

Abstract
The mechanism underlying thrombosis in atrial fibrillation (AF) is not yet clearly understood. Oncostatin M (OSM), as a member of IL-6 family, is involved in atherosclerosis-mediated thrombosis. The present study hypothesizes that OSM and its downstream factors play a role in thrombogenesis in AF.
The specimens of left atrial appendages collected from patients with rheumatic mitral stenosis who underwent valve replacement were divided into 3 groups: sinus rhythm, AF(+)/thrombus(−), and AF(+)/thrombus(+) group. The macrophage infiltration in atrial tissue was assessed by immunohistochemistry, and the amount of OSM, tissue factor (TF), and tissue factor pathway inhibitors (TFPIs) was detected by Western blot.
The infiltration of the M1 macrophages was significantly increased in the AF with thrombus group compared with the sinus rhythm group (P = .03). Moreover, the expression of OSM and TF was much higher in the AF with thrombus group compared with the sinus rhythm group (P = .02, .009, respectively) while the TFPI was decreased in the AF with thrombus group (P = .04).
OSM might be correlated with thrombosis in patients with AF mediated by TF and TFPI.
Keywords: atrial fibrillation, oncostatin M, thrombosis
1. Introduction
Atrial fibrillation (AF) is a commonly occurring cardiac dysrhythmia in clinical practice, which has a high risk of thrombosis and cardioembolic stroke.[1,2] Inflammation is known to play a vital role in the pathogenesis of AF.[3,4] Several inflammatory cytokines, such as C-reactive protein (CRP), heat shock protein (HSP) β1, interleukin (IL)-6, IL-8, and tumor necrosis factor (TNF) have been reported to be involved in AF.[5,6] The increased CRP levels were observed to be a prediction of the new AF onset in several large, prospective cohort studies.[1] These data suggested that a persistent inflammatory state can promote AF.
Oncostatin M (OSM) is a member of the IL-6 family, which acts as a proinflammatory cytokine in some cardiovascular diseases.[7] OSM is secreted by several immune cells[8] and detected in myocardium.[9] Recent studies reported that OSM affects the healing of injured tissue via recruiting M1 and M2 macrophages in myocardial infarction.[10] In the event of heart failure, continued activation of OSM leads to the remodeling of cardiomyocytes, thereby reducing contractility.[11]
Several studies reported that the immunological cells participated in the pathogenesis of AF.[12] Whether the immunological cell dependent-AF is critical for thrombogenesis in AF is not clearly known; however, OSM is a crucial cytokine secreted by M1 macrophages. In addition to its role in proinflammatory response, OSM also promotes thrombogenesis by breaking the balance between the tissue factor (TF) and tissue factor pathway inhibitors (TFPIs).[13] Hence, whether OSM from macrophages in involved in thrombogenesis during the AF needs to be elucidated further. In this present study, we analyzed the macrophage distribution and OSM expression in the atrium of patients with rheumatic mitral stenosis with or without thrombus formation to explore the alleged role of OSM in thrombogenesis of AF.
2. Materials and methods
2.1. Human subjects and samples
Patients with rheumatic mitral stenosis were selected who underwent valve replacement and were hospitalized between January 2014 and May 2015 at Nanjing Drum Tower Hospital Affiliated to Nanjing University Medical School and Huaian First People's Hospital. All patients were divided into 3 groups: group A (n = 14, sinus rhythm): AF(−)/thrombus(−), group B (n = 43): AF(+)/thrombus(−), and group C (n = 14): AF(+)/thrombus(+). Pre- and postoperative 2-dimensional echocardiography was performed in each patiently. Patients with hyperthyreosis, sick sinus syndrome, and renal disease were excluded from the study. All medications, with an exception for warfarin, were continued before surgery until the morning of surgery. The operations were performed as described previously.[14] After the specimens of the left atrial appendages obtained, a section of the tissue was fixed in 4% paraformaldehyde and used for immunohistochemistry (IHC) assay; the remaining tissue was frozen in liquid nitrogen and preserved at −80°C.
Human tissue collection and analyses strictly abided by the principles outlined in the Declaration of Helsinki. All such procedures were approved by Nanjing Drum Tower Hospital Affiliated to Nanjing University Medical School and Huaian First People's Hospital Ethics Committees. All participants enrolled in the present study provided written informed consent.
2.2. IHC analysis
The IHC staining was performed as described previously.[15] A series of 5 μm thick paraffin sections from human left atrial appendages were blocked with 2% BSA (bovine serum albumin) in PBS (phosphate-buffered saline) for 30 minutes and then stained with antibodies, anti-HLA-DR (Abcam, Cambridge,UK) and anti-CD 163 (Abcam) overnight at 4°C.[16] The slides were washed with PBS, incubated with biotinylated secondary antibody for 20 minutes, counterstained with hematoxylin, and mounted with glycerol gelatin. The staining was visualized using the VectaStain ABC-AP Kit (Alkaline Phosphatase, Standard, Burlingame) as per the protocols. PBS served as a negative control.
2.3. Western blot
Whole protein extract was prepared using Total Protein Isolation Kit (KGP2100; KeyGEN BioTech, Jiangsu, China), according to the manufacturer's instructions. Briefly, the tissue samples were washed with PBS and homogenized in lysis buffer containing 1% proteinase inhibitors, 5% phosphatase inhibitors, and 5% phenylmethanesulfonyl fluoride on an ice rotator. Then, the homogenates were centrifuged at 12,000 × g for 5 minutes at 4°C to precipitate the cell debris. The supernatants were subjected to estimate protein concentrations using BCA protein assay (Pierce, Rockford). Subsequently, the supernatants was mixed with 5× protein loading buffer (Beyotime Biotechnology, Shanghai, China) and then heated for denaturation for 10 minutes.
Western blot was performed as described previously.[17] Briefly, the protein lysates were resolved on 12% SDS–PAGE and transferred to polyvinylidene difluoride membranes. After blocking with 5% nonfat milk in Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBST) for 1 hour at room temperature, the membranes were probed at 4°C for overnight with the following primary antibodies: anti-OSM (1:500, R&D Systems, Minneapolis), anti-TF (1:500, R&D Systems), and anti-TFPI (1:1000, R&D Systems); anti-GAPDH (1:10,000, Bioworld) and anti-β-actin (1:5000, Bioworld) were used as loading controls. After washing with TBST, the membranes were incubated with horseradish peroxidase-conjugated goat antimouse (1:10,000, Bioworld) and mouse antigoat (1:15,000, Bioworld) at room temperature for 1 hour, respectively. The reactions were detected with enhanced chemiluminescence reagents (Millipore, Darmstadt, Germany) and images obtained by film exposure. The bands were quantified using Image J software. All the quantification of the proteins was normalized using GAPDH or β-actin.
2.4. Statistical analysis
Data were expressed as mean ± SEM. All analyses were performed using SPSS 13.0 software (SPSS, Inc, Chicago, IL). The statistical significance was defined as P < .05 (2-tailed). The differences between groups were compared using Kruskal–Wallis test. The χ2 test or Fisher exact test was used for the analysis of percentage differences among groups.
3. Results
3.1. Clinical and echocardiographic characteristics
We recruited 71 patients with mitral stenosis undergoing mitral valve replacement surgery. All patients were divided into 3 groups based on AF and thrombus formation. No differences were observed in the clinical characteristics among the 3 groups, including age, sex bias, smoke, alcohol abuse, anamnesis of hypertension, diabetes mellitus, coronary heart disease, pulmonary hypertension, and congestive heart failure.
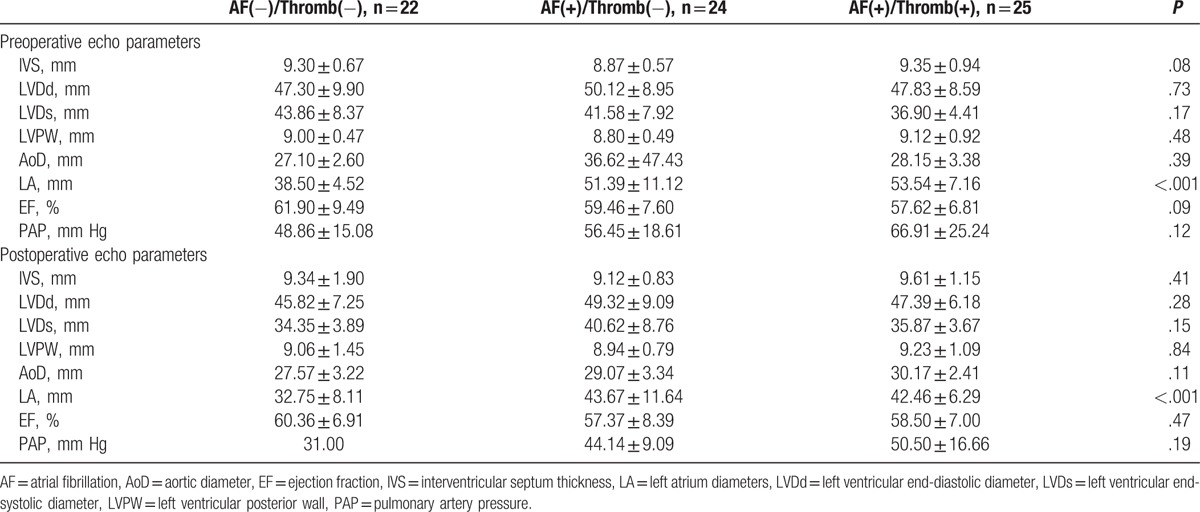
Next, the echocardiography parameters[18] among the 3 groups, either pre- or postoperation, were found to be similar regarding interventricular septum thickness (IVS), left ventricular end-systolic diameter (LVDs), left ventricular end-diastolic diameter (LVDd), left ventricular posterior wall (LVPW), aortic diameter (AoD), ejection fraction (EF), pulmonary artery pressure (PAP), and left atrium diameter (LA). The diameter of LA was significantly increased in patients with AF and thrombus compared with patients with or without AF (Table 1).
Table 1.
Clinical data.
3.2. Infiltration of M1 macrophages was increased significantly in the tissue
First, we detected the phenotype of macrophages in the atrial tissue with thrombosis. We investigated the infiltration of macrophages in the atrial tissue by IHC, with antibodies against HLADR and CD163. HLADR was the marker of M1 macrophages and CD163 was the marker of M2. The number of HLADR-positive cells in tissue with AF and thrombus was more than that in tissue with sinus rhythm and that with only AF (Fig. 1A). No visible difference was observed between group B and C, as designated in the Materials and Methods Section (Fig. 1B and C). The number of M2 macrophages in each group was similar (Fig. 1D and F). These results suggested that M1 macrophages might participate in thrombogenesis in the patients with mitral stenosis and AF.
Figure 1.
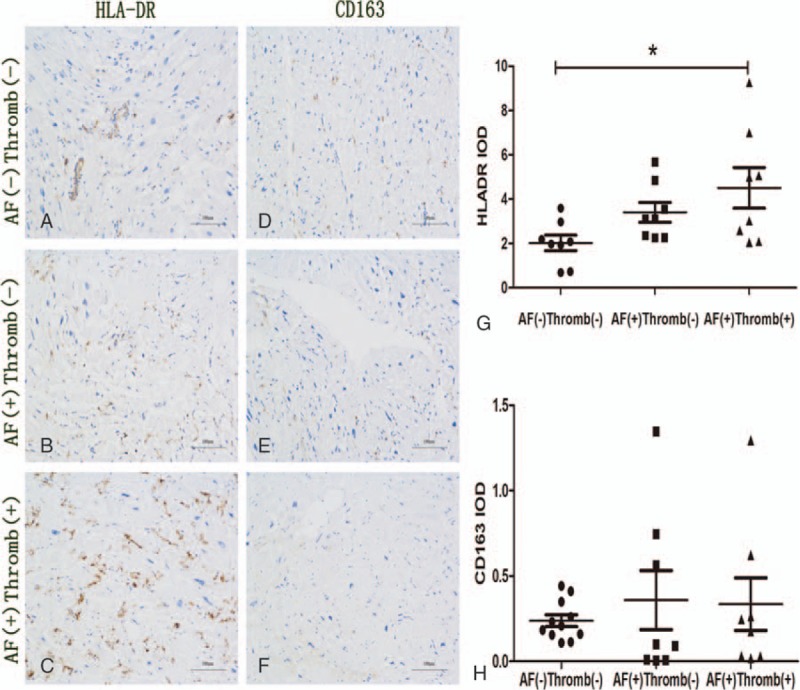
Immunostaining analysis of infiltration M1 and M2 macrophages. (A–C) Infiltration of M1 macrophages. (D–F) Infiltration of M2 macrophages. (G and H) The result of statistical analysis for the 3 groups; ∗P = .03. AF = atrial fibrillation.
3.3. Increased expression of OSM in AF with thrombus
We assessed the OSM expression in each set using Western blot. It has been postulated that OSM could express at the presence of macrophages. According to IHC results, it can be proposed that the augmentative M1 macrophages could induce the upregulation of OSM. OSM expression is similar between AF without thrombus and sinus rhythm. However, in the group of AF and thrombus, the amount of OSM was higher than that in the other 2 groups (Fig. 2A and B).
Figure 2.
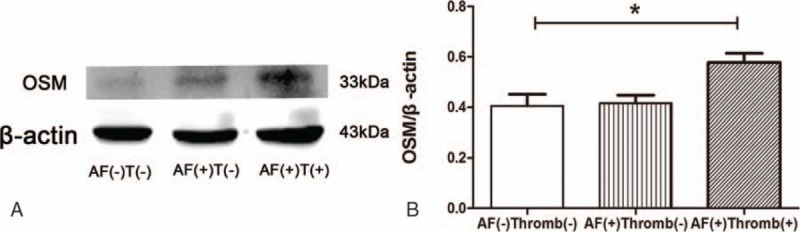
Increased expression of OSM in the AF with thrombus. (A) Western blot analysis for the expression of OSM among the 3 groups, β-actin is the loading control. (B) Increased amount of OSM in the AF and thrombus group. All the results are compared with the sinus rhythm, ∗P = .02. AF = atrial fibrillation, T = thrombus, OSM = oncostatin M.
3.4. Quantitation of TF and TFPI
TF, that functions as a cofactor for plasma factor VIIa, could initiate the exogenous coagulation cascade.[19,20] TF is known to play a vital role in mediating thrombosis associated with the progression of atherosclerosis. A previous study demonstrated that OSM could induce TF expression.[13] In our study, TF expression in the tissue from AF with thrombus group was increased significantly compared with other groups (Fig. 3A and C). TFPI was a critical anticoagulant protein present in endothelium and platelets, which targeted to TF. The expression of TFPI was decreased in AF with thrombus group as compared to the sinus rhythm group (Fig. 3B and D). These results indicated that the TF and TFPI played pivotal roles in the process of thrombogenesis in AF.
Figure 3.
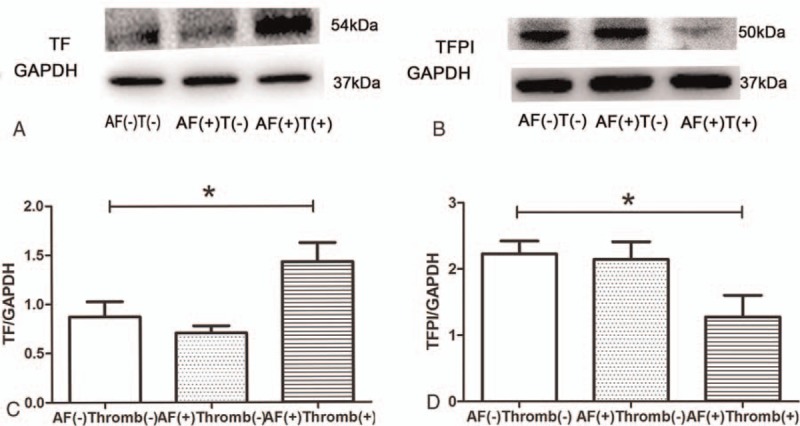
Western blot analysis of TF and TFPI. (A) Result of TF for 3 groups. (B) Image of TFPI. (C) Increased quantity of TF in AF with thrombus group, ∗P = .009. (D) TFPIs were decreased in AF with thrombus group, ∗P = .04. AF = atrial fibrillation, T = thrombus, TF = tissue factor, TFPIs = tissue factor pathway inhibitors.
4. Discussion
In the present study, we first validated that in AF with thrombus, the atrial tissue infiltration of M1 macrophages increased significantly; the OSM expression was also found to increase simultaneously. Next, we detected that the downstream TF increased and TFPI decreased, leading to an imbalance between TF and TFPI eventually.
AF is one of the most common and complex arrhythmias.[21] In the past years, several investigators have reported that inflammation played a major role in AF,[22] especially, in the initiation and development of AF. Aviles et al[3] reported that elevated CRP in patients with AF at baseline was a predicted independent risk factor for the future development of AF. Moreover, Sata et al[23] reported that the levels of IL-6 were significantly higher in AF patients with cardioversion than patients with sinus arrhythmia. As a member of IL-6 family, OSM was also involved in the pathogenesis of AF.
OSM, a monocyte and T-lymphocyte-specific cytokine, is present in atherosclerotic lesions that can induce the proliferation and migration of smooth muscle cells and remodeling of extracellular matrix.[24] Several reports demonstrated that OSM was involved in thrombogenesis in situ atherosclerotic lesions via TF-dependent pathway.[25] Mirshahi et al[13] further reported that OSM could adjust the balance between TF and TFPI, which is critical in blood clotting.
AF with thrombosis is a process carried out by complex influencing factors. Hitherto, the underlying mechanism for immunological cell-induced thrombosis is less known. The lack of a suitable animal model is the primary cause of poor research progress in AF-induced thrombosis. Here, we enrolled 71 patients with rheumatic mitral stenosis who underwent valve replacement and explored the OSM change in left atrial appendages. In this study, we observed the macrophage infiltration and OSM expression in AF with thrombosis. In addition, the fluctuation of balance between TF and TFPI was observed. Hence, we speculate that OSM was the key molecule in thrombosis in AF. In the left atria, in situ infiltrating macrophages could secrete OSM and others cytokines. Subsequently, OSM promotes Smooth Muscle Cells in atrial endometrial secretion of TF and inhibits the expression of TFPI, thereby inducing an increased exogenous coagulation cascade activity that may eventually induce thrombosis. On the other hand, some studies have found that OSM could induce the infiltration of macrophages that may convert to the M1 phenotype.[10] The infiltrated M1 macrophages can again secrete OSM to attract more monocytes from circulation, contributing to a positive feedback loop. Therefore, we proposed that OSM could induce thrombosis in patients with AF, which is mediated by TF and TFPI.
4.1. Limitations
First, all the experiments in the present study were performed in human atrial specimens. We could not use gain- or loss-of-function models to demonstrate a causal relationship between OSM and thrombosis. Second, all patients enrolled were diagnosed with rheumatic mitral stenosis and were undergoing valve replacement. These patients might not represent a majority of the AF patients. A further study of nonvalvular AF patients is essential to verify these hypotheses.
5. Conclusion
OSM might be related to thrombosis in patients with AF mediated by TF and TFPI.
Footnotes
Abbreviations: AF = atrial fibrillation, AoD = aortic diameter, BSA = bovine serum albumin, CRP = C-reactive protein, EF = ejection fraction, HSP = heat shock protein, IL = interleukin, IVS = interventricular septum thickness, LA = left atrium diameter, LVDd = left ventricular end-diastolic diameter, LVDs = left ventricular end-systolic diameter, LVPW = left ventricular posterior wall, OSM = oncostatin M, PAP = pulmonary artery pressure, PBS = phosphate-buffered saline, TF = tissue factor, TFPI = tissue factor pathway inhibitor, TNF = tumor necrosis factor.
JX and SZ contributed equally to this article.
Funding: This study was supported by grants from the Funds for Peak of Six Personnel in Jiangsu Province (2013-WSN-008), the Funds for Distinguished Young Scientists in Nanjing (JQX13006), the Jiangsu Provincial Commission of Health and Family Planning Funds for Young Scholar (Q201610), and the Programs of the Science Foundation in Nanjing (ZKX13023 and 201605015).
The authors have no conflicts of interest to disclose.
References
- [1].Galea R, Cardillo MT, Caroli A, et al. Inflammation and C-reactive protein in atrial fibrillation: cause or effect? Tex Heart Inst J 2014;41:461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ferro JM. Atrial fibrillation and cardioembolic stroke. Minerva Cardioangiol 2004;52:111–24. [PubMed] [Google Scholar]
- [3].Aviles RJ, Martin DO, Apperson-Hansen C, et al. Inflammation as a risk factor for atrial fibrillation. Circulation 2003;108:3006–10. [DOI] [PubMed] [Google Scholar]
- [4].Harada M, Van Wagoner DR, Nattel S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ J 2015;79:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hu YF, Chen YJ, Lin YJ, et al. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol 2015;12:230–43. [DOI] [PubMed] [Google Scholar]
- [6].Deng XT, Jiang MH, Zhu JH, et al. The association of interleukin 6-634C/G polymorphism with left atrial thrombus and severe spontaneous echocontrast in patients with atrial fibrillation. Clin Appl Thromb Hemost 2013;19:673–8. [DOI] [PubMed] [Google Scholar]
- [7].Richards CD. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm 2013;8:512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zarling JM, Shoyab M, Marquardt H, et al. Oncostatin M: a growth regulator produced by differentiated histiocytic lymphoma cells. Proc Natl Acad Sci USA 1986;83:9739–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sun D, Li S, Wu H, et al. Oncostatin M (OSM) protects against cardiac ischaemia/reperfusion injury in diabetic mice by regulating apoptosis, mitochondrial biogenesis and insulin sensitivity. J Cell Mol Med 2015;19:1296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lorchner H, Poling J, Gajawada P, et al. Myocardial healing requires Reg3beta-dependent accumulation of macrophages in the ischemic heart. Nat Med 2015;21:353–62. [DOI] [PubMed] [Google Scholar]
- [11].Kubin T, Poling J, Kostin S, et al. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell 2011;9:420–32. [DOI] [PubMed] [Google Scholar]
- [12].He G, Tan W, Wang B, et al. Increased M1 macrophages infiltration is associated with thrombogenesis in rheumatic mitral stenosis patients with atrial fibrillation. PLoS One 2016;11:e0149910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mirshahi F, Vasse M, Tedgui A, et al. Oncostatin M induces procoagulant activity in human vascular smooth muscle cells by modulating the balance between tissue factor and tissue factor pathway inhibitor. Blood Coagul Fibrinolysis 2002;13:449–55. [DOI] [PubMed] [Google Scholar]
- [14].Saint LL, Damiano RJ., Jr Surgical treatment of atrial fibrillation. Mo Med 2012;109:281–7. [PMC free article] [PubMed] [Google Scholar]
- [15].Rudnicka C, Mochizuki S, Okada Y, et al. Overexpression and knock-down studies highlight that a disintegrin and metalloproteinase 28 controls proliferation and migration in human prostate cancer. Medicine (Baltimore) 2016;95:e5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang H, Ye H, Huang W. A meshfree method for simulating myocardial electrical activity. Comput Math Methods Med 2012;2012:936243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cai XY, Ni XC, Yi Y, et al. Overexpression of CD39 in hepatocellular carcinoma is an independent indicator of poor outcome after radical resection. Medicine (Baltimore) 2016;95:e4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Petersen M, Roehrich A, Balzer J, et al. Left atrial appendage morphology is closely associated with specific echocardiographic flow pattern in patients with atrial fibrillation. Europace 2015;17:539–45. [DOI] [PubMed] [Google Scholar]
- [19].Honda Y, Morishima Y. Thrombin generation induced by tissue factor plus ADP in human platelet rich plasma: a potential new measurement to assess the effect of the concomitant use of an oral factor Xa inhibitor edoxaban and P2Y12 receptor antagonists. Thromb Res 2015;135:958–62. [DOI] [PubMed] [Google Scholar]
- [20].Luo ZQ, Hao XH, Li JH, et al. Left atrial endocardial dysfunction and platelet activation in patients with atrial fibrillation and mitral stenosis. J Thorac Cardiovasc Surg 2014;148:1970–6. [DOI] [PubMed] [Google Scholar]
- [21].Corradi D. Atrial fibrillation from the pathologist's perspective. Cardiovasc Pathol 2014;23:71–84. [DOI] [PubMed] [Google Scholar]
- [22].Kourliouros A, Savelieva I, Kiotsekoglou A, et al. Current concepts in the pathogenesis of atrial fibrillation. Am Heart J 2009;157:243–52. [DOI] [PubMed] [Google Scholar]
- [23].Sata N, Hamada N, Horinouchi T, et al. C-reactive protein and atrial fibrillation. Is inflammation a consequence or a cause of atrial fibrillation? Jpn Heart J 2004;45:441–5. [DOI] [PubMed] [Google Scholar]
- [24].Albasanz-Puig A, Murray J, Preusch M, et al. Oncostatin M is expressed in atherosclerotic lesions: a role for oncostatin M in the pathogenesis of atherosclerosis. Atherosclerosis 2011;216:292–8. [DOI] [PubMed] [Google Scholar]
- [25].Nishibe T, Parry G, Ishida A, et al. Oncostatin M promotes biphasic tissue factor expression in smooth muscle cells: evidence for Erk-1/2 activation. Blood 2001;97:692–9. [DOI] [PubMed] [Google Scholar]