stress. J Biol Chem. 2010;285(37):28596-28603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng X, Xu ZY, Liu QD, Li XM, Li XY, Liu J. Purification and characterization of a platelet agglutinating inhibiting protein (Agkisacutacin) from Agkistrodon acutus venom. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai). 2000;32(6):653-656. [PubMed] [Google Scholar]
- 32.Girma JP, Takahashi Y, Yoshioka A, Diaz J, Meyer D. Ristocetin and botrocetin involve two distinct domains of von Willebrand factor for binding to platelet membrane glycoprotein Ib. Thromb Haemost. 1990;64(2):326-332. [PubMed] [Google Scholar]
- 33.Maita N, Nishio K, Nishimoto E, et al. Crystal structure of von Willebrand factor A1 domain complexed with snake venom, bitiscetin: insight into glycoprotein Ibalpha binding mechanism induced by snake venom proteins. J Biol Chem. 2003;278(39):37777-37781. [DOI] [PubMed] [Google Scholar]
- 34.Fukuda K, Doggett TA, Bankston LA, Cruz MA, Diacovo TG, Liddington RC. Structural basis of von Willebrand factor activation by the snake toxin botrocetin. Structure. 2002;10(7):943-950. [DOI] [PubMed] [Google Scholar]
- 35.Motto DG, Chauhan AK, Zhu G, et al. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. J Clin Invest. 2005;115(10):2752-2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin SY, Xiao J, Bao J, Zhou S, Wright JF, Zheng XL. AAV-mediated expression of an ADAMTS13 variant prevents shigatoxin-induced thrombotic thrombocytopenic purpura. Blood. 2013;121(19):3825-3829. [DOI] [PMC free article] [PubMed]
- 37.Huang J, Motto DG, Bundle DR, Sadler JE. Shiga toxin B subunits induce VWF secretion by human endothelial cells and thrombotic microangiopathy in ADAMTS13-deficient mice. Blood. 2010;116(18):3653-3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Navdaev A, Dörmann D, Clemetson JM, Clemetson KJ. Echicetin, a GPIb-binding snake C-type lectin from Echis carinatus, also contains a binding site for IgMkappa responsible for platelet agglutination in plasma and inducing signal transduction. Blood. 2001;97(8):2333-2341. [DOI] [PubMed] [Google Scholar]
- 39.Xu G, Ulrichts H, Vauterin S, et al. How does agkicetin-C bind on platelet glycoprotein Ibalpha and achieve its platelet effects? Toxicon. 2005;45(5):561-570. [DOI] [PubMed] [Google Scholar]
- 40.Zheng XL, Kaufman RM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103(11):4043-4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujimura Y, Matsumoto M, Isonishi A, et al. Natural history of Upshaw-Schulman syndrome based on ADAMTS13 gene analysis in Japan. J Thromb Haemost. 2011;9(suppl 1):283-301. [DOI] [PubMed] [Google Scholar]
- 42.Lotta LA, Wu HM, Mackie IJ, et al. Residual plasmatic activity of ADAMTS13 is correlated with phenotype severity in congenital thrombotic thrombocytopenic purpura. Blood. 2012;120(2):440-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.