

Abstract
ADAM17 is implicated in several debilitating diseases. However, drug discovery efforts targeting ADAM17 have failed due to the utilization of zinc-binding inhibitors. We previously reported discovery of highly selective nonzinc-binding exosite-targeting inhibitors of ADAM17 that exhibited not only enzyme isoform selectivity but synthetic substrate selectivity as well (J. Biol. Chem. 2013, 288, 22871). As a result of SAR studies presented herein, we obtained several highly selective ADAM17 inhibitors, six of which were further characterized in biochemical and cell-based assays. Lead compounds exhibited low cellular toxicity and high potency and selectivity for ADAM17. In addition, several of the leads inhibited ADAM17 in a substrate-selective manner, which has not been previously documented for inhibitors of the ADAM family. These findings suggest that targeting exosites of ADAM17 can be used to obtain highly desirable substrate-selective inhibitors. Additionally, current inhibitors can be used as probes of biological activity of ADAM17 in various in vitro and, potentially, in vivo systems.
Graphical abstract
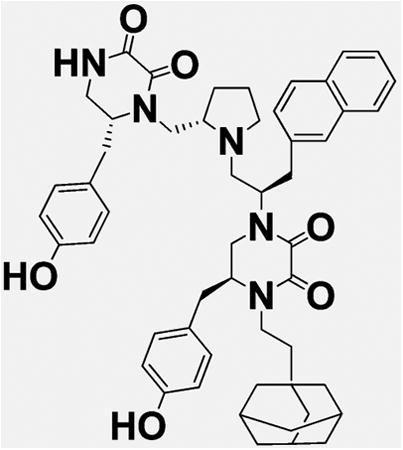
Introduction
ADAM17 is a prototypical member of a disintegrin and metalloproteinase family of metzincin proteases1 implicated in several aggressive forms of cancer,2,3 rheumatoid arthritis,2 and Alzheimer's disease.4 Not surprisingly, multiple pharmaceutical companies attempted to develop inhibitors of ADAM17 as potential drug candidates for the above-mentioned therapeutic indications. From 2001 to 2009, more than a hundred ADAM17 inhibitors were patented.5 Even though some of these molecules inhibit ADAM17 with subnanomolar potency and several of them reached clinical trials,2 they ultimately failed as drugs. It is believed that the main reason for these failures was the zinc-binding mechanism of action leading to a broad-spectrum inhibition of many zinc-dependent metalloproteases leading to in vivo side effects most frequently manifesting as a musculoskeletal syndrome (MSS).6 Additionally, many of the zinc-binding moieties, such as hydroxamates, are metabolically unstable, which limits their bioavailability.7 There are several comprehensive reviews available on collagenase inhibitors,8 TACE inhibitors,9 metzincin inhibitors,10 and metalloenzyme inhibitors11,12 that address the drawbacks of zinc-binding inhibitors.
To circumvent the problems of zinc-binding inhibitors of ADAM17 and metzincins in general, several groups, including ours, pursued discovery and development of inhibitors that act via a nonzinc-binding mechanism and target secondary substrate binding sites (also known as exosites).13–16 For example, Aventis discovered a pyrimidine dicarboxamide that had low nanomolar potency for MMP-13 and no activity against other MMPs when tested at 100 μM.13 Pfizer reported discovery of highly selective nanomolar range MMP-13 inhibitors based on pyrimidinedione and quinazolinone scaffolds acting via binding to the S1′ exosite.14,17 Similarly, Alantos Pharmaceuticals identified a new class of highly selective nonzinc-binding MMP-13 inhibitors.15,16 Most recently, Takeda Pharmaceutical Company reported yet another nonzinc-binding inhibitor of MMP-13 that acts via binding to the S1′ site.18 The lead from the Takeda series, compound 26c, exhibited subnanomolar activity against MMP-13 and good oral bioavailability. Our group reported the discovery of a MMP-13 exosite inhibitor that binds to two different exosites.19–21 Several of these inhibitors were efficacious in in vivo models of arthritis without causing MSS,14,15,22 suggesting that nonzinc-binding inhibition can indeed overcome the drawbacks of zinc-binding inhibitors of metzincins.
In addition to potentially being drug candidates, selective inhibitors of ADAM17 could represent extremely useful tools for dissecting the highly complex role of ADAM17 in various diseases and normal processes. Such probes have become the staple of chemical biology for studying cellular processes.23 The development of exosite inhibitors of ADAM17 and ADAM in general have been hampered by the lack of knowledge about exosites in the structures of ADAM. However, development of an ADAM17-selective low nanomolar inhibitory antibody24 was reported in 2011. This antibody was shown to work via binding across the catalytic and noncatalytic domains of ADAM17, thereby obstructing access of the substrate to the catalytic cleft of ADAM17.
Our group has previously reported the discovery of the first small molecule exosite-binding selective inhibitor of ADAM17.25 In the present study. we report results of structure–activity relationship studies and cellular characterization of this novel class of ADAM17 inhibitors that could potentially be used as probes for the biological activity of ADAM17.
Results
SAR Studies
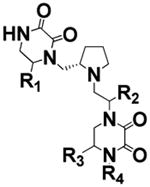
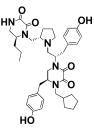
We previously conducted initial exploration of structure–activity relationship of pyrrolidine diketopiperazine analogues using S-4-hydroxybenzyl and S-propyl in the R1 position.25 In both cases, we tried S- and R-hydroxybenzyl and S-2-naphthylmethyl in position R2, S- and R-hydroxybenzyl in position R3, and 2-phenylbutyl, 2-adamantan-1-yl-ethyl, and cyclopentyl-methyl in position R4. Analogues with S-propyl in R1 position did not produce highly potent compounds (best compound IC50 value 12 μM) but were selective for ADAM17 as compared to ADAM10. In the present study, we expanded the SAR of the R1 S-propyl analogues in order to determine whether greater activity can be achieved without loss of selectivity. Thus, compounds 1–7 have S-propyl in the R1 position, R-hydroxybenzyl in position R2, and S-hydroxybenzyl in position R3, whereas residues in position R4 are varied (Table 1). All compounds of this series produced complete inhibition at 100 μM, with IC50 values ranging from 94 to 5.9 μM. Two out of three three best compounds (2 and 6, IC50 = 10 ± 1.3 and 5.9 ± 0.9 μM, respectively) contained 4-adamantan-1-yl-methyl and 2-adamantan-1-yl-ethyl in the R4 position, respectively. From our previous study, the best compound reported by us (compound 15 in ref 25 and compound 17 in present work) had 2-adamantan-1-yl-ethyl moiety in the R4 position. Overall, when 2-adamantan-1-yl-ethyl was present in position R4, it produced the greatest inhibition without loss of selectivity regardless of which functionalities were present in positions R1–3. On the basis of this consideration, the next series of analogues all have S-propyl in the R1 position, R-hydroxybenzyl in R2, and 2-adamantan-1-yl-ethyl in R4, while position R3 is varied to produce compounds 8–11 (Table 2). The functionalities used in position R3 were selective in the positional scan study during the deconvolution stage as described previously.25 Stereo-isomers with S- and R-hydroxybenzyl in position R3, compounds 6 and 8, exhibited similar IC50 values (IC50 = 5.9 ± 0.9 and 5.4 ± 0.5 μM for 6 and 8, respectively). R- and S-5-Naphthalen-2-ylmethyl (compounds 9 and 10) were also equipotent (IC50 = 7.9 ± 0.8 and 10 ± 0.8 μM for 10 and 9, respectively). Finally, compound 11 with S-butyl in R3 had IC50 of 6.5 ± 0.9 μM. These results suggested that the potency of compounds of this scaffold is largely determined by the presence of 2-adamantan-1-yl-ethyl in R4. To confirm this hypothesis, we introduced R-6-naphthalen-2-ylmethyl in position R1. Activity of R-6-naphthalen-2-ylmethyl was greater than S-propyl based on the positional scan.25 Additionally, positions R4 and R2 were fixed with 2-adamantan-1-yl-ethyl and S-4-hydroxybenzyl, respectively, while R3 was varied (Table 3). Introduction of R-6-naphthalen-2-ylmethyl in position R1 resulted in 2–3-fold loss of potency (Table 3, IC50 compound 8 = 5.4 ± 0.5 μM versus 15 ± 1.2 μM for compound 13; 5.9 ± 0.9 μM for 6 and 8.1 ± 0.9 μM for compound 12) when hydroxybenzyl was present in position R3. When 5-naphthalen-2-ylmethyl was present in R3, introduction of R-6-naphthalen-2-ylmethyl into R1 did not have a significant effect (Table 3, IC50 compound 9 = 10 ± 0.8 μM versus 8.4 ± 1.0 μM for 14; 7.9 ± 0.8 μM for 10 versus 5.7 ± 1.0 μM for 15). Similarly, the presence of S-5-butyl in R3 resulted in comparable IC50 values whether R1 contained either S-propyl or R-6-naphthalen-2-ylmethyl (Table 3, IC50 compound 11 = 6.5 ± 0.9 μM versus 5.7 ± 0.8 for 16).
Table 1. SAR Study Results of Individual Compounds Synthesized Based on Kinetic Studies of Mixture Samplesa.
| Sample # | STRUCTURE | R4 | ADAM10 IC50, μM | ADAM17 IC50, μM |
|---|---|---|---|---|
| Core |
|
N/A | N/A | N/A |
| 1 |
|
4-tert-Butyl-Cyclohexyl | >100 | 10 ± 1.0 |
| 2 |

|
4-adamantan-l-yl-methyl | >100 | 10 ± 1.3 |
| 3 |

|
Cyclohexylmethyl | >100 | 29 ± 2.0 |
| 4 |

|
4-Cyclobutylmethyl | >100 | 94 ± 8.1 |
| 5 |

|
2-Phenylbutyl | >100 | 15 ± 2.1 |
| 6 |

|
2-adamantan-l-yl-ethyl | >100 | 5.9 ± 0.9 |
| 7 |
|
4-Cyclopentylmethyl | >100 | 35 ± 3.3 |
Positions R1-3 are fixed, R4 is scanned. IC50 values reported as a mean of three experiments ± standard deviation.
Table 2. SAR Study Results of Individual Compounds Synthesized Based on Positional Scan of Mixture Samplesa.
| Sample # | STRUCTURE | R3 | ADAM10 IC50, μM | ADAM17 IC50, μM |
|---|---|---|---|---|
| Core |

|
N/A | N/A | N/A |
| 6 |

|
S-4-hydroxybenzyl | >100 | 5.9 ± 0.9 |
| 8 |

|
R-4-hydroxybenzyl | >100 | 5.4 ± 0.5 |
| 9 |

|
S-5-naphthalen-2-ylmethyl | >100 | 10 ± 0.8 |
| 10 |
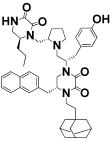
|
R-5-naphthalen-2-ylmethyl | >100 | 7.9 ± 0.8 |
| 11 |

|
S-5-butyl | >100 | 6.5 ± 0.9 |
R3 is scanned, and R1, R2, and R4 are fixed. IC50 values data reported as a mean of three experiments ± standard deviation.
Table 3. SAR Study Results of Individual Compounds Synthesized Based on Positional Scan of Mixture Samplesa.
| Sample # | STRUCTURE | R3 | ADAM10 IC50, μM | ADAM 17 1C50, μM |
|---|---|---|---|---|
| Core |

|
N/A | N/A | N/A |
| 6 |
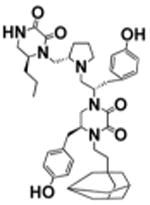
|
S-4-hydroxybenzyl | >100 | 5.9 ± 0.9 |
| 8 |

|
R-4-hydroxybenzyl | >100 | 5.4 ± 0.5 |
| 12 |

|
S-4-hydroxybenzyl | >100 | 8.1 ±0.9 |
| 13 |

|
R-4-hydroxybenzyl | >100 | 15 ±1.2 |
| 9 |

|
S-5-naphthalen-2-ylmethyl | >100 | 10 ± 0.8 |
| 10 |

|
R-5-naphthalen-2-ylmethyl | >100 | 7.9 ± 0.8 |
| 14 |
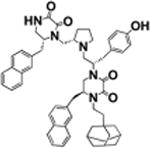
|
S-5-naphthalen-2-ylmethyl | >100 | 8.4 ± 1.0 |
| 15 |

|
R-5-naphthalen-2-ylmethyl | >100 | 5.7 ±1.0 |
| 11 |
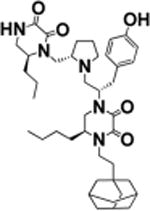
|
S-5-butyl | >100 | 6.5 ± 0.9 |
| 16 |
|
S-5-butyl | >100 | 5.7 ± 0.8 |
R3 is scanned, and R1, R2, and R4 are fixed. IC50 values data reported as a mean of three experiments ± standard deviation.
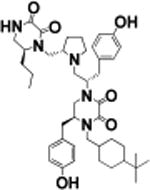
As a result of the structure–activity relationship study, several compounds with low micromolar IC50 values were identified. We chose compounds exhibiting the greatest potency for ADAM17 containing 2-adamantan-1-yl-ethyl in R4 (20, 16, and 17 described in ref 25) to be tested against an extended panel of zinc metalloproteinases. Additionally, we chose the most potent compounds without 2-adamantan-1-yl-ethyl (18, 19, and 21) for comparison in selectivity testing and cell-based studies.
Compounds 21, 19, 20, and 16 exhibited the best selectivity profile, inhibiting only ADAM17 and sparing five other metzincins. Compound 17 exhibited limited inhibition of MMP-2, MMP-9, and MMP-14/MT1-MMP (Table 4) at 100 μM, most likely due to nonspecific binding. This biochemical inhibition profile suggested that compounds of this series could potentially be used as selective in vitro and in vivo probes of ADAM17.
Table 4. Summary of Selectivity Testing of Novel ADAM17 Inhibitors against a Panel Zinc of Metalloproteasesa.
| ID | Structure | MMP2 | MMP8 | MMP9 | MMP14 | ADAM10 | ADAM17 |
|---|---|---|---|---|---|---|---|
| 16 |
|
>100 | >100 | >100 | >100 | >100 | 5.7 (100) |
| 17 |

|
NT | > 100 (13) | NT | > 100 (25) | >100 | 4.2 (95) |
| 18 |
|
>100 | > 100 (28) | > 100 (13) | > 100 (27) | >100 | 5.4 (100) |
| 19 |

|
>100 | >100 | >100 | >100 | >100 | 4.3 (100) |
| 20 |
|
>100 | >100 | >100 | >100 | >100 | 11 (100) |
| 21 |

|
>100 | >100 | >100 | >100 | >100 | 8.5 (100) |
All results are IC50, μM (%inhibition at 100 μM). No parentheses indicates 0% inhibition at 100 μM. NT: not tested.
Cell-Based Studies: Cell Toxicity and Cell Surface Targets
Before testing the ADAM17 inhibitors for abrogation of shedding of cell surface proteins that are generally considered to be physiologically relevant substrates of ADAM17, the compounds were tested for toxicity in cell viability assays. All six leads were tested with healthy and lung cancer cell lines (CHO-K1 and A549, respectively). Additionally, 18, 17, and 20 were tested with liver and breast cancer cell lines (HEPG2 and MDA-MB-231, respectively). CHO-K1 and A549 cells were viable for 72 h in the presence of up to 33 μM of lead compounds (Figure 1). 18, 17, and 20 did not inhibit viability of HEPG2 and MDA-MB-231 cells for 72 h up to 33 μM (data not shown). These results suggested that the lead compounds could be utilized as probes of ADAM17 activity in cell-based assays without compromising the health of the cellular system under investigation.
Figure 1.

Results of cell viability assay with ADAM17 inhibitors. (A) Viability assay with CHO-K1 cells. (B) Viability assay with A549 cells.
ADAM17 has been implicated as either the main or, in some cases, the only protease responsible for the cleavage of more than 70 cell surface proteins.26 To assess activity of the lead compounds against ADAM17 in a cell-based setting, several cell lines and cell surface proteins were examined.
Cytokines
The ability of compounds to protect of TNFα from shedding by ADAM17 was assessed. TNFα is a canonical substrate of ADAM17.27,28 The present ADAM17 inhibitors were discovered using substrate based on the TNFα cleavage sequence;25 therefore, we were interested to see whether TNFα shedding would be inhibited in a cell-based assay. We tested compounds at 25 μM and used Marimastat (a broad spectrum metzincin inhibitor) at 10 μM as a control. Five out of six test compounds and Marimastat inhibited approximately 30–50% of TNFα cleavage in THP1 cells (Figure 2A). To assess the potency of this inhibition, we performed a dose–response study with compounds 17 and 19, whereby they exhibited ∼100 and 28 μM IC50 values, respectively (Figure 2B).
Figure 2.
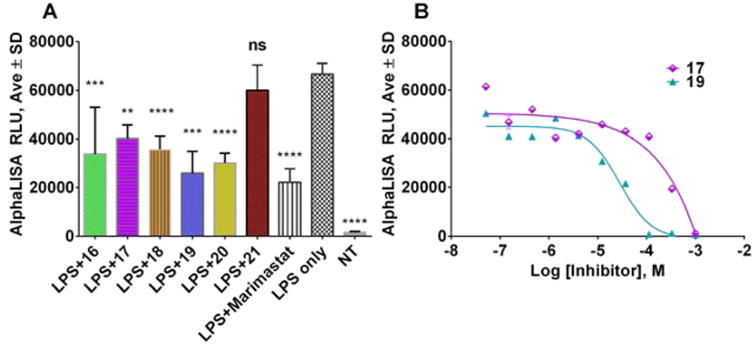
Results of TNFα shedding assay in THP-1 cells with ADAM17 inhibitors. (A) Single point assay. One-way analysis of variance (ANOVA) was used followed by Dunnett posthoc test. The data shown are the mean ± SEM, n = 5. *****p < 0.0001; ***p < 0.001, **p < 0.01, *p < 0.05. (B) Dose–response assay with compounds 17 and 19.
PMA - or LPS-induced production of chemokine IL-8 by epithelial cells of human airway (NCI-H292) or human trachel smooth muscle cells (HTSMC) was reported to be mediated by ADAM17's cleavage of TGFα and subsequent trans-activation of ErB/EGFR29 and was inhibited by ADAM17 siRNA. In human tracheal smooth muscle cells (HTSMC), IL-8 was shed in response to LPS, acid, and IL-1 stimulation30 and was decreased as a result of ADAM17 shRNA application. Thus, IL-8 production by these cells can be used as read out for TGFα shedding. We tested compounds 18, 17, 20, and 16 for inhibition of LPS- and IL-1-stimulated IL-8 release from HTSMCs. Ten μM of compounds 18, 17, 20, and 16 inhibited 18–62% of LPS-stimulated IL-8 shedding (Figure 3A). Interestingly, none of the tested compounds were able to inhibit IL-1-mediated production of IL-8 (Figure 3B). It is known that IL-1 stimulates production of IL-8 via inducing cleavage of neuregulins, which can be cleaved by multiple proteases of which ADAM17 is the critical sheddase in the cell system that was used here.29,30 Lack of inhibition of IL-1-induced IL-8 release in combination with inhibition of LPS-induced IL-8 release suggests that our lead compounds target TGFα cleavage by ADAM17 but not cleavage of neuregulins. Our compounds may have significant utility for dissecting of complex proteolytic pathways where ADAM17 is implicated in a substrate-specific manner.
Figure 3.
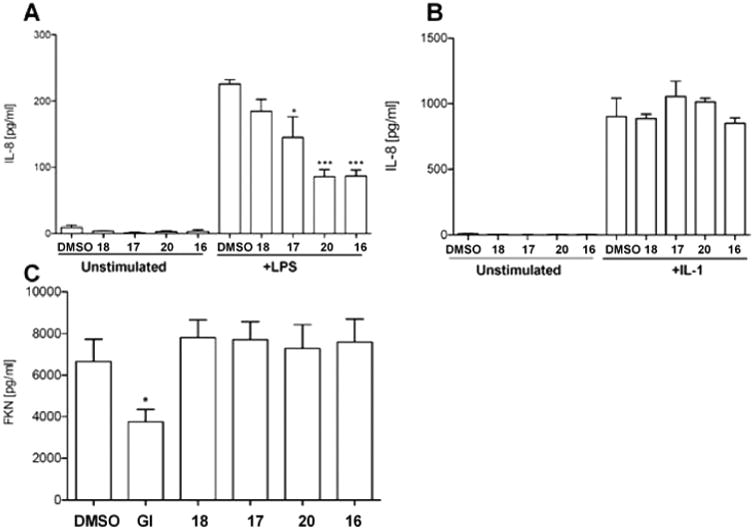
Effect of ADAM17 inhibitors on cytokine shedding in HTMS cells. (A) LPS-stimulated IL-8 shedding inhibition assay. (B) IL-1-stimulated IL-8 shedding inhibition assay. (C) Fractalkine constitutive shedding inhibition assay. One-way analysis of variance (ANOVA) was used followed by Dunnett posthoc test. The data shown are the mean ± SEM, n = 3 from three independent experiments. *****p value <0.0001, ***p value <0.001, **p value <0.01, *p value <0.05. GI: control ADAM10 selective inhibitor GI254023X.
To test for selectivity of compounds, we conducted assays for inhibition of shedding of fractalkine/CX3CL1 and CXCL16 using HTSMC. Fractalkine/CX3CL1 is shed both constitutively and in response to PMA stimulation. ADAM17 was shown to be responsible for PMA-induced cleavage,31 whereas constitutive shedding is ascribed to ADAM10.32 None of the lead compounds were able to abrogate constitutive ADAM10-mediated release of fractalkine/CX3CL1 (Figure 3C), suggesting that the compounds are selective for ADAM17. Additionally, compounds 18, 17, 20, and 16 did not inhibit the shedding of CXCL16 ascribed to ADAM10,33 further suggesting their selectivity for ADAM17 in HTSMC (data not shown).
Receptors
Shedding of discoidin domain receptor 1 (DDR1) in HCC1806 breast cancer cells is mediated by cell surface-bound metalloproteases.34 Membrane type 1 matrix metalloprotease (MT1-MMP, also known as MMP-14) was determined to be one of the enzymes responsible for the constitutive cleavage of DDR1. However, MT1-MMP knockdown or inhibition by pharmacological agents did not result in the abrogation of DDR1 shedding. These data suggested the existence of a compensatory mechanism whereby another metalloprotease cleaves DDR1 in the event of MT1-MMP inhibition. Because ADAM10 and ADAM17 mRNAs were detected in HCC1806 cells,34 the hypothesis of ADAM proteases involvement in DDR1 cleavage was formed and it was decided to investigate whether shedding of DDR1 ectodomain could be affected by the application of ADAM17 selective inhibitors. Compound 17 was tested using 10, 20, and 40 μM concentrations. DDR1 shedding was inhibited in a dose-dependent manner (Figure 4), however, even at the highest tested dosage (40 μM), soluble DDR1 was detectable. This suggests that both ADAM17 and MT1-MMP have to be inhibited to completely abrogate the shedding of DDR1. Additionally, compound 17 did not inhibit ADAM10 in the in vitro assay (Table 4), suggesting that this enzyme does not appear to be involved in constitutive shedding of DDR1 despite the presence of its mRNA in HCC1806 cells.
Figure 4.
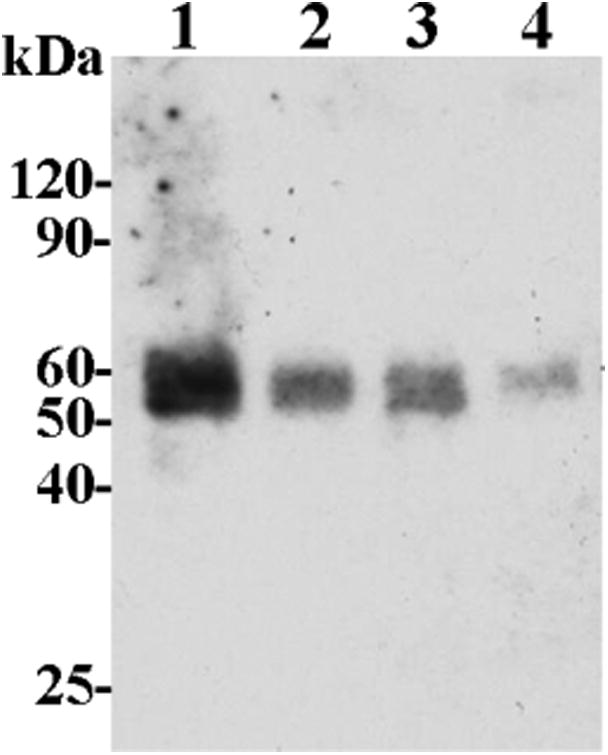
Results of DDR1 release assay in HCC1806 breast cancer cells with compound 17. One, untreated control; 2, 10 μM compound 17; 3, 20 μM compound 17, 4–40 μM compound 17.
Another cell surface protein with importance in cancer progression that is cleaved by MT1-MMP and ADAM17 is protein tyrosine kinase 7 (PTK7). PTK7 proteolysis controls cell migration in early embryogenesis and regulates cancer cell directional motility, invasion, and metastasis.35–39 The full-length PTK7 efficiently inhibits cell invasion and downregulates myosin light chain (MLC) phosphorylation, a downstream event in the Wnt/PCP pathway.36 Proteolysis of PTK7 reverses the inhibitory signal of the full-length PTK7 and subsequently promotes cell invasion35 and metastasis.38 Thus, PTK7 proteolysis by MT1-MMP and ADAM17 is a proteolytic master switch that turns on the motility of cells. We tested lead compounds (18, 17, 19, and 16) at 25, 50, and 100 μM concentrations using a human fibrosarcoma cell line that expressed a PTK7 mutant uncleavable by MT1-MMP (HT1080-L622D) and colorectal cancer cells (SW480) expressing wild-type PTK7 endogenously (wtPTK7). Compound 17 inhibited shedding of L622D-PTK7 and wtPTK-7 at 50 and 100 μM (Figure 5A) when both HT1080-L622D and SW480 cells were stimulated with PMA. 18, 19, and 16 did not exhibit inhibition of L622D-PTK7 and wtPTK-7 up to 100 μM (data not shown).
Figure 5.
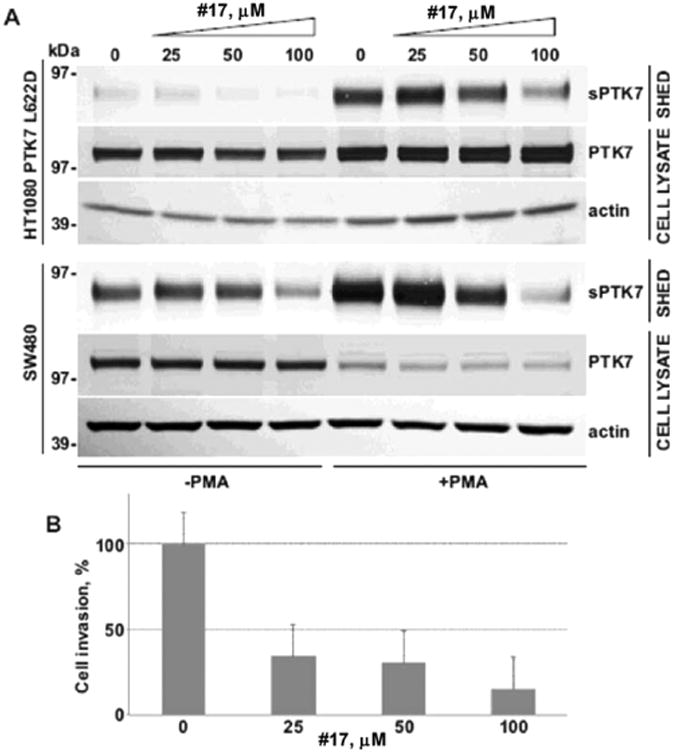
Effect of compound 17 on PTK7 shedding and invasion. (A) PTK7 shedding assay in HT1080 (fibrosarcoma) and SW480 (colorectal cancer) cells. (B) Invasion assay in HT1080 cells. sPTK7: soluble PTK7.
A dual inhibitor of ADAM10 and ADAM17, INCB3619 (ADAM17 IC50 = 14 nM, ADAM10 IC50 = 22 nM, MT1-MMP IC50 = 772 nM)40 was able to inhibit both shedding of PTK7 and invasion of HT1080 cells.37 On the basis of this evidence, we hypothesized that selective inhibition of ADAM17 should result in a decrease of invasiveness of HT1080 cells. Indeed, we observed a dose-dependent inhibition of invasion of HT1080 cells in a Transwell assay (Figure 5B).
Growth Factors
ADAM10 and ADAM17 are believed to be the main sheddases of EGFR ligands.41 Moreover, the expression of ADAM17 was shown to be up-regulated in lung cancer,42 where it has been shown to shed heregulin43 leading to activation of EGFR/ERK/Akt pathways and resulting in cancer cell proliferation,44 resistance to EGFR-based therapies,40 and poor prognosis.40,42 On the basis of these considerations, we were interested to see whether exosite inhibition of ADAM17 can be an effective tool to study the role of ADAM17 in lung cancer. We used A549 nonsmall cell lung cancer cells, which were shown to have high expression levels of heregulin40 to determine the ability of our lead compounds to inhibit its shedding. Heregulin is specifically cleaved by ADAM1745 and therefore represents a suitable end point in determining cellular efficacy of ADAM17 inhibitors. Interestingly, despite very similar IC50 values for ADAM17 inhibition in the biochemical assay (Table 4), only compound 17 completely inhibited shedding of heregulin in the cell-based assay (Figure 6A, all leads tested at 40 μM). To assess enzyme selectivity of compound 17, we tested its ability to inhibit shedding of another EGFR ligand, betacellulin, which is cleaved exclusively by ADAM10.41,45 Marimastat and compounds 21, 18, and 16 were also tested. None of tested selective ADAM17 inhibitors affected the shedding of betacellulin from A549 cell surface at 40 μM, whereas Marimastat exhibited almost complete inhibition at 10 μM. This suggests that 17 selectively inhibits ADAM17 not only in biochemical but in a cell-based environment and could be a useful tool to study the role of ADAM17 in cells.
Figure 6.
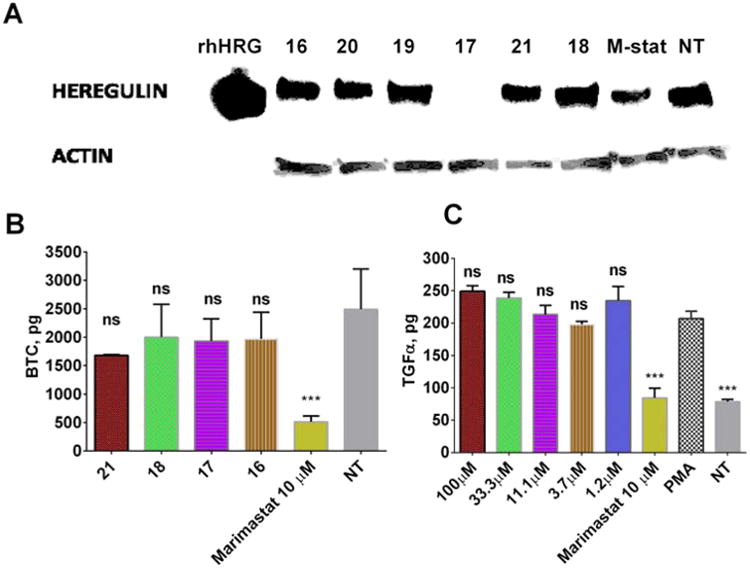
Effect of ADAM17 inhibitors on growth factors shedding in A549 cells. (A) Effect on shedding of heregulin. M-stat: marimastat. (B) Effect on shedding of betacellulin. One-way analysis of variance (ANOVA) was used followed by Dunnett posthoc test. The data shown are the mean ± SEM, n = 3. ***p <0.001. (C) Effect on shedding of TGFα. One-way analysis of variance (ANOVA) was used followed by Dunnett posthoc test. The data shown are the mean ± SEM, n = 3. ***p <0.001.
We previously reported that 17 was able to preferentially inhibit ADAM17 hydrolysis of a glycosylated synthetic substrate based on the TNFα sequence (15 in ref 46). ADAM17 is known to be able to cleave more than 70 cell surface proteins,47 creating concern that indiscriminate inhibition of all ADAM17-mediated shedding could have unpredictable side effects. In turn, inhibitors that can selectively inhibit hydrolysis of certain substrates or a subset of substrates could be of interest for drug discovery. To assess whether 17 inhibits heregulin with some degree of selectivity, we tested it for the inhibition of shedding of TGFα, another EGFR ligand with importance in lung cancer. Indeed, 17 did not affect shedding of TGFα in A549 cells up to 100 μM, whereas 10 μM Marimastat decreased soluble TGFα in the supernatant of A549 cells to the level of non-PMA-treated control (Figure 6C).
Synergy Studies
It was shown that ADAM17 inhibition by broad spectrum metalloprotease inhibitor INCB3619 led to abrogation of heregulin shedding in A549 cells, which potentiated the activity of the EGFR tyrosine kinase inhibitor gefitinib.40 As mentioned above, 17 inhibited shedding of heregulin in A549 cells (Figure 6A); therefore, we used it to test for synergy with gefitinib. Then 40 μM compound 17 potentiated gefitinib activity against A549 cells in the viability assay (Figure 1). The IC50 value for gefitinib inhibition of A549 cell growth was 8.4 μM, but in the presence of 40 μM compound 17 the IC50 value decreased 4-fold (1.9 μM). For comparison, 10 μM INCB3619 shifted gefitinib's IC50 value from ∼8 to ∼1 μM in A549 cells.40 Because 17 exhibited no intrinsic activity against A549 cell in viability assay (Figure 1), the improvement of gefitinib's potency can be ascribed to the synergy between 17 and gefitinib. For comparison, compound 17 did not potentiate the activity of two other FDA-approved lung cancer drugs etoposide or doxorubicin (Figure 7B,C). Etoposide and doxorubicin act via topoisomerase inhibition, distinct from the inhibition of EGFR tyrosine kinase activity of gefitinib.
Figure 7.
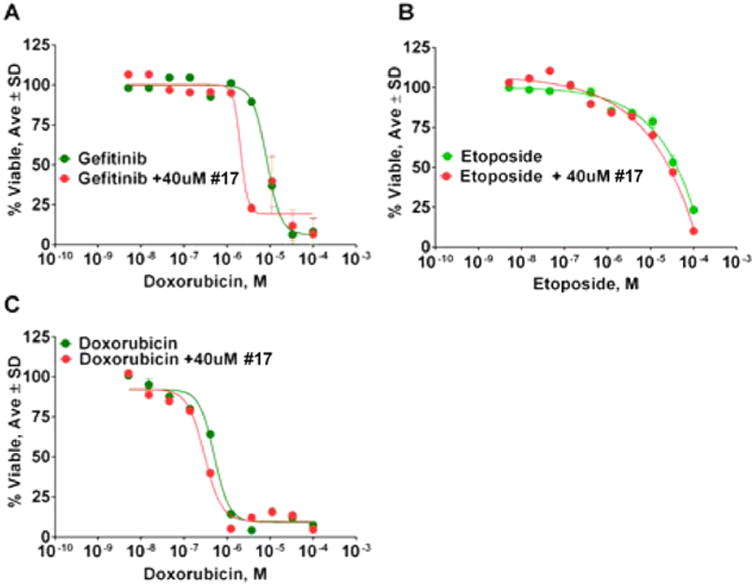
Results of synergy studies of compound 17 with FDA-approved lung cancer drugs. (A) Synergy study with gefitinib. (B) Synergy study with etoposide. (C) Synergy study with doxorubicin.
Discussion
Despite the large number of ADAM17 inhibitors developed by various academic and industrial entities, there is a paucity of ADAM17 inhibitors that work by binding to so-called exosites. Therefore, there is limited understanding of the biological and biochemical effects of inhibition by such molecules. Indeed, what can be expected from molecules that bind to an exosite rather to an active site of ADAM17 or any other enzyme? For example, the anti-BACE1 exosite-binding antibody inhibited hydrolysis of a longer APP-based synthetic substrate 10-fold more potently than a shorter version of the same substrate,48 suggesting that exosite-based inhibition can lead to substrate selectivity. Substrate-selective inhibition is much more rare than enzyme-selective inhibition but has been demonstrated for different classes of enzymes. In the case of COX-2 substrate-selective inhibitors, binding to an allosteric subunit results in noncompetitive inhibition of endocannabinoid oxygenation, but no inhibition of arachidonic acid oxygenation was observed.49 Substrate-selective inhibition of protein kinase PDK1 was also achieved as a result of binding of small molecule to an allosteric site.50
To our knowledge, substrate-selective inhibition of ADAM17 has not been reported so far. While metzincin isoform selectivity was addressed in many studies, the possibility of substrate selectivity of metzincin inhibitors still remains largely unexplored. It is now understood that some metzincins should not be inhibited in certain disease scenarios (i.e., MMP-8 and MMP-14 in skin and breast cancer51–53). However, it is much less known about whether hydrolysis of any of the cognate substrates should be spared from inhibition. As demonstrated in the case of γ-secretase inhibitor discovery for Alzheimer's disease, inhibition of γ-secretase abrogated release of β-amyloid but also prevented a cleavage of Notch leading to toxicity (reviewed in ref 54). It is well-known that metzincins have broad substrate repertoires, therefore, it is possible that total abrogation of activity of a target enzyme might be unwanted.
Our data suggest that exosite inhibition of ADAM17 can lead to a substrate-selective inhibition (Table 5). Data presented herein demonstrate that compound 17 inhibits ADAM17 selectively in cell-based assays, which in turn suggests that it can be used as a probe of ADAM17 activity in in vitro and, potentially, in vivo, systems. In addition to enzyme selectivity, compound 17 also exhibited unusual substrate selectivity by sparing ADAM17-mediated cleavage of TGFα. For comparison, both Marimastat (a broad spectrum ADAM and MMP zinc-binding inhibitor) and INCB84298 (a moderately ADAM17-selective zinc-binding inhibitor) inhibited shedding of each tested EGFR ligand (heregulin, TGFα, HB-EGF, amphiregulin, and EGF) in A549 cells almost equipotently.40 Even though limited enzyme selectivity might be achieved by targeting the zinc of the ADAM17 active site, the zinc-binding inhibitors cannot selectively inhibit proteolysis of subset of ADAM17 substrates. Further studies characterizing the substrate inhibition profile of exosite inhibitor 17 are needed to determine which substrates of ADAM17 are protected from shedding and what is the underlying mechanism of this unusual substrate selectivity. One possibility is that different ADAM17 substrates interact with different exosites or sets of exosites in ADAM17, while compound 17 potentially interacts with just one exosite and, therefore, cannot inhibit binding of substrates that interact with exosites different from the one that compound 17 binds to. This possible explanation is consistent with the fact that ADAM17-selective exosite-binding antibody D1(A12)24 inhibits shedding of all tested ADAM17 substrates (i.e., TNFα, TGFα, amphiregulin, HB-EGF-AP, and TNFR1a) while sparing ADAM10. D1(A12) interacts with a significantly greater surface area of ADAM17 as compared to 17 bridging catalytic and noncatalytic domains and, therefore, potentially interacting with many more exosites than 17. The limited exosite interaction is possibly due to the fact that inhibitors of the 17 chemotype were discovered using a TNFα-based glycosylated substrate, creating a bias toward inhibition of TNFα (Table 5) over other ADAM17 substrates (Table 5, TNFα versus TGFα, heregulin). It will be interesting to see whether a similar bias toward inhibiting certain other ADAM17 substrates can be achieved by using HTS substrates derived from different cognate substrates of these adamalysins or other metzincins. Conversely, this ability to “program” or bias discovery toward substrate-selective inhibitors can be highly desirable for drug discovery for enzymes with complex substrate repertoires.
Table 5. Summary of Cell-Based Testing of Novel ADAM17 Inhibitors for Suppression of Shedding of Cell-Surface Proteinsa.
| target ID | cell line | [C] tested (μM) | 16 | 17 | 18 | 19 | 20 | 21 |
|---|---|---|---|---|---|---|---|---|
| TNFα | THP1 | 25 | 57 | 43 | 36 | 60 | 44 | 0 |
| PTK7 | HT1080 | 25–100 | 0 | 25–50 | 0 | 0 | NT | NT |
| PTK7 | SW480 | 25–100 | 0 | 0–82 | 0 | 0 | NT | NT |
| DDR1 | HCC1806 | 10–40 | NT | 59–81 | NT | NT | NT | NT |
| IL-8 | HTSMC | 10 | 62 | 36 | 18 | NT | 62 | NT |
| FKN | HTSMC | 10 | 0 | 0 | 0 | NT | 0 | NT |
| CXCL16 | HTSMC | 10 | 0 | 0 | 0 | NT | 0 | NT |
| Heregulin | A549 | 40 | 0 | 100 | 0 | 28 | 0 | 34 |
| TGFα | A549 | 40 | 0 | 0 | 0 | 0 | 0 | 0 |
| BTC | A549 | 40 | 0 | 0 | 0 | NT | 0 | 0 |
| Notchb | SKMEL-28 | 25 | NT | 0 | NT | NT | NT | NT |
| Neprilysin63 | Ea.hy926 | 15 | NT | NT | 62 | 38 | NT | NT |
Data reported as inhibition, %. NT: not tested.
Personal communication from Dr. Kaushik (Kansas University Medical Center).
Conclusion
The findings presented herein suggest that targeting exosites of ADAM17 can be used to obtain highly desirable enzyme isoform and substrate-selective inhibitors. Additionally, current inhibitors can be used as probes of the biological activity of ADAM17 in various in vitro and, potentially, in vivo systems.
Experimental Procedures
General Synthesis Procedure for Pyrrolidine-bis-diketopiperazine
All compounds were synthesized via solid-phase methodology (Scheme 1) on 4-methylbenzhydrylamine hydrochloride resin (MBHA) (1.1 mmol/g, 100–200 mesh) using the “tea-bag” approach55 as previously described elsewhere.56 Boc-amino acids were coupled utilizing standard coupling procedures (6 equiv) with hydroxybenzotriazole hydrate (HOBt, 6 equiv) and N,N′-diisopropylcarbodiimide (DIC, 6 equiv) in dimethylformamide (DMF, 0.1 M) for 120 min. Boc protecting groups were removed with 55% trifluoroacetic acid (TFA)/45% dichloromethane (DCM) (1×, 30 min) and subsequently neutralized with 5% diisopropylethylamine (DIEA)/95% DCM (3×, 2 min). Carboxylic acids (10 equiv) were coupled utilizing standard coupling procedures with HOBt (10 equiv) and DIC (10 equiv) in DMF (0.1 M) for 120 min. Completion of all couplings was monitored with a ninhydrin test. Compounds were reduced to polyamines (Scheme 1) using a 40× excess of borane (1.0 M in tetrahydrofuran (THF)) over each amide bond in a glass vessel under nitrogen at 65 °C for 72 h. The solution was then poured off, the reaction was quenched with methanol (MeOH), and the bags were washed with THF (1×, 1 min) and MeOH (4×, 1 min) and allowed to air-dry. Once dry, the bags were treated with piperidine overnight at 65 °C in a glass vessel. The solution was poured off, and the bags were washed with DMF (2×, 1 min), DCM (2×, 1 min), MeOH (1×, 1 min), DMF (2×, 1 min), DCM (2×, 1 min), and MeOH (1×, 1 min) and allowed to air-dry. Completion of reduction was checked by cleaving a control sample and analyzing using LCMS. As previously reported by our group and others, the reduction of polyamides with borane is free of racemization (refs 1–3). Diketopiperazine cyclization (Scheme 1) was performed under anhydrous conditions (<22% humidity). The dry bags were washed with anhydrous DMF (2×, 1 min), then added to a solution of 1,1′-oxalyldiimidazole (5-fold excess for each cyclization site) in anhydrous DMF (0.1 M) and shaken at room temperature overnight. The solution was poured off, and the bags were rinsed with DMF (3×, 1 min) and DCM (3×, 1 min). Completion of cyclization was checked by cleaving a control sample and analyzing by LCMS. The compounds were then cleaved from the resin with hydrofluoric acid (HF) in the presence of anisole in an ice bath at 0 °C for 90 min (Scheme 1) and extracted using 95% acetic acid (AcOH)/5% H2O (2×, 5 mL). Final crude products were purified using HPLC as described below. All chirality was generated from the corresponding amino acids. Under the reaction conditions described, no epimerization was observed, and for those compounds with multiple chiral centers, a single diastereomer was obtained
Scheme 1. General Synthesis Procedure for Leads of Pyrrollidine-bis-diketopiperazine Series Used in Present Studies.

Compound Purification and Characterization
The final compounds were purified using preparative HPLC with a dual pump Shimadzu LC-20AB system equipped with a Luna C18 preparative column (21.5 mm × 150 mm, 5 μm) at λ = 214 nm, with a mobile phase of (A) H2O (+0.1% formic acid)/(B) acetonitrile (ACN) (+0.1% formic acid) at a flow rate of 13 mL/min; gradients varied by compound based on hydrophobicity. 1H NMR and 13C NMR spectra were recorded in DMSO-d6 on a Bruker Ascend 400 MHz spectrometer at 400.14 and 100.62 MHz, respectively, and MALDITOF mass spectra were recorded using an Applied Biosystems Voyager DE-PRO Biospectrometry workstation. The purities of synthesized compounds were confirmed to be greater than 95% by liquid chromatography and mass spectrometry on a Shimadzu LCMS-2010 instrument with ESI Mass Spec and SPD-20A liquid chromatograph with a mobile phase of (A) H2O (+0.1% formic acid)/(B) ACN (+0.1% formic acid) (5–95% over 6 min with a 4 min rinse).
(S)-4-((4-(tert-Butyl)cyclohexyl)methyl)-1-((S)-1-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)-5-(4-hydroxybenzyl)piperazine-2,3-dione (1)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 1 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-tyrosine(2-Br-Z)-OH (R3), and 4-(tert-butyl)-cyclohexanecarboxylic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): (400 MHz, DMSO-d6): δ ppm 8.5 (br s, 1 H) 7.6 (br s, 1 H) 7.0–7.1 (m, 1 H) 6.9 (br s, 1 H) 6.7 (br s, 1 H) 6.5 (br s, 1 H) 6.3 (br s, 1 H) 3.6 (d, J = 12.5 Hz, 1 H) 3.5 (br s, 1 H) 3.3 (br s, 19H) 3.2 (br s, 1 H) 2.9 (br s, 1 H) 2.7–2.8 (m, 1 H) 2.7 (br s, 13 H) 2.3 (br s, 4 H) 2.1 (br s, 1 H) 1.9 (br s, 1 H) 1.8 (br s, 1 H) 1.7 (br s, 1 H) 1.5 (br s, 1 H) 1.4 (d, J = 12.3 Hz, 1 H) 1.3 (br s, 1 H) 1.2 (br s, 1 H) 0.9 (br s, 1 H) 0.7–0.9 (m, 2 H). m/z calcd C43H61N5O6 [M + H]+, 744.46; found (MS ESI), 744.35. Purity LCMS: 95.5% (254 nm, peak height).
(S)-4-((3S,5S,7S)-Adamantan-1 -ylmethyl)-1-((S)-1-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)-5-(4-hydroxybenzyl)piperazine-2,3-dione (2)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 2 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-tyrosine(2-Br-Z)-OH (R3), and 1-adamantanecarboxylic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.5 (br s, 1 H) 7.3 (d, J = J = 6.8 Hz, 1 H) 7.1 (br s, 1 H) 7.0 (br s, 1 H) 6.9 (br s, 1 H) 6.7 (br s, 1 H) 6.5 (br s, 1 H) 6.3 (br s, 1 H) 3.6 (br s, 3 H) 3.3 (br s, 18 H) 3.1 (br s, 2 H) 2.8 (d, J = 11.7 Hz, 2 H) 2.7 (br s, 9 H) 2.3 (br s, 5 H) 2.0 (br s, 1 H) 1.9 (br s, 1 H) 1.6 (br s, 1 H) 1.5 (br s, 2 H) 1.3 (br s, 1 H) 1.2 (br s, 1 H) 0.9 (br s, 1 H). m/z calcd C43H57N5O6 [M + H]+, 740.43; found (MS ESI), 740.40. Purity LCMS: 95.1% (254 nm, peak height).
(S)-4-(Cyclohexylmethyl)-1-((S)-1-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)-5-(4-hydroxybenzyl)piperazine-2,3-dione (3)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 3 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-Tyrosine-(2-Br-Z)-OH (R3), and cyclohexanecarboxylic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.4 (br s, 1 H) 7.6–7.7 (m, 2 H) 7.3 (d, J = 5.8 Hz, 1 H) 7.1 (br s, 1 H) 7.0 (br s, 1 H) 6.9 (br s, 1 H) 6.6 (br s, 1 H) 6.5 (br s, 1 H) 4.8 (br s, 1 H) 3.8 (br s, 2 H) 3.6 (br s, 3 H) 3.3 (br s, 13 H) 3.0 (br s, 3 H) 2.8 (d, J = 11.7 Hz, 2 H) 2.7 (br s, 6 H) 2.2–2.5 (m, 3 H) 2.0 (br s, 1 H) 1.8 (br s, 1 H) 1.7 (br s, 2 H) 1.5 (br s, 2 H) 1.2 (br s, 1 H) 1.1 (d, J = 16.9 Hz, 1 H) 0.9 (br s, 1 H) 0.8 (br s, 1 H). m/z calcd C39H53N5O6 [M + H]+, 688.40; found (MS ESI), 688.20. Purity LCMS: 96.1% (254 nm, peak height).
(S)-4-(Cyclobutylmethyl)-1-((S)-1-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)-5-(4-hydroxybenzyl)piperazine-2,3-dione (4)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 4 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-tyrosine-(2-Br-Z)-OH (R3), and cyclobutanecarboxylic acid (R4). 1H NMR (400 MHz, DMSO-d6): 8.5 (br s, 1 H) 6.9–7.0 (m, 5 H) 6.6–6.7 (m, 4 H) 3.6 (d, J = 12.47 Hz, 1 H) 3.3 (br s, 13 H) 2.8 (d, J = 10.86 Hz, 2 H) 2.7 (br s, 8 H) 2.3 (br s, 4 H) 1.9 (br s, 1 H) 1.8 (br s, 1 H) 1.7 (br s, 2 H) 1.6 (d, J = 11.30 Hz, 4 H) 1.3 (br s, 1 H) 1.2 (br s, 1 H) 0.9 (br s, 1 H). m/z calcd C37H49N5O6 [M + H]+, 660.37; found (MS ESI), 660.30. Purity LCMS: 97.3% (254 nm, peak height).
(5S)-1-((S)-1-((S)-2-(((S)-2,3-Dioxo-6-propylpiperazin-1-yl)-methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)-5-(4-hydroxybenzyl)-4-(2-phenylbutyl)piperazine-2,3-dione (5)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 5 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-Tyrosine(2-Br-Z)-OH (R3), and 2-phenylbutanoic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.5 (br s, 1 H) 7.6 (br s, 1 H) 6.9 (br s, 1 H) 6.7–6.8 (m, 1 H) 6.5 (br s, 1 H) 6.3 (br s, 1 H) 3.3 (br s, 17 H) 2.8 (br s, 2 H) 2.7 (br s, 9 H) 2.3 (br s, 1 H) 2.1–2.2 (m, 1 H) 2.1 (br s, 1 H) 2.0 (br s, 1 H) 1.9 (br s, 1 H) 1.8 (br s, 1 H) 1.7 (br s, 2 H) 1.5 (br s, 2 H) 1.5– 1.6 (m, 3 H) 1.3 (br s, 1 H) 1.2 (br s, 1 H) 0.9 (br s, 1 H) 0.7 (br s, 1 H). m/z calcd C44H59N5O6 [M + H]+, 724.40; found (MS ESI), 724.30. Purity LCMS: 97.0% (254 nm, peak height).
(S)-4-(2-((3S,5S,7S)-Adamantan-1-yl)ethyl)-1-((S)-1-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)-5-(4-hydroxybenzyl)piperazine-2,3-dione (6)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 6 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-tyrosine(2-Br-Z)-OH (R3), and 1-adamantaneacetic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.5 (br s, 1 H) 7.0 (br s, 1 H) 6.9 (br s, 1 H) 6.7 (br s, 1 H) 6.5 (br s, 1 H) 6.3 (br s, 1 H) 3.4 (br s, 22 H) 2.8 (br s, 2 H) 2.7 (br s, 14 H) 2.3 (br s, 3 H) 2.1 (br s, 1 H) 2.0 (br s, 1 H) 1.9 (br s, 1 H) 1.8 (br s, 1 H) 1.7 (br s, 1 H) 1.6 (br s, 1 H) 1.4 (br s, 1 H) 1.3 (br s, 1 H) 1.2 (br s, 1 H) 0.9 (br s, 1 H). m/z calcd C44H59N5O6 [M + H]+, 754.45; found, (MS ESI), 754.30. Purity LCMS: 99.3% (254 nm, peak height).
(S)-4-(Cyclopentylmethyl)-1-((S)-1-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)-5-(4-hydroxybenzyl)piperazine-2,3-dione (7)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 7 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-tyrosine-(2-Br-Z)-OH (R3), and cyclopentylcarboxylic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.4 (br s, 1 H) 7.00 (br s, 1 H) 6.9 (d, J = 6.28 Hz, 2 H) 6.7–6.8 (m, 1 H) 6.5 (br s, 1 H) 3.6 (br s, 3 H) 3.3 (br s, 25 H) 2.8 (br s, 2 H) 2.7 (br s, 13 H) 2.3 (br s, 3 H) 1.4–1.6 (m, 1 H) 1.3 (br s, 1 H) 1.2 (br s, 1 H) 0.9 (br s, 1 H). m/z calcd C38H51N5O6 [M + H]+, 674.38; found (MS ESI), 674.25. Purity LCMS: 95.8% (254 nm, peak height).
(R)-4-(2-((3R,5R,7R)-Adamantan-1-yl)ethyl)-1-((S)-1-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)-5-(4-hydroxybenzyl)piperazine-2,3-dione (8)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 8 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-d-tyrosine(2-Br-Z)-OH (R3), and 1-adamantaneacetic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.4 (br s, 1 H) 7.3 (d, J = 5.8 Hz, 1 H) 6.9–7.0 (m, 1 H) 6.9 (br s, 1 H) 6.6–6.7 (m, 3 H) 6.5 (br s, 1 H) 3.8 (d, J = 10.64 Hz, 1 H) 3.5–3.7 (m, 3 H) 3.4–3.5 (m, 3 H) 3.3 (br s, 7 H) 3.1–3.3 (m, 3 H) 3.0 (br s, 2 H) 2.7–2.9 (m, 4 H) 2.3–2.5 (m, 2 H) 2.2 (d, J = 7.8 Hz, 1 H) 1.8–2.0 (m, 3 H) 1.8 (br s, 1 H) 1.7 (d, J = 11.74 Hz, 4 H) 1.8 (d, J = 11.00 Hz, 4 H) 1.4 (br s, 5 H) 1.3 (br s, 1 H) 1.1–1.3 (m, 2 H) 0.8–1.0 (m, 3 H). m/z calcd C44H59N5O6 [M + H]+, 754.45; found (MS ESI), 754.30. Purity LCMS: 96.3% (254 nm, peak height).
(S)-4-(2-((3S,5S,7S)-Adamantan-1-yl)ethyl)-1-((S)-1-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)-5-(naphthalen-2-ylmethyl)piperazine-2,3-dione (9)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 9 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-Ala(2-naphthyl)-OH (R3), and 1-adamantaneacetic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.4 (br s, 1 H) 7.8–8.0 (m, 2 H) 7.7 (br s, 1 H) 7.5 (br s, 1 H) 7.4 (br s, 1 H) 6.9 (d, J = 6.4 Hz, 2 H) 6.6 (d, J = 7.5 Hz, 2 H) 6.5 (br s, 1 H) 4.0 (br s, 1 H) 3.8 (d, J = 12.8 Hz, 1 H) 3.5 (d, J = 13.4 Hz, 4 H) 3.3 (br s, 6 H) 3.0–3.2 (m, 3 H) 2.9 (d, J = 14.1 Hz, 2 H) 2.8 (br s, 2 H) 2.7 (d, J = 12.8 Hz, 4 H) 2.2–2.5 (m, 3 H) 1.9 (br s, 1 H) 1.8 (br s, 3 H) 1.7–1.8 (m, 2 H) 1.6 (d, J = 11.9 Hz, 3 H) 1.4–1.5 (m, 4 H) 1.3 (br s, 4 H) 1.0–1.2 (m, 4 H) 0.7–1.0 (m, 2 H). m/z calcd C48H61N5O5 [M + H]+, 788.47; found (MS ESI), 788.30. Purity LCMS: 98.9% (254 nm, peak height).
(R)-4-(2-((3R,5R,7R)-Adamantan-1-yl)ethyl)-1-((S)-1-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)-5-(naphthalen-2-ylmethyl)piperazine-2,3-dione (10)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 10 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-d-Ala(2-naphthyl)-OH (R3), and 1-adamantaneacetic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.4 (br s, 1 H) 7.7–7.9 (m, 2 H) (br s, 1 H) 7.5 (br s, 1 H) 7.3 (d, J = 5.75 Hz, 1 H) 7.0 (br s, 1 H) (d, J = 7.0 Hz, 1 H) 3.9 (br s, 1 H) 3.8 (d, J = 9.0 Hz, 1 H) 3.6 (d, J = 14.9 Hz, 4 H) 3.4 (br s, 9 H) 3.2 (br s, 3 H) 3.0 (br s, 2 H) 2.9 (br s, 2 H) 2.8 (d, J = 11.9 Hz, 2 H) 2.7 (br s, 3 H) 2.4 (br s, 2 H) 2.2 (d, J =6.7 Hz, 1 H) 2.1 (br s, 1 H) 1.9 (br s, 1 H) 1.8 (br s, 3 H) 1.7 (br s, 2 H) 1.5–1.6 (m, 3 H) 1.4–1.5 (m, 3 H) 1.3 (br s, 5 H) 1.2 (br s, 1 H) 1.1 (br s, 1 H) 0.9 (br s, 2 H). m/z calcd C48H61N5O5 [M + H]+, 788.47; found (MS ESI), 788.35. Purity LCMS: 98.3% (254 nm, peakheight).
(S)-4-(2-((3S,5S,7S)-adamantan-1-yl)ethyl)-5-butyl-1-((S)-1-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-(4-hydroxyphenyl)propan-2-yl)piperazine-2,3-dione (11)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 11 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-norleucine-OH (R3), and 1-adamantaneacetic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ ppm 9.2 (br s, 1 H) 8.4 (br s, 1 H) 6.9 (d, J = 7.3 Hz, 1 H) 6.6 (d, J = 7.3 Hz, 1 H) 3.1 (br s, 1 H) 3.8 (d, J = 12.8 Hz, 1 H) 3.5–3.7 (m, 3 H) 3.3 (br s, 12 H) 3.2 (d, J = 10.9 Hz, 1 H) 2.9 (d, J = 11.6 Hz, 2 H) 2.8 (br s, 1 H) 2.8 (br s, 1 H) 2.7 (br s, 3 H) 2.4 (br s, 1 H) 2.3 (d, J = 7.8 Hz, 1 H) 1.9 (br s, 3 H) 1.7–1.9 (m, 1 H) 1.5–1.7 (m, 6 H) 1.5 (br s, 6 H) 1.4 (br s, 5 H) 1.3 (br s, 6 H) 0.9 (d, J = 8.6 Hz, 2 H). m/z calcd C41H61N6O5 [M + H]+, 704.47; found (MS ESI), 704.35. Purity LCMS: 99.8% (254 nm, peak height).
(S)-4-(2-((3S,5S,7S)-Adamantan-1-yl)ethyl)-5-(4-hydroxybenzyl)-1-((S)-1-(4-hydroxyphenyl)-3-((S)-2-(((R)-6-(naphthalen-2-ylmethyl)-2,3-dioxopiperazin-1-yl)methyl)pyrrolidin-1-yl)propan-2-yl)-piperazine-2,3-dione (12)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 12 was synthesized using the following reagents: Boc-d-Ala(2-naphthyl)-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-tyrosine(2-Br-Z)-OH (R3), and 1-adamantaneacetic acid (R4).). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/30, 40/65. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.5 (br s, 1 H) 7.9–7.9 (m, 3 H) 7.6–7.7 (d, J = 6.5 Hz, 1 H) 7.5 (br s, 2 H) 7.0 (d, J = 7.4 Hz, 1 H) 6.9 (d, J = 7.3 Hz, 2 H) 6.6–6.7 (m, 1 H) 6.5 (br s, 1 H) 3.8 (br s, 4 H) 3.7 (br s, 1 H) 3.4 (br s, 19 H) 23.0 (d, J = 11.5 Hz, 3 H) 2.8 (br s, 2 H) 2.7 (br s, 8 H) 2.3 (br s, 2 H) 1.9 (br s, 1 H) 1.6 (br s, 1 H) 1.6 (br s, 1 H) 1.4 (br s, 1 H) 1.2 (br s, 2 H) 1.0 (br s, 2 H). m/z calcd C52H61N5O6 [M + H]+, 852.46; found (MS ESI),852.25. Purity LCMS: 95.1% (254 nm, peak height).
(R)-4-(2-((3R,5R,7R)-Adamantan-1-yl)ethyl)-5-(4-hydroxybenzyl)-1-((S)-1-(4-hydroxyphenyl)-3-((S)-2-(((R)-6-(naphthalen-2-ylmethyl)-2,3-dioxopiperazin-1-yl)methyl)pyrrolidin-1-yl)propan-2-yl)-piperazine-2,3-dione (13)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 13 was synthesized using the following reagents: Boc-d-Ala(2-naphthyl)-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-d-tyrosine(2-Br-Z)-OH (R3), and 1-adamantaneacetic acid (R4).). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/30, 40/65. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.6 (br s, 1 H) 7.8–7.9 (m, 3 H) 7.6 (d, J = 7.6 Hz, 1 H) 7.6 (br s, 2 H) 7.0 (d, J = 7.5 Hz, 1 H) 6.9 (d, J = 7.3 Hz, 2 H) 6.5–6.7 (m, 3 H) 3.8 (br s, 1 H) 3.7 (br s, 1 H) 3.4–3.6 (m, 4 H) 3.2–3.4 (m, 3 H) 3.2 (br s, 2 H)2.9–3.1 (m, 3 H) 2.8 (br s, 1 H) 2.5–2.7 (m, 5 H) 2.3–2.5 (m, 3 H)2.2 (br s, 1 H) 2.4 (br s, 1 H) 1.9 (br s, 3 H) 1.6 (br s, 5 H) 1.6 (d, J =11.0 Hz, 4 H) 1.4 (br s, 5 H) 1.3 (br s, 1 H) 1.2 (br s, 2 H) 1.0 (br s, 1 H). m/z calcd C52H61N5O6 [M + H]+, 852.46; found (MS ESI), 852.35. Purity LCMS: 96.6% (254 nm, peak height).
(S)-4-(2-((3S,5S,7S)-Adamantan-1-yl)ethyl)-1-((S)-1-(4-hydroxyphenyl)-3-((S)-2-(((R)-6-(naphthalen-2-ylmethyl)-2,3-dioxopiperazin-1-yl)methyl)pyrrolidin-1-yl)propan-2-yl)-5-(naphthalen-2-ylmethyl)piperazine-2,3-dione (14)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 14 was synthesized using the following reagents: Boc-d-Ala(2-naphthyl)-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-Ala(2-naphthyl)-OH (R3), and 1-adamantaneacetic acid (R4).). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/30, 40/65. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.0 (br s, 1 H) 7.9–8.1 (m, 2 H) 7.8 (d, J = 13.0 Hz, 1 H) 7.6–7.7 (m, 2 H) 7.5–7.6 (m, 2 H) 7.4–7.5 (m, 2 H) 7.4 (br s, 1 H) 6.9–7.1 (m, 1 H) 6.7–6.8 (m, 1 H) 6.6 (d, J = 7.5 Hz, 2 H) 4.1 (br s, 1 H) 3.4–3.7 (m, 5 H) 3.3 (br s, 8 H) 3.1–3.3 (m, 3 H) 3.0–3.1 (m, 2 H) 2.9 (br s, 1 H) 2.8 (br s, 1 H) 2.6 (br s, 4 H) 2.4 (d, J = 8.4 Hz, 2 H) 2.2 (br s, 1 H) 2.1 (d, J = 11.3 Hz, 1 H) 1.2 (br s, 1 H) 1.99 (br s, 2 H) 1.7–1.8 (m, 2 H) 1.6 (d, J = 11.4 Hz, 3 H) 1.5 (d, J = 11.1 Hz, 3 H) 1.3 (br s, 4 H) 1.3 (d, J = 14.6 Hz, 1 H) 1.2 (br s, 1 H). m/z calcd C56H63N5O5 [M + H]+, 886.48; found (MS ESI), 886.40. Purity LCMS: 99.8% (254 nm, peak height).
(R)-4-(2-((3R,5R,7R)-Adamantan-1-yl)ethyl)-1-((S)-1-(4-hydroxyphenyl)-3-((S)-2-(((R)-6-(naphthalen-2-ylmethyl)-2,3-dioxopiperazin-1-yl)methyl)pyrrolidin-1-yl)propan-2-yl)-5-(naphthalen-2-ylmethyl)piperazine-2,3-dione (15)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 15 was synthesized using the following reagents: Boc-d-Ala(2-naphthyl)-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-d-Ala(2-naphthyl)-OH (R3), and 1-adamantaneacetic acid (R4).). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/30, 40/65. 1H NMR (400 MHz, DMSO-d6): δ ppm 8.6 (br s, H) 7.8–7.9 (m, 6 H) 7.5–7.6 (m, 2 H) 7.5 (br s, 2 H) 7.4 (br s, 1 H) 7.3 (d, J = 8.2 Hz, 1 H) 7.0 (d, J = 6.7 Hz, 2 H) 6.7 (d, J = 7.2 Hz, H) 4.0 (br s, 1 H) 3.8 (br s, 1 H) 3.5–3.7 (m, 1 H) 3.5 (d, J = 12.0 Hz, 3 H) 3.3 (d, J = 12.2 Hz, 3 H) 3.2 (br s, 2 H) 2.8–3.1 (m, 3 H) 2.8 (br s, 3 H) 2.7 (br s, 2 H) 2.6 (d, J = 14.31 Hz, 3 H) 2.4–2.5 (m, 1 H) 2.1–2.4 (m, 1 H) 1.8 (br s, 3 H) 1.9 (br s, 1 H) 1.5–1.7 (m, 5 H) 1.4–1.5 (m, 5 H) 1.3 (br s, 5 H) 1.2 (d, J = 11.7 Hz, 1 H) 1.1 (br s, 1 H). m/z calcd C56H63N5O5 [M + H]+, 886.48; found (MS ESI), 886.30. Purity LCMS: 99.6% (254 nm, peak height).
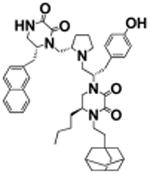
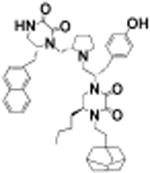
(S)-4-(2-(Adamantan-1-yl)ethyl)-5-butyl-1-((S)-1-(4-hydroxyphenyl)-3-((S)-2-(((R)-6-(naphthalen-2-ylmethyl)-2,3-dioxopiperazin-1-yl)methyl)pyrrolidin-1-yl)propan-2-yl)piperazine-2,3-dione (16)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 16 was synthesized using the following reagents: Boc-d-Ala(2-naphthyl)-OH (R1), Boc-l-tyrosine(2-Br-Z)-OH (R2), Boc-l-norleucine-OH (R3), and 1-adamantaneacetic Acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/30, 40/65. 1H NMR (400 MHz, DMSO-d6): δ 8.56 (d, J = 5.01 Hz, 1 H) 7.8–8.0 (m, 3 H) 7.5–7.6 (m, 2 H) 7.0 (m, J = 8.1 Hz, 2 H) 6.6 (m, J = 8.1 Hz, 2 H) 3.8 (br s, 1 H) 3.6–3.8 (m, 3 H) 3.5 (d, J = 10.4 Hz, 3 H) 3.2–3.5 (m, 8 H) 2.9–3.2 (m, 4 H) 2.7–2.9 (m, 4 H) 2.7 (d, J = 6.5 Hz, 1 H) 2.6 (d, J = 11.6 Hz, 1 H) 2.2–2.5 (m, 2 H) 1.9–2.1 (m, 3 H) 1.5–1.8 (m, 10H) 1.5 (br s, 5 H) 1.3 (br s, 5 H) 0.9 (br s, 3 H) 13C NMR (100 MHz, DMSO-d6) δ 158.3, 157.9, 1557.5, 156.4, 156.1, 135.7, 133.6, 132.4, 129.9, 128.7, 128.5, 128.3, 128.2, 127.9, 126.7, 126.2, 115.5, 56.1, 54.8, 53.7, 42.1, 41.5, 40.7, 40.5, 37.0, 34.7, 32.0, 30.8, 28.4, 23.4, 22.5, 14.3. m/z calcd C49H63N5O5 [M + H]+, 802.49; found (MS ESI), 802.3. Purity LCMS: 99% (254 nm, peak height)
(5R)-4-(2-((1R,3R)-Adamantan-1-yl)ethyl)-5-(4-hydroxybenzyl)-1-((2R)-1-(2-(((R)-6-(4-hydroxybenzyl)-2,3-dioxopiperazin-1-yl)-methyl)pyrrolidin-1-yl)-3-(naphthalen-2-yl)propan-2-yl)piperazine-2,3-dione. (17)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 17 was synthesized using the following reagents: Boc-d-tyrosine(2-Br-Z)-OH (R1), Boc-d-2-naphthylalanine-OH (R2), Boc-d-tyrosine(2-Br-Z)-OH (R3), and 1-adamantaneacetic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ 8.5 (br s, 2 H) 7.8–7.9 (m, 5 H) 7.4–7.5 (m, 5 H) 7.2 (d, J = 8.1 Hz, 2H) 7.0 (br s, 2H) 6.9 (d, J = 7.2 Hz, 4 H) 6.7 (d, J = 6.5 Hz, 7H) 3.9 (d, J = 13.6 Hz, 2 H) 3.8 (br s, 2 H) 3.7 (br s, 2 H) 3.5 (s, 3 H) 3.5 (s, 2 H) 3.4 (br s, 3 H) 3.3 (br s, 2 H) 3.2 (br s, 3 H) 3.0 (t, J = 14.6 Hz, 4 H) 2.9 (d, J = 13.1 Hz, 3 H) 2.7–2.9 (m, 7 H) 2.3–2.5 (m, 3 H) 2.3 (d, J = 8.19 Hz, 2 H) 1.8 (br s, 7 H) 1.6–1.8 (m 10 H) 1.4–1.6 (m, 6 H) 1.3 (br s, 10 H) 1.2 (br s, 2 H) 1.1 (br s, 2 H). 13C NMR (100 MHz, DMSO-d6) δ 158.4, 157.9, 157.1, 156.8, 156.4, 133.5, 132.2, 130.7, 130.4, 128.3, 127.9, 127.7, 127.7, 127.1, 126.5, 125.9, 116.0, 115.9, 64.2, 58.7, 56.3, 54.8, 53.6, 41.9, 41.1, 40.7, 40.5, 37.0, 35.9, 34.7, 31.8, 28.3, 27.8, 23.3. m/z calcd C44H65N5O5 [M + H]+, 744.51; found (MS ESI), 744.25. Purity LCMS: 98% (254 nm, peak height).
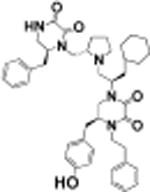
(S)-1((R)-1((S)-2-(((S)-6-Benzyl-2,3-dioxopiperazin-1-yl)methyl)-pyrrolidin-1-yl)-3-cyclohexylpropan-2-yl)-5-(4-hydroxybenzyl)-4-phenethylpiperazine-2,3-dione (18)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 18 was synthesized using the following reagents: Boc-l-phenylalanine-OH (R1), Boc-d-cyclohexylalanine-OH (R2), Boc-l-tyrosine(2-Br-Z)-OH (R3), and phenylacetic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6) δ ppm 0.80–1.05 (m, 2 H) 1.08–1.20 (m, 3 H) 1.24 (br s, 2 H) 1.50–1.71 (m, 6 H) 1.78–1.96 (m, 2 H) 1.97–2.13 (m, 2 H) 2.55–2.65 (m, 2 H) 2.67–2.88 (m, 6 H) 2.90–3.04 (m, 3 H) 3.11 (d, J = 12.96 Hz, 1 H) 3.19–3.32 (m, 2 H) 3.37 (br s, 1 H) 3.52 (dd, J = 13.08, 3.30 Hz, 2 H) 3.65 (d, J = 10.03 Hz, 2 H) 3.70–3.94 (m, 2 H) 6.73 (m, J = 8.07 Hz, 2 H) 7.01 (m, J = 8.07 Hz, 2 H) 7.15–7.31 (m, 9 H) 8.50 (d, J = 5.01 Hz, 1 H) 13C NMR (101 MHz, DMSO-d6) δ ppm 22.74 (s, 1 C) 26.13 (s, 1 C) 26.30 (s, 1 C) 26.56 (s, 1 C) 29.54 (s, 1 C) 32.83 (s, 1 C) 33.55 (s, 1 C) 33.63 (s, 1 C) 34.51 (s, 1 C) 37.14 (s, 1 C) 37.51 (s, 1 C) 39.40 (s, 1 C) 39.61 (s, 1 C) 39.82 (s, 1 C) 40.03 (s, 1 C) 40.23 (s, 1 C) 40.45 (s, 1 C) 40.66 (s, 1 C) 48.41 (s, 1 C) 55.86 (s, 1 C) 58.51 (s, 1 C) 62.76 (s, 1 C) 115.89 (s, 1 C) 126.79 (s, 1 C) 127.11 (s, 1 C) 127.76 (s, 1 C) 128.81 (s, 1 C) 128.98 (s, 1 C) 129.13 (s, 1 C) 129.77 (s, 1 C) 130.35 (s, 1 C) 137.72 (s, 1 C) 139.29 (s, 1 C) 156.65 (s, 1 C) 156.72 (s, 1 C) 157.34 (s, 1 C) 157.80 (s, 1 C) 157.95 (s, 1 C). m/z calcd C44H55N5O5 [M + H]+, 734.42; found, 734.15 (MALDI), 734.15 (MS ESI) Purity LCMS: 99.0% (254 nm, peak height).
(S)-5-Benzyl-1-((R)-1-cyclohexyl-3-((S)-2-(((S)-2,3-dioxo-6-propylpiperazin-1-yl)methyl)pyrrolidin-1-yl)propan-2-yl)-4-phenethylpiperazine-2,3-dione. (19)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 19 was synthesized using the following reagents: Boc-l-norvaline-OH (R1), Boc-d-cyclohexylalanine-OH (R2), Boc-l-phenylalanine-OH (R3), and phenylacetic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ 8.4 (br s, 1 H) 7.3 (br s, 2 H) 7.3 (br s, 3 H) 7.2 (s, 2 H) 7.2 (s, 3 H) 3.7 (br s, 3 H) 3.4–3.7 (m, 2 H) 3.2 (d, J = 11.86 Hz, 2 H) 3.0 (br s, 1 H) 2.9 (br s, 1 H) 2.8 (br s, 1 H) 2.7 (br s, 3 H) 2.6 (br s, 2 H) 2.1 (br s, 2 H) 1.9 (br s, 2 H) 1.6 (br s, 5 H) 1.6 (br s, 4 H) 1.3 (br s, 4 H) 1.2 (br s, 4 H) 1.0 (br s, 1 H) 0.8–1.0 (m, 4 H) 0.8 (br s, 1 H) 13C NMR (100 MHz, DMSO-d6) δ 158.0, 157.4, 156.8, 139.3, 137.9, 129.4, 129.1, 128.8, 127.2, 126.7, 62.9, 56.8, 55.6, 53.9, 51.2, 48.3, 41.0, 40.7, 38.0, 37.7, 34.5, 33.7, 33.6, 32.8, 29.6, 26.6, 26.3, 26.1, 22.9, 19.4, 14.2. m/z calcd C40H55N5O4 [M + H]+, 670.43; found (MS ESI), 670.20. Purity LCMS: 99% (254 nm, peak height).
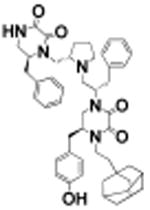
(S)-4-(2-(Adamantan-1yl)ethyl)-1-((S)-1-((S)-2-(((S)-6-benzyl-2,3-dioxopiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-phenylpropan-2-yl)-5-(4-hydroxybenzyl)piperazine-2,3-dione (20)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 20 was synthesized using the following reagents: Boc-l-phenylalanine-OH (R1), Boc-l-phenylalanine-OH (R2), Boc-l-tyrosine(2-Br-Z)-OH (R3), and 1-adamantaneacetic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/30, 40/65. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.54 (d, J = 5.13 Hz, 1 H) 7.09–7.34 (m, 6 H) 7.02 (d, J = 6.97 Hz, 3 H) 6.76 (d, J = 8.07 Hz, 2 H) 3.88 (d, J = 10.51 Hz, 1 H) 3.66–3.83 (m, 2 H) 3.42–3.63 (m, 4 H) 3.29–3.42 (m, 7 H) 3.22 (d, J = 12.10 Hz, 2 H) 3.12 (br s, 1 H) 2.75–3.02 (m, 6 H) 2.61 (br s, 2 H) 2.38 (d, J = 7.46 Hz, 2 H) 2.25 (q, J = 8.15 Hz, 1 H) 1.86–2.02 (m, 3 H) 1.74–1.86 (m, 1 H) 1.61–1.73 (m, 4 H) 1.48–1.60 (m, 3 H) 1.38 (br s, 5 H) 1.06–1.32 (m, 2 H). 13C NMR (101 MHz, DMSO-d6) δ ppm 176.94 (s, 1 C) 176.50 (s, 1 C) 174.17 (s, 1 C) 165.03 (s, 1 C) 158.36 (s, 1 C) 157.84 (s, 1 C) 156.77 (s, 1 C) 156.40 (s, 1 C) 144.44 (s, 1 C) 138.69 (s, 1 C) 137.91 (s, 1 C) 137.20 (s, 1 C) 130.46 (s, 1 C) 129.78 (s, 1 C) 129.08 (s, 1 C) 128.79 (s, 1 C) 128.72 (s, 1 C) 127.73 (s, 1 C) 127.20 (s, 1 C) 126.74 (s, 1 C) 115.97 (s, 1 C) 114.23 (s, 1 C) 63.97 (s, 1 C) 58.48 (s, 1 C) 56.35 (s, 1 C) 54.85 (s, 1 C) 41.96 (s, 1 C) 41.13 (s, 1 C) 40.66 (s, 1 C) 40.45 (s, 1 C) 40.23 (s, 1 C) 40.03 (s, 1 C) 39.82 (s, 1 C) 39.61 (s, 1 C) 39.40 (s, 1 C) 36.98 (s, 1 C) 36.89 (s, 1 C) 31.86 (s, 1 C) 28.37 (s, 1 C) 23.36 (s, 1 C). m/z calcd C48H59N5O5 [M + H]+, 786.46; found, 787.0037 (MALDI), 786.15 (MS ESI). Purity LCMS: 98.0% (254 nm, peak height).
(S)-5-Benzyl-1-((R)-1-((S)-2-(((S)-6-benzyl-2,3-dioxopiperazin-1-yl)methyl)pyrrolidin-1-yl)-3-cyclohexylpropan-2-yl)-4-phenethylpiperazine-2,3-dione (21)
Using general scheme (Scheme 1) for the synthesis of bis-cyclic diketopiperazines, compound 21 was synthesized using the following reagents: Boc-l-phenylalanine-OH (R1), Boc-d-cyclohexylalanine-OH (R2), Boc-l-phenylalanine-OH (R3), and phenylacetic acid (R4). The final crude product was purified using HPLC as described above, with a gradient of (B) 0/5, 2/5, 4/20, 40/55. 1H NMR (400 MHz, DMSO-d6): δ 8.5 (br s, 2 H) 7.4 (d, J = 7.1 Hz, 4 H) 7.2–7.3 (m, 17 H) 7.2 (d, J = 7.2 Hz, 6 H) 3.7–3.9 (m, 5 H) 3.7 (d, J = 9.4 Hz, 2 H) 3.5 (d, J = 12.7 Hz, 3 H) 3.3 (d, J = 12.6 Hz, 4 H) 3.1 (d, J = 12.8 Hz, 2 H) 2.9–3.0 (m, 7 H) 2.7–2.9 (m, 11 H) 2.6 (br s, 4 H) 2.0–2.2 (m, 4 H) 1.9 (d, J = 11.74 Hz, 4 H) 1.5–1.7 (m, 12 H) 1.2 (br s, 5 H) 1.2 (br s, 6 H) 0.8–1.1 (m, 4 H). 13C NMR (100 MHz, DMSO-d6) δ 158.0, 157.8, 157.3, 156.8, 139.2, 137.9, 137.7, 129.8, 129.4, 129.1, 129.0, 128.8, 127.3, 127.1, 126.8, 62.8, 58.5, 55.6, 53.8, 51.2, 48.4, 40.7, 40.5, 37.9, 37.8, 37.5, 34.5, 33.6, 33.5, 32.8, 29.6, 26.6, 26.3, 26.1, 22.8; m/z calcd C44H55N5O4 [M + H]+, 718.43; found (MS ESI), 718.20. Purity LCMS: 98.0% (254 nm, peak height).
SAR Studies
Test compounds57,58 were solubilized in 3% DMSO/H2O and added to polypropylene 384-well plates (Greiner catalogue no. 781280). ADAM10 and 17 glycosylated substrate assays followed the same general protocol. Then 5 μL of 3× enzyme solution (30 nM) in assay buffer (10 mM Hepes, 0.001% Brij-35, pH 7.5) were added to solid bottom white 384 low volume plates (Nunc catalogue no. 264706). Test compounds and pharmacological assay control were prepared as 10-point, 1:3 serial dilutions starting at 300 μM, then added to the cells (5 μL per well) using the Biomek NXP. After 30 min incubation at RT the reactions were started by addition of 5 μL of 3× solutions of substrate (30 μM). Fluorescence was measured every 30 min for 2 h using the multimode microplate reader Synergy H4 (Biotek Instruments, Winooski, VT) using λexcitation = 360 nm and λemission = 460 nm. Rates of hydrolysis were obtained from plots of fluorescence versus time, and inhibition was calculated using rates obtained from wells containing substrates only (100% inhibition) and substrates with enzyme (0% inhibition). Three parameters were calculated on a per-plate basis, (a) the signal-to-background ratio (S/B), (b) the coefficient for variation [CV; CV = (standard deviation/mean) × 100)] for all compound test wells, and (c) the Z′-factor.59 The IC50 value of the pharmacological control ((N-hydroxy-1-(4-methoxyphenyl)sulfonyl-4-(4-biphenylcarbonyl)piperazine-2-carboxamide, Calbiochem catalogue no. 444252) was also calculated to ascertain the assay robustness.
MMP assays
MMP assays followed the same general protocol. First 5 μL of 3×enzyme solution (3 nM for MMP-8 and -14, 6 nM for MMP-2 and -9) in TCB assay buffer (50 mM Tricine, 50 mM NaCl, 10 mM CaCl2, 0.005% Brij-35, pH 7.5) were added to solid bottom white 384 low volume plates (Nunc catalogue no. 264706). Test compounds and pharmacological assay control (marimastat) were prepared as 10-point, 1:3 serial dilutions starting at 300 μM, then added to the cells (5 μL per well) using the Biomek NXP. After 30 min incubation at RT, the reactions were started by addition of 5 μL of 3× solutions of Mca-KLPGL-Dnp-AR substrate (30 μM, R&D Systems catalogue no. ES010). Fluorescence was measured after 1 h incubation at RT using the multimode microplate reader Synergy H4 (Biotek Instruments, Winooski, VT) using λexcitation = 324 nm and λemission = 393 nm. Inhibition was calculated using RFU values obtained from wells containing substrate only (100% inhibition) and substrates with enzyme (0% inhibition). Three parameters were calculated on a per-plate basis: (a) the signal-to-background ratio (S/B), (b) the coefficient for variation [CV; CV = (standard deviation/mean) × 100)] for all compound test wells, and (c) the Z′-factor.59 The IC50 value of the pharmacological control marimastat ((2S,3R)-N4-[(1S)-2,2-dimethyl-1-[(methylamino)carbonyl]propyl]-N1,2-dihydroxy-3-(2-methylpropyl)butanediamide, Tocris catalogue no. 2631) was also calculated to ascertain the assay robustness.
CellTiter Glo Cell Viability Assay
Compounds were solubilized in 3% DMSO/H2O and added to polypropylene 384-well plates (Greiner catalogue no. 781280). Then 1250 of CHO-K1, HEPG2, MDA-MB-438, M14, or A549 cells were plated in 384-well plates in 5 μL of serum-free media (EMEM for HEPG2, DMEM/F12 for MDA-MB-438, F12 for CHO-K1 and A549). Test compounds and gefitinib (pharmacological assay control) were prepared as 10-point, 1:3 serial dilutions starting at 300 μM, then added to the cells (5 μL per well) using the Biomek NXP. Plates were incubated for 72 h at 37 °C, 5% CO2, and 95% RH. After incubation, 5 μL of CellTiter-Glo (Promega catalogue no. G7570) were added to each well and incubated for 15 min at room temperature. Luminescence was recorded using a Biotek Synergy H4 multimode microplate reader. Viability was expressed as a percentage relative to wells containing cells treated with 100 μM gefitinib (0%) and wells containing cells treated with 1% DMSO only (100%). Three parameters were calculated on a per-plate basis: (a) the signal-to-background ratio (S/B), (b) the coefficient for variation [CV; CV = (standard deviation/mean) × 100)] for all compound test wells, and (c) the Z′-factor (18). The IC50 value of the pharmacological control (gefitinib, LC Laboratories no. G-4408) was also calculated to ascertain the assay robustness.
Synergy Studies
First, 1250 of A549 cells were plated in 384-well plates in 5 μL of serum-free F12 media. Next, compound 17 was added in 5 μL of serum-free F12 media such that its final concentration in the assay was 40 μM and incubated for 1 h at 37 °C, 5% CO2 and 95% RH. Gefitinib, etoposide, and doxorubicin were prepared as 10-point, 1:3 serial dilutions starting at 300 μM, then added to the cells (5 μL per well) using the Biomek NXP. Plates were incubated for 72 h at 37 °C, 5% CO2 and 95% RH. After incubation, 5 μL of CellTiter-Glo (Promega catalogue no. G7570) were added to each well and incubated for 15 min at room temperature. Luminescence was recorded using a Biotek Synergy H4 multimode microplate reader. Viability was expressed as a percentage relative to wells containing cells treated with 100 μM gefitinib (0%) and wells containing cells treated with 1% DMSO only (100%). Three parameters were calculated on a per-plate basis: (a) the signal-to-background ratio (S/B), (b) the coefficient for variation [CV; CV = (standard deviation/mean) × 100)] for all compound test wells, and (c) the Z′-factor (18). The IC50 value of the pharmacological control (gefitinib, LC Laboratories no. G-4408) was also calculated to ascertain the assay robustness.
Inhibition of TNFα Shedding
THP-1 cells were cultured in RPMI 1640 (ATCC) medium supplemented with 10% fetal bovine serum (FBS, Thermo Scientific HyClone, Logan, UT, USA), 50 U/mL penicillin, and 50 μg/mL streptomycin (Invitrogen, CA), and 0.05 mM 2-mercaptoethanol (Invitrogen). The cell cultures were maintained in a 37 °C incubator with 5% CO2 and under a humidified atmosphere. Protocol described in60 was followed. Briefly, THP-1 cells were dispensed at 3000 cells/4 μL/well in 384-well white solid bottom plates, followed by an addition of 4 μL compound in 3% DMSO solution or 3% DMSO only. After 1 h incubation, 1 μL LPS in assay medium at 5 μg/mL LPS (final concentration of 1 μg/mL) was dispensed into the assay plates and plates were incubated overnight at 37 °C with 5% CO2 and under a humidified atmosphere. 1.5 μL of the mixture containing acceptor beads conjugated with anti-TNFα antibody was added into each well The assay plates were incubated in the dark at RT for 1 h, followed by the addition of 1.5 μL of donor bead coated with streptavidin which captures the biotinylated anti-TNFα antibody. After the assay plates were incubated in the dark at RT for another 1 h, the assay plates were read in the AlphaScreen mode using Biotek Synergy H4 multimode microplate reader (Biotek Inc., VT).
Inhibition of DDR1 Shedding
Briefly, HCC1806 cells, seeded in 6-well plates, were treated overnight without (control) or with various concentrations of inhibitor 17 in serum-free media. Then 400 μL/well of conditioned media was collected, TCA precipitated, and the resultant pellets were resolved by reducing 12% SDS-PAGE gel followed by immunoblot analyses. The blot was probed with N-terminal DDR1 antibody (R&D Systems no. AF2396).
Inhibition of PTK7 Shedding
Cells were grown to reach a 60–90% confluence in wells of a 12-well tissue culture plate (BD Biosciences). Cells were washed with and then transferred to fresh serum-free DMEM (0.5 mL/well). PMA (50 ng/mL) and the inhibitors were added to the cells at the indicated concentrations, and incubation was continued for an additional 16 h. The medium aliquots were then withdrawn and centrifuged to remove cell debris. The supernatant was concentrated until dryness using a Speed-Vac. To remove salt, the pellet was washed using acetone, dissolved in 1% SDS, and centrifuged to remove insoluble material (14000g, 10 min). The supernatant aliquots were separated by SDS-gel electrophoresis in 4–12% NuPAGE gels (Invitrogen). The shed PTK7 was detected by Western blotting with a goat polyclonal PTK7 antibody.
Western Blot Densitometry
The immunoblots were scanned and digitized. Pixel density of the protein bands was quantified using the ImageJ software. Inhibition of shedding (%) with compound was calculated.
Inhibition is demonstrated as a percentage relative to the density of protein bands of untreated samples containing no compound (0%) and the density of background intensity (no protein band, complete inhibition, 100%).
Inhibition of Shedding of Cytokines
Tracheal smooth muscle cells (HTSMC, Promocell no. C-12565) were grown to 70% confluency in 24 well-plates. Cells were starved with growth factor free medium (smooth muscle cell basal medium, C-22262, PromoCell, Germany) supplemented with 0.5% FBS and 1% penicillin/streptavidin 24 h before stimulation. Cells were washed twice to remove growth factors and preincubated with 10 μm inhibitor or DMSO (0.1%) or 10 μM of GI254023X61 as inhibition control for 30 min. Supernatant was removed, and cells were washed once and then stimulated with or without 100 ng/mL LPS or 10 ng/mL of IL-1 in the absence or presence of inhibitors. Release of cytokines was measured 4 and 24 h after stimulation using ELISA kits for IL-8, FKN, and CXCL16 (R&D Systems, D8000C, DX310, and DCX160, respectively).Percent inhibition was calculated as a percentage relative to the untreated control samples containing no compound (0%).
Inhibition of Heregulin Shedding
Briefly, A549 cells were cultured in T175 flasks in F12K media supplemented by 10% FBS. For assay, cells were passaged and seeded at 5 × 106 A549 cells in T175 flasks in 20 mL of serum-free F12K media. All test and control inhibitors were added and incubated for 1 h, after which PMA was added at 500 ng/mL final concentration to stimulate heregulin shedding. All leads were tested at 40 μM, and marimastat was tested at 5 μM. A549 cells were incubated with PMA for 1 h, after which supernatant was collected and concentrated using Amicon Ultra-15. Cells were harvested and lysed using mammalian protein extraction buffer (GE Healthcare no. 28-9412-79). Protein concentration was determined in both supernatants and lysates using NanoDrop (ThermoFisher), and all samples were run on SDS-PAGE using recombinant Heregulin (R&D Systems no. 5898-NR) as a control. Gels were transferred and probed by Heregulin mAb (R&D Systems no. MAB377). Lysates were probed for actin to control for protein concentration.
Inhibition of TGFα and Betacellulin Shedding
A549 cells were cultured in T175 flasks in F12K media supplemented by 10% FBS. For assay, cells were passaged and seeded at 1.3 × 104 in 24-well plates in serum-free F12K media. All test and control inhibitors were added and incubated for 1 h, after which PMA was added at 500 ng/mL final concentration to stimulate TGFα shedding. In the case of betacellulin, PMA was not added because betacellulin is cleaved constitutively. All leads were tested at 40 μM, and marimastat was tested at 10 μM. A549 cells were incubated with PMA for 1 h, after which supernatant was collected and concentrated using Amicons. TGFα and betacellulin were detected in supernatant by ELISA (R&D Systems catalogue nos. DTGA00 and DY261, respectively).
Invasion Studies
Invasion assay protocol has been extensively published.35,36,62 Briefly, we used 6.5 mm, 24-well Transwell plates with the 8 μm pore size membranes. The membrane was coated with Matrigel. Cells (1 × 105/well) were seeded in the serum-free media (0.1 mL) into the upper chamber. The 10% serum-containing media (0.6 mL) was added to the lower chamber as a chemoattractant. Serum-free media in both upper and lower chambers was used as a negative control. Compound 17 was added to both chambers. Cells were allowed to invade for 5–24 h. The cells on the upper membrane surface were removed with a cotton swab. The cells that migrated onto the membrane's lower surface were stained for 10 min using 0.2% crystal violet in 20% methanol/water solution (0.3 mL). The dye was extracted using 1% SDS (0.25 mL). The OD570 nm values were measured using a plate reader.
Statistics
One-way analysis of variance (ANOVA) was used followed by Dunnett posthoc test.
Supplementary Material
Acknowledgments
This work was supported by the James and Esther King Biomedical Research Program (2KN05 to D.M.), the National Institutes of Health (DA033985-01 to D.M., CA098799 to G.B.F., and DA031370 to R.A.H.), and by the State of Florida, Executive Office of the Governor's Office of Tourism, Trade, and Economic Development. We thank Angela Morales for her technical help.
Abbreviations Used
- ADAM
a disintegrin and metalloprotease
- MMP
matrix metalloprotease
- TACE
TNFα converting enzyme
- DDR1
discoidin domain receptor 1
- PTK7
protein tyrosine kinase 7
- PMA
phorbol myristoic acid
- LPS
lipopolysaccharide
Footnotes
Supporting Information: Molecular formula strings (CSV). The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00354.
Author Contributions: The manuscript was written by D.M. and edited by G.B.F. All authors have given approval to the final version of the manuscript.
Notes: The authors declare no competing financial interest.
References
- 1.Edwards DR, Handsley MM, Pennington CJ. The ADAM metalloproteinases. Mol Aspects Med. 2008;29(5):258–89. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moss ML, Sklair-Tavron L, Nudelman R. Drug insight: tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat Clin Pract Rheumatol. 2008;4(6):300–9. doi: 10.1038/ncprheum0797. [DOI] [PubMed] [Google Scholar]
- 3.Kataoka H. EGFR ligands and their signaling scissors, ADAMs, as new molecular targets for anticancer treatments. J Dermatol Sci. 2009;56(3):148–53. doi: 10.1016/j.jdermsci.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Asai M, et al. Putative function of ADAM9, ADAM10, andADAM17 as APP alpha-secretase. Biochem Biophys Res Commun. 2003;301(1):231–5. doi: 10.1016/s0006-291x(02)02999-6. [DOI] [PubMed] [Google Scholar]
- 5.Murumkar PR, et al. Novel TACE inhibitors in drug discovery: a review of patented compounds. Expert Opin Ther Pat. 2010;20(1):31–57. doi: 10.1517/13543770903465157. [DOI] [PubMed] [Google Scholar]
- 6.Renkiewicz R, et al. Broad-spectrum matrix metalloproteinase inhibitor marimastat-induced musculoskeletal side effects in rats. Arthritis Rheum. 2003;48(6):1742–9. doi: 10.1002/art.11030. [DOI] [PubMed] [Google Scholar]
- 7.Murumkar PR, Giridhar R, Yadav MR. Novel methods and strategies in the discovery of TACE inhibitors. Expert Opin Drug Discovery. 2013;8(2):157–81. doi: 10.1517/17460441.2013.744745. [DOI] [PubMed] [Google Scholar]
- 8.Yadav MR, Murumkar PR, Zambre VP. Advances in studies on collagenase inhibitors. EXS. 2012;103:83–135. doi: 10.1007/978-3-0348-0364-9_4. [DOI] [PubMed] [Google Scholar]
- 9.DasGupta S, et al. Current perspective of TACE inhibitors: a review. Bioorg Med Chem. 2009;17(2):444–59. doi: 10.1016/j.bmc.2008.11.067. [DOI] [PubMed] [Google Scholar]
- 10.Georgiadis D, Yiotakis A. Specific targeting of metzincin family members with small-molecule inhibitors: progress toward a multifarious challenge. Bioorg Med Chem. 2008;16(19):8781–94. doi: 10.1016/j.bmc.2008.08.058. [DOI] [PubMed] [Google Scholar]
- 11.Fisher JF, Mobashery S. Recent advances in MMP inhibitor design. Cancer Metastasis Rev. 2006;25(1):115–36. doi: 10.1007/s10555-006-7894-9. [DOI] [PubMed] [Google Scholar]
- 12.Yiotakis A, Dive V. Synthetic active site-directed inhibitors of metzincins: achievement and perspectives. Mol Aspects Med. 2008;29(5):329–38. doi: 10.1016/j.mam.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Engel CK, et al. Structural basis for the highly selective inhibition of MMP-13. Chem Biol. 2005;12(2):181–9. doi: 10.1016/j.chembiol.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Johnson AR, et al. Discovery and characterization of a novel inhibitor of matrix metalloprotease-13 that reduces cartilage damage in vivo without joint fibroplasia side effects. J Biol Chem. 2007;282(38):27781–91. doi: 10.1074/jbc.M703286200. [DOI] [PubMed] [Google Scholar]
- 15.Baragi VM, et al. A new class of potent matrix metalloproteinase 13 inhibitors for potential treatment of osteoarthritis: Evidence of histologic and clinical efficacy without musculoskeletal toxicity in rat models. Arthritis Rheum. 2009;60(7):2008–18. doi: 10.1002/art.24629. [DOI] [PubMed] [Google Scholar]
- 16.Piecha D, et al. Novel selective MMP-13 inhibitors reduce collagen degradation in bovine articular and human osteoarthritis cartilage explants. Inflammation Res. 2010;59(5):379–89. doi: 10.1007/s00011-009-0112-9. [DOI] [PubMed] [Google Scholar]
- 17.Li JJ, et al. Quinazolinones and pyrido[3,4-d]pyrimidin-4-ones as orally active and specific matrix metalloproteinase-13 inhibitors for the treatment of osteoarthritis. J Med Chem. 2008;51(4):835–41. doi: 10.1021/jm701274v. [DOI] [PubMed] [Google Scholar]
- 18.Nara H, et al. Thieno[2,3-d]pyrimidine-2-carboxamides bearing a carboxybenzene group at 5-position: Highly potent, selective, and orally available MMP-13 inhibitors interacting with the S1″ binding site. Bioorg Med Chem. 2014;22(19):5487–505. doi: 10.1016/j.bmc.2014.07.025. [DOI] [PubMed] [Google Scholar]
- 19.Spicer TP, et al. Characterization of selective exosite-binding inhibitors of matrix metalloproteinase 13 that prevent articular cartilage degradation in vitro. J Med Chem. 2014;57(22):9598–611. doi: 10.1021/jm501284e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth J, et al. Identification of novel, exosite-binding matrixmetalloproteinase-13 inhibitor scaffolds. Bioorg Med Chem Lett. 2011;21(23):7180–4. doi: 10.1016/j.bmcl.2011.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lauer-Fields JL, et al. High throughput screening of potentially selective MMP-13 exosite inhibitors utilizing a triple-helical FRET substrate. Bioorg Med Chem. 2009;17(3):990–1005. doi: 10.1016/j.bmc.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jungel A, et al. Effect of the oral application of a highly selective MMP-13 inhibitor in three different animal models of rheumatoid arthritis. Ann Rheum Dis. 2010;69(5):898–902. doi: 10.1136/ard.2008.106021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wijdeven RH, Neefjes J, Ovaa H. How chemistry supports cell biology: the chemical toolbox at your service. Trends Cell Biol. 2014;24(12):751–760. doi: 10.1016/j.tcb.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Tape CJ, et al. Cross-domain inhibition of TACE ectodomain. Proc Natl Acad Sci U S A. 2011;108(14):5578–83. doi: 10.1073/pnas.1017067108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minond D, et al. Discovery of Novel Inhibitors of a Disintegrin and Metalloprotease 17 (ADAM17) Using Glycosylated and Non-glycosylated Substrates. J Biol Chem. 2012;287(43):36473–87. doi: 10.1074/jbc.M112.389114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rose-John S. ADAM17, shedding, TACE as therapeutic targets. Pharmacol Res. 2013;71:19–22. doi: 10.1016/j.phrs.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 27.Moss ML, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385(6618):733–6. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- 28.Black RA, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385(6618):729–33. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 29.Nakanaga T, et al. Regulation of interleukin-8 via an airway epithelial signaling cascade. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1289–96. doi: 10.1152/ajplung.00356.2006. [DOI] [PubMed] [Google Scholar]
- 30.Dreymueller D, et al. Smooth muscle cells relay acute pulmonary inflammation via distinct ADAM17/ErbB axes. J Immunol. 2014;192(2):722–31. doi: 10.4049/jimmunol.1302496. [DOI] [PubMed] [Google Scholar]
- 31.Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW, et al. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J Biol Chem. 2001;276(41):37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- 32.Hundhausen C, et al. The disintegrin-like metalloproteinaseADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102(4):1186–95. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 33.Abel S, et al. The transmembrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAM10. J Immunol. 2004;172(10):6362–72. doi: 10.4049/jimmunol.172.10.6362. [DOI] [PubMed] [Google Scholar]
- 34.Fu HL, et al. Shedding of discoid in domain receptor 1 by membrane-type matrix metalloproteinases. J Biol Chem. 2013;288(17):12114–29. doi: 10.1074/jbc.M112.409599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golubkov VS, Aleshin AE, Strongin AY. Potential relation of aberrant proteolysis of human protein tyrosine kinase 7 (PTK7) chuzhoiby membrane type 1 matrix metalloproteinase (MT1-MMP) to congenital defects. J Biol Chem. 2011;286(23):20970–6. doi: 10.1074/jbc.M111.237669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golubkov VS, et al. The Wnt/planar cell polarity protein-tyrosine kinase-7 (PTK7) is a highly efficient proteolytic target of membrane type-1 matrix metalloproteinase: implications in cancer and embryogenesis. J Biol Chem. 2010;285(46):35740–9. doi: 10.1074/jbc.M110.165159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Golubkov VS, Strongin AY. Insights into Ectodomain Shedding and Processing of Protein-tyrosine Pseudokinase 7 (PTK7) J Biol Chem. 2012;287(50):42009–18. doi: 10.1074/jbc.M112.371153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golubkov VS, et al. Protein-tyrosine pseudokinase 7 (PTK7) directs cancer cell motility and metastasis. J Biol Chem. 2014;289(35):24238–49. doi: 10.1074/jbc.M114.574459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Golubkov VS, Strongin AY. Downstream signaling and genome-wide regulatory effects of PTK7 pseudokinase and its proteolytic fragments in cancer cells. Cell Commun Signaling. 2014;12:15. doi: 10.1186/1478-811X-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou BB, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10(1):39–50. doi: 10.1016/j.ccr.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horiuchi K, Schulte M, Le Gall S, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P, Blobel CP, et al. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007;18(1):176–88. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ni SS, et al. ADAM17 is overexpressed in non-small cell lung cancer and its expression correlates with poor patient survival. Tumor Biol. 2013;34(3):1813–8. doi: 10.1007/s13277-013-0721-3. [DOI] [PubMed] [Google Scholar]
- 43.Hynes NE, Schlange T. Targeting ADAMS and ERBBs in lung cancer. Cancer Cell. 2006;10(1):7–11. doi: 10.1016/j.ccr.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 44.Rocks N, et al. Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie. 2008;90(2):369–79. doi: 10.1016/j.biochi.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Horiuchi K, et al. Evaluation of the contributions of ADAMs 9, 12, 15, 17, and 19 to heart development and ectodomain shedding of neuregulins beta1 and beta2. Dev Biol. 2005;283(2):459–71. doi: 10.1016/j.ydbio.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 46.Stawikowska R, et al. Activity of ADAM17 (a disintegrin and metalloprotease 17) is regulated by its noncatalytic domains and secondary structure of its substrates. J Biol Chem. 2013;288(31):22871–9. doi: 10.1074/jbc.M113.462267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scheller J, et al. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol. 2011;32(8):380–7. doi: 10.1016/j.it.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 48.Wang W, Liu Y, Lazarus RA. Allosteric inhibition of BACE1 by an exosite-binding antibody. Curr Opin Struct Biol. 2013;23(6):797–805. doi: 10.1016/j.sbi.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 49.Hermanson DJ, et al. Substrate-selective COX-2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation. Trends Pharmacol Sci. 2014;35(7):358–67. doi: 10.1016/j.tips.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Busschots K, et al. Substrate-selective inhibition of protein kinase PDK1 by small compounds that bind to the PIF-pocket allosteric docking site. Chem Biol. 2012;19(9):1152–63. doi: 10.1016/j.chembiol.2012.07.017. [DOI] [PubMed] [Google Scholar]
- 51.Balbin M, et al. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35(3):252–7. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 52.Palavalli LH, et al. Analysis of the matrix metalloproteinase family reveals that MMP8 is often mutated in melanoma. Nat Genet. 2009;41(5):518–20. doi: 10.1038/ng.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szabova L, et al. MT1-MMP is required for efficient tumor dissemination in experimental metastatic disease. Oncogene. 2008;27(23):3274–81. doi: 10.1038/sj.onc.1210982. [DOI] [PubMed] [Google Scholar]
- 54.Wolfe MS. gamma-Secretase inhibitors and modulators for Alzheimer's disease. J Neurochem. 2012;120(Suppl S1):89–98. doi: 10.1111/j.1471-4159.2011.07501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Houghten RA. General method for the rapid solid-phase synthesis of large numbers of peptides: specificity of antigen-antibody interaction at the level of individual amino acids. Proc Natl Acad Sci U S A. 1985;82(15):5131–5. doi: 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinilla C, et al. Selective agonists and antagonists of formylpeptide receptors: duplex flow cytometry and mixture-based positional scanning libraries. Mol Pharmacol. 2013;84(3):314–24. doi: 10.1124/mol.113.086595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pinilla C, et al. Advances in the use of synthetic combinatorial chemistry: mixture-based libraries. Nat Med. 2003;9(1):118–22. doi: 10.1038/nm0103-118. [DOI] [PubMed] [Google Scholar]
- 58.Houghten RA, et al. Strategies for the use of mixture-based synthetic combinatorial libraries: scaffold ranking, direct testing in vivo, and enhanced deconvolution by computational methods. J Comb Chem. 2008;10(1):3–19. doi: 10.1021/cc7001205. [DOI] [PubMed] [Google Scholar]
- 59.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screening. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 60.Leister KP, et al. Two High Throughput Screen Assays for Measurement of TNF-alpha in THP-1 Cells. Curr Chem Genomics. 2011;5:21–9. doi: 10.2174/1875397301105010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ludwig A, Hundhausen C, Lambert M, Broadway N, Andrews R, Bickett D, Leesnitzer M, Becherer J, et al. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb Chem High Throughput Screening. 2005;8(2):161–171. doi: 10.2174/1386207053258488. [DOI] [PubMed] [Google Scholar]
- 62.Golubkov VS, et al. Membrane type-1 matrix metalloproteinase confers aneuploidy and tumorigenicity on mammary epithelial cells. Cancer Res. 2006;66(21):10460–5. doi: 10.1158/0008-5472.CAN-06-2997. [DOI] [PubMed] [Google Scholar]
- 63.Kuruppu S, et al. Production of soluble Neprilysin by endothelial cells. Biochem Biophys Res Commun. 2014;446(2):423–7. doi: 10.1016/j.bbrc.2014.01.158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.