

Abstract
Although mechanisms of immune activation against liver ischemia reperfusion injury (IRI) have been studied extensively, questions regarding liver resident macrophages, i.e., Kupffer cells, remain controversial. Recent progress in the biology of tissue resident macrophages implicates homeostatic functions of KCs. This study aims to dissect responses and functions of KCs in liver IRI. In a murine liver partial warm ischemia model, we analyzed liver resident vs. infiltrating macrophages by fluorescence-activated cell sorting (FACS) and immunofluorescence staining. Our data showed that liver immune activation by IR was associated with not only infiltrations/activations of peripheral macrophages (iMØ), but also necrotic depletion of KCs. Inhibition of Receptor Interacting Protein 1 (RIP1) by necrostatin-1s protected KCs from ischemia-induce depletion, resulting in the reduction of iMØ infiltration, suppression of pro-inflammatory immune activation and protection of livers from IRI. The depletion of KCs by clodronate-liposomes abrogated these effects of Nec-1s. Additionally, liver reconstitutions with KCs post-ischemia exerted anti-inflammatory/cytoprotective effects against IRI. These results reveal a unique response of KCs against liver IR, i.e., RIP-1-dependent necrosis, which constitutes a novel mechanism of liver inflammatory immune activation in the pathogenesis of liver IRI.
Introduction
Liver ischemia reperfusion injury (IRI) is one of the primary cause of organ dysfunction and failure after surgery, including tumor resection and transplantation. The pro-inflammatory immune response is the driving force of the disease pathogenesis (1–3). Prolonged ischemia causes initial tissue damages to release danger associated molecular pattern, which activates innate immune cells via pattern recognition receptors, such as Toll-Like-Receptor (TLR) 4. The resultant tissue inflammation promotes reperfusion injury. Major progress has been made in the last decade in our understanding of the molecular mechanisms of liver innate immune activation. However, cellular basis of liver inflammation in IRI has been ambiguous, particularly regarding roles of liver resident macrophages, Kupffer cells (KCs). Both pro- and anti-inflammatory properties have been associated with these liver macrophages (4–6).
Liver is a unique organ in our body with high content of resident macrophages in the homeostatic state. KCs have been assumed to be the major responding cells against IR. However, majority of studies measures liver inflammatory responses only at the tissue level without cell type-specific analysis. Liver IRI is characterized by the infiltration of large amounts of circulating monocyte-derived macrophages. The question of whether and how tissue resident vs. infiltrating macrophages are involved in liver IRI has yet to be fully addressed. As liver resident KCs are CD11b negative and relatively radiation-resistant (7, 8), experiments using bone marrow chimeras or CD11b- diphtheria toxin receptor mice document functions of infiltrating, but not resident, macrophages (9–11). Although clodronate-liposomes target mainly KCs, effects of KC depletion on liver IRI have been controversial in literature (4–6). Most importantly, these studies focused primarily on the outcome of liver inflammation/injury following KC depletion. The actual response of KCs vs. infiltrating macrophages in the disease process was not defined. The recent seminal finding in macrophage biology reveals that tissue resident and monocyte-derived macrophages are distinctive in their lineages and functions (12–17). Tissue resident macrophages are embryonically derived from yolk sac and play fundamental roles in tissue homeostasis (13–15). This further prompts us to differentially define functional roles of KCs vs.iMΦs in liver IRI.
In current study, we employed both FACS and immunofluorescent staining to analyze liver macrophages in vitro and in situ at different stages of liver IRI. Distinctive responses of KCs vs. iMΦs were identified and functional significance of KC necrosis in response to liver IR was determined.
Materials and Methods
Animals
Male wide-type (WT) C57BL/6 mice (6–8 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME), and housed in the UCLA animal facility under specific pathogen-free conditions, and received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institute of Health.
Mouse liver partial warm ischemia model
After a midline laparotomy, mice were injected with heparin (100 μg/kg) and an atraumatic clip was used to interrupt arterial/portal venous blood supply to the cephalad-liver lobes. After 90 min of ischemia, the clip was removed initiating hepatic reperfusion. Sham controls underwent the same procedure, but without vascular occlusion. Mice were sacrificed after 0h or 6h or 24h post-reperfusion, and liver and serum samples were collected. 7-Cl-O-Nec-1 (Nec-1s, 4mg/kg) or zDEVD-fmk (3.3mg/kg) was administered 1 hour prior to the ischemia, i.p.(EMD Millipore, Billerica, MA); Macrophage Depletion Kit (Encapsula NanoSciences LLC, Brentwood, TN) was used to deplete KCs according to manufacturer’s protocols. In brief, 100–200μl/mouse Clodronate encapsulated liposomes or control liposomes were injected i.p., 48 hours prior to the onset of liver ischemia.
Serum alanine aminotransferase (sALT) levels were measured with an autoanalyzer by ANTECH Diagnostics (Los Angeles, CA). Part of liver specimens was fixed in 10% buffered formalin and embedded in paraffin. Liver sections (4μm) were stained with hematoxylin and eosin (HE). The severity of liver IRI was graded blindly using Suzuki’s criteria on a scale from 0 to 4. No necrosis, congestion/ centrilobular ballooning is given a score of 0, while severe congestion and >60% lobular necrosis are given a score of 4.
Liver NPC/Kupffer cell isolation
Liver NPC/KCs were isolated from normal or IR livers of B6 mice by in situ collagenase perfusion. In brief, livers were perfused via the portal vein with calcium- and magnesium-free HBSS supplemented with 2% heat-inactivated FBS, followed by 0.27% collagenase IV (Sigma, St Louis, MO). Perfused livers were dissected, and teased through 70 μm nylon mesh cell strainers (BD Biosciences, San Diego, CA). Non-parenchymal cells (NPCs) were separated from hepatocytes by centrifuging at 50g for 2 min three times. NPCs were stained with fluorescence-labeled Abs and analyzed by FACS.
To purify KCs, NPCs were suspended in HBSS and layered onto a two-layer 25%–50% Percoll gradient (Sigma-Aldrich, St. Louis, MO) in a 50-ml conical centrifuge tube and centrifuged at 1800g at 4°C for 15 min. KCs in the middle layer were collected and allowed to attach to cell culture plates in DMEM with 10% FBS, 10 mM HEPES, 2 mM GlutaMax, 100 U/ml penicillin, and 100 mg/ml streptomycin for 15 min at 37°C. Nonadherent cells were removed by replacing the culture medium. These cells were trypsinized and washed twice with cold PBS and injected immediately in mice: 2×106 cells were injected into ischemic livers through portal vein prior to the onset of reperfusion. The purity of KCs in the adherent cells was determined by immunofluorescent staining with anti-F4/80 (Supplement Fig.1). 80–90% adherent cells were F4/80 positive.
Flow Cytometric Analysis
Liver non-parenchymal cells (NPCs) were isolated from sham or IR livers, as described above. 1×106 cells were incubated with purified rat anti-mouse CD16/32 for 10 minutes and stained with rat anti-mouse F4/80-PeCy5/PE, CD11b-FITC or -APC, Gr1-FITC (Clone: RB6–8C5), CD40-PE and isotype-matched negative control antibody (eBioscience, San Diego, CA) were added to the cell suspension. After 20 minutes of incubation in the dark, the cells were washed with PBS and subjected to flow cytometric analysis with FACS Calibur (BD Biosciences). For intracellular staining of CD206 and iNOS, cells were fixed in 4% formaldehyde for 20 minutes after the staining of F4/80 and CD11b, and washed twice with 1×permeabilization buffer (eBioscience, San Diego, CA). After incubation with CD206-APC (Biolegend, San Diego, CA) and iNOS-PE (eBioscience, San Diego, CA) in 1×permeabilization buffer for 20 minutes in the dark, the cells were washed with PBS and subjected to flow cytometric analysis.
Immunofluorescence staining
Liver samples were embedded in TissueTek OCT compound (Electron Microscopy Sciences, Japan). Five micrometers of the frozen section were fixed in 4% paraformaldehyde and blocked with 2% bovine serum albumin, then incubated with anti-mouse F4/80- eFluor® 570, or anti-mouse CD11b Alexa Fluor® 488 (eBioscience, San Diego, CA) for 2 hours at room temperature. After washing the sections were covered with Vectashield mounting medium containing DAPI and observed under a microscope (Leica M165 FC). In a blinded fashion, liver sections were first observed under low power (40×), 10–20 representative areas were selected and positive cells were counted under high power (×400). Quantitation was based on average numbers of positive cells/high power field. 3 mice/group/time point were analyzed.
To stain LSECs, liver sections (5μm) were stained with rat anti-mouse CD31 antibody (BD Biosciences, San Jose, CA) for 2 hours at room temperature, followed by FITC -labeled goat anti-rat IgG (Vector, Burlingame, CA). DAPI (blue) was used for nuclear counterstaining.
Quantitative RT-PCR
Total RNA (2.0μg) was reverse-transcribed into cDNA using SuperScriptTM III First-Strand Synthesis System (Invitrogen, Carlsbad, CA). Quantitative-PCR was performed using the DNA Engine with Chromo 4 Detector (MJ Research, Waltham, MA). In a final reaction volume of 20 μl, the following were added: 1xSuperMix (Platinum SYBR Green qPCR Kit, Invitrogen, Carlsbad, CA), cDNA and 0.5 mM of each primer. Amplification conditions were: 50°C (2 min), 95°C (5 min) followed by 50 cycles of 95 °C (15 s), 60 °C (30 s). The primers for mouse gene fragments, including TNF-a, IL-1b, IL-6, IL-10, IL-12p40 were as described previously (18). Additional primers to study LSECs are the followings: von Willebrand factor (vWF) F: GGGTTTTCTCTCCCTGGCTC; R: ACAGAGCCACAAAGGA CTCG; CD31 F:CAAGGCCAAACAGAAACCCG; R:TCGACCTTCCGGATCTCACT; vascular cell adhesion molecule 1 (VCAM-1) F:CTGGGAAGCTGGAACGAAGT; R:GCCAAACACTTGACCGTGAC.
Western Blots
Tissue or cellular proteins were extracted with ice-cold lysis buffer (1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 10% glycerol, 137mM sodium chloride, 20mM Tris, pH 7.4). Proteins (20 ug) were subjected to 12% SDS-PAGE electrophoresis and transferred to PVDF nitrocellulose membrane. Antibodies against RIP1 and RIP3 (Novus Biologicals, LLC, Littleton, CO) were used for Western blot analysis. Membranes were probed with primary antibody in 10 ml blocking buffer with gentle agitation overnight at 4°C. After washing, membranes were further probed with an appropriate HRP-conjugated secondary antibody (Cell Signaling Technology, San Diego, CA) in 10 ml of blocking buffer for 1 h at room temperature. SuperSignal® West Pico Chemiluminescent Substrates (Thermo Fisher Scientific, Rockford, IL) were used for chemo-luminescence development.
Cell Cultures
Bone marrow-derived macrophages (BMMs), obtained from femoral bones of 6–10-week old C57B/6 mice, were cultured in DMEM w/ 10% FBS and 20% L929 conditioned medium for 6–7 days. The cell purity was assayed to be 94–99% CD11b+. BMMs were trypsinized and washed twice with cold PBS. 2×106 BMMs were injected into ischemic livers similarly as above.
BMMs were stimulated with LPS (1μg/ml, Invivogen, San Diego, CA) in the absence or presence of caspase inhibitor zVAD-fmk (50μM), or Nec-1s (10μM). Culture supernatants were taken at 6–24h post- stimulation for measurements of LDH (Biochain Institute, Hayward, CA) and cytokines.
ELISA
Cytokines (TNF-α, IL-6, and IL-10) levels in cell culture supernatants or serum was measured by ELISA, according to manufacturer’s standard protocols (eBioscience, San Diego, CA). OD was measured in a Multiscan FC plate reader and analyzed with SkanIt for Multiscan FC software (Thermo Scientific).
Statistical Analysis
Results are shown as mean±SD. Statistical analyses were performed using unpaired Student’s t-test with p<0.05 (two-tailed) considered as significant.
Results
IR triggers distinctive changes in KCs vs. infiltrating macrophages
We isolated liver macrophages from both normal (sham) and IR livers at 0h or 6h post- reperfusion (peak of liver injury and immune response) and performed FACS analysis to determine their compositions and functional status. These cells were separated into resident and infiltrating subsets, based on F4/80 and CD11b staining (Fig.1a). The relative % of KCs/iMՓs in the whole liver macrophage population (F4/80+) was calculated to quantitate their changes during the course of IR (Fig.1b, n=3–4/time point). IR triggered a significant increase in iMՓs (67.7±0.5% vs. 2.2 ±1.2% in sham), as well as a drastic decrease in KCs (32.3±0.5% vs. 97.5 ±1.3% in sham) in livers at 6h post-reperfusion when liver inflammatory immune activation peaked in our model (18). The neutrophil population with F4/80−CD11b+ phenotype was absent in sham livers, and increased in IR livers at 6h reperfusion (Fig.1b).
Figure 1.
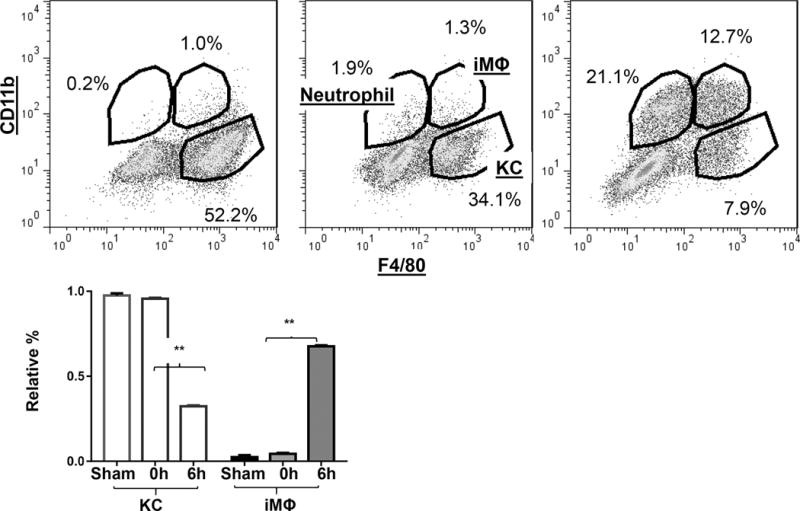
Dynamic changes of KC/macrophage subsets in liver IRI. NPCs were isolated from sham liver, or ischemic livers after 0h or 6h of reperfusion, as described in the Material and Methods. Cells were stained with fluorochrome-conjugated anti-F4/80, -CD11b. Granulocytes and monocytes were gated first based on their sizes and granularities, and then separated into resident and infiltrating subsets based on F4/80 and CD11b staining (a) F4/80 positive and CD11b negative cells were identified as KCs, F4/80 and CD11b double-positive were infiltrating MΦs and F4/80 negative and CD11b positive cells were neutrophils. Quantitation of dynamic changes of liver macrophage subsets in IR were plotted (b). Average percentages of KCs and iMΦs in total F4/80+ liver macrophages were calculated in sham, or ischemic livers after 0h or 6h of reperfusion based on FACS analysis. Representative of 3 independent experiments; n=3–6/time point. *p<0.05
As we calculated the relative frequencies of KCs and iMՓs in the total liver macrophage population, the decrease of KC percentages could result from a massive increase of iMՓs, rather than their physical “depletion”. To further examine IR-induced alterations in liver macrophage compositions, we performed immunofluorescence staining of liver sections with anti-F4/80 and -CD11b antibodies. As shown in Fig.2a, b, numbers of F4/80+ cells in ischemic livers at 6h post-reperfusion were significantly lower than those in sham controls. In contrast, numbers of CD11b+ cells (include both iMՓ and neutrophils) increased significantly in the same ischemic liver sections, as compared with those of sham. These results, derived from both FACS and immunofluorescence staining, reveal that liver macrophage subsets underwent distinctive changes in response to IR, that both iMՓ infiltration as well as KC depletion were associated with liver immune activation.
Figure 2.
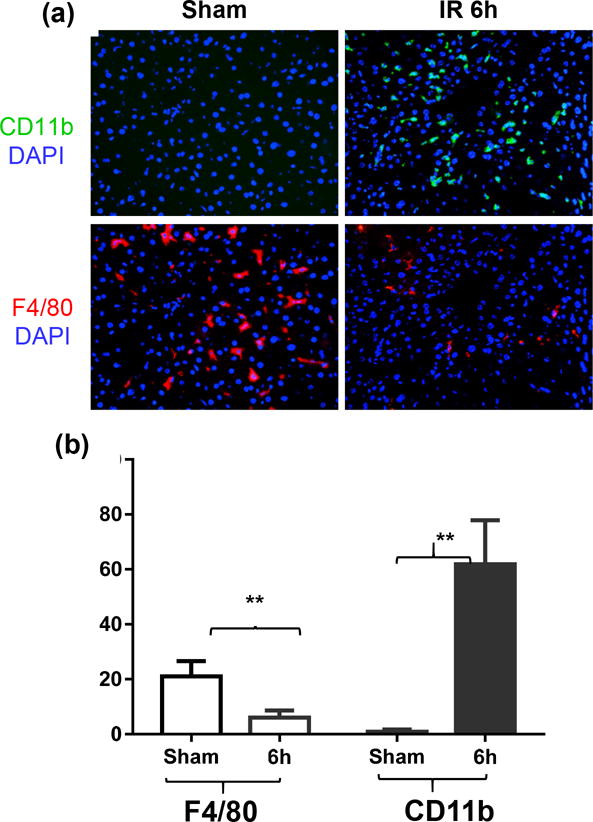
Immunofluorescence staining of liver macrophages. Frozen liver tissue sections were stained with fluorochrome-labeled anti-F4/80 or –CD11b and DAPI as described in the Materials and Methods. (a) Representative images (×400) of sham, IR livers at 6h post-reperfusion were shown. Positively stained cells were counted in at least 10 fields in each tissue section and (b) average numbers/field in each group of tissue section were charted. **p<0.01
Liver IR induces KC necrotic depletion
To determine mechanisms of KC depletion by IR, we analyzed cell death in KCs after 90 min. of ischemia prior to the onset of reperfusion. Liver macrophages at this time point were mostly KCs, similar to those in sham controls (Fig.3a). However, percentages of necrotic KCs, detected by cell-viable dyeTopro3-positive staining, were significantly increased (Fig.3a, b, 58.27±1.62% in ischemic livers, vs. 35.13±2.78% in sham, n=4/group, p<0.01). Transient liver ischemia for 30m did not induce significant increases in necrotic KCs (data not shown). To determine whether necroptosis was involved in ischemia-induced KC death, we measured expression levels of receptor-interacting protein 1 and 3 (RIP1/3) by Western blotting in KCs isolated from sham and ischemic livers. Clearly, ischemia (30m to 90m) triggered an upregulation of RIP1/3 expressions in KCs (Fig.3c). Functionally, necroptosis inhibitor Nec-1s (19, 20), the second generation of necrostatin with higher in vivo specificity and stability, was administered in test animals prior to the onset of liver ischemia. Compared with vehicle controls, pre-treatment with Nec-1s resulted in significant reductions of Topro3-positive KCs in ischemic livers at 0h of reperfusion (32.7±4.54%, Fig.3a, b). The protection of KCs from ischemia-induced necrosis led to alterations in macrophage compositions at 6h post-reperfusion: the relative percentage of KC increased (86.7% vs. Ctl. 51.7%) and that of iMՓs decreased (13.3% vs. Ctl. 48.3%), as well as inhibitions of neutrophil infiltration (Fig.3a). Importantly, liver inflammatory immune activation was suppressed: pro-inflammatory gene inductions, including TNF-α, IL-1β, IL-6, IL-12p40 and CXCL10, were reduced, while anti-inflammatory IL-10 induction was enhanced (measured by qRT-PCR, Fig.4a). All of these resulted in the protection of livers from IRI (lower sALT levels and better preserved liver architectures with lower Suzuki scores at both 6h and 24h post reperfusion, Fig.4b). As iMՓs were present only after IR, necrosis was not detected in this liver macrophage subset, Topro3-positive iMΦs in IR-livers were similar to those KCs from sham livers, and were not affected by the Nec-1s treatment (data not shown). As a control, caspase 3-specific inhibitor zDEVD-fmk was also tested in a separate group of mice. Liver IRI was in fact aggravated by the caspase inhibitor (Fig.4a).
Figure 3.
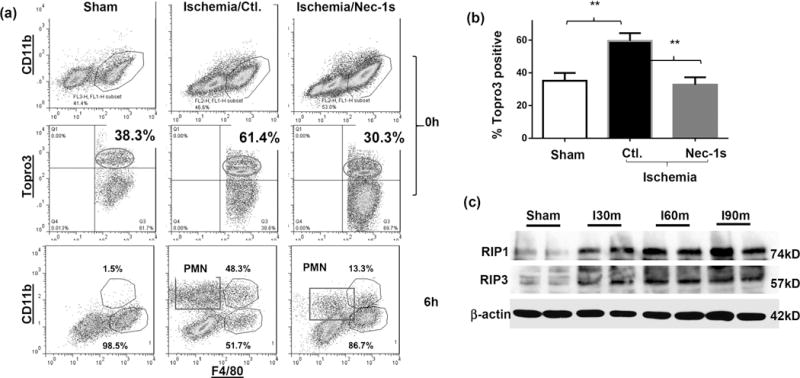
KC necroptosis in liver IRI. Groups of B6 mice were treated with either vehicle or Nec-1s, prior to the onset of liver ischemia, as described in the Materials and Methods. (a) Liver macrophages were isolated from different groups of mice without the attachment step. Total NPCs were stained with CD11b-FITC, F4/80-PE, Topro3. Topro3-positive KCs (F4/80+CD11b-) in sham or ischemic livers at 0h post-reperfusion were analyzed, as well as liver macrophage subsets and neutrophils in sham or ischemic livers at 6h post-reperfusion. (b) Average percentages of Topro3-positive KCs in sham, vehicle- or Nec-1s treated ischemic livers at 0h post-reperfusion were plotted. (c) Western blot analysis of RIP1 and 3 expressions in KCs. KCs were isolated from sham or ischemic livers at 0h post-reperfusion (after 30m or 60m or 90m ischemia). Cellular proteins were prepared and separated by SDS-PAGE, followed by Western blotting with anti-RIP1/3 Abs. Representative results of 2 independent experiments; n=3–6/group, **p<0.01
Figure 4.
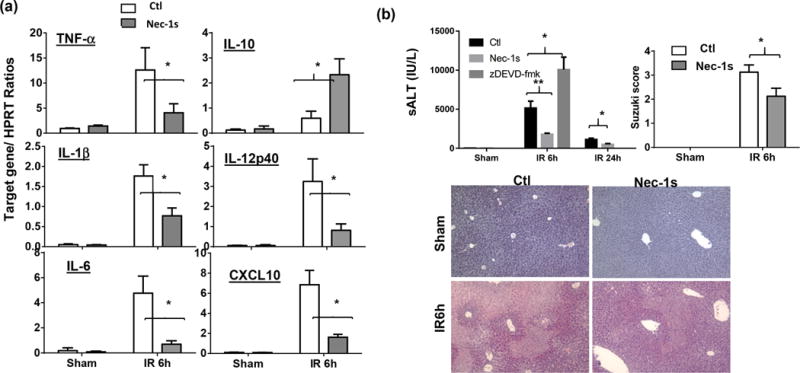
Nec-1s inhibits liver inflammatory immune activation and protects livers from IRI. Groups of B6 mice were treated with either vehicle or Nec-1s, prior to the onset of liver ischemia, as described in the Materials and Methods. (a) Liver inflammatory gene inductions by IR (6h post-reperfusion) in different groups of mice were measured by qRT-PCR. Average ratios of target gene/HPRT were plotted. (b) Liver IRI in different groups of mice (treated with vehicle or Nec-1s or zDEVD-fmk) were measured at 6h and/or 24h post-reperfusion by sALT levels, liver histological analysis with Suzuki scores. Representative results of 2 independent experiments; n=3–6/group, *p<0.05, **p<0.01
To determine cellular targets of the anti-inflammatory/cytoprotective effect of Nec-1s in liver IRI, we treated mice with a single dose of clodronate-liposomes 48h. prior to the onset of liver ischemia to deplete KCs/macrophages. Liver macrophages, inflammatory immune activation, and IRI were analyzed at 6h post-reperfusion. FACS results showed that clodronate preferentially depleted KCs in livers. Relative percentages of iMՓs, vs. KCs at 6h of reperfusion increased significantly in IR livers of clodronate-liposome treated mice, as compared with those of blank-liposome treated ones (91.9% vs. 65.8%, Fig.5a). This led to an increase of liver IRI (Fig.5b, c. sALT and liver histological analysis with Suzuki scores) and elevated pro-inflammatory, but suppressed anti-inflammatory IL-10, gene inductions in these animals (Fig.5d. qRT-PCR). Importantly, the Nec-1s treatment was no longer able to protect livers from IRI, or inhibit liver inflammatory immune response in these KC-depleted mice (Fig.5b, c, d). The chlodronate dose we used did not deplete liver sinusoidal endothelia cells (LSEC). Immunofluorescent staining (anti-CD31) showed intact LSEC structures (central/portal veins and sinusoids) in treated livers; and RT-PCR analysis documented similar expression levels of CD31, VCAM and Von Willebrand Factor (vWF) in vehicle and clodronate-treated livers pre- and post-reperfusions (Supplement Fig.2). These results indicate that Nec-1s protected livers against IRI in KC-dependent manner. KCs, rather than parenchymal cells, underwent Nec-1s-dependent necrosis which was critical for the liver inflammatory immune activation against IR.
Figure 5.
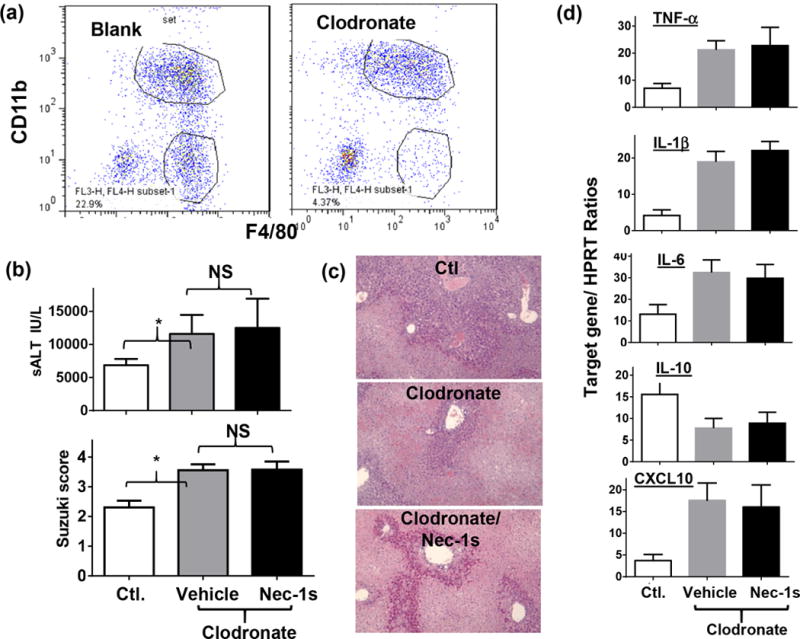
KC depletion and Nec-1s effects in liver IRI. Groups of mice were treated with either control- or clodronate-liposomes to deplete KCs. Vehicle or Nec-1s was administered 1h prior to the onset of liver ischemia, as described in the Material and Methods. Liver macrophages, IRI and inflammatory gene inductions were measured after reperfusion. (a) Liver macrophage constitutions were analyzed at 6h post reperfusion by FACS. (b) Liver IRI was evaluated at 6h post reperfusion by sALT and liver histological analysis with Suzuki scores. (c) Liver inflammatory gene expressions were measured by qRT-PCR. Target gene/HPRT ratios were plotted. Representative results of 2 independent experiments; n=3, *p<0.05
KC reconstitution protects ischemic livers from IRI
To directly test the potential KC’s homeostatic function in liver IRI, we reconstituted mice with exogenous KCs after liver ischemia prior to the onset of reperfusion. As controls, bone marrow-derived macrophages (BMMs) were also used in a separate group of mice. Liver inflammation and IRI were evaluated at 6h post-reperfusion. Indeed, KC reconstituted mice were protected from liver IRI, as shown by reduced sALT levels and better-preserved liver architecture with lower Suzuki scores (Fig.6a). Furthermore, these exogenous KCs inhibited liver pro-inflammatory immune response against IR, as documented by lower levels of pro-inflammatory, but a higher level of IL-10, gene induction in livers measured by qRT-PCR (Fig.6b). Reconstitution of ischemic livers with BMMs, on the other hand, increased liver IRI and enhanced local inflammatory gene inductions (Fig.6a, b). These results indicate that KCs, but not BMMs, were indeed capable of protecting livers from inflammatory tissue injury against IR.
Figure 6.
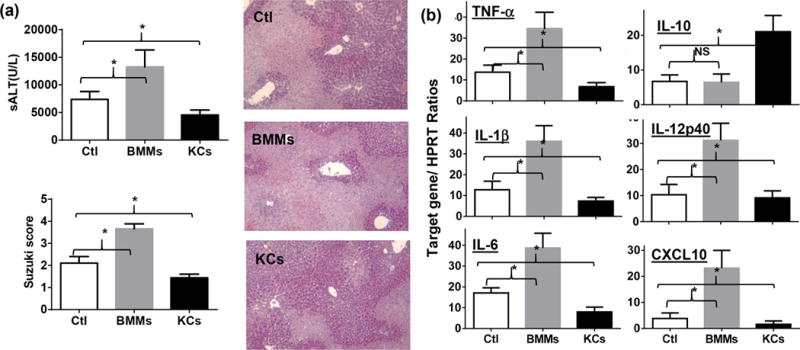
KC reconstitution in liver IRI. KCs and BMMs were obtained as described in the Materials and Methods. These cells were injected in separate groups of mice via portal vein after liver ischemia prior to the onset of reperfusion. Liver IRI was evaluated at 6h post-reperfusion. (a) Serum ALT and liver histological analysis with Suzuki scores. (b) Liver inflammatory gene expressions measured by target gene/HPRT ratios. Representative results of 2 independent experiments; n=3–4/group, *p<0.05
Discussion
Our study documented for the first time that liver IR triggered necrotic depletion of liver resident macrophages, which occurred in parallel with infiltration/activations of circulating monocyte-derived macrophages, to facilitate liver pro-inflammatory immune activation against IR. Nec-1s protected KCs from IR-induced necrosis, leading to diminished liver inflammatory immune activation and reduced liver IRI. We provided direct evidence in support of homeostatic functions of KCs that intrahepatic infusion of exogenous KCs post-ischemia inhibited liver immune response and protected livers from IRI. Thus, despite that total numbers of liver macrophages were increased during IR, resident vs. infiltrating macrophages had very distinctive fates. Opposite to what we had been assumed, liver resident macrophages underwent inflammatory cell death in response to IR, which disrupted tissue homeostatic balance and facilitated inflammatory macrophage/neutrophil infiltration/activation. This novel mechanism of liver IRI adds a new aspect to our understanding of tissue innate immune activation that tissue resident macrophage depletion may in fact promotes inflammation.
Roles of KCs in liver IRI have been controversial. Studies using clodronate-liposomes (CL) to deplete KCs have shown both aggravation of liver IRI with enhancement of liver inflammatory immune response (5, 6), as well as protection of livers from IRI (4). The expression of heme oxygenase-1 (HO-1) and production of IL-10 have been proposed as mechanisms of KC-mediated liver protection (5, 6). We have observed in our study that IR condition and CL doses/timing were two key factors in determining effects of CL treatments in liver IRI. As KCs were in fact depleted by prolonged but not transient liver IR, CL would not do much to the already depleted pool after prolonged ischemia. However, it would help to eliminate KCs from transient ischemic livers and aggravate liver inflammation and injury. Depending on the dose and timing, CL could impact types and activation of iMՓs, due possibly to the depletion of peripheral or bone marrow monocytes/macrophages (21) (data not shown). This would reduce liver inflammation and injury in the prolonged ischemia condition. In our current study, the prolonged ischemia resulted in the depletion of approximately 60% KCs. The lower dose of CL and 48h prior to the start of liver ischemia preferentially depleted KCs, but not iMΦs. Therefore, the CL treatment resulted in the deterioration of liver IRI. Importantly, our calibrated experimental setting enabled us to determine the functional mechanism and significance of KC necrosis in response to liver IR, which facilitated iMΦ accumulation in livers post reperfusion. Thus, liver infiltration/activation of peripheral macrophages is inhibited by functional KCs.
KCs are heterogeneous (7, 8, 22, 23). Although their responses, as a whole, to innate immune stimulation are relatively more immune regulatory than those of BMMs (higher IL-10/lower TNF-α, data not shown), KCs are capable of producing pro-inflammatory cytokines/chemokines (24). As KC depletion by IR is partial, it remains to be determined whether and how residual KCs are activated and functioning in inflammatory response against IR. We have shown in a previous study that KCs isolated from ischemic livers did respond differently from those in sham livers by selectively increasing pro- but decreasing anti-inflammatory cytokine productions (24). Only prolonged (90m), but not transient (30m), ischemia triggered KC necrotic depletion. This is correlated with the level of liver pro-inflammatory immune activation and hepatocellular injury, as 30m liver ischemia causes minimal IRI (sALT at 6h post reperfusion, 142.8±22.6 vs. 7079±1132 after 90m ischemia and 6h reperfusion, n=4, p<0.0001) and little inflammatory gene induction (25).
KC depletion has been reported in an acute liver injury model. An overdose of N-acetyl-p-aminophenol triggered a significant reduction in the frequency of resident KCs along with massive infiltration of circulating monocytes (26). Drug-induced direct cytotoxicities were probably responsible for KC death. However, mechanisms and functional significance of KC depletion in liver inflammation and injury were not studied in the model. Macrophage necroptosis was found during S. Typhimurium infection and constituted a key immune evading mechanism of the intracellular bacterial (27). KC necroptosis has been found recently to play key roles in orchestrating type-I microbicidal inflammation in Listeria infected livers (28). Type I interferon signaling pathway was critically involved in the death of bacteria-infected macrophages/KCs (27, 29, 30), as well as in inflammatory macrophage necroptosis in vitro (31). Either type I interferon receptor deficiency, or RIP1 inhibition or RIP3 knock-down/deficiency prevented this type of macrophage death, improved control of bacteria and prolonged survival of infected mice in vivo (27). Our study is the first in a non-infectious disease model to document the functional significance of RIP1-dependent necrosis of KCs.
Although we have detected upregulations of RIP1 and RIP3 in KCs from ischemic livers, details of their activation mechanisms remain to be elucidated. Inflammatory stimuli, including TNF-α, TLRs and interferon, are classical inducers of macrophage necroptosis and clearly relevant in liver IRI. Metabolic stress due to ischemia is another possible trigger (32). Oxidative stress and ER stress are both implicated in programed necrosis mediated by both RIP1/3 dependent and independent pathways (32, 33). We have shown that ER stress plays a critical role in both hepatocellular damages and inflammatory immune activation in our model (34, 35). We have tested roles of ER stress in KC depletion by using chemical chaperon 4-PBA to pretreat mice and preliminary results did show protections of these tissue resident macrophages from necrosis (data not shown). Inflammatory and metabolic stress signals can trigger apoptosis via extrinsic and intrinsic pathways, respectively. As ischemia deprives organs of oxygen and nutrients, ATP-dependent apoptosis is inhibited early post-reperfusion (36–38). Programed necrosis, as a caspase-independent regulated cell death mechanism, may become the alternative death response against IR. Necroptosis has been shown as a major cellular injury mechanism in parenchymal cells of several organ types in their response to IR (19, 39–43). Its contribution to inflammatory immune activation has been hypothesized to be indirect by the release of DAMPs (44). Our current study provides a unique case that necrosis directly regulates innate immune activation in vivo by both selectively depleting homeostatic tissue resident macrophages and augmenting inflammatory macrophage TLR activation. Our data that Nec-1s failed to protect livers in the absence of KCs implicated that liver parenchymal cells were damaged via RIP1-independent mechanism by IR. This cell-type specific differences in necroptotic pathways have been noticed in an in vitro study that RIP1 is required only for macrophages, but not fibroblasts, in their necroptosis upon TLR stimulation (45).
In summary, our differential analysis of liver macrophage subset reveals the distinctive fate of KCs and their functional role in the pathogenesis of liver IRI. The finding of KC homeostatic function and of their depletion mechanism by IR not only furthers our understanding of the disease mechanism but also provides us a novel therapeutic rationale to ameliorate liver IRI, i.e., preservation of liver resident macrophages from inflammatory cell death.
Supplementary Material
Acknowledgments
Grant Support: The study is supported by NIH Grants R21AI126516 (YZ), The Dumont Research Foundations; and grants from National Nature Science Foundation of China 81100270, 1310108001, 81210108017 (XW)
List of abbreviations
- IRI
Ischemia reperfusion injury
- KC
Kupffer cell
- FACS
Fluorescence-activated cell sorting
- RIP1/3
Receptor-interacting protein 1/3
- iMՓ
Infiltrating macrophage
- BMM
Bone marrow derived macrophage
- TLR4
Toll-like-receptor 4
- Nec-1s
necrostatin 1 analogue or 7-Cl-O-necrostatin 1
- sALT
Serum alanine aminotransferase
- H/E
hematoxylin and eosin
- NPCs
Non-parenchymal cells
- qRT-PCR
quantitative reverse transcription-polymerase chain reaction
- BMMs
Bone marrow derived macrophages
- LPS
Lipopolysaccharide
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2011;11:1563–1569. doi: 10.1111/j.1600-6143.2011.03579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eltzschig HK, Eckle T. Ischemia and reperfusion–from mechanism to translation. Nat Med. 2011;17:1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaczorowski DJ, Tsung A, Billiar TR. Innate immune mechanisms in ischemia/reperfusion. Front Biosci (Elite Ed) 2009;1:91–98. doi: 10.2741/E10. [DOI] [PubMed] [Google Scholar]
- 4.Raptis DA, Limani P, Jang JH, Ungethüm U, Tschuor C, Graf R, Humar B, Clavien P-A. GPR120 on Kupffer cells mediates hepatoprotective effects of ω3-fatty acids. J Hepatol. 2014;60:625–632. doi: 10.1016/j.jhep.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Devey L, Ferenbach D, Mohr E, Sangster K, Bellamy CO, Hughes J, Wigmore SJ. Tissue-resident macrophages protect the liver from ischemia reperfusion injury via a heme oxygenase-1-dependent mechanism. Molecular therapy : the journal of the American Society of Gene Therapy. 2009;17:65–72. doi: 10.1038/mt.2008.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellett JD, Atkinson C, Evans ZP, Amani Z, Balish E, Schmidt MG, van Rooijen N, Schnellmann RG, Chavin KD. Murine Kupffer cells are protective in total hepatic ischemia/reperfusion injury with bowel congestion through IL-10. J Immunol. 2010;184:5849–5858. doi: 10.4049/jimmunol.0902024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, Pierce RH, Crispe IN. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110:4077–4085. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikarashi M, Nakashima H, Kinoshita M, Sato A, Nakashima M, Miyazaki H, Nishiyama K, Yamamoto J, Seki S. Distinct development and functions of resident and recruited liver Kupffer cells/macrophages. Journal of leukocyte biology. 2013;94:1325–1336. doi: 10.1189/jlb.0313144. [DOI] [PubMed] [Google Scholar]
- 9.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. The Journal of clinical investigation. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ji H, Liu Y, Zhang Y, Shen XD, Gao F, Busuttil RW, Kuchroo VK, Kupiec-Weglinski JW. T-cell immunoglobulin and mucin domain 4 (TIM-4) signaling in innate immune-mediated liver ischemia-reperfusion injury. Hepatology. 2014;60:2052–2064. doi: 10.1002/hep.27334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, Lotze MT, Geller DA, Billiar TR. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175:7661–7668. doi: 10.4049/jimmunol.175.11.7661. [DOI] [PubMed] [Google Scholar]
- 12.Perdiguero EG, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2014 doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulz C, Perdiguero E Gomez, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SEW, Pollard JW, Frampton J, Liu KJ, Geissmann F. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 14.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38:79–91. doi: 10.1016/j.immuni.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38:792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14:986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies LC, Taylor PR. Tissue-resident macrophages: then and now. Immunology. 2015;144:541–548. doi: 10.1111/imm.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhai Y, Shen XD, Gao F, Zhao A, Freitas MC, Lassman C, Luster AD, Busuttil RW, Kupiec-Weglinski JW. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology. 2008;47:207–214. doi: 10.1002/hep.21986. [DOI] [PubMed] [Google Scholar]
- 19.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature chemical biology. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, Declercq W, Callewaert N, Prendergast GC, Degterev A, Yuan J, Vandenabeele P. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sunderkötter C, Nikolic T, Dillon MJ, Rooijen NVan, Stehling M, Drevets DA, Leenen PJM. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 22.Kinoshita M, Uchida T, Sato A, Nakashima M, Nakashima H, Shono S, Habu Y, Miyazaki H, Hiroi S, Seki S. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol. 2010;53:903–910. doi: 10.1016/j.jhep.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 23.Movita D, Kreefft K, Biesta P, van Oudenaren A, Leenen PJ, Janssen HL, Boonstra A. Kupffer cells express a unique combination of phenotypic and functional characteristics compared with splenic and peritoneal macrophages. Journal of leukocyte biology. 2012;92:723–733. doi: 10.1189/jlb.1111566. [DOI] [PubMed] [Google Scholar]
- 24.Rao J, Yue S, Fu Y, Zhu J, Wang X, Busuttil RW, Kupiec-Weglinski JW, Lu L, Zhai Y. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2014;14:1552–1561. doi: 10.1111/ajt.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou H, Zhu J, Yue S, Lu L, Busuttil RW, Kupiec-Weglinski JW, Wang X, Zhai Y. The Dichotomy of Endoplasmic Reticulum Stress Response in Liver Ischemia-Reperfusion Injury. Transplantation. 2016;100:365–372. doi: 10.1097/TP.0000000000001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, Varol C. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. 2014;193:344–353. doi: 10.4049/jimmunol.1400574. [DOI] [PubMed] [Google Scholar]
- 27.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. 2012;13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-Resident Macrophage Necroptosis Orchestrates Type 1 Microbicidal Inflammation and Type-2-Mediated Tissue Repair during Bacterial Infection. Immunity. 2014 doi: 10.1016/j.immuni.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 29.Stockinger S, Materna T, Stoiber D, Bayr L, Steinborn R, Kolbe T, Unger H, Chakraborty T, Levy DE, Muller M, Decker T. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J Immunol. 2002;169:6522–6529. doi: 10.4049/jimmunol.169.11.6522. [DOI] [PubMed] [Google Scholar]
- 30.Di Paolo NC, Doronin K, Baldwin LK, Papayannopoulou T, Shayakhmetov DM. The transcription factor IRF3 triggers “defensive suicide” necrosis in response to viral and bacterial pathogens. Cell Rep. 2013;3:1840–1846. doi: 10.1016/j.celrep.2013.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, Startek JB, Gamero AM, Mossman KL, Sad S. Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc Natl Acad Sci U S A. 2014;111:E3206–3213. doi: 10.1073/pnas.1407068111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanlangenakker N, Vanden Berghe T, Vandenabeele P. Many stimuli pull the necrotic trigger, an overview. Cell death and differentiation. 2012;19:75–86. doi: 10.1038/cdd.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saveljeva S, Mc Laughlin SL, Vandenabeele P, Samali A, Bertrand MJM. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015;6:e1587. doi: 10.1038/cddis.2014.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rao J, Yue S, Fu Y, Zhu J, Wang X, Busuttil RW, Kupiec-Weglinski JW, Lu L, Zhai Y. ATF6 Mediates a Pro-Inflammatory Synergy Between ER Stress and TLR Activation in the Pathogenesis of Liver Ischemia-Reperfusion Injury. Am J Transplant. 2014 doi: 10.1111/ajt.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Ren F, Cheng Q, Bai L, Shen X, Gao F, Busuttil RW, Kupiec-Weglinski JW, Zhai Y. Endoplasmic reticulum stress modulates liver inflammatory immune response in the pathogenesis of liver ischemia and reperfusion injury. Transplantation. 2012;94:211–217. doi: 10.1097/TP.0b013e318259d38e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: apoptosis or necrosis? Hepatology. 2001;33:397–405. doi: 10.1053/jhep.2001.22002. [DOI] [PubMed] [Google Scholar]
- 37.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 38.Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- 39.Linkermann A, Brasen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, Krautwald S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012;81:751–761. doi: 10.1038/ki.2011.450. [DOI] [PubMed] [Google Scholar]
- 40.Xu X, Chua KW, Chua CC, Liu CF, Hamdy RC, Chua BH. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res. 2010;1355:189–194. doi: 10.1016/j.brainres.2010.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenbaum DM, Degterev A, David J, Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J, Savitz SI. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia-reperfusion injury model. J Neurosci Res. 2010;88:1569–1576. doi: 10.1002/jnr.22314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lau A, Wang S, Jiang J, Haig A, Pavlosky A, Linkermann A, Zhang ZX, Jevnikar AM. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am J Transplant. 2013;13:2805–2818. doi: 10.1111/ajt.12447. [DOI] [PubMed] [Google Scholar]
- 43.Liu J, van Mil A, Vrijsen K, Zhao J, Gao L, Metz CH, Goumans MJ, Doevendans PA, Sluijter JP. MicroRNA-155 prevents necrotic cell death in human cardiomyocyte progenitor cells via targeting RIP1. J Cell Mol Med. 2011;15:1474–1482. doi: 10.1111/j.1582-4934.2010.01104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 45.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, Mocarski ES. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–31279. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.