

Abstract
BACKGROUND
The incidence of human papillomavirus (HPV)‐associated (HPV‐positive) head and neck squamous cell carcinoma (HNSCC) of the oropharynx has dramatically increased over the last decade and continues to rise. Newly diagnosed HPV‐positive HNSCCs in the United States currently outnumber any other HPV‐associated cancers, including cervical cancer. Despite introduction of the HPV vaccine, the epidemic of HPV‐positive HNSCC is expected to continue for approximately 60 years. Compared with patients who have tobacco‐associated HNSCC, those who have HPV‐positive HNSCC have better overall survival and response to treatment. Current treatment, including chemotherapy and radiation therapy, is associated with lifelong morbidity, and there are limited treatments and no curative options for patients who develop recurrent metastatic disease. Therapeutic de‐escalation (decreased radiation dose) is being tested through clinical trials; however, those studies select patients based solely on tumor and patient smoking characteristics. Mechanisms of HPV‐driven carcinogenesis in HNSCC are not well understood, which limits new therapeutic strategies and hinders the appropriate selection of patients for de‐escalation therapy.
METHODS
The authors analyzed HNSCC data from The Cancer Genome Atlas to identify molecular characteristics that correlate with outcomes and integration status of the HPV genome.
RESULTS
The current investigations identified a subset of HPV‐positive HNSCCs with mutations in the genes TRAF3 (tumor necrosis factor receptor‐associated factor 3) and CYLD (cylindromatosis lysine 63 deubiquitinase). Defects in TRAF3 and CYLD correlated with the activation of transcriptional factor nuclear factor κB, episomal HPV status of tumors, and improved patient survival.
CONCLUSIONS
Defects in TRAF3/CYLD were accompanied with the activation of nuclear factor κB signaling and maintenance of episomal HPV in tumors, suggesting that these mutations may support an alternative mechanism of HPV tumorigenesis in head and neck tumors. Cancer 2017;123:1778–1790. © 2017 The Authors. Cancer published by Wiley Periodicals, Inc. on behalf of American Cancer Society. This is an open access article under the terms of the Creative Commons Attribution NonCommercial License, which permits use, distribution and reproduction in any medium, provided the original work is properly cited and is not used for commercial purposes.
Keywords: head and neck squamous cell carcinoma, human papillomavirus (HPV), nuclear factor κB (NF‐κB), prognosis, tumor necrosis factor receptor‐associated factor 3 (TRAF3)
Short abstract
Human papillomavirus‐associated head and neck cancer tends to respond better to treatment compared with tobacco‐associated tumors; however, patients suffer severe and long‐lasting side effects. Somatic mutations in the genes TRAF3 and CYLD identified in The Cancer Genome Atlas data set are correlated with the activation of nuclear factor‐κB, define a distinct etiologic subset of head and neck cancers, and will be useful as biomarkers for predicting improved prognosis and selecting patients with human papillomavirus‐positive head and neck cancer who may be successfully treated with de‐escalating therapy.
See also pages 1695‐98.
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer in the world and carries a grim prognosis, with a 5‐year survival rate of only 63%.1, 2 In recent years, decreased tobacco consumption has paralleled falling rates of HNSCC in the United States; however, oropharyngeal squamous cell carcinoma, a major subset of HNSCC driven largely by infection with human papillomavirus (HPV) type 16 (HPV‐16), has been rapidly increasing.3 HPV‐positive and HPV‐negative HNSCC are distinct diseases with different molecular drivers, distinct risk factors and patient demographics, and different rates of response to therapy and cure.4, 5 HPV‐positive HNSCC is distinguished from HPV‐negative tumors by its occurrence in younger patients with less tobacco exposure and its association with improved treatment response and survival. Current therapy for advanced HNSCC includes combinations of primary surgical resection, cervical lymphadenectomy, radiation, or radiation given with platin drugs or cetuximab, depending on the stage, location, and pathologic features of the tumor; however, National Comprehensive Cancer Network guidelines state that HPV status should not change treatment strategies outside of clinical trials.6 Treatment for advanced, HPV‐positive HNSCC includes chemotherapy and radiation, which causes long‐term side effects, such as swallowing and speech dysfunction, neck muscle fibrosis, xerostomia, accelerated dental decay, and lymphedema. To decrease the morbidity associated with therapy, 1 goal of the head and neck oncology community is to de‐escalate therapy for HPV‐associated tumors. Currently, identification of appropriate patients for de‐escalation depends only on the absence of aggressive histologic tumor characteristics and history of minimal tobacco use. Conversely, up to 25% of patients with HPV‐positive tumors suffer recurrent or metastatic disease despite aggressive and morbid therapy. Therefore, identifying subsets of patients who have HPV‐positive head and neck cancer with better or worse prognoses will help select patients appropriate for de‐escalation therapy as well as those who need more effective therapy. Because the overall prevalence of oral HPV‐16 infection is 1% of the US population ages 14 to 69 years (6.9% prevalence of all HPV subtypes) and the majority of adults have been infected with HPV at some point in their lives, understanding the molecular mechanisms of HPV‐induced carcinogenesis is of utmost importance.7 Research defining HPV‐induced carcinogenesis has focused on cervical cancer and on the roles of the major HPV oncogenes, E6 and E7, which bind and promote the degradation of tumor suppressors p53 and retinoblastoma (Rb), respectively.8, 9 In the canonical model of HPV‐induced cervical carcinogenesis, integration of the HPV genome is a critical step required for malignant transformation. HPV integration commonly disrupts the viral E2 gene, which relieves the repression of E6 and E7 expression, resulting in much higher levels of these oncoproteins.10, 11 The role of viral integration in HPV‐positive HNSCC is less clear. Analyses of specimens in The Cancer Genome Atlas (TCGA) unexpectedly indicated that up to 30% of HPV‐positive HNSCCs lack HPV integration and instead maintain episomal HPV. HPV‐positive HNSCCs that lack integration have different gene‐expression profiles and DNA methylation patterns compared with tumors that have HPV integration12, 13; however, the clinical significance of tumors with integrated HPV compared with those that maintain episomal HPV has yet to be elucidated.
A seminal finding of the TCGA head and neck study identified tumor necrosis factor receptor‐associated factor 3 (TRAF3) as 1 of the most commonly mutated genes in HPV‐positive HNSCC.12 It is noteworthy that TRAF3 mutations are not described in HPV‐negative HNSCC, suggesting that TRAF3 inactivation is required only for HPV‐driven carcinogenesis in HNSCC. TRAF3 is a member of the TRAF family of proteins, which serve as both crucial intracellular adaptors and E3 ubiquitin ligases that mediate signaling after the activation of various receptors. Receptors that signal through TRAF proteins include those involved in inflammation, innate immune responses, and cell death, most notably: tumor necrosis factor receptors (TNFR), Toll‐like receptors (TLRs), RIG‐1‐like receptors (RLRs), and interleukin‐1 receptors (IL‐1Rs).14 These receptors initiate signaling that ultimately leads to activation of the innate immune response and nuclear factor‐κB (NF‐κB), a potent transcription factor central to the cell's control of apoptosis, inflammation, and several aspects of the immune response.14, 15 It has long been noted that NF‐κB signaling is activated in many HNSCCs.16, 17, 18, 19 It has been demonstrated that constitutive NF‐κB activation is induced by carcinogens or oncogenic viruses in patients with head and neck cancer or in cell lines.20, 21 TRAF3 is unique, in that it plays a role in negatively regulating canonical and noncanonical NF‐κB pathways while simultaneously stimulating a potent antiviral response, which is mediated through type I interferon (IFN) signaling.14, 22 Here, we report that the gene cylindromatosis lysine 63 deubiquitinase (CYLD), which, like TRAF3, inhibits NF‐κB pathways,23 is also mutated in a subset of HPV‐positive HNSCC. Mutations in TRAF3 and CYLD leading to constitutive activation of NF‐κB have been identified in other cancers, such as multiple myeloma; however, among solid tumors, inactivating TRAF3/CYLD gene defects were most common in HPV‐positive HNSCC. The absence of frequent mutations in uterine cervical cancer24, 25 provides yet another difference between these HPV‐associated tumor types.
In the current report, through in‐depth analysis of the TCGA HNSCC data set, we propose that inactivating mutations in TRAF3 or CYLD identify a distinct subset of HPV‐positive HNSCC and that this subset has constitutive activation of NF‐κB signaling. This previously undescribed subtype of HPV‐positive HNSCC marked by TRAF3 or CYLD mutations is associated with the absence of integrated HPV and improved patient survival.
MATERIALS AND METHODS
Analysis of TRAF3, CYLD, and PIK3CA Mutations
To analyze TRAF3, CYLD, and phosphatidylinositol‐4,5‐bisphosphae 3‐kinase catalytic subunit α (PIK3CA) genetic alterations in HNSCC and other types of cancer, we obtained data using the cBioPortal for Cancer Genomics (available at: www.cbioportal.org).26, 27 Figures 1 through 3 and Supporting Figure 1 (see online supporting information) were downloaded and adapted from the portal.
Figure 1.
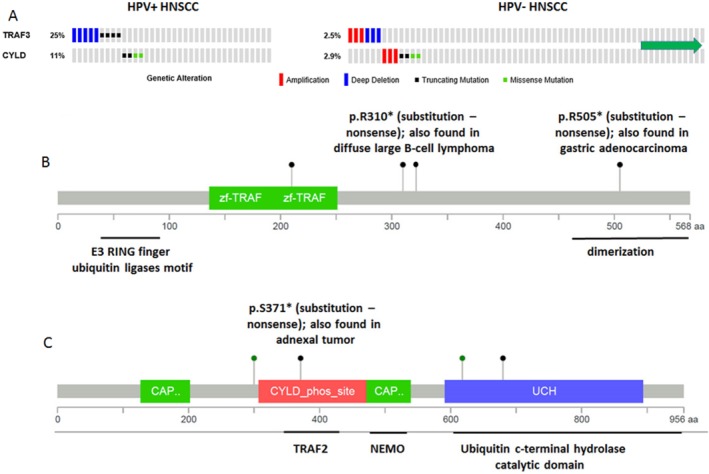
Tumor necrosis factor receptor‐associated factor 3 (TRAF3) or cylindromatosis lysine 63 deubiquitinase (CYLD) is mutated in 36% of human papillomavirus‐positive (HPV+) head and neck squamous cell carcinomas (HNSCCs). (A) Genetic alterations in the TRAF3 and CYLD genes were identified in patients with HPV‐positive (n = 36) and HPV‐negative (n = 243) HNSCC. Columns represent individual tumors. The green arrow indicates that several tumors without alterations were omitted to fit the figure. (A,B) Schematic representations of missense point (green) and truncating (black) mutations in the (B) TRAF3 and (C) CYLD genes identified in HPV‐positive HNSCC are shown. NEMO indicates NF‐κB essential modulator; phos_site, phosphorylation site; UCH, ubiquitin C‐terminal hydrolase; zf, zinc finger domain; CAP, cytoskeletal‐associated protein domain. Adapted from the cBioPortal for Cancer Genomics (available at: www.cbioportal.org).26, 27
Figure 3.
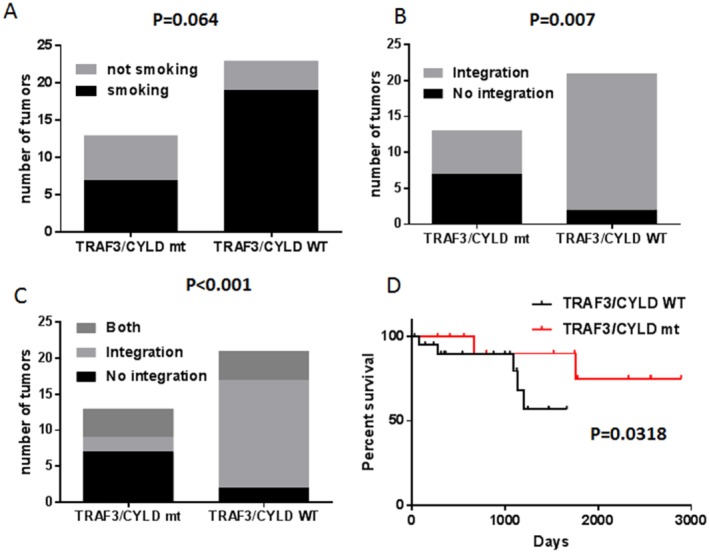
Tumor necrosis factor receptor‐associated factor 3 (TRAF3) or cylindromatosis lysine 63 deubiquitinase (CYLD) mutations correlate with the absence of human papillomavirus (HPV) integration and improved survival in HPV‐associated head and neck squamous cell carcinoma (HNSCC). Contingency graphs represent (A) the distribution of smokers or nonsmokers in the TRAF3/CYLD wild‐type (WT) and mutant (mt) groups (the P value was calculated using a 2‐sided Fisher exact test); (B) the distribution of tumors with or without HPV integration in the TRAF3/CYLD WT and mt groups (the P value was calculated using a 2‐sided Fisher exact test); and (C) the distribution of tumors with episomal HPV only, with HPV integrations only, or containing both integrated and episomal HPV DNA in the TRAF3/CYLD WT and mt groups (the P value was calculated using a 2‐sided chi‐square test). (D) Kaplan‐Meier curves illustrate the overall survival of patients who had HPV‐associated HNSCC with or without alterations in TRAF3/CYLD. Statistics were calculated using the log‐rank (Mantel‐Cox) test. Adapted from the cBioPortal for Cancer Genomics (available at: www.cbioportal.org).26, 27
Association of TRAF3, CYLD, and PIK3CA, Mutations With Smoking and HPV Integration
Contingency graphs representing the distribution of patients with HPV‐positive HNSCC among smokers or nonsmokers (Fig. 3A) and among those with HPV‐positive HNSCC with or without HPV integration (Fig. 3B,C), in the TRAF3/CYLD or PIK3CA wild‐type and mutant groups were produced using GraphPad Prism 6 software, and P values were calculated using Fisher exact tests and chi‐square tests. Patients’ smoking status and the absence or presence of HPV integration (HPV and human breakpoints) for each TCGA sample were obtained from the Table S2 in reference 13.
Survival
Kaplan‐Meier curves illustrating the overall survival of patients who had HPV‐associated HNSCC with or without alterations in the TRAF3/CYLD or PIK3CA genes were produced using GraphPad Prism 6 software, and the log‐rank (Mantel‐Cox) test was used for statistical analyses. Overall patient survival data were obtained from the Table S2 in reference 13.
Gene Set Enrichment Analysis
The list of genes that had significantly different expression (P < .05) in HPV‐positive TRAF3/CYLD or PIK3CA mutant and wild‐type HNSCC was downloaded from the cBioPortal for Cancer Genomics.26, 27 Gene set enrichment analysis (GSEA) (Figs. 5 and 6; Supporting Fig. 3; see online supporting information) was performed using software available from The Broad Institute (available at: http://software.broadinstitute.org/gsea/index.jsp).28, 29
Figure 5.
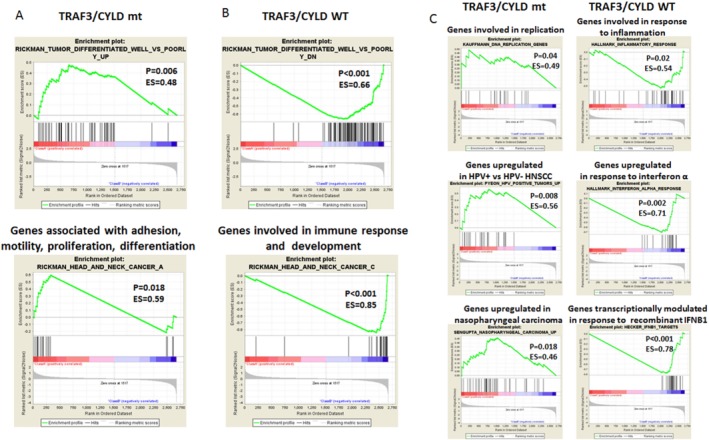
Classification of head and neck squamous cell carcinoma (HNSCC) based on tumor necrosis factor receptor‐associated factor 3 (TRAF3) and cylindromatosis lysine 63 deubiquitinase (CYLD) mutation status is illustrated. Results are shown from gene set enrichment analysis using data sets of (A,B) oncogenic and (C) hallmark and immunologic signatures for genes had significantly different expression in the TRAF3/CYLD mutant (mt) group versus the wild‐type (WT) group. ES indicates enrichment score; HPV−, human papillomavirus‐negative; HPV+, human papillomavirus‐positive; IFNB1, interferon β1; P, nominal P value.
Figure 6.
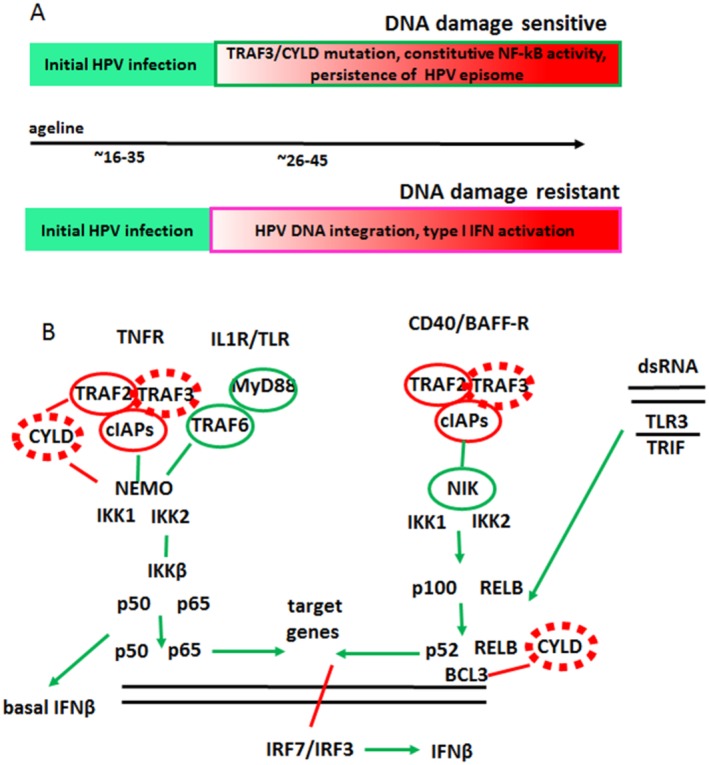
The etiology of human papillomavirus (HPV)‐positive head and neck squamous cell carcinoma (HNSCC) is illustrated. (A) This is a model for the etiology of HPV‐positive HNSCC. (Top) After initial HPV infection, mutations in tumor necrosis factor receptor‐associated factor 3 (TRAF3) and cylindromatosis lysine 63 deubiquitinase (CYLD), or in other genes, lead to constitutively active nuclear factor κB (NF‐κB), which supports persistence of the HPV episome; these tumors will be sensitive to DNA damage. (Bottom) Initial HPV infection activates type I interferon (IFN) signaling that selects for DNA damage‐resistant tumors with HPV DNA integrated into the human genome. (B) This is a schematic model of (left) canonical and (right) noncanonical NF‐κB pathways. Inactivating mutations in TRAF3/CYLD (dashed red ovals) identified in HPV‐positive HNSCC result in the activation of both NF‐κB pathways. Inactivating (solid red ovals) or activating (green ovals) mutations that activate NF‐κB identified in other types of human cancer are shown. BAFF‐R indicates B‐cell activating factor receptor; BCL3, B‐cell leukemia/lymphoma 3; CD40, cluster of differentiation 40 (costimulatory protein on antigen‐presenting cells); cIAPs, cellular inhibitor of apoptosis proteins; dsRNA, double‐stranded RNA; IKK, inhibitor of NF‐κB kinase; IL1R, interleukin 1 receptor; MyD88, myeloid differentiation primary response 88; NEMO indicates NF‐κB essential modulator; NIK, NF‐κB B‐inducing kinase; RELB, RELB proto‐oncogene, NF‐κB subunit; Toll‐like receptor; TNFR, tumor necrosis factor receptor; TRIF, Toll/interleukin‐1 receptor (TIR)‐domain–containing adapter‐inducing interferon β.
RESULTS AND DISCUSSION
One‐Third of HPV‐Positive Head and Neck Tumors Harbor TRAF3 or CYLD Gene Defects
One unexpected discovery from TCGA head and neck cancer project was the finding of deletions and/or mutations of TRAF3 in 25% of HPV‐associated HNSCCs.12 The majority of TRAF3 alterations were homozygous gene deletions (55%), whereas the remainder were truncating mutations (45%), consistent with TRAF3 inactivation in these tumors (Fig. 1A, left). Notably, 2 mutations, namely, p.R310*/c.928C>T and p.R505*/c.1513C>T, have been reported previously in diffuse large‐B cell (DLBC) lymphoma30 and gastric adenocarcinoma, respectively (Fig. 1B). In contrast, alterations in the TRAF3 gene were infrequently detected (2%) in HPV‐negative HNSCCs, and amplification and deletion/truncating mutations were equally represented, suggesting no functional requirement for TRAF3 activation or inhibition in HNSCCs lacking HPV (Fig. 1A, right). TRAF3 is an important component of both innate and acquired immune responses against many viruses,31 including Epstein‐Barr virus (EBV),32 human immunodeficiency virus,33 and HPV.34 It is noteworthy that antiviral functions of TRAF3 are unique and cannot be substituted by other members of the TRAF family. Because the antiviral activity of TRAF3 requires a full‐length, wild‐type TRAF3 protein,35, 36 alterations in the TRAF3 gene observed in HPV‐positive HNSCC most likely result in the loss of TRAF3 antiviral activity.
TRAF3 functions as a ubiquitin ligase to activate the type I IFN response while simultaneously inhibiting noncanonical and canonical NF‐κB activation.14, 22, 37 Type I IFN signaling is a potent and first‐line cellular defense against viral infection, and this response relies on pattern recognition receptors (PRRs) that also activate NF‐κB signaling. Although NF‐κB is required for constitutive IFN expression, it is not required for IFN expression late in viral infection, in which a major role for NF‐κB is to promote cell survival.38, 39, 40
To determine whether additional genes regulating IFN and NF‐κB responses may also be altered to promote HPV‐positive HNSCC, TCGA data were queried. These analyses revealed that the tumor suppressor CYLD, which inhibits NF‐κB pathways at various steps, was mutated in 11% of HPV‐positive HNSCCs (Fig. 1A, left). One‐half of identified CYLD genetic variations were truncating mutations, whereas the other one‐half included missense point mutations affecting either its ubiquitin hydrolase catalytic domain or situated in close proximity to the TRAF2 binding site (Fig. 1C). The p.S371*/ c.1112C>A, nonsense substitution, has been described previously in adnexal skin tumors.41 In addition, another amino acid 618 substitution in the CYLD gene, an aspartic acid‐to‐asparagine substitution at codon 618 (D618N) (identified in HPV‐positive HNSCC) has been recently reported in cutaneous squamous cell carcinoma.42 It is noteworthy that, the copy number status of mutated CYLD indicates a shallow loss (a possible heterozygous deletion) in all 4 CYLD‐mutant samples (Table 1). Notably, CYLD mutations have been identified in another cohort of HPV‐associated head and neck cancer.43 Like the findings in TRAF3, few CYLD mutations occurred in HPV‐negative HNSCC, and there was roughly equal representation of amplification and truncating/missense mutations, suggesting no striking functional consequences in HPV‐negative tumors (Fig. 1A, right). It is also worth noting that TRAF3 mutations were not observed in CYLD‐mutant tumors (Fig. 1A), revealing strong, mutual exclusivity (log odds ratio, < −3), although the difference was not statistically significant because of the relatively small number of events (P = .29). A tendency toward mutual exclusivity suggests that TRAF3 and CYLD mutations may be affecting the same downstream activities. Like TRAF3, CYLD inhibits NF‐κB pathways at various steps including, binding, deubiquitinating, and inhibiting the NF‐κB essential modulator (NEMO). It was initially believed that CYLD inhibited IFN signaling by deubiquitinating the PRR, RIG‐1, and downstream kinases TANK‐binding kinase 1 (TBK‐1) and inhibitor of NF‐κB kinase Ε (IKKΕ)44; but, surprisingly, IFN response to vesicular stomatitis virus in CYLD knockout mice or cells from these mice was abrogated.45 On the basis of these reports, TRAF3 and CYLD may serve similar functions after viral infection, namely, to inhibit NF‐κB and activate IFN. These findings suggest that up to 36% of HPV‐positive HNSCCs harboring defective TRAF3 or CYLD may rely on overactive NF‐κB and defective innate immunity.
Table 1.
CYLD Gene Alterations in Human Papillomavirus‐Associated Head and Neck Cancer
| Amino Acid Change | Type | Copy No. |
|---|---|---|
| K680* | Nonsense | Shallow deletion |
| S371* | Nonsense | Shallow deletion |
| D618A | Missense | Shallow deletion |
| N300S | Missense | Shallow deletion |
TRAF3 and CYLD Genetic Abnormalities are Not Frequent in HPV‐Positive Uterine Cervical Cancer
The above data suggest that a portion of HPV‐positive HNSCCs may rely on TRAF3 or CYLD mutations. Because high‐risk HPV is the primary etiologic agent of uterine cervical cancer, sequencing data from these tumors was examined to determine whether they also contained TRAF3 or CYLD gene defects. Surprisingly, TRAF3 defects detected in uterine cervical cancer, which is associated with high‐risk HPVs in about 93% of cases, were reminiscent of the pattern observed in HPV‐negative HNSCCs. Uterine cervical cancers had infrequent TRAF3 alterations that included both amplifications and deletions (Fig. 2A). Alterations of the CYLD gene in uterine cervical cancer were even less frequent (2%), comprising 1 missense mutation and 2 deletions (Fig. 2A). Compared with HPV‐positive HNSCC, in which TRAF3 and CYLD defects were mutually exclusive, TRAF3 and CYLD defects overlapped in 2 of the 9 uterine cervical cancers that had alterations in either gene.
Figure 2.

Tumor necrosis factor receptor‐associated factor 3 (TRAF3) or cylindromatosis lysine 63 deubiquitinase (CYLD) genetic abnormalities are not present in human papillomavirus (HPV)‐positive uterine cervical cancer. (A) Alterations in the TRAF3 and CYLD genes are illustrated in cervical cancer (n = 191). The green arrow indicates several tumors without alterations that were omitted to fit the figure. (B) Cross‐cancer alterations are summarized for the TRAF3 and CYLD genes (combined) from 126 studies; studies that included < 5% alterations in genes were omitted to fit the figure. Adeno indicates adenocarcinoma; BCCRC, breast cancer patient xenografts; ACyC, adenoid cystic carcinoma; CCLE, Cancer Cell Line Encyclopedia; chRCC, chromophobe renal cell carcinoma; CNA, copy number alteration; CS, carcinosarcoma; DLBC, diffuse large B‐cell lymphoma; LUAD, lung adenocarcinoma; MICH, University of Michigan; MSKCC, Memorial Sloan Kettering Cancer Center; NCI, National Cancer Institute; NEPC, neuroendocrine prostate cancer; TCGA, The Cancer Genome Atlas; squ, squamous; UTSW, University of Texas Southwestern Medical Center. Adapted from the cBioPortal for Cancer Genomics (available at: www.cbioportal.org).26, 27
To extend these analyses, deep‐sequencing data from multiple tumor types was queried for mutations in the TRAF3 and CYLD genes. Tumor types with > 10% alterations of TRAF/CYLD were: breast (data from xenografted tumors), neuroendocrine prostate cancer (NEPC), DLBC lymphoma, and lung squamous cell carcinoma. Breast cancer and NEPC contained only amplifications of TRAF3/CYLD, suggesting that, unlike HPV‐positive HNSCC, TRAF and CYLD or some adjacent genes, are activated in these tumor types. DLBC lymphoma and lung squamous cancer each had a significant proportion of total TRAF/CYLD alterations (mutations and deletions) and relatively few samples had amplification. It is noteworthy that, although lung squamous cancer is not connected to viral infection, DLBC lymphoma can be associated with EBV, suggesting that TRAF3/CYLD inactivation may be serving a similar role in DLBC lymphoma and HPV‐positive HNSCC. A recent study of another EBV‐associated cancer, nasopharyngeal carcinoma, identified numerous inactivating mutations in NF‐κB–negative regulators, including CYLD, NF‐κB inhibitor α (NFKBIA), and tumor necrosis factor α‐induced protein 3 (TNFAIP3), in 17% of these tumors.46
Across all solid tumor types, frequent inactivating TRAF3 and CYLD alterations, which are predicted to constitutively activate NF‐κB pathways while inhibiting innate immunity, occur only in HPV‐positive HNSCC. This is surprising, because 36% of HPV‐positive HNSCCs harbored defects in these genes, and it is particularly striking that HPV‐associated uterine cervical cancer did not exhibit a similar pattern of TRAF3/CYLD mutation, suggesting that HPV‐driven carcinogenesis in the uterine cervix may be distinct from HPV‐driven carcinogenesis in the head and neck.
We and others previously reported that HPV‐associated head and neck cancer more frequently, although not exclusively, contained alterations in the PIK3CA gene47, 48 In contrast to TRAF3/CYLD mutations, PIK3CA mutations or amplifications are commonly detected in cervical and head and neck cancers regardless of HPV status (Supporting Fig. 1A‐C; see online supporting information). It is noteworthy that, among 20 HPV‐positive head and neck tumors with alterations in PIK3CA gene, 4 samples harbored mutations in TRAF3/CYLD; whereas, among 16 tumors without PIK3CA mutations, 9 contained alterations in TRAF3/CYLD, indicating a tendency toward mutual exclusivity (log odds ratio, −1.1; P = .25) (Supporting Fig. 1A; see online supporting information).
TRAF3 and CYLD Gene Defects in HPV‐Positive HNSCC Correlate With the Absence of HPV Integration and Improved Survival
In addition to high‐risk HPV infection, several cofactors are associated with uterine cervical tumorigenesis, including alcohol consumption, coinfection with other infectious agents, and smoking.49, 50, 51 In contrast, tobacco smoking is not etiologically implicated in HPV‐positive head and neck cancer, but it is associated with decreased survival in patients who have HPV‐positive head and neck cancer.4, 52 It is noteworthy that there were relatively high numbers of patients who had a history of smoking in the HPV‐positive TCGA cohort.12 Querying the TCGA cohort for TRAF3/CYLD mutations and smoking history revealed a correlation between the absence of smoking and TRAF3/CYLD gene defects, but this association did not reach statistical significance (P = .064) (Fig. 3A).
Although both head and neck cancer and uterine cervical cancer are driven by high‐risk HPV types, these tumor types have many differences, including gene and protein expression profiles.53 The classic cervical HPV tumorigenesis model is based on enhanced expression of the E6 and E7 oncogenes, which depends on HPV genome integration and loss of HPV E2 and other HPV genes.10 In opposition to this classic HPV carcinogenesis model, HPV integration into the cellular genome was not identified in approximately 30% of HPV‐positive HNSCCs.13 The subset of HPV‐positive HNSCCs that lacked integration had a distinct methylation profile as well as unambiguous differences in cellular and HPV gene expression compared with tumors that had integration. DNA methylation and somatic gene expression profiles of integration‐positive tumors were analogous to those of HPV‐negative HNSCCs and normal tissues, whereas tumors that lacked integration were different from the other groups.13 The expression of HPV genes was also different between tumors with and without integration, because HPV integration‐negative tumors had lower expression of the HPV E6 and E7 oncogenes and increased expression of HPV E2, E4, and E5.13 Expression of the early genes E2, E4, and E5 with lower expression of E6 and E7 is a pattern observed in the HPV lifecycle, in which HPV exists as an episome in basal, undifferentiated cells.54
Although HPV uses defenses to dampen immune recognition and to suppress innate immunity in infected cells, episomal HPV activates innate immune responses.55, 56, 57 To determine whether the maintenance of episomes in HPV‐positive HNSCC was associated with inhibition of immune responses or NF‐κB activation, HPV integration was associated with TRAF3/CYLD gene defects. The majority of TRAF3/CYLD mutant tumors (54%) had no HPV integration, whereas only 10% of TRAF3/CYLD wild‐type samples lacked HPV integration (Fig. 3B). These data suggest that defects in TRAF3/CYLD may not be required if tumors contain integrated HPV or, alternatively, that the maintenance of episomal HPV pressures cells to mutate TRAF3/CYLD. To begin distinguishing these possibilities, cells with both episomal and integrated HPV would be ideal to correlate with TRAF3/CYLD defects. Although there are no direct sequencing data available that identify episomal and integrated forms in HPV‐positive HNSCC, a subset of tumors with HPV DNA integrated into the human genome were identified as having expression profiles reminiscent of tumors lacking integration. These data led the authors of a previous publication to propose that a subset of HPV‐positive HNSCC tumors contain both episomal and integrated HPV DNA.13 The incorporation of tumors suspected to harbor both integrated and episomal forms of HPV into the TRAF3/CYLD mutation analyses revealed that 11 of 13 tumors (85%) with mutant TRAF3/CYLD contained episomal HPV, whereas 6 of 21 (29%) TRAF3/CYLD wild‐type tumors harbored episomal HPV DNA (Fig. 3C). These data suggest that the presence of episomal HPV in HNSCC, regardless of the presence of integrated copies, may drive the dependence of these tumors on TRAF3 or CYLD gene defects. Mechanistically, mutations in TRAF3 or CYLD may create a cellular microenvironment needed to support the maintenance of the HPV episome as an alternative to classic HPV carcinogenesis, which that depends on the integration and loss of episomal HPV.
Studies from EBV‐related cancers or hepatocellular carcinoma, along with HNSCC, suggest that PIK3CA mutations are common in virally associated cancers and may be mediated by viral infection. However, in contrast to TRAF3/CYLD mutants, altered PIK3CA did not significantly correlate with the presence or absence of HPV integration sites (Supporting Fig. 2A‐C; see online supporting information), suggesting no requirement for PIK3CA activation in HPV episome maintenance.
Irrespective of HPV status, current therapy for advanced HNSCC includes concomitant radiation therapy (66‐70 grays) and chemotherapy as a radiation sensitizer. Although HPV‐positive head and neck tumors respond better to conventional chemotherapy and radiotherapy than HPV‐negative HNSCCs, patients are prone to short‐term and long‐term, deleterious side effects of this aggressive treatment. In addition, up to 25% of HPV‐positive HNSCCs recur at regional or distant metastatic sites. To begin assessing the possible biologic or prognostic potential of TRAF3/CYLD genetic alterations in HPV‐positive tumors, Kaplan‐Meier survival analyses of 34 patients with HPV‐positive HNSCC who had available data was performed based on the presence or absence of TRAF3 or CYLD mutations (Fig. 3D). Remarkably, TRAF3/CYLD alterations significantly correlated with improved overall survival. These data suggest that somatic defects in the CYLD or TRAF3 gene may be used to identify a subset of patients who have HPV‐positive HNSCC with a significantly improved prognosis. In contrast, PIK3CA mutations did not influence the overall survival of patients in this cohort (Supporting Fig. 2D; see online supporting information).
Mutations in TRAF3 or CYLD Activate NF‐κB Signaling in HPV‐Positive HNSCC
Inactivating abnormalities in TRAF3 and CYLD lead to constitutive activation of both canonical and alternative NF‐κB signaling, as reported in multiple myeloma.24, 25 Because of its ability to induce expression of immune response genes, NF‐κB signaling was initially viewed as a protector against viral infection; however, it became evident that viruses co‐opt NF‐κB activity to increase viral replication, elevate expression of viral genes, promote virion assembly, and block apoptosis in infected cells, allowing completion of the viral life cycle.58, 59, 60 To assess whether the genetic alterations in TRAF3 and CYLD genes identified in HPV‐positive HNSCC were associated with activation of NF‐κB, we performed GSEA using 615 data sets focusing on transcription factor target genes and a list of genes with significantly different expression in TRAF3/CYLD‐mutant compared versus wild‐type tumors. Genes that were highly expressed in tumors with TRAF3/CYLD mutations were enriched for those that shared NF‐κB or epidermal growth factor 1 (EGR‐1) binding sites (Fig. 4A). Although the role of EGR‐1 in relation to NF‐κB is not well defined, similar to NF‐κB, it has been demonstrated that EGR‐1 promotes cell survival and proliferation in B‐cell lymphoma and prostate cancer cells.61 Consistent with these data, previous gene expression analyses revealed that HPV‐positive HNSCC with episomal HPV DNA had differential expression of genes involved in NF‐κB signaling.13
Figure 4.
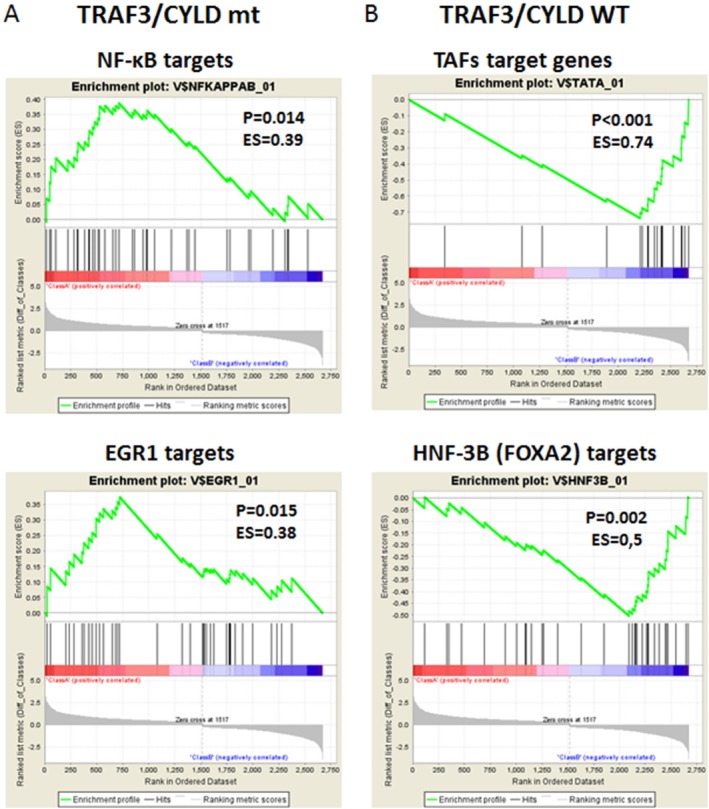
Mutations in tumor necrosis factor receptor‐associated factor 3 (TRAF3) and cylindromatosis lysine 63 deubiquitinase (CYLD) are associated with activated nuclear factor κB (NF‐κB) in human papillomavirus (HPV)‐positive head and neck squamous cell carcinoma. The results from transcription factor target genes gene set enrichment analysis are illustrated for genes that had significantly different expression in (A) the TRAF3/CYLD‐mutant (mt) group versus (B) the wild‐type (WT) group. EGR1 indicates Early Growth Response 1; ES, enrichment score; TAF, TATA box binding protein associated factor; HNF‐3B (FOXA2), hepatocyte nuclear factor 3β; hits, genes from the dataset; P, nominal P value.
In contrast to the TRAF3/CYLD‐mutant group, genes commonly expressed in TRAF3/CYLD wild‐type tumors encode for TATA‐binding proteins or contain a forkhead box A2 (FOXA2) binding site in the promoter region (Fig. 4B), whereas genes that are highly expressed in tumors with altered PIK3CA are enriched for those that share androgen receptor or hypoxia‐inducible factor 1α binding sites (Supporting Fig. 3A; see online supporting information).
These data reveal that HPV‐positive HNSCC with TRAF3 or CYLD gene defects have increased transcription of targets on the NF‐κB pathway and are consistent with the known roles of TRAF3 and CYLD as negative regulators of NF‐κB. Combined with data correlating the maintenance of episomal forms with TRAF3 and CYLD gene alterations, these data suggest that NF‐κB activation may be required for episomal maintenance in HPV‐positive HNSCC.
Classification of Tumors Based on TRAF3/CYLD Mutation Status
The differences between the transcriptomes of HNSCC with mutant versus wild‐type TRAF3/CYLD may lead to diverse clinical and biologic features, such as improved survival (Fig. 3D). To identify additional biologic functions or pathways associated with TRAF3/CYLD mutations, we ran GSEA using data sets of oncogenic and immunologic signatures as well as hallmark gene sets. Remarkably, HPV‐positive HNSCCs that harbored TRAF3 or CYLD defects expressed genes that were up‐regulated in well versus poorly differentiated, HPV‐negative HNSCC,62 whereas TRAF3/CYLD wild‐type tumors were enriched in genes that were down‐regulated in well differentiated, HPV‐negative HNSCC (Fig. 5A,B). In addition, TRAF3/CYLD‐mutant tumors were characterized by up‐regulation of a gene cluster associated with adhesion, motility, proliferation, and differentiation; whereas TRAF3/CYLD wild‐type HNSCCs were linked to genes involved in the immune response and development62 (Fig. 5A,B). Confirming these observations, we observed a substantial correlation between genes expressed in the TRAF3/CYLD wild‐type group with several data sets containing immune and inflammatory response genes. Conversely, mutant tumors were enriched in DNA replication genes,53, 63 in genes that distinguished HPV‐positive from HPV‐negative HNSCC,53 and in genes that were up‐regulated in nasopharyngeal carcinoma compared with normal tissue64 (Fig. 5C). Notably, the majority of TRAF3‐mutant and CYLD‐mutant tumors did not contain HPV integration (Fig. 3B), and tumors without integration had a gene expression profile different from that of HPV‐negative HNSCC and normal tissue. Conversely, expression profiles of HPV integration‐positive HNSCC were similar to HPV‐negative HNSCC and normal samples.13 In addition, genes expressed in nasopharyngeal carcinoma that were enriched in TRAF3‐defective or CYLD‐defective, HPV‐positive HNSCC (Fig. 5C) had a high representation of DNA repair and replication genes.64
Confirming our previous finding that mutant PIK3CA in HPV‐positive tumors correlated with activation of the mechanistic target of rapamycin (mTOR) pathway, tumors with altered PIK3CA in the TCGA cohort overexpressed genes that were up‐regulated by activation of the mTORC1 complex or during the unfolded protein response (Supporting Fig. 3B; see online supporting information).
Many reports have highlighted differences between HPV‐positive and HPV‐negative HNSCC. The data presented here suggest that, within HPV‐positive tumors, there are 2 major subgroups that can be distinguished based on TRAF3 or CYLD mutations. Comparison of HPV‐positive tumors with and without TRAF3 or CYLD mutations revealed that mutant tumors had improved survival and expressed genes that were enriched in HPV‐positive gene sets, suggesting that the tumors harboring TRAF3 or CYLD defects are the drivers of differences in gene expression between HPV‐positive versus HPV‐negative HNSCC and contribute significantly to the improved survival of patients with HPV‐positive tumors. Our data also suggest that TRAF3 or CYLD mutations harbor episomal HPV and have activated NF‐κB, which may be required for episomal maintenance.
The normal life cycle of HPV is linked to epithelial differentiation, with viral DNA maintained at a low copy number in basal cells, amplified and encapsidated only in more differentiated cells, and finally shed from the epithelial surface. It is noteworthy that the transcriptome of tumors with altered TRAF3/CYLD is highly enriched in genes that are overexpressed in well differentiated squamous cells (Fig. 5A). Because the majority of these tumors contained episomal HPV DNA (Fig. 3B,C), the data suggest that they may maintain replicating viral DNA and that differentiation signals possibly may trigger encapsidation. Supporting our hypothesis, tumors with episomal HPV, regardless of the presence or absence of integration sites, overexpress the HPV E2, E4, and E5 genes,13 which control viral replication.
Treatment of cells with IFN causes resistance to radiation,65, 66 and an IFN‐related DNA damage‐resistance gene signature (IRDS) is associated with resistance to chemotherapy and radiation in various cancer cells and in patients with breast cancer.67, 68 A significant feature of TRAF3/CYLD wild‐type tumors was the expression of genes involved in the type I interferon and inflammatory response. We observed that patients who had wild‐type TRAF3 and CYLD tumors had worse survival, which may implicate IFN signaling in therapeutic resistance (Fig. 3D).
The major limitation of our study is the relatively small number of HPV‐positive HNSCCs in the data set. Further profiling of HPV‐associated head and neck samples is needed to confirm the significance of TRAF3/CYLD mutations in HNSCC and to determine whether additional regulators of NF‐κB, which is known to be mutated in human cancer (Fig. 6B), are genetically altered in HNSCC. Although additional data may improve biomarker selection for predicting prognosis, these studies suggest that TRAF3 and CYLD mutations are useful for selecting patients who have HPV‐positive HNSCC with an improved outcome and suggest that these patients may be most appropriate for trials of therapeutic de‐escalation. Targeted therapy, such as NF‐κB inhibitors, may also be a rational option for preclinical studies.
In summary, using TCGA data we identified and characterized 2 groups of HPV‐positive HNSCCs (Fig. 6A). These groups are distinguished by the presence or absence of TRAF3/CYLD mutations, which are associated with the presence of HPV episomes and a lack of HPV genome integration. Future studies are needed, but these data suggest that the activation of NF‐κB signaling and the inhibition of innate immunity through TRAF3/CYLD mutations may be required for episomal maintenance. The presence of HPV episomes is not well described in other HPV‐associated cancers, and uterine cervical cancers do not have frequent mutations in TRAF3 or CYLD. The co‐occurrence of episomal HPV with TRAF3 or CYLD mutations suggests an alternative route of HPV carcinogenesis based on selection for HPV integration and high levels of HPV E6 and E7 expression (classic) or for TRAF3 or CYLD mutations with low HPV oncogene expression (alternative). The major difference between the classic and alternative HPV carcinogenesis pathways is how the innate immune reaction to episomal HPV is relieved. In the classic pathway, HPV integrates to avoid recognition by PRR; whereas, in the alternative pathway, episomal HPV can be recognized, but somatic mutations that block innate immune response while activating NF‐κB to promote cell survival are selected. Significantly, HNSCC with defects in the TRAF3/CYLD genes have a better prognosis compared with tumors that lack mutations (Fig. 3D). One of the major goals of head and neck oncology is personalization of treatment so that resistant tumors are aggressively treated and sensitive tumors can be less aggressively treated. Our data suggest that constitutively active NF‐κB resulting from somatic mutations in TRAF3 or CYLD may serve as a biomarker to predict an improved prognosis for patients with HPV‐positive head and neck cancer.
FUNDING SUPPORT
This work was supported by the Yale Department of Surgery Ohse Award. Michael Hajek was supported by grant TL1TR000141 from the National Institutes of Health, from the National Center for Advancing Translational Science, a component of the National Institutes of Health.
CONFLICT OF INTEREST DISCLOSURES
The authors made no disclosures.
AUTHOR CONTRIBUTIONS
Michael Hajek: Study concept and design, acquisition of data, and article preparation. Andrew Sewell: Acquisition of data. Susan Kaech: Analysis and interpretation of data. Barbara Burtness: Analysis and interpretation of data and article preparation. Wendell G. Yarbrough: Study concept and design, analysis and interpretation of data, and article preparation. Natalia Issaeva: Study concept and design, acquisition of data; analysis and interpretation of data, and article preparation.
Supporting information
Additional supporting information may be found in the online version of this article.
Supporting Information Figures.
See related editorial on pages 1695‐8.
The contents of this work are solely the responsibility of the authors and do not necessarily represent the official view of National Institutes of Health.
Contributor Information
Wendell G. Yarbrough, Email: wendell.yarbrough@yale.edu.
Natalia Issaeva, Email: natalia.issaeva@yale.edu.
REFERENCES
- 1. Hunter KD, Parkinson EK, Harrison PR. Profiling early head and neck cancer. Nat Rev Cancer. 2005;5:127–135. [DOI] [PubMed] [Google Scholar]
- 2. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66:271–289. [DOI] [PubMed] [Google Scholar]
- 3. Sturgis EM, Cinciripini PM. Trends in head and neck cancer incidence in relation to smoking prevalence: an emerging epidemic of human papillomavirus‐associated cancers? Cancer. 2007;110:1429–1435. [DOI] [PubMed] [Google Scholar]
- 4. Ang KK, Harris J, Wheeler R, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. N Engl J Med. 2010;363:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gillison ML, Chaturvedi AK, Anderson WF, Fakhry C. Epidemiology of human papillomavirus‐positive head and neck squamous cell carcinoma. J Clin Oncol. 2015;33:3235–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pfister DG, Spencer S, Brizel DM, et al. Head and neck cancers, version 1.2015. J Natl Compr Canc Netw. 2015;13:847–855; quiz 856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gillison ML, Broutian T, Pickard RK, et al. Prevalence of oral HPV infection in the United States, 2009‐2010. JAMA. 2012;307:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. [DOI] [PubMed] [Google Scholar]
- 9. Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hudelist G, Manavi M, Pischinger KI, et al. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: different levels of viral integration are correlated with lesion grade. Gynecol Oncol. 2004;92:873–880. [DOI] [PubMed] [Google Scholar]
- 11. Yu T, Ferber MJ, Cheung TH, Chung TK, Wong YF, Smith DI. The role of viral integration in the development of cervical cancer. Cancer Genet Cytogenet. 2005;158:27–34. [DOI] [PubMed] [Google Scholar]
- 12. The Cancer Genome Atlas Network . Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517:576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parfenov M, Pedamallu CS, Gehlenborg N, et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc Natl Acad Sci U S A. 2014;111:15544–15549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hacker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri‐faced immune regulator. Nat Rev Immunol. 2011;11:457–468. [DOI] [PubMed] [Google Scholar]
- 15. Ghosh S, Karin M. Missing pieces in the NF‐kappaB puzzle. Cell. 2002;109(suppl):S81–S96. [DOI] [PubMed] [Google Scholar]
- 16. Stadler ME, Patel MR, Couch ME, Hayes DN. Molecular biology of head and neck cancer: risks and pathways. Hematol Oncol Clin North Am. 2008;22:1099–1124, vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dong G, Chen Z, Kato T, Van Waes C. The host environment promotes the constitutive activation of nuclear factor‐kappaB and proinflammatory cytokine expression during metastatic tumor progression of murine squamous cell carcinoma. Cancer Res. 1999;59:3495–3504. [PubMed] [Google Scholar]
- 18. Loercher A, Lee TL, Ricker JL, et al. Nuclear factor‐kappaB is an important modulator of the altered gene expression profile and malignant phenotype in squamous cell carcinoma. Cancer Res. 2004;64:6511–6523. [DOI] [PubMed] [Google Scholar]
- 19. Allen C, Duffy S, Teknos T, et al. Nuclear factor‐kappaB‐related serum factors as longitudinal biomarkers of response and survival in advanced oropharyngeal carcinoma. Clin Cancer Res. 2007;13:3182–3190. [DOI] [PubMed] [Google Scholar]
- 20. Chen Z, Malhotra PS, Thomas GR, et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin Cancer Res. 1999;5:1369–1379. [PubMed] [Google Scholar]
- 21. Zhang PL, Pellitteri PK, Law A, et al. Overexpression of phosphorylated nuclear factor‐kappa B in tonsillar squamous cell carcinoma and high‐grade dysplasia is associated with poor prognosis. Mod Pathol. 2005;18:924–932. [DOI] [PubMed] [Google Scholar]
- 22. Hoebe K, Beutler B. TRAF3: a new component of the TLR‐signaling apparatus. Trends Mol Med. 2006;12:187–189. [DOI] [PubMed] [Google Scholar]
- 23. Harhaj EW, Dixit VM. Regulation of NF‐kappaB by deubiquitinases. Immunol Rev. 2012;246:107–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Annunziata CM, Davis RE, Demchenko Y, et al. Frequent engagement of the classical and alternative NF‐kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keats JJ, Fonseca R, Chesi M, et al. Promiscuous mutations activate the noncanonical NF‐kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data [abstract]. Cancer Discov. 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal [serial online]. Sci Signal. 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mootha VK, Lindgren CM, Eriksson KF, et al. PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. [DOI] [PubMed] [Google Scholar]
- 30. Pasqualucci L, Trifonov V, Fabbri G, et al. Analysis of the coding genome of diffuse large B‐cell lymphoma. Nat Genet. 2011;43:830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oganesyan G, Saha SK, Guo B, et al. Critical role of TRAF3 in the Toll‐like receptor‐dependent and ‐independent antiviral response. Nature. 2006;439:208–211. [DOI] [PubMed] [Google Scholar]
- 32. Eliopoulos AG, Dawson CW, Mosialos G, et al. CD40‐induced growth inhibition in epithelial cells is mimicked by Epstein‐Barr virus‐encoded LMP1: involvement of TRAF3 as a common mediator. Oncogene. 1996;13:2243–2254. [PubMed] [Google Scholar]
- 33. Imbeault M, Ouellet M, Giguere K, et al. Acquisition of host‐derived CD40L by HIV‐1 in vivo and its functional consequences in the B‐cell compartment. J Virol. 2011;85:2189–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karim R, Tummers B, Meyers C, et al. Human papillomavirus (HPV) upregulates the cellular deubiquitinase UCHL1 to suppress the keratinocyte's innate immune response [serial online]. PLoS Pathog. 2013;9:e1003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guven‐Maiorov E, Keskin O, Gursoy A, et al. TRAF3 signaling: competitive binding and evolvability of adaptive viral molecular mimicry. Biochim Biophys Acta 1860(11 pt B):2646–2655, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang P, Reichardt A, Liang H, et al. Single amino acid substitutions confer the antiviral activity of the TRAF3 adaptor protein onto TRAF5 [serial online]. Sci Signal. 2012;5:ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guven‐Maiorov E, Keskin O, Gursoy A, et al. The architecture of the TIR domain signalosome in the toll‐like receptor‐4 signaling pathway [serial online]. Sci Rep. 2015;5:13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chan YK, Gack MU. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol. 2016;14:360–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Balachandran S, Beg AA. Defining emerging roles for NF‐κB in antivirus responses: revisiting the interferon‐β enhanceosome paradigm [serial online]. PLoS Pathog. 2011;7:e1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3‐dependent innate immune response. Immunity. 2006;24:93–103. [DOI] [PubMed] [Google Scholar]
- 41. Sima R, Vanecek T, Kacerovska D, et al. Brooke‐Spiegler syndrome: report of 10 patients from 8 families with novel germline mutations: evidence of diverse somatic mutations in the same patient regardless of tumor type. Diagn Mol Pathol. 2010;19:83–91. [DOI] [PubMed] [Google Scholar]
- 42. Pickering CR, Zhou JH, Lee JJ, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. 2014;20:6582–6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seiwert TY, Zuo Z, Keck MK, et al. Integrative and comparative genomic analysis of HPV‐positive and HPV‐negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015;21:632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Friedman CS, O'Donnell MA, Legarda‐Addison D, et al. The tumour suppressor CYLD is a negative regulator of RIG‐I‐mediated antiviral response. EMBO Rep. 2008;9:930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang M, Lee AJ, Wu X, Sun SC. Regulation of antiviral innate immunity by deubiquitinase CYLD. Cell Mol Immunol. 2011;8:502–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zheng H, Dai W, Cheung AK, et al. Whole‐exome sequencing identifies multiple loss‐of‐function mutations of NF‐kappaB pathway regulators in nasopharyngeal carcinoma. Proc Natl Acad Sci U S A. 2016;113:11283–11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yarbrough WG, Whigham A, Brown B, Roach M, Slebos R. Phosphoinositide kinase‐3 status associated with presence or absence of human papillomavirus in head and neck squamous cell carcinomas. Int J Radiat Oncol Biol Phys. 2007;69:S98–S101. [DOI] [PubMed] [Google Scholar]
- 48. Sewell A, Brown B, Biktasova A, et al. Reverse‐phase protein array profiling of oropharyngeal cancer and significance of PIK3CA mutations in HPV‐associated head and neck cancer. Clin Cancer Res. 2014;20:2300–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mzarico E, Gomez‐Roig MD, Guirado L, Lorente N, Gonzalez‐Bosquet E. Relationship between smoking, HPV infection, and risk of cervical cancer. Eur J Gynaecol Oncol. 2015;36:677–680. [PubMed] [Google Scholar]
- 50. Austin DF. Smoking and cervical cancer. JAMA. 1983;250:516–517. [PubMed] [Google Scholar]
- 51. Fonseca‐Moutinho JA. Smoking and cervical cancer. ISRN Obstet Gynecol 2011:847684, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. D'Souza G, Kreimer AR, Viscidi R, et al. Case‐control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. 2007;356:1944–1956. [DOI] [PubMed] [Google Scholar]
- 53. Pyeon D, Newton MA, Lambert PF, et al. Fundamental differences in cell cycle deregulation in human papillomavirus‐positive and human papillomavirus‐negative head/neck and cervical cancers. Cancer Res. 2007;67:4605–4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nakahara T, Kiyono T. Interplay between NF‐κB/interferon signaling and the genome replication of HPV. Future Virol. 2016;11:141–155. [Google Scholar]
- 55. Amador‐Molina A, Hernandez‐Valencia JF, Lamoyi E, Contreras‐Paredes A, Lizano M. Role of innate immunity against human papillomavirus (HPV) infections and effect of adjuvants in promoting specific immune response. Viruses. 2013;5:2624–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stanley M. Immune responses to human papillomavirus. Vaccine. 2006;24(suppl 1):S16–S22. [DOI] [PubMed] [Google Scholar]
- 57. Scott M, Nakagawa M, Moscicki AB. Cell‐mediated immune response to human papillomavirus infection. Clin Diagn Lab Immunol. 2001;8:209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schwarz EM, Badorff C, Hiura TS, et al. NF‐kappaB‐mediated inhibition of apoptosis is required for encephalomyocarditis virus virulence: a mechanism of resistance in p50 knockout mice. J Virol. 1998;72:5654–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Amici C, Belardo G, Rossi A, Santoro MG. Activation of I kappa b kinase by herpes simplex virus type 1. A novel target for anti‐herpetic therapy. J Biol Chem. 2001;276:28759–28766. [DOI] [PubMed] [Google Scholar]
- 60. Santoro MG, Rossi A, Amici C. NF‐kappaB and virus infection: who controls whom. EMBO J. 2003;22:2552–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Parra E, Ferreira J, Ortega A. Overexpression of EGR‐1 modulates the activity of NF‐kappaB and AP‐1 in prostate carcinoma PC‐3 and LNCaP cell lines. Int J Oncol. 2011;39:345–352. [DOI] [PubMed] [Google Scholar]
- 62. Rickman DS, Millon R, De Reynies A, et al. Prediction of future metastasis and molecular characterization of head and neck squamous‐cell carcinoma based on transcriptome and genome analysis by microarrays. Oncogene. 2008;27:6607–6622. [DOI] [PubMed] [Google Scholar]
- 63. Kauffmann A, Rosselli F, Lazar V, et al. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene. 2008;27:565–573. [DOI] [PubMed] [Google Scholar]
- 64. Dodd LE, Sengupta S, Chen IH, et al. Genes involved in DNA repair and nitrosamine metabolism and those located on chromosome 14q32 are dysregulated in nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15:2216–2225. [DOI] [PubMed] [Google Scholar]
- 65. Kita K, Sugaya S, Zhai L, et al. Involvement of LEU13 in interferon‐induced refractoriness of human RSa cells to cell killing by x‐rays. Radiat Res. 2003;160:302–308. [DOI] [PubMed] [Google Scholar]
- 66. Sirota NP, Bezlepkin VG, Kuznetsova EA, et al. Modifying effect in vivo of interferon alpha on induction and repair of lesions of DNA of lymphoid cells of gamma‐irradiated mice. Radiat Res. 1996;146:100–105. [PubMed] [Google Scholar]
- 67. Weichselbaum RR, Ishwaran H, Yoon T, et al. An interferon‐related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105:18490–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A. 2004;101:1714–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article.
Supporting Information Figures.