

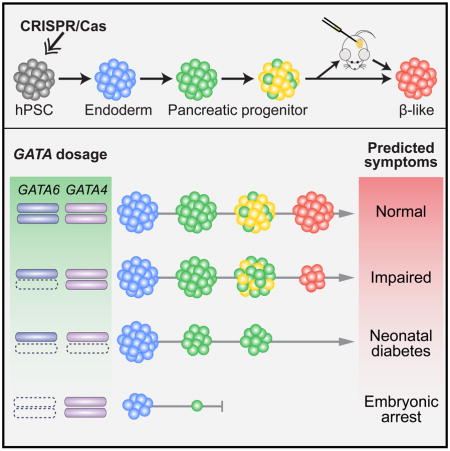
Summary
Human disease phenotypes associated with haploinsufficient gene requirements are often not recapitulated well in animal models. Here, we have investigated the association between human GATA6 haploinsufficiency and a wide-range of clinical phenotypes that include neonatal and adult-onset diabetes using CRISPR/Cas9-mediated genome editing coupled with human pluripotent stem cell (hPSC) directed differentiation. We found that loss of one GATA6 allele specifically affects the differentiation of human pancreatic progenitors from the early PDX1+ stage to the more mature PDX1+NKX6.1+ stage, leading to impaired formation of glucose-responsive β-like cells. In addition to this GATA6 haploinsufficiency, we also identified dosage-sensitive requirements for GATA6 and GATA4 in the formation of both definitive endoderm and pancreatic progenitor cells. Our work expands the application of hPSCs from studying the impact of individual gene loci to investigation of multigenic human traits, and establishes an approach for identifying genetic modifiers of human disease.
eTOC
Huangfu, Chen and colleagues model human pancreatic disease by step-wise differentiation of genetically modified hPSCs to characterize phenotypic effects of GATA6 haploinsufficiency not evident in mouse models plus genetic interaction with GATA4.
Introduction
Haploinsufficiency is increasingly recognized as an important contributor to human disease. Several hundred haploinsufficient genes, many of which encode transcription factors, have been reported to cause diverse disorders affecting the pancreas, heart, brain and other organs (Dang et al., 2008; Seidman and Seidman, 2002). For example, heterozygous inactivation of TBX5, NKX2.5 or GATA4 are associated with dominantly inherited congenital heart defects (Ang et al., 2016; Bruneau, 2008), while HNF1A, HNF4A and HNF1B heterozygous loss-of-function mutations are each found in patients with a monogenic form of diabetes known as maturity onset diabetes of the young (MODY) (Ryffel, 2001). Notably, haploinsufficiency is typically associated with a wide spectrum of phenotypic manifestations, suggesting a significant contribution of modifier genes and/or non-genetic factors such as lifestyle and diet (Seidman and Seidman, 2002). This multifactorial contribution of genetic and environmental components is typical of complex traits, and the relatively simple genetics of haploinsufficiency presents a unique opportunity for dissecting the molecular basis of the disease and the roles of potential modifying factors thus facilitating the development of treatments.
Despite its recognized importance in human disease, how a reduced dosage of a transcription factor affects downstream target genes to cause a disease is poorly understood. A main hurdle lies in the difficulty of modeling human haploinsufficiency in mice: inactivating both alleles of the mouse ortholog is almost always necessary in order to fully recapitulate a disease phenotype that appears to only require the loss of one allele in humans (Seidman and Seidman, 2002; Veitia, 2002). For example, mice with heterozygous loss-of-function mutations in Hnf1a, Hnf4a or Hnf1b are not diabetic in contrast to the phenotypes observed in MODY patients with similar mutations (Ryffel, 2001). To investigate such a genetic discrepancy between mice and humans, one promising approach is to use directed differentiation of human pluripotent stem cells (hPSCs) to recapitulate the developmental context, and to further employ recently developed precision genome-editing tools to dissect the genetic context of the disease. This approach is beginning to elucidate cellular mechanisms underlying human diseases (Musunuru, 2013), and our recent work has extended this approach to understanding the roles of lineage determinants in the more complex multistep differentiation processes for studying developmental mechanisms underlying congenital disorders (Zhu et al., 2016). However, it remains uncertain whether hPSC-based assays would be sensitive enough to detect dosage-dependent phenotypes associated with a haploinsufficient disease, and whether the hPSC system could be used to further explore complex genetic interactions and identify potential disease modifying factors.
GATA6 encodes a zinc finger transcription factor that shares homology with 5 additional GATA factors all known to bind the consensus (A/T)GATA(A/G) sequence (Patient and McGhee, 2002). A recent study identified GATA6 heterozygous inactivating mutations in a large number of patients with pancreatic agenesis, a rare birth defect marked by a complete absence of the pancreas or an extreme reduction in its size (Lango Allen et al., 2012). Consequently, these patients suffer from severe exocrine pancreatic insufficiency and neonatal diabetes due to the absence of insulin-secreting endocrine β cells. Further studies have identified heterozygous GATA6 patients with a wide spectrum of phenotypes ranging from non-diabetic, mildly diabetic in adults, to severely diabetic with no pancreas in newborns, and marked phenotypic variability is observed even among affected members of the same family (Bonnefond et al., 2012; Catli et al., 2013; Chao et al., 2015; De Franco et al., 2013; Eifes et al., 2013; Gong et al., 2013; Stanescu et al., 2015; Suzuki et al., 2014; Yorifuji et al., 2012; Yu et al., 2014). However, as with other cases of haploinsufficient disease genes, inactivation of one Gata6 allele does not cause apparent defects in mice (Morrisey et al., 1998). Instead, simultaneous deletion of all 4 murine alleles of Gata6 and the sister gene Gata4 is needed to recreate the pancreatic agenesis phenotype (Carrasco et al., 2012; Xuan et al., 2012). Thus a mechanistic understanding of how GATA6 haploinsufficiency affects human pancreatic development is hindered by the lack of appropriate model systems.
Utilizing efficient genome editing tools we have established (González et al., 2014), we generated a large array of isogenic GATA6, GATA4, and compound GATA6/4 mutant lines to investigate the influence of GATA6/4 dosage on pancreatic differentiation. Directed differentiation assays reveal a previously unknown requirement for GATA6 in the formation of definitive endoderm (DE), and a dosage-sensitive requirement for GATA6 in the formation of pancreatic progenitors (PP) and subsequently glucose-responsive β cells. Supporting the influence of genetic modifiers of GATA6 haploinsufficiency, we find that the formation of PP cells is highly sensitive to the GATA6 and GATA4 gene dosage. A better understanding of human pancreatic development will provide critical information for improving hPSC differentiation into functional β cells to be used as a cell replacement therapy for insulin-requiring diabetes. Our investigation of complex genetic interactions between GATA6 and GATA4 also supports the broader application of hPSC-based models for investigating genetic or environmental modifiers involved in multifactorial traits such as type 2 diabetes and for the discovery of therapeutic targets.
Results
GATA6 and GATA4 are expressed during human endoderm and pancreas differentiation
The appearance of PDX1-expressing (PDX1+) multipotent PP cells marks the onset of pancreatic specification at around embryonic day 8.5 in mice and 4 weeks post-conception in humans (Jennings et al., 2013). Originating from the DE germ layer, PP cells expand and ultimately give rise to exocrine acini and ducts as well as endocrine islets consisting of the five main endocrine cell types including insulin-secreting β cells (Pan and Wright, 2011). As both endocrine and exocrine tissue development is affected in GATA6 heterozygous patients, GATA6 haploinsufficiency is likely to affect early differentiation events involving the specification of PP cells. Thus we first analyzed the expression of GATA6 and GATA4 during these initial stages of pancreatic differentiation to test if the distinct GATA6 heterozygous phenotypes observed between humans and mice could be explained simply by species-specific gene expression patterns.
In mice, Gata6 and Gata4 are both expressed in DE and its derivatives including the pancreas (Decker et al., 2006; Walker et al., 2014; Watt et al., 2007). In human embryos, GATA4 is expressed at the onset of pancreatic development (Jennings et al., 2013). However, it is unknown whether GATA4 is also expressed in DE cells prior to pancreas formation, nor is the expression of GATA6 reported at comparable developmental stages. In hPSC differentiation cultures, both GATA4 and GATA6 are expressed in DE cells (McLean et al., 2007; Pan et al., 2011; Vallier et al., 2009), but their expression patterns are not well characterized in PP cells. We adapted an established directed differentiation protocol (D’Amour et al., 2006; Kroon et al., 2008) to generate PDX1+NKX6.1− cells that resemble the nascent pancreatic progenitor cells, which will be referred to as PP1 cells henceforth (Figure 1A). The PP1 cells can further differentiate into pancreatic endocrine precursors and ultimately mature into functional insulin-secreting β cells through further in vitro differentiation or after transplantation into immunocompromised mice (Kroon et al., 2008; Pagliuca et al., 2014; Rezania et al., 2014; Russ et al., 2015). RT-qPCR detected significant increase of GATA4 and GATA6 expression levels upon differentiation to the DE and PP1 stages (Figure 1B). Immunofluorescence staining and fluorescence-activated cell sorting (FACS) showed that GATA4 and GATA6 were first co-expressed with DE markers SOX17 and CXCR4, and later at the PP1 stage they were both co-expressed with PDX1 (Figures 1C–1E and S1A). These data suggest that similar to mice, GATA4 and GATA6 are co-expressed in endoderm and nascent pancreatic progenitor cells during human development.
Figure 1. GATA6 and GATA4 expression during early pancreatic progenitor differentiation in hPSCs.

(A) Schematic of directed endoderm and PDX1+ early pancreatic progenitor differentiation from HUES8 hPSCs using the 1st-generation protocol. The cell types and key markers at each stage are shown. Chemicals and durations for each differentiation stage are indicated. PS: undifferentiated hPSC stage; DE: definitive endoderm stage; PP1: early pancreatic progenitor stage (cells expressing PDX1 but not NKX6.1); Activin A, a TGF-β superfamily ligand; BIO: BIO-acetoxime, a GSK3 inhibitor that activates WNT signaling; FGF10: fibroblast growth factor 10; SANT-1, a Hedgehog pathway antagonist; RA: retinoic acid; LDN: LDN-193189, a BMP type 1 receptor (ALK2/3) inhibitor. The day when differentiation is initiated is designated as day 0 (d0).
(B) The mRNA expression patterns of GATA6 and GATA4 during hPSC differentiation to early pancreatic progenitor. The mRNA levels were measured by RT-qPCR (n=4) and normalized to internal control ACTB. Student’s t-test with two-tailed distribution and two-sample equal variance was used for statistics analysis. P values <0.05, 0.01, and 0.001 were indicated with 1, 2, and 3 asterisks, respectively, in all figures.
(C) Representative immunostaining of GATA6 or GATA4 with other stage-specific markers including OCT4, SOX17, and PDX1. Scale bar, 100 μm for all images throughout.
(D) Representative FACS dot plots of cells stained for CXCR4, SOX17, GATA6 and GATA4 factors at the DE stage. The percentage of each cell population is indicated in the corresponding quadrant for all FACS plots.
(E) Representative FACS dot plots for co-expression of GATA6 or GATA4 with PDX1 at the PP1 stage.
All data in this figure were generated from HUES8 line using 1st-generation differentiation protocol. See also Method Details.
GATA6 is required for efficient definitive endoderm formation and pancreatic progenitor specification
We next generated clonal GATA6 mutant hPSC lines using our iCRISPR system (González et al., 2014). To control for potential CRISPR off-target effects and line-to-line variations, we analyzed 4 heterozygous and 6 homozygous HUES8 mutant lines carrying frameshift mutations generated using two CRISPRs targeting different sequences, and compared them to isogenic GATA6+/+ wild-type (WT) control HUES8 lines (Figure 2A, Tables S1 and S2). Western blotting verified the absence of WT GATA6 proteins in homozygous mutant lines (Figures 2B and S1B). We also detected reduced GATA6 expression in GATA6 heterozygous cells by both Western blotting (Figure 2B) and FACS analysis (Figure S1C), showing that we can test a possible haploinsufficient role of GATA6. Off-target analysis on multiple clonal lines revealed no modifications in the top 5 potential off-target exomic regions (Figure S1G). Thus we designated heterozygous and homozygous GATA6 mutant lines as the GATA6−/+ and GATA6−/− genotypes respectively henceforth.
Figure 2. The role of GATA6 in human definitive endoderm and pancreatic progenitor specification.

(A) CRISPR gRNA design for generating GATA6 mutants from the HUES8 parental line. Note that the GATA6 gene uses two initiation codons. Shown here is the schematic of the long isoform. The short isoform starts from amino acid 147 of the long isoform. To control for potential off-target effects associated with CRISPR/Cas, two CRISPRs were used to target different sequences corresponding to the conserved C-terminal zinc finger domain critical for the DNA binding function of GATA6. The target sequences of the two CRISPR gRNAs (GATA6-Cr1 and Cr2) and the corresponding protospacer adjacent motif (PAM) sequences are indicated in green and red, respectively. The corresponding sequences of two representative GATA6 homozygous mutant lines (m37 and m45) are shown underneath the WT reference sequence.
(B) Western blotting for GATA6 protein expression at the DE stage with the corresponding CRISPR gRNA and genotype indicated above each hPSC clonal line. ACTB (β-Actin) was used as a loading control. Solid arrowheads indicate the WT proteins of GATA6; unfilled arrowheads indicate the truncated mutant proteins. Sizes of molecular weight marker shown on the right side. G6: GATA6.
(C) Representative immunostaining images of SOX17 and FOXA2 at the DE stage (d5).
(D) Representative FACS dot plots of cells co-stained for SOX17 and CXCR4 (d5).
(E) FACS quantification of DE differentiation efficiency based on the percentages of CXCR4+SOX17+ cells at the DE stage. Each bar indicates the mean with SEM for a clonal line. Data was generated from three independent experiments (n=6 for WT and G6−/+ and n=9 for G6−/−).
(F) Representative immunostaining images for pancreatic progenitor marker PDX1 at the PP1 stage.
(G) Representative FACS dot plots for PP1 marker PDX1.
(H) FACS quantification of differentiation efficiency based on the percentage of PDX1+ PP1 cells (n=6 for WT and G6−/+ and n=9 for G6−/−). t-test with two-tailed distribution and two-sample equal variance was used for statistics analysis in panels E and H to compare with WT controls.
All data in this figure were generated from HUES8 lines using the 1st-generation differentiation protocol. See also Figure S1 and Tables S1 and S2.
Both WT and GATA6−/+ hPSCs could be differentiated into DE cells co-expressing SOX17, FOXA2 and CXCR4 with comparable efficiencies as determined by immunofluorescence staining and FACS analysis (Figures 2C–2E). However, a significantly lower number of SOX17+CXCR4+ DE cells were formed from all 6 GATA6−/− mutant lines examined; and subsequently at the PP1 stage, PDX1+ cells failed to form as determined by immunostaining and FACS analysis likely due to the earlier requirement for GATA6 at the DE stage (Figures 2F–2H). Consistent with this notion, in a parallel study, Gadue and colleagues largely rescued the PP1 phenotype through isolating and expanding the small number of DE cells formed from GATA6−/− hPSCs (Tiyaboonchai et al., 2017). Although truncated GATA6 proteins were detected in some homozygous mutant lines, their phenotypes were indistinguishable from mutant lines that had no detectable GATA6 protein (Figures 2B, 2E and S1B). This indicates that the truncated GATA6 protein lacking the C-terminal zinc finger domain is not functional, consistent with its known biochemical characteristics (Bates et al., 2008; Molkentin, 2000). These data demonstrate that GATA6 is required for efficient formation of definitive endoderm and subsequently pancreatic progenitor cells in hPSC differentiation, a phenotype not observed in mouse studies (Izumi et al., 2007; Koutsourakis et al., 1999; Morrisey et al., 1998; Zhao et al., 2008). In the absence of functional GATA6 proteins, a human embryo would likely fail to form a pancreas due to a primary defect in definitive endoderm formation.
Different from GATA6−/− hPSCs, all 4 GATA6−/+ mutant lines formed a significant number of PDX1+ PP1 cells with efficiencies comparable to those of WT lines (Figures 2F–2H), though RT-qPCR analysis showed a reduction in PDX1 mRNA expression (Figure S1H). No other progenitor markers, with the exception of GATA6 itself, showed a statistically significant difference in expression levels between GATA6−/+ and WT cells, although a trend towards reduced levels was observed for some genes. Apoptosis assays based on cleaved caspase-3 expression revealed a small increase in apoptotic rate in PDX1+ cells in GATA6−/+ mutants and there was no significant change in the proliferation rate based on phospho-histone H3 staining (Figures S1I–S1L). Thus our analysis of GATA6−/+ hPSC lines did not reveal a major defect in the formation or proliferation of PP1 cells. However, GATA6−/+ PP1 cells exhibited subtle phenotypes including increased apoptosis and reduced PDX1 mRNA expression, which could lead to more pronounced defects at later differentiation stages.
A patient-specific GATA6 allele behaves as a null mutation
To more precisely mimic the disease condition, we focused on the GATA6Arg456Cys point mutation (GATA6R456C for simplicity), which is the most common GATA6 mutation reported in unrelated patients (Lango Allen et al., 2012; Yu et al., 2014). After creating GATA6R456C/R456C and GATA6R456C/+ hPSC lines through seamless genome editing using the iCRISPR system (Figures 3A and 3B), we differentiated two independent lines of each genotype to the DE and PP1 stages. Western blotting analysis detected comparable levels of GATA6 protein in GATA6R456C/R456C, GATA6R456C/+ and WT controls at the DE stage (Figure 3C), indicating that the resulting mutant protein was stable. At the PP1 stage, the GATA6 expression levels declined in GATA6R456C/R456C cells and PDX1 expression was not detected (Figure 3C), similar to the phenotypes observed for GATA6−/− mutants (Figures 2F–2H). FACS quantification showed that GATA6R456C/R456C mutant lines formed a reduced number of DE cells co-expressing CXCR4 and SOX17, and no significant numbers of PDX1+ cells were subsequently detected at the PP1 stage (Figures 3D and 3E). As no phenotypic differences were detected between GATA6R456C/R456C and GATA6−/− mutants at both DE and PP1 stages, these findings indicate that GATA6R456C is a null mutation, and further confirm an absolute requirement for GATA6 in human pancreatic development.
Figure 3. Characterization of the patient-specific GATA6R456C mutation.

(A) Schematic of CRISPR targeting for generating hPSC lines carrying heterozygous and homozygous GATA6R456C mutations. The GATA6R456C missense disease mutation (c.1366C>T, shown in red) was introduced through homology direct repair using a ssDNA donor template (110-nt long). To minimize potential secondary cutting by Cas9 after homologous recombination, a silent mutation (G>A, shown in orange) was introduced in the PAM sequence. The blue arrow indicates the predicted Cas9 cleavage site.
(B) Representative sequencing graphs of generated heterozygous and homozygous mutants carrying the disease mutation. The red * indicates the C>T switch; the orange # indicates the G>A change.
(C) Western blotting showed that GATA6R456C mutant protein was still expressed and detected by the same GATA6 antibody used elsewhere. The cells were differentiated to the DE stage. Western blotting was also used to detect PDX1 and GATA6 expression at the PP1 stage. ACTB was used as a loading control.
(D) FACS quantification of percentages of DE and PP1 cells at the corresponding stages (n=6). For direct comparison and easy visualization, we also included data shown in Figures 2E and 2H for WT, −/+ and −/− genotypes (obtained from the same differentiation experiments). One-way ANOVA was used followed by multiple comparisons with Bonferroni correction for statistics test.
(E) Representative immunostaining images for PDX1 at the PP1 stage.
All data in this figure were generated from HUES8 lines using the 1st-generation differentiation protocol. See also Tables S1 and S2 and Figure S1.
In contrast to the clear differentiation defect of GATA6R456C/R456C hPSCs, GATA6R456C/+ lines exhibited the same efficiencies of forming both DE and PP1 cells compared to WT and GATA6−/+ lines (Figure 3D), and no dominant negative effects were observed. The PDX1+ cells formed from GATA6R456C/+ lines displayed similar proliferation and apoptotic rates as GATA6−/+ mutant cells (Figures S1I and S1J). Notably, the GATA6R456C/+ genotype was reported in two unrelated patients with pancreatic agenesis, and a third individual with no indication of hyperglycemia or exocrine pancreatic insufficiency suggesting the presence of a functional pancreas (Lango Allen et al., 2012; Yu et al., 2014). Taken together with our findings, losing one functional GATA6 allele on its own does not significantly affect the formation of early pancreatic progenitors and thus is unlikely to cause the extreme pancreatic agenesis condition in humans.
A haploinsufficient requirement for GATA6 in the formation of PDX1+NKX6.1+ pancreatic progenitor cells
Recent studies have developed protocols to further differentiate early PDX1+NKX6.1− PP1 cells into PDX1+NKX6.1+ progenitor cells (designated as PP2), a key intermediate step necessary for the generation of glucose-responsive β-like cells (Kelly et al., 2011; Nostro et al., 2015; Pagliuca et al., 2014; Rezania et al., 2014; Russ et al., 2015). Based on these findings, we applied the 2nd-generation differentiation protocol (Zhu et al., 2016) on HUES8 and H1 lines (Figure 4A). Consistent with findings using the 1st-generation protocol (Figures 1B–1E), GATA4 and GATA6 were co-expressed at the DE stage with SOX17 (Figures 4B and S2A). At the PP1 stage, >80% PDX1+ cells were detected, and they co-expressed GATA4 and GATA6 but NKX6.1 was not detected (Figures 4B and S2A). At the PP2 stage, GATA4 and GATA6 were still co-expressed in almost all cells, though the expression levels were decreased (especially for GATA4); many cells co-expressed NKX6.1 (~55% in H1 and ~35% in HUES8, Figures 4B and S2A). At the β-like stage, GATA4 and GATA6 expression both decreased (Figure S2B); FACS analysis and confocal microscopy revealed no expression of GATA4 or GATA6 in C-peptide (CPEP)+ cells; and no significant co-expression of GATA4 and GATA6 was detected (Figure 4B and S2A). Consistent with these findings, Gata4/6 expression is excluded from β cells at comparable stages in mice, and GATA4 expression is restricted to tip progenitor and later to acinar cells in humans (Decker et al., 2006; Jennings et al., 2013; Pan and Wright, 2011).
Figure 4. GATA6 haploinsufficiency in the specification of PDX1+NKX6.1+ pancreatic progenitor cells.

(A) Schematic of modified 2nd-generation differentiation protocol toward β-like stage from hPSCs. The key lineage markers at each stage are shown. Chemicals and durations for each differentiation stage are indicated. hPS: undifferentiated hPSC stage; DE: definitive endoderm stage; PP1: early pancreatic progenitor stage; PP2: pancreatic endoderm progenitor stage; β: β-like stage. From PP2 to β-like stage, the cells were cultured at air-liquid interface. See also Table S3 for detailed differentiation medium recipes.
(B) Representative FACS dot plots of GATA6 co-staining with stage-specific lineage markers or GATA4 for each stage.
(C) Representative FACS dot plots for PDX1 and NKX6.1 co-staining at the PP2 stage.
(D) Quantification of PDX1+ cells and PDX1+NKX6.1+ cells at the PP2 stage based on FACS analysis from 3 independent experiments. The statistics was done by comparing the mutant group with the WT group. The hPSC lines with the same genotypes were treated as one group (n=6).
(E) Representative images for PDX1 and NKX6.1 expression at the PP2 stage.
(F) RT-qPCR analysis of pancreatic progenitor marker expression at the PP2 stage (n=4). Genes labeled in red were not expressed in the PP1 cells. t-test with two-tailed distribution and two-sample equal variance was used to determine the significance in this figure panels D and F to compare with WT group.
All data in this figure were generated from H1 lines using the 2nd-generation differentiation protocol (Table S3). See also Figures S2 and S3.
Using the 2nd-generation differentiation protocol, we verified that GATA6−/+, GATA6R456C/+ and GATA6−/− cells exhibited similar phenotypes at the DE and PP1 stages as observed with the 1st-generation protocol (Figures S3B–S3D). In the H1 background, GATA6−/+ and GATA6R456C/+ mutant hPSCs formed PDX1+ cells as efficiently as WT cells at both PP1 and PP2 stage (Figures 4C–4E and S3D). However, there was a significant reduction of NKX6.1+PDX1+ PP2 cells formed from GATA6−/+ hPSCs (~20% compared to ~55% in WT cells, Figures 4C–4E and S3E), indicating a specific defect in the transition of PP1 to PP2 cells. PP2 cells showed relatively low proliferation and apoptosis rates, and no significant differences were detected between WT and GATA6−/+ cells (Figures S3F and S3G). Reduction of PP2 cells was also observed with GATA6−/+ hPSCs in the HUES8 background (Figure S3H). Thus losing one functional GATA6 allele impairs the formation of PP2 cells from hPSCs. A reduced number of pancreatic progenitor cells during embryonic development would predict a smaller pancreas (Stanger et al., 2007), a phenotype observed in some GATA6 heterozygous patients even in the absence of overt pancreatic insufficiency (Bui et al., 2013; Yorifuji et al., 2012).
We further performed RNA-seq analysis to identify differentially expressed genes between GATA6−/+ and WT cells at the PP2 stage. The majority of top downregulated transcription factors are known to play key roles in pancreatic development (Figure S3I). Gene ontology analysis showed that top downregulated genes were enriched with factors involved in developmental processes, with the pancreas being the top affected lineage (Figure S3J). RT-qPCR further confirmed the significant downregulation of both pan-pancreatic and proendocrine transcription factors in GATA6−/+ mutant cells (Figure 4F). Among the severely affected genes were a number of pro-endocrine progenitor markers detected at the PP2 but not at the PP1 stage in WT cells including NKX6.1, NGN3, NEUROD1 and NKX2.2 (Figure S2C). This later wave of gene expression appears to correlate with differences in timing between human and mouse pancreatic development. For example, NKX2.2 is expressed in early pancreatic progenitors in mice, but it is detected in human progenitors only after endocrine commitment (Jennings et al., 2013). These differences may underlie species-specific sensitivity to GATA6 haploinsufficiency.
A haploinsufficient requirement for GATA6 in the formation of glucose-responsive β-like cells
To investigate the functional consequences of GATA6 haploinsufficiency, we differentiated the cells further to the β-like stage (Figures 4A and 5A). Immunostaining and FACS analyses showed that GATA6−/+ hPSCs formed CPEP+ monohormonal β-like cells, but the percentage was greatly reduced compared to WT cells (~3.5% vs 15%, Figures 5B–5D and S4A). Consistent with this finding, GATA6−/+ cells showed reduced expression of all endocrine markers, including genes encoding key transcription factors, endocrine hormones, and proteins important for β cell function (Figure S4B). The majority of CPEP+ cells were monohormonal for both WT and GATA6−/+ cells, but fewer GATA6−/+ cells among CPEP+ cells co-expressed NKX6.1 (Figures 5B–5E and S4A), which is known to play important roles in maintaining adult β cell function (Taylor et al., 2013).
Figure 5. GATA6 haploinsufficiency in the formation of functional β-like cells.

(A) Schematic of hPSC-derived PP2 cell differentiation to β-like cells in vitro and in vivo (after transplantation).
(B) Representative images for CPEP co-staining with glucagon (GCG) or NKX6.1 at the β-like stage in vitro.
(C) Representative FACS dot plots for CPEP co-staining with GCG or NKX6.1 at the β-like stage.
(D) Quantification of FACS analysis for CPEP+ and GCG+ or - cells from 3 independent experiments.
(E) Quantification of FACS analysis for percentage of NKX6.1+ cells within CPEP+ cells from 3 independent experiments.
(F) In vitro GSIS at the β-like stage (n=6). See Figure S4C for associated data.
(G) Representative images for CPEP co-staining with GCG or NKX6.1 of the grafts removed from mouse kidney capsules 4 months after transplantation.
(H) Mouse GSIS experiment at one month after transplantation. Box and whiskers plot for human insulin secretion ratio (post glucose/fasting) in mouse sera. The analysis was done using unpaired two-tailed t test with Welch’s correction (unequal variance) (n=8–10). See raw data for each mouse in Figure S4E.
All data in this figure were generated from H1 lines using the 2nd-generation differentiation protocol. See also Table S3 and Figures S4.
We further performed functional assays on hPSC-derived β-like cells. Both WT and GATA6−/+ cells exhibited similar glucose-stimulated insulin secretion (GSIS); the ratio of C-peptide secreted in high glucose (16.7 mM) to low glucose (2.8 mM) was ~2-fold (Figure 5F). However, the total C-peptide secreted from GATA6−/+ cells was reduced (Figure S4C), consistent with the reduced number of β-like cells (Figures 5B–5E and S4A). We also transplanted WT and GATA6−/+ cells at ~10 days after the PP2 stage under kidney capsules of immunocompromised SCID-beige mice (Figure 5A and Table S3). CPEP+NKX6.1+PDX1+ cells were identified in grafts derived from both WT and GATA6−/+ cells (Figure 5G and S4D), with CPEP+ cells more frequently observed in WT compared to GATA6−/+ cells. Similar to in vitro GSIS, significantly lower levels of human insulin were detected in mice receiving GATA6−/+ cells compared to those receiving WT cells (Figure S4E). Nevertheless, upon glucose challenge, 75% of mice receiving GATA6−/+ mutant cells (6/8 animals) showed increased human insulin secretion at one month post-transplantation (Figures 5H and S4E). The lower levels of circulating C-peptide is likely a result of reduced β-like cell number in the grafts, and a reduced percentage of NKX6.1+ β-like cells may also contribute to the phenotype. These findings support a predisposition of GATA6 heterozygous patients to diabetes due to a decrease in β cell number, and possibly also β cell function.
Human pancreatic differentiation is exquisitely sensitive to GATA6 and GATA4 gene dosage
A broad range of phenotypes has so far been reported for ~100 GATA6 heterozygous patients (Table S4, Figures 6A, 6B and S5). Among the 56 cases with clear documentation of the presence or absence of pancreatic phenotypes, 23 individuals showed no apparent diabetes or exocrine pancreatic insufficiency at birth: 6 patients developed diabetes later in life, yet 17 remained non-diabetic with no apparent pancreatic defects when the case was reported. Although this could be explained by hypomorphic mutations in some patients, in a number of cases the same mutation was identified in multiple patients with divergent phenotypes (Table S4, Figure S5). Similarly divergent phenotypes have also been observed with GATA4 heterozygous loss-of-function mutations: 2 patients with specific GATA4 heterozygous mutations have so far been diagnosed with pancreatic agenesis, yet a large number (>140) of GATA4 heterozygous patients exhibit cardiac defects without pancreatic agenesis (D’Amato et al., 2010; Shaw-Smith et al., 2014). These patient data support the involvement of genetic or environmental modifying factors.
Figure 6. GATA6 and GATA4 control pancreatic specification in a dosage-sensitive manner.

(A) Table summarization of diabetes phenotypes in reported GATA6 heterozygous humans. See Table S4 and Figure S5 for detailed information.
(B) Pie chart summary of wide-range of diabetes phenotypes in reported GATA6 heterozygous humans with clear documentation regarding phenotype of the pancreas.
(C) Representative FACS dot plots for each genotype at DE, PP1 and PP2 stages.
(D) Quantification of DE differentiation by FACS. Mean values from multiple clonal lines with the same genotypes from multiple independent experiments were used for graph plotting and statistics analysis (n=6–10). All clonal lines were generated in the HUES8 hPSC background, and one-way ANOVA was used followed by multiple comparisons with Bonferroni correction for all the statistics tests in this figure (panels D to G).
(E) FACS quantification of percentages of PDX1+ cells at PP1 stage. Mean values from multiple clonal lines with the same genotypes from multiple experiments were used for graph plotting and statistics analysis (n=6–10).
(F) Quantification of PDX1+ cells at PP2 stage (n=6–10).
(G) Quantification of PDX1+NKX6.1+ cells at PP2 stage (n=6–10).
(H) Model showing GATA gene dosage affects early and late pancreatic progenitor formation.
All differentiation data in this figure were generated from HUES8 lines using the 2nd-generation differentiation protocol. See also Figures S1, S5, S6 and Tables S2 and S4.
In both H1 and HUES8 backgrounds, GATA6−/+ hPSCs formed reduced numbers of NKX6.1+PDX1+ PP2 cells compared to the corresponding isogenic WT control cells (Figures 4E and S3H). These findings predict that GATA6 heterozygous patients would form a smaller pancreas with reduced number of functional β cells. To explore potential genetic modifying factors, we investigated how GATA4 gene dosage could influence the GATA6 haploinsufficiency phenotype. This is based on the co-expression of GATA4 and GATA6 at the DE and PP stages (Figures 1, 4 and S2), as well as the reported GATA4 and GATA6 heterozygous patient phenotypes mentioned above. We first analyzed clonal GATA4−/− and GATA4−/+ HUES8 mutant lines (2 of each genotype, Tables S1 and S2, Figures S1D–S1F). Unlike the DE phenotype observed in GATA6−/− mutants, homozygous deletion of GATA4 had no significant impact on endoderm formation based on FACS and RT-qPCR analysis (Figures 6C, 6D and S6A), but it led to a dramatic reduction in the number of PDX1+ PP1 cells (Figures 6C and 6E). These findings support a direct requirement for GATA4 in the differentiation of endoderm to nascent pancreatic progenitor cells. Heterozygous deletion of GATA4 had similar effects as observed in GATA6−/+ cells: fewer PDX1+NKX6.1+ PP2 cells were formed compared to WT cells. These results suggest that additional factors likely contribute to the development of pancreatic agenesis in heterozygous GATA4 patients as with the heterozygous GATA6 patients.
Based on the reduction of PDX1+ PP1 cells formed from GATA4−/− hPSCs, we postulated that reducing the dosage of GATA4 could modify the GATA6−/+ mutant phenotype. Deleting one or both GATA4 alleles in GATA6−/+ hPSCs did not significantly affect the formation DE cells compared to WT (Figures 6C, 6D and S6A). However, GATA6−/+GATA4−/+ hPSCs formed a reduced number of PDX1+ cells at both PP1 and PP2 stages, a phenotype not observed in either GATA6−/+ or GATA4−/+ mutants (Figures 6C, 6E and 6F); and GATA6−/+GATA4−/− hPSCs failed to form a significant number of PDX1+ cells (Figures 6C, 6E and 6F). Corresponding defects were observed in the formation of PDX1+NKX6.1+ PP2 cells (Figures 6C and 6G), likely resulting from earlier defects at the PP1 stage. We also observed similar dosage sensitive effects at the PP1 stage using the 1st-generation protocol (Figures S6B–S6D), supporting the utility of both protocols for studying early differentiation events. These data demonstrate an exquisite sensitivity of human pancreas development to GATA6/4 dosage (Figure 6H), and suggest that the severity of the disease phenotype in heterozygous GATA6 or GATA4 patients could be influenced by genetic, epigenetic or environmental factors that affect the abundance or activity of the remaining WT GATA4 or GATA6 proteins.
Discussion
hPSCs offer a unique opportunity for studying disease phenotypes that are not readily recapitulated in model organisms. This is particularly relevant for haploinsufficient disease genes. The current work has revealed dosage-sensitive requirements for GATA6 in lineage decisions leading to the formation of pancreatic progenitors and functional β cells. Although our work is focused on the pancreatic lineage, it is likely that additional endoderm-derived cell types are affected. Some GATA6 heterozygous patients show defects in endodermal organs such as the gall bladder, intestine and liver (Lango Allen et al., 2012), and these phenotypes could be studied through similar hPSC-based approaches. On the other hand, there are limitations to the current strategy. The differentiation system may mask non-cell autonomous requirements for GATA6. For example, Sussel and colleagues find that GATA6 inhibits the expression of secreted Hedgehog ligands in mice (Xuan and Sussel, 2016), whereas the application of Hedgehog inhibitor in the hPSC differentiation media will probably mask any consequence of such an effect. Future studies will likely benefit from developing 3D organoid culture systems, which may enable the study of the interactions between different pancreatic cell types including the interplay with possible niche signals. Along this line, additional roles of GATA6 remain to be investigated. For instance, GATA6 haploinsufficiency may impair the expansion and maintenance of pancreatic progenitor cells during the long embryonic development time window, a process not yet fully recapitulated in the current study. GATA6 dosage may influence the β cell mass, function, and its susceptibility to adverse environmental stress, which could be further investigated in xenograft models after transplantation of hPSC-derived β-like cells into mice.
There has been much speculation concerning why mouse models do not recapitulate phenotypes of human mutations, especially for haploinsufficient disease genes (Bruneau, 2008; Seidman and Seidman, 2002; Theodoris et al., 2015; Wilkie, 2003). In hPSCs, deleting GATA6 impaired DE formation, deleting GATA4 impaired PP1 formation, and deleting one allele of either GATA6 or GATA4 reduced the number of PDX1+NKX6.1+ PP2 cells. These findings highlight overlapping and distinct roles of GATA4 and GATA6 during human endoderm and pancreatic development, and demonstrate phenotypes not previously reported in mice (Carrasco et al., 2012; Izumi et al., 2007; Morrisey et al., 1998; Xuan et al., 2012; Zhao et al., 2008). For instance, no pancreatic defects have been uncovered in Gata6−/+ or Gata4−/+ mouse embryos or adults. In addition to distinct gene dosage sensitivities between humans and mice, a number of other factors may also contribute to the different observations between mouse and hPSC models. In contrast to inbred laboratory mouse strains, the human genetic diversity may underlie the diagnosis of extreme conditions such as pancreatic agenesis in some but not all GATA6 or GATA4 heterozygous patients. The different timing of the deletion could also contribute to some of the different phenotypes observed between the mouse and hPSC systems. Due to early lethality of Gata4 and Gata6 knockout embryos associated with extraembryonic defects (Kuo et al., 1997; Morrisey et al., 1998), the Pdx1-Cre or Foxa3-Cre driver was used to conditionally inactivate Gata6/4 in early PP cells or the gut endoderm just before PP specification (Carrasco et al., 2012; Xuan et al., 2012). In contrast, embryonic lethality is not a concern in hPSC differentiation cultures, enabling the deletion of GATA6 or GATA4 before any differentiation is initiated. Despite these differences, GATA6/4 interactions are observed in both mouse and hPSC models, supporting the complementary utility of both approaches for investigating genetic and environmental modifiers of human disease.
Thus far hPSCs have been used for study of either global genetic effects by comparing patient-derived hPSCs to control cells, or the effects of a single genetic locus on disease phenotypes through gene editing. Our findings support an important yet less appreciated role of hPSCs for interrogating complex genetic interactions, which has broad implications for investigation of multigenic human traits. Unlike in mice, there is no allelic segregation in hPSCs, thus it is possible to interrogate complex genetic interactions with relative ease. Our GATA6/4 compound mutant analysis suggests that patients with GATA6 or GATA4 heterozygous mutations are likely susceptible to even subtle decreases in the expression or activity of the remaining GATA6/4 factors that could be caused by the environment, background genetic modifiers or other stochastic events. The findings offer a plausible explanation for the wide range of clinical phenotypes observed in individuals carrying GATA6 or GATA4 heterozygous mutations, and suggest potential treatment options through augmenting the expression or activity of GATA6/4 or their downstream targets, a strategy that may be extended to diabetic patients with WT GATA6/4 sequences but misregulated gene expression. For instance, retinoic acid has been shown to increase Gata6 expression in macrophages (Okabe and Medzhitov, 2014), and FGF signaling is important for maintaining GATA6 expression in primitive endoderm (Kang et al., 2013). Independent work from Gadue and colleagues suggests that the retinoic acid dosage influences GATA6/4 expression in GATA6−/+ cells and affects the formation of pancreatic progenitor cells (Tiyaboonchai et al., 2017). Thus one may explore these pathways for the development of a therapeutic approach such as in the form of vitamin A supplements. The GATA6 or GATA4 heterozygous patient phenotypes may also be influenced by yet-unidentified factors, and the platform established in the current study may be further utilized for discovering novel disease-causing or -modifying factors through a candidate approach or unbiased chemical or genetic screens.
STAR METHODS
KEY RESOURCES TABLE
See the Table in a separate file.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to the corresponding author, Dr. Danwei Huangfu (huangfud@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Line and Maintenance
Human embryonic stem cell lines HUES8 and H1 (NIH approval number NIHhESC-09-0021 and NIHhESC-10-0043) were maintained in KSR/iMEF condition and/or E8 condition and used for genome editing and pancreatic differentiation. All experiments involving hPSCs were approved by the Tri-SCI Embryonic Stem Cell Research Oversight Committee (ESCRO).
Mouse Strain
Immunocompromised SCID-Beige male mice (CB17.Cg-PrkdcscidLystbg-J/Crl) purchased from Charles River Laboratories were used for transplantation studies under a protocol approved by the MSKCC Institutional Animal Care and Use Committee (IACUC).
METHOD DETAILS
hPSC Culture
Experiments in this study were performed using two well-characterized human embryonic stem cell lines HUES8 and H1 (NIH approval number NIHhESC-09-0021 and NIHhESC-10-0043). Cells were maintained at 37 °C with 5% CO 2, and regularly confirmed to be mycoplasma-free by the MSKCC Antibody & Bioresource Core Facility.
Undifferentiated hPSCs were maintained in either feeder-dependent condition or chemically defined E8 culture condition. Briefly, for the feeder-dependent condition, hPSCs were cultured on irradiated mouse embryonic fibroblast (iMEF) feeder cells (seeded ~3.75×104 cells/cm2) in hPSC medium consisting of DMEM/F12 (Invitrogen, #11320-082), 20% KnockOut Serum Replacement (KSR) (Invitrogen, #10828028), 1X GlutaMAX (GIBCO, #35050-061), 1X non-essential amino acids (NEAA) (GIBCO, #11140-050), 0.05 mM β-mercaptoethanol (Invitrogen, #21985023) and penicillin/streptomycin (Gemini, #400-109) supplemented with 10 ng/ml bFGF (Invitrogen, #PHG0263). The medium was changed every day, and cells were passaged every ~4 days using TrypLE Select (1X, Invitrogen, #12563-029). For E8 culture condition, hPSCs were cultured on vitronectin (VTN-N, GIBCO, # A14700) pre-coated plates in complete Essential 8 (E8) medium (GIBCO, # A1517001). Medium was changed every day, and cells were passaged every ~4 days using 0.5 mM EDTA (KD Medical, #RGE-3130) to dissociate cells. In all normal hPSC cultures, 5 μM Rho-associated protein kinase (ROCK) inhibitor Y-27632 (Selleck Chemicals, #S1049) was only added into the culture media when passaging or thawing hPSCs.
Generation of Clonal hPSC Mutant Lines
gRNA or gRNA+ssDNA transfection in iCas9 hPSCs
All mutant lines were generated using the iCRISPR platform we previously established (González et al., 2014). The generation of HUES8 iCas9 hPSCs was described previously. H1 iCas9 hPSCs were generated using a modified approach (Figure S3A). iCas9 hPSCs support doxycycline-inducible Cas9 expression for efficient gene editing. One day before gRNA transfection, iCas9 hPSCs were treated with doxycycline (2 μg/ml) (Sigma, #D9891). Cells were then dissociated using Accutase (STEMCELL Technologies, #07920) or TrypLE select, replated onto iMEF feeder plates for HUES8 lines and VTN-N-coated plates for H1 lines and transfected in suspension with gRNAs or gRNA+ssDNA using Lipofectamine RNAiMAX (Invitrogen, #13778-150) following manufacturer’s instructions. gRNA and ssDNA were added at 10 nM and 20 nM final concentration, respectively. gRNAs or gRNA+ssDNA and Lipofectamine RNAiMAX were diluted separately in Opti-MEM (GIBCO, #31985070), mixed together, incubated for 5 min at room temperature (R.T.), and added dropwise to the iCas9 hPSCs just plated. A second transfection was performed the next day. The gRNA transfection efficiency is estimated to be ~70–80% based on transfection of control fluorescence-labeled dsRNA (Zhu et al., 2015).
Establishment of clonal hPSC mutant lines
Two days after the last gRNA or gRNA+ssDNA transfection, hPSCs were dissociated into single cells and replated at a low density (1,000–2,000 cells per 10cm dish). The rest of cells were collected and genomic DNA was extracted using DNeasy kit (Qiagen, #69506). T7 endonuclease I assay was then performed to assess the CRISPR targeting efficiency. The seeded cells were allowed to grow and form colonies from single cells. Medium was changed every 2–3 days. ~10 days later, individual colonies were picked, mechanically disaggregated and replated into individual wells of 96-well plates pre-seeded with the iMEF feeder or coated with VTN-N for hPSCs maintained in KSR/iMEF or E8 condition, respectively. Clonal lines were expanded. A portion of the cells was lysed by 1 mg/ml proteinase K (Roche, #50-720-3027) in 1X JumpStart PCR buffer (Sigma, #P2192) overnight at 56 °C to release genomic DNA followed by 10 min at 99 °C to inactivate proteinase K. Then PCR was performed using Herculase II Fusion DNA Polymerase (Agilent Technologies, #600679) followed by Sanger sequencing (primers provided in Table S1) to identify mutant clones. For biallelic frameshift mutants, we chose homozygous mutants over compound heterozygous mutants since homozygous mutations are easier to characterize. Clonal cell lines carrying desired mutations were further expanded and frozen down. For heterozygous mutants and compound heterozygous mutants, sub-cloning of cells and TOPO cloning (Invitrogen, #450245) of PCR products were performed to exclude potential contamination of cells with different genotypes (e.g. mixing of homozygous mutant cells with WT cells). WT clonal lines from the corresponding targeting experiments were included as WT controls to account for potential nonspecific effects associated with the targeting process. All clonal lines used in this study were listed in Table S2. During differentiation experiments, all lines were re-sequenced to confirm the correct genotypes.
Specifics on the targeting of various loci
gRNA target sequences are listed in Table S1. For GATA6, we targeted sequences corresponding to the C-terminal zinc finger DNA binding domain, which is shared by both the long and short isoforms of GATA6. For GATA4, we targeted sequences corresponding to the transactivation domain. GATA6 and GATA4 single mutants were generated using the same approach as previously described (González et al., 2014). The efficiencies for generating GATA6 mutants were 82% for biallelic and 6.4% for monoallelic. The efficiencies for generating GATA4 mutants were 67% for biallelic and 6.8% for monoallelic. The GATA6 and GATA4 compound mutants used in this study were generated through sequential targeting, which allowed us to efficiently generate all desired genotype configurations through screening a relatively small number of clones. WT clonal lines from the corresponding targeting experiments were used as controls.
We have previously also generated GATA6R456C/R456C mutant lines, but those experiments failed to generate GATA6R456C/+ mutants, as all mutant lines carrying only one GATA6R456C allele harbor additional mutations in the GATA6R456C allele or the other allele. Here we used a 110-nt ssDNA HDR template with a silent mutation (G>A) in the PAM sequence in addition to the c.1366C>T mutation corresponding to the R456C amino acid substitution (Table S1). This change should prevent additional cleavage of the correctly edited allele with the GATA6R456C mutation after homologous recombination, and we were able to generate both GATA6R456C/R456C and GATA6R456C/+ mutant lines with high efficiencies (8 and 4 clonal lines, respectively, out of 96 clones screened in HUES8 cells, similar efficiencies were obtained for H1 cells) (Figure 3 and Table S2).
hPSC Definitive Endoderm and Pancreatic Lineage Differentiation
In all differentiation assays, mutants were analyzed in parallel with isogenic WT controls (generated from the same targeting experiments). All differentiation experiments were repeated at least 3 times independently on at least 2 lines of the same genotype. In the 1st-generation differentiation protocol, hPSCs were differentiated to the definitive endoderm (DE) stage and PDX1+ early pancreatic progenitor (PP1) stage as previously described (Zhu et al., 2016). Briefly, hPSCs from KSR/iMEF condition were plated on plates pre-seeded with the iMEF feeder (~9×104 cells/cm2) and grown for 2 days to reach ~50% confluence at which point directed differentiation to DE was initiated (designated as d0). The cells were first treated with 100 ng/ml of activin A (PeproTech, #120-14E) and 2 μM of BIO-acetoxime (Tocris, #3874) in Advanced RPMI (A-RPMI) or RPMI (GIBCO, #12633020 or #11875-093) medium containing 1X GlutaMAX and 0.005% BSA (GIBCO, #15260-037). On day 1 and day 3, the medium was changed to activin A in A-RPMI or RPMI containing 1X GlutaMAX and 0.2% FBS (Sigma, #F2442). The efficiency of DE differentiation was analyzed at day 4 or day 5 (the DE stage) by flow cytometry, immunostaining and/or RT-qPCR. Differentiation to PDX1+ PP1 differentiation was performed following a 4-day DE differentiation. Briefly, cells were treated with 50 ng/ml FGF10 and 0.25 μM SANT-1 with 2% FBS in A-RPMI medium followed by 50 ng/ml FGF10, 0.25 μM SANT-1, 2 μM retinoic acid and 250 nM LDN-193189 in DMEM medium with 1% B27, 0.005% BSA and 1X GlutaMAX and maintained in the same medium condition for 4 days to reach the PP1 stage. The medium was change every two days during PP1 differentiation.
In the 2nd-generation differentiation protocol, hPSCs were differentiated to DE, PP1, and subsequently the PDX1+NKX6.1+ pancreatic progenitor (PP2) cells and β-like cells following published studies (Pagliuca et al., 2014; Rezania et al., 2014) with some modifications as described in details in our recent publication (Zhu et al., 2016). To initiate differentiation, 70–80% confluent hPSCs in E8 condition were dissociated into single cells using 1X TrypLE Select and replated at ~75,000 cells/cm2 in E8 medium supplemented with 5 μM Y-27632 in VTN-N-coated plates. Media were changed after 24 hours and differentiation was initiated 48 hours after plating when the culture was ~75% in confluency (designated as “d0”). The detailed differentiation stages and media recipes are presented in Figure 4A and Table S3.
Immunofluorescence Staining
For immunofluorescence staining, 2-dimensional planar cultured cells were fixed in 4% paraformaldehyde in PBS for 10 min at R.T., washed once with PBS and permeabilized in PBS with 0.2% Triton (PBST) for 30 min. Blocking was done for 30 min at R.T. with blocking solution (5% donkey serum in PBST). For the cell aggregates cultured on air-liquid interface, at β-like stage, the aggregates were picked and fixed overnight at 4 °C and washed with PBS. Some of aggregates were embedded in optimum cutting temperature (O.C.T.) for further analysis using cryostat sectioning. For transplants from mouse kidney capsules, graft samples were harvested ~4 months after kidney capsule injection. After fixation with 4% paraformaldehyde in PBS for overnight, samples embedded in O.C.T reagent and cryostat sectioning was performed. Frozen blocks were sectioned in 12-μm thickness and stored in −20C. Before staining, all samples were permeabilized followed by blocking for 30 min. Antibody incubation for sectioned samples on slides was performed in humid chamber. Primary and second antibodies were diluted in the blocking solution. Primary antibodies were incubated at R.T. for 2 hours or overnight at 4 °C, and secondary antibodies at R.T. for 1 hour. The following primary antibodies and dilutions were used: goat anti-OCT4 (Santa Cruz, #sc-8628, 1:100), rabbit anti-FOXA2 (Millipore, #07-633, 1:200), goat anti-SOX17 (R&D, #AF1924, 0.4 μg/ml), rabbit anti-GATA6 (Cell Signaling Technology, #5851S, 1:2,000), mouse anti-GATA4 (BD, #560327, 1:100), goat anti-PDX1 (R&D, #AF2419, 0.4 μg/ml), mouse anti-NKX6.1 (DSHB # F55A12-c, 1:500), rat anti-C-peptide (DSHB, #GN-ID4-c, 1:2,000), mouse anti-Glucagon (Sigma, #G2654, 1:1000). Alexa Fluro secondary antibodies from Molecular Probes were used at 1:500. Cell nuclei were counter-stained with DAPI (Sigma, #32670-5MG-F, 0.2 μg/ml). Images were taken using a Zeiss Axio Observer microscope for cell culture plates Confocal Laser Scanning Platform Leica TCS SP8 for cell aggregates and Leica TCS SP5 for tissue slides. Z-stacks were collected with a 0.3 micron step size and the projections images are shown. Scale bars indicate 100 μm for all the images in this study.
Flow Cytometry
Cells in 2D adherent cultures were dissociated using 1X TrypLE Select. Cell aggregates cultured in air-liquid interface were picked and dissociated using Collagenase IV (10 mg/ml) for 5–10 min followed by 1X TrypLE for 5–10 min at 37°C. Cells were then collected and washed with cold FACS buffer (2–5% FBS in 1x PBS). Cell surface marker (CXCR4-APC from R&D, #FAB170A, 1:25) staining and live/dead staining (LIVE/DEAD Fixable Violet Dead Cell Stain Kit from Molecular Probes, #L34955, 1:1000) were then performed for 30 min on ice in FACS buffer. Intracellular staining was performed with Foxp3 Staining Buffer Set (eBioscience, #00-5523-00). Fixation and permeabilization was carried out on ice for 1 hour and followed by antibody staining: rabbit anti-GATA6 (Cell Signaling Technology, #5851S, 1:250), mouse anti-GATA4 (BD 560327, 1:100), mouse anti-GATA4-AF647 (BD 560400, 1:50), SOX17-PE (BD Biosciences, #561591, 1:50), goat anti-PDX1 (R&D, #AF2419, 1:250), mouse anti-NKX6.1 (DSHB # F55A12-c, 1:250), rat anti-C-peptide (DSHB, #GN-ID4-c, 1:500), mouse anti-Glucagon (Sigma, #G2654, 1:250), rabbit anti-Somatostatin (DAKO, #A0566, 1:250). Cleaved Caspase-3 (Cell Signaling Technology, #9661, 1:250), Phospho-Histone H3 (Cell Signaling Technology, #9701, 1:50). For quantification of median fluorescence intensity (MFI) of GATA6 or GATA4, the GATA6+ or GATA4+ population was first gated and then MFI was calculated using FlowJo. The mean of MFI value of each sample was normalized to the mean of all the WT controls processed in the same FACS experiment. Normalized MFI data from clonal lines of the same genotype from multiple independent experiments were then combined for calculation of the average MFI.
Western Blotting
Cells were harvested at DE and PP stages and lysed using cell lysis buffer (Cell Signaling Technology, #9803) containing protease inhibitor (Roche, #05892791001) and 1mM PMSF (MP Biomedicals, #ICN19538105). Samples preparation complied with NuPAGE® Novex protocol and samples were separated by 10% Bis-Tris Gel (Novex, #NP0301BOX). The protein samples were then transferred to Nitrocellulose Pre-Cut Blotting Membranes (Novex, #LC2001) followed by blocking with 5% milk in Tris-based saline with Tween 20 (0.1% TBST) buffer for 1 hour at R.T.. The membrane was incubated with primary antibodies overnight at 4°C, followed by incubation with HRP conjugated secondary antibodies at R.T. for 1 hour. ECL western blotting detection reagent (Amersham, #RPN2236) was used to visualize the protein bands. The following antibodies were used with the dilution ratio noted: rabbit anti-GATA6 (Cell Signaling Technology, #5851S, 1:1,000), mouse anti-GATA4 (BD, #560327, 1:100), goat anti-PDX1 (R&D, #AF2419, 1:1,000), mouse anti-ACTB (β-actin) (Cell Signaling Technology, #3700S, 1:10,000)
RNA Isolation and RT-qPCR
Cells were lysed in TRIzol (Invitrogen, #15596018) and total RNA was isolated using the miRNeasy Mini Kit (Qiagen, #217004). Then cDNA was produced using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, #4368814). Quantitative real-time PCR was performed in triplicate using Power SYBR® Green PCR Master Mix (Applied Biosystems, #4368708) or ABsolute Blue QPCR SYBR Green Mix with low ROX (Thermo Scientific, # AB4322B) on the ABI PRISM® 7500 Real Time PCR System (Applied Biosystems) using the following protocol: 12 minutes at 95°C followed by 40 cycles of 20 seconds at 95°C, 30 seconds at 55°C, and 30 seconds at 70°C. The signal was det ected at 70°C. All primers used for RT-qPCR were listed in Table S5.
RNA-Seq and Functional Annotation
RNA samples from two GATA6−/+ mutant lines and two wild-type lines were collected at the PP2 stage using the above-mentioned RNA isolation method. RNA sequencing was performed by the MSKCC Integrated Genomics Operation Core and data processing and differential expression analysis was done by the MSKCC Bioinformatics Core. The differential expressed genes were determined based on both the adjusted P values (Padj.<0.05, using the Benjamini-Hochberg correction to account for the number of genes) and the mean coverage (at least one group (WT or GATA6−/+) in the comparison must have a mean count per gene equals to at least 15). Transcription functional annotation was performed with DAVID, using Gene Ontology (GO) biological process. We sorted GO terms by Benjamini value.
Glucose-Stimulated Insulin Secretion
Glucose stimulated insulin secretion (GSIS) assay was performed following previously described protocols (Zhu et al., 2016). Briefly, ~8 cell aggregates (~5 million cells in total) at β-like stage (S7) were picked using wide orifice pipet tip into an Eppendorf tube and rinsed three times with the KRBH buffer (129 mM NaCl, 4.8 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 5 mM NaHCO3, 10 mM HEPES, 0.1% BSA in ddH2O and sterile filtered). Cell aggregates were equilibrated in KRBH at 37°C for 1 hour. Cell aggregates were then incubated in KRBH spiked with 2.8 mM Glucose at 37°C for 30 m in and supernatants were collected. Next, cell aggregates were rinsed three times with KRBH, incubated in KRBH spiked with 16.7 mM Glucose at 37°C for 30 min and supernatants were co llected again. This sequence was then repeated again. At the end of the experiment, cell aggregates were dissociated into single cells using collagenase IV and TrypLE Select treatment, and the cell numbers were counted. Mercodia Ultrasensitive Human C-peptide ELISA kit (Mercodia, # 10-1141-01) was used to measure the C-peptide content in supernatant samples following manufacturer’s instruction.
Transplantation Studies
hPSC-derived cell aggregates at ~10 days after PP2 stage were loaded into catheter for cell delivery under the kidney capsule of the SCID-Beige mice (~3 million cells per animal). To perform in vivo GSIS, mice were fasted for 16–18 hours overnight one month after transplantation. Mouse blood was collected from mouse tail veins under fasting condition and at 30 min after intraperitoneal (IP) injection with 3 g D-(+)-glucose/1 kg body weight. Human insulin levels in the mouse sera were quantified right after blood collection using the Human Ultrasensitive Insulin ELISA (ALPCO Diagnostics, # 80-INSHUU-E01.1). The grafts were removed from mouse kidney capsules at ~ 4 months after transplantation for immunostaining analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification data are presented as mean ± SEM for all the pooled data. Data from clonal lines of the same genotype were combined for calculating the significance of the differences between different genotypes. To directly compare two groups, t-test with two-tailed distribution and two-sample equal variance was used in Microsoft Excel and the equal variance was checked by F test. When more than two groups are compared, one-way ANOVA is used followed by multiple comparisons with Bonferroni correction using Prism 6. Exceptions are noted in the corresponding figure legends. P <0.05, 0.01 and 0.001 are indicated with 1, 2, and 3 asterisks, respectively.
DATA AND SOFTWARE AVAILABILITY
RNA-Seq data has been deposited in GEO under accession number GSE92581.
Supplementary Material
Highlights.
GATA6 deletion impairs differentiation of definitive endoderm from hPSCs
A patient-specific GATA6 Arg456Cys mutation has similar effects to GATA6 deletion
GATA6 haploinsufficiency impairs pancreatic progenitor formation
GATA4 dosage affects the GATA6+/− phenotype highlighting disease-modifying effects
Acknowledgments
We thank Drs. Licia Selleri, Wenjun Guo and members of the Huangfu laboratory for insightful discussions and critical reading of the manuscript. The mouse anti-NKX6.1 and rat anti-C-Peptide antibodies were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242. This study was funded in part by New York State Stem Cell Science (NYSTEM C029567 and C029156), NIH/NIDDK (R01DK096239), NIH/NIDDK (1 DP2 DK098093-01), Tri-Institutional Stem Cell Initiative (#2016-004), and the MSKCC Cancer Center Support Grant (P30 CA008748). K.L. was supported by the Korean Government Scholarship Program for Study Overseas and Mogam Science Scholarship Foundation. N.V. was supported by the Howard Hughes Medical Institute (HHMI) Medical Research. Z.Z. was supported by the NYSTEM postdoctoral fellowship from the Center for Stem Cell Biology of the Sloan Kettering Institute.
Footnotes
Supplemental information includes 6 figures and 5 tables and can be found with this article online at (to be filled by the journal editor)
Author Contributions
D.H., Z.-D.S., and K.L. designed experiments, analyzed and interpreted results. Z.-D.S., K.L. and D.Y. generated mutant lines and performed key experiments. Z.-D.S., K.L., S.A., and S.C. performed mouse transplantation and analysis. N.V., Q.V.L., Z.Z., C.-L.S., and R.K. performed additional necessary experiments. D.H., S.C. and T.E. supervised the work. D.H., Z.-D.S. and K.L. wrote the manuscript; all other authors provided editorial advice.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ang YS, Rivas RN, Ribeiro AJ, Srivas R, Rivera J, Stone NR, Pratt K, Mohamed TM, Fu JD, Spencer CI, et al. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell. 2016;167:1734–1749. doi: 10.1016/j.cell.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DL, Chen Y, Kim G, Guo L, Chen L. Crystal structures of multiple GATA zinc fingers bound to DNA reveal new insights into DNA recognition and self-association by GATA. J Mol Biol. 2008;381:1292–1306. doi: 10.1016/j.jmb.2008.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefond A, Sand O, Guerin B, Durand E, De Graeve F, Huyvaert M, Rachdi L, Kerr-Conte J, Pattou F, Vaxillaire M, et al. GATA6 inactivating mutations are associated with heart defects and, inconsistently, with pancreatic agenesis and diabetes. Diabetologia. 2012;55:2845–2847. doi: 10.1007/s00125-012-2645-7. [DOI] [PubMed] [Google Scholar]
- Bruneau BG. The developmental genetics of congenital heart disease. Nature. 2008;451:943–948. doi: 10.1038/nature06801. [DOI] [PubMed] [Google Scholar]
- Bui PH, Dorrani N, Wong D, Perens G, Dipple KM, Quintero-Rivera F. First report of a de novo 18q11.2 microdeletion including GATA6 associated with complex congenital heart disease and renal abnormalities. Am J Med Genet A. 2013;161A:1773–1778. doi: 10.1002/ajmg.a.35974. [DOI] [PubMed] [Google Scholar]
- Carrasco M, Delgado I, Soria B, Martin F, Rojas A. GATA4 and GATA6 control mouse pancreas organogenesis. J Clin Invest. 2012;122:3504–3515. doi: 10.1172/JCI63240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catli G, Abaci A, Flanagan SE, De Franco E, Ellard S, Hattersley A, Guleryuz H, Bober E. A novel GATA6 mutation leading to congenital heart defects and permanent neonatal diabetes: a case report. Diabetes Metab. 2013;39:370–374. doi: 10.1016/j.diabet.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Chao CS, McKnight KD, Cox KL, Chang AL, Kim SK, Feldman BJ. Novel GATA6 mutations in patients with pancreatic agenesis and congenital heart malformations. PLoS One. 2015;10:e0118449. doi: 10.1371/journal.pone.0118449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amato E, Giacopelli F, Giannattasio A, D’Annunzio G, Bocciardi R, Musso M, Lorini R, Ravazzolo R. Genetic investigation in an Italian child with an unusual association of atrial septal defect, attributable to a new familial GATA4 gene mutation, and neonatal diabetes due to pancreatic agenesis. Diabet Med. 2010;27:1195–1200. doi: 10.1111/j.1464-5491.2010.03046.x. [DOI] [PubMed] [Google Scholar]
- D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. 2006;24:1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- Dang VT, Kassahn KS, Marcos AE, Ragan MA. Identification of human haploinsufficient genes and their genomic proximity to segmental duplications. Eur J Hum Genet. 2008;16:1350–1357. doi: 10.1038/ejhg.2008.111. [DOI] [PubMed] [Google Scholar]
- De Franco E, Shaw-Smith C, Flanagan SE, Shepherd MH, International NDMC, Hattersley AT, Ellard S. GATA6 mutations cause a broad phenotypic spectrum of diabetes from pancreatic agenesis to adult-onset diabetes without exocrine insufficiency. Diabetes. 2013;62:993–997. doi: 10.2337/db12-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker K, Goldman DC, Grasch CL, Sussel L. Gata6 is an important regulator of mouse pancreas development. Dev Biol. 2006;298:415–429. doi: 10.1016/j.ydbio.2006.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eifes S, Chudasama KK, Molnes J, Wagner K, Hoang T, Schierloh U, Rocour-Brumioul D, Johansson S, Njolstad PR, de Beaufort C. A novel GATA6 mutation in a child with congenital heart malformation and neonatal diabetes. Clin Case Rep. 2013;1:86–90. doi: 10.1002/ccr3.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong M, Simaite D, Kuhnen P, Heldmann M, Spagnoli F, Blankenstein O, Hubner N, Hussain K, Raile K. Two novel GATA6 mutations cause childhood-onset diabetes mellitus, pancreas malformation and congenital heart disease. Horm Res Paediatr. 2013;79:250–256. doi: 10.1159/000348844. [DOI] [PubMed] [Google Scholar]
- González F, Zhu Z, Shi ZD, Lelli K, Verma N, Li QV, Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell stem cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi N, Era T, Akimaru H, Yasunaga M, Nishikawa S. Dissecting the molecular hierarchy for mesendoderm differentiation through a combination of embryonic stem cell culture and RNA interference. Stem Cells. 2007;25:1664–1674. doi: 10.1634/stemcells.2006-0681. [DOI] [PubMed] [Google Scholar]
- Jennings RE, Berry AA, Kirkwood-Wilson R, Roberts NA, Hearn T, Salisbury RJ, Blaylock J, Piper Hanley K, Hanley NA. Development of the human pancreas from foregut to endocrine commitment. Diabetes. 2013;62:3514–3522. doi: 10.2337/db12-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Piliszek A, Artus J, Hadjantonakis AK. FGF4 is required for lineage restriction and salt-and-pepper distribution of primitive endoderm factors but not their initial expression in the mouse. Development. 2013;140:267–279. doi: 10.1242/dev.084996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly OG, Chan MY, Martinson LA, Kadoya K, Ostertag TM, Ross KG, Richardson M, Carpenter MK, D’Amour KA, Kroon E, et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat Biotechnol. 2011;29:750–756. doi: 10.1038/nbt.1931. [DOI] [PubMed] [Google Scholar]
- Koutsourakis M, Langeveld A, Patient R, Beddington R, Grosveld F. The transcription factor GATA6 is essential for early extraembryonic development. Development. 1999;126:723–732. [PubMed] [Google Scholar]
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, Soudais C, Leiden JM. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:1048–1060. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- Lango Allen H, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, Ferrer J, Hattersley AT, Ellard S. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2012;44:20–22. doi: 10.1038/ng.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean AB, D’Amour KA, Jones KL, Krishnamoorthy M, Kulik MJ, Reynolds DM, Sheppard AM, Liu H, Xu Y, Baetge EE, et al. Activin a efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3-kinase signaling is suppressed. Stem Cells. 2007;25:29–38. doi: 10.1634/stemcells.2006-0219. [DOI] [PubMed] [Google Scholar]
- Molkentin JD. The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem. 2000;275:38949–38952. doi: 10.1074/jbc.R000029200. [DOI] [PubMed] [Google Scholar]
- Morrisey EE, Tang Z, Sigrist K, Lu MM, Jiang F, Ip HS, Parmacek MS. GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev. 1998;12:3579–3590. doi: 10.1101/gad.12.22.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K. Genome editing of human pluripotent stem cells to generate human cellular disease models. Dis Model Mech. 2013;6:896–904. doi: 10.1242/dmm.012054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nostro MC, Sarangi F, Yang C, Holland A, Elefanty AG, Stanley EG, Greiner DL, Keller G. Efficient generation of NKX6-1+ pancreatic progenitors from multiple human pluripotent stem cell lines. Stem cell reports. 2015;4:591–604. doi: 10.1016/j.stemcr.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe Y, Medzhitov R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell. 2014;157:832–844. doi: 10.1016/j.cell.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240:530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- Pan Y, Ouyang Z, Wong WH, Baker JC. A new FACS approach isolates hESC derived endoderm using transcription factors. PLoS One. 2011;6:e17536. doi: 10.1371/journal.pone.0017536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patient RK, McGhee JD. The GATA family (vertebrates and invertebrates) Curr Opin Genet Dev. 2002;12:416–422. doi: 10.1016/s0959-437x(02)00319-2. [DOI] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. 2014;32:1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Russ HA, Parent AV, Ringler JJ, Hennings TG, Nair GG, Shveygert M, Guo T, Puri S, Haataja L, Cirulli V, et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015;34:1759–1772. doi: 10.15252/embj.201591058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryffel GU. Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor (HNF)1 and HNF4 families: functional and pathological consequences. J Mol Endocrinol. 2001;27:11–29. doi: 10.1677/jme.0.0270011. [DOI] [PubMed] [Google Scholar]
- Seidman JG, Seidman C. Transcription factor haploinsufficiency: when half a loaf is not enough. J Clin Invest. 2002;109:451–455. doi: 10.1172/JCI15043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw-Smith C, De Franco E, Lango Allen H, Batlle M, Flanagan SE, Borowiec M, Taplin CE, van Alfen-van der Velden J, Cruz-Rojo J, Perez de Nanclares G, et al. GATA4 mutations are a cause of neonatal and childhood-onset diabetes. Diabetes. 2014;63:2888–2894. doi: 10.2337/db14-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanescu DE, Hughes N, Patel P, De Leon DD. A novel mutation in GATA6 causes pancreatic agenesis. Pediatr Diabetes. 2015;16:67–70. doi: 10.1111/pedi.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanger BZ, Tanaka AJ, Melton DA. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature. 2007;445:886–891. doi: 10.1038/nature05537. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Nakao A, Sarhat AR, Furuya A, Matsuo K, Tanahashi Y, Kajino H, Azuma H. A case of pancreatic agenesis and congenital heart defects with a novel GATA6 nonsense mutation: evidence of haploinsufficiency due to nonsense-mediated mRNA decay. Am J Med Genet A. 2014;164A:476–479. doi: 10.1002/ajmg.a.36275. [DOI] [PubMed] [Google Scholar]
- Taylor BL, Liu FF, Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. 2013;4:1262–1275. doi: 10.1016/j.celrep.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoris CV, Li M, White MP, Liu L, He D, Pollard KS, Bruneau BG, Srivastava D. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell. 2015;160:1072–1086. doi: 10.1016/j.cell.2015.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallier L, Touboul T, Chng Z, Brimpari M, Hannan N, Millan E, Smithers LE, Trotter M, Rugg-Gunn P, Weber A, et al. Early cell fate decisions of human embryonic stem cells and mouse epiblast stem cells are controlled by the same signalling pathways. PLoS One. 2009;4:e6082. doi: 10.1371/journal.pone.0006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitia RA. Exploring the etiology of haploinsufficiency. Bioessays. 2002;24:175–184. doi: 10.1002/bies.10023. [DOI] [PubMed] [Google Scholar]
- Walker EM, Thompson CA, Battle MA. GATA4 and GATA6 regulate intestinal epithelial cytodifferentiation during development. Dev Biol. 2014;392:283–294. doi: 10.1016/j.ydbio.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, Zhao R, Li J, Duncan SA. Development of the mammalian liver and ventral pancreas is dependent on GATA4. BMC Dev Biol. 2007;7:37. doi: 10.1186/1471-213X-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie AO. Why study human limb malformations? J Anat. 2003;202:27–35. doi: 10.1046/j.1469-7580.2003.00130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan S, Borok MJ, Decker KJ, Battle MA, Duncan SA, Hale MA, Macdonald RJ, Sussel L. Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J Clin Invest. 2012;122:3516–3528. doi: 10.1172/JCI63352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan S, Sussel L. GATA4 and GATA6 regulate pancreatic endoderm identity through inhibition of hedgehog signaling. Development. 2016;143:780–786. doi: 10.1242/dev.127217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorifuji T, Kawakita R, Hosokawa Y, Fujimaru R, Yamaguchi E, Tamagawa N. Dominantly inherited diabetes mellitus caused by GATA6 haploinsufficiency: variable intrafamilial presentation. J Med Genet. 2012;49:642–643. doi: 10.1136/jmedgenet-2012-101161. [DOI] [PubMed] [Google Scholar]
- Yu L, Bennett JT, Wynn J, Carvill GL, Cheung YH, Shen Y, Mychaliska GB, Azarow KS, Crombleholme TM, Chung DH, et al. Whole exome sequencing identifies de novo mutations in GATA6 associated with congenital diaphragmatic hernia. J Med Genet. 2014;51:197–202. doi: 10.1136/jmedgenet-2013-101989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev Biol. 2008;317:614–619. doi: 10.1016/j.ydbio.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Li QV, Lee K, Rosen BP, González F, Soh CL, Huangfu D. Genome Editing of Lineage Determinants in Human Pluripotent Stem Cells Reveals Mechanisms of Pancreatic Development and Diabetes. Cell stem cell. 2016;18:755–768. doi: 10.1016/j.stem.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Verma N, González F, Shi ZD, Huangfu D. A CRISPR/Cas-Mediated Selection-free Knockin Strategy in Human Embryonic Stem Cells. Stem cell reports. 2015;4:1103–1111. doi: 10.1016/j.stemcr.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.