

Abstract
Alterations in proteins that regulate endoplasmic reticulum morphology are common causes of hereditary spastic paraplegia (SPG1‐78, plus others). Mutations in the REEP1 gene that encodes an endoplasmic reticulum‐shaping protein are well‐known causes of SPG31, a common autosomal dominant spastic paraplegia. A closely‐related gene, REEP2, is mutated in SPG72, with both autosomal and recessive inheritances. Here, we report a patient with a pure hereditary spastic paraplegia due to a de novo missense mutation (c.119T > G, p.Met40Arg) in REEP2 at a highly‐conserved residue very close to another known pathogenic missense change. This represents only the second autosomal dominant SPG72 missense mutation reported.
Introduction
The hereditary spastic paraplegias (HSPs) are a heterogeneous group of disorders united by the common feature of a prominent, progressive, length‐dependent axonopathy of the corticospinal motor neurons, giving rise to lower extremity spasticity and gait impairment. Classically, the HSPs have been described as “pure” or “complex” based on the absence or presence, respectively, of other significant clinical features. More recently a genetic classification scheme has predominated, with HSPs typically referred to by their numbered genetic loci, in order of identification. In fact, the HSPs are among the most genetically‐diverse neurologic disorders, with mutations known in over 90 genes (SPG1‐78, plus others).1, 2, 3, 4 Functional analyses of these gene products support convergence within a relatively small number of cellular pathogenic themes, including abnormalities of endoplasmic reticulum (ER) morphology, axonal transport, lipid/sterol metabolism, mitochondrial function, myelination, microtubule dynamics, nucleotide metabolism, and endolysosomal membrane trafficking and degradation pathways.1
Mutations in the spastin (SPAST), atlastin‐1 (ATL1), and receptor expression‐enhancing protein 1 (REEP1) genes account for the three most common forms of HSP–SPG4, SPG3A, and SPG31, respectively. Together these account for a preponderance of autosomal dominant HSP cases, most of which appear to be “pure.” In humans, there are at least six family members in the REEP superfamily, REEP1‐6, and these can be structurally and functionally divided into two groups, comprising REEP1‐4 and REEP5‐6.5 All REEPs are membrane‐bound ER proteins harboring hydrophobic hairpin domains. REEP1‐4 proteins have been investigated for their ability to enhance the surface expression of G protein‐coupled olfactory and taste receptors as well as to shape the tubular ER network, bind microtubules, interact with mitochondria, and affect lipid droplet size.5, 6, 7, 8, 9, 10
Both the SPG31 protein REEP1 and its most closely‐related ortholog REEP2 are preferentially expressed in neuronal and exocytotic tissues.11 Furthermore, REEP2 mutations have been identified as a cause of “pure” HSP, SPG72, in two families.12 A c.107T > A (p.Val36Glu) mutation segregated in an autosomal dominant manner in numerous members of a French family across three generations, while in a Portuguese family those affected were compound heterozygous for c.215T > A (p.Phe72Tyr) and c.105 + 3G>T.12 The overall prevalence of REEP2 mutations is unknown, but they are likely very rare. In addition to the families described above, just two members of one other family with “pure” HSP have been described harboring REEP2 mutation, a likely pathogenic homozygous mutation in the canonical start codon of REEP2, p.Met1Thr.13 The “pure” phenotypes in these cases are reminiscent of those of many patients with SPG31 due to autosomal dominant REEP1 mutations.14, 15
Case Report
The subject is a 9‐year old boy (Fig. 1A, II.1) who was the product of an uncomplicated full‐term pregnancy, with delivery via Cesarean section. His parents provided informed consent for him to participate in a clinical research protocol (00‐N‐0043) approved by the NIH Combined NeuroScience Institutional Review Board. The subject walked independently at 11 months, though from the start he preferred walking on his toes. At 3 years, his gait began to decline noticeably; he waddled and appeared off‐balance and “pigeon‐toed.” He received ankle foot orthosis at 4 years. In school, he receives extracurricular assistance with writing, but he otherwise does well. Examination revealed a cheerful, well‐developed child with markedly increased tone in the lower extremities, but more mild distal lower extremity weakness (MRC scale 5‐/5 and 4‐/5 for plantar flexion and dorsiflexion, respectively). Sensory examination was normal, as was coordination. Reflexes were very brisk with spread at the biceps and knees; there was sustained clonus at the ankles. Gait was very spastic, and he scissored while walking. He is the only child of his parents, although he has a paternal half‐sister (Fig 1A, II.2) who is unaffected at 7 years of age. There is no family history of neurological disorders.
Figure 1.
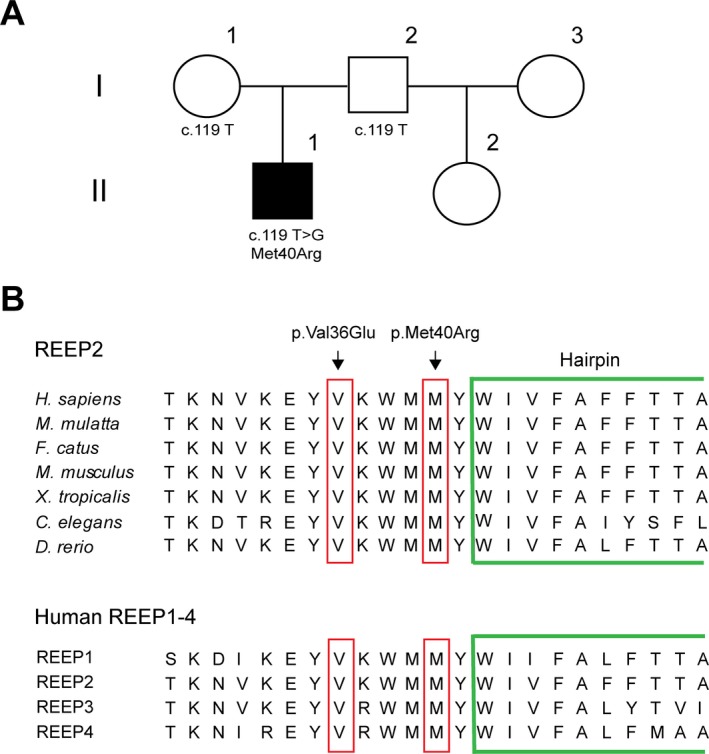
Pedigree and REEP2 mutation. (A) Family pedigree. The index patient (II.1; black‐filled square) carries a de novo p.Met40Arg missense mutation in REEP2. (B) Conservation of the mutated REEP2 residue across species (top) and human REEP1‐4 proteins (bottom). The single letter amino acid code is shown. A portion of the putative hydrophobic hairpin domain is surrounded in green. The novel missense mutation described here as well as the previously reported p.Val36Glu SPG72 mutation are indicated at the top.
MRI of the brain and the spine were unremarkable, and EMG with nerve conduction studies was also normal. Commercial genetic testing (Medical Neurogenetics, Atlanta, GA) for the following HSP genes failed to reveal any disease variants: ABCD1, ACOX1, AP4B1, AP4E1, AP4M1, AP4S1, AP5Z1, ATL1, B4GALNT1, BSCL2, C12orf65, CCT5, CLPP, CYP2U1, CYP7B1, DDHD1, DDHD2, ERLIN2, FA2H, FBXO7, GAD1, GAN, GBA2, GJC2, HARS2, HSPD1, KDM5C, KIAA0196, KIF1A, KIF5A, LARS2, MARS2, NIPA1, OPA3, PLP1, PNPLA6, PSEN1, REEP1, RTN2, SLC16A2, SLC19A3, SLC2A1, SLC33A1, SPAST, SPG11, SPG20, SPG21, SPG7, STXBP1, TECPR2, TFG, TTR, VAMP1, VPS37A, ZFYVE26, ZFYVE27. A variant of unknown significance in L1CAM (c.436G > A, p.Val146Met) was reported. However, this X‐linked variant is predicted to be benign by SIFT and MutationTaster, it occurs at a divergent residue, and it was found once in 85,777 alleles in the ExAC browser (January, 2017).16 Next, we performed whole‐exome sequencing and filtered for variants in all known HSP genes as well as inherited leukodystrophies, ataxias, and motor neuron diseases. This analysis revealed a heterozygous c.119T>G (p.Met40Arg) mutation in REEP2 (Fig. 1B), which is mutated in SPG72. We confirmed the presence of this variant in the index patient via Sanger sequencing. This variant is not found in the ExAC browser and has a Combined Annotation‐Dependent Depletion (CADD)17 score of 21.79. This change is predicted to be possibly damaging by PolyPhen2, deleterious by SIFT, and disease‐causing by MutationTaster.18 Met40 is also a very highly‐conserved amino acid residue (Fig. 1B). To evaluate the segregation of this variant further, we tested the subject's unaffected parents using Sanger sequencing. Neither parent carries the mutation, suggesting that it appeared de novo in the index subject (Fig. 1A).
Discussion
We present a novel de novo missense variant in REEP2 that is causative for HSP. The mutation occurs very close to a hydrophobic membrane hairpin domain, and it is in close proximity to the only other reported autosomal dominant REEP2 mutant (Fig. 1A).12 The fact that this is a de novo mutation emphasizes that the possibility of sporadic cases of dominant HSP must be considered in clinical practice. Whether the disorder in this case results from partial loss‐of‐function, haploinsufficiency or a dominant‐negative effect of the mutant REEP2 protein remains unclear. Along these lines, siRNA‐mediated REEP2 depletion in COS7 cells leads to altered ER morphology, and the pathogenic p.Val36Glu mutant REEP2 protein not only has impaired association with membranes, but also inhibits the normal binding of wild‐type REEP2 to membranes. These data could be compatible with both loss‐of‐function and dominant‐negative pathogenic mechanisms.
The families already published presented with different inheritance patterns. The description of their symptoms suggests that those carrying the heterozygous, missense variant were less severely afflicted; those affected longest with the disease were still able to ambulate without assistance at the ages of 43, 55 and 61. In contrast, the compound heterozygotes needed assistance in ambulation at ages ranging from 6 to 23 years. Similarly, two young children with pure HSP born of consanguineous parents (family 1967) had homozygous p.Met1Thr mutations in REEP2 predicted to destroy the protein translation initiation site;13 this would very likely result in the absence of REEP2 protein. Interestingly, though these children could still walk unaided at the ages of 3 and 4 years (as of when their cases were reported in 2014), they had symptom onset in infancy.13 Taken together, these data suggest that complete loss of protein activity from biallelic mutations may be more deleterious overall than the heterozygous missense changes. As more SPG72 cases are identified, the functional sequelae of these different types of alterations will become more apparent. Finally, this case also highlights the importance of a comprehensive approach in diagnosing genetically‐heterogeneous disorders such as HSP in patients without clear family history or known consanguinity.
Conflict of Interest
None declared.
Acknowledgments
We thank Elizabeth Hartnett for help with patient scheduling. The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
References
- 1. Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci 2012;35:25–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. de Souza PVS, de Rezende Pinto WBV, de Rezende Batistella GN, et al. Hereditary spastic paraplegia: clinical and genetic hallmarks. Cerebellum 2017;16:525–551. [DOI] [PubMed] [Google Scholar]
- 3. Fink JK. Hereditary spastic paraplegia: clinical principles and genetic advances. Semin Neurol 2014;34:293–305. [DOI] [PubMed] [Google Scholar]
- 4. Tesson C, Koht J, Stevanin G. Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum Genet 2015;134:511–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park SH, Zhu P‐P, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin‐1 coordinate microtubule interactions with the tubular ER network. J Clin Invest 2010;120:1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Falk J, Rohde M, Bekhite MM, et al. Functional mutation analysis provides evidence for a role of REEP1 in lipid droplet biology. Hum Mutat 2014;35:497–504. [DOI] [PubMed] [Google Scholar]
- 7. Mainland J, Matsunami H. RAMP like proteins: RTP and REEP family of proteins. Adv Exp Med Biol 2012;744:75–86. [DOI] [PubMed] [Google Scholar]
- 8. Renvoisé B, Malone B, Falgairolle M, et al. Reep1 null mice reveal a converging role for hereditary spastic paraplegia proteins in lipid droplet regulation. Hum Mol Genet 2016;25:5111–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schlaitz A‐L, Thompson J, Wong CCL, et al. REEP3/4 ensure endoplasmic reticulum clearance for metaphase chromatin and proper nuclear envelope architecture. Dev Cell 2013;26:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lim Y, Cho I‐T, Schoel LJ, et al. Hereditary spastic paraplegia‐linked REEP1 modulates endoplasmic reticulum/mitochondria contacts. Ann Neurol 2015;78:679–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hurt CM, Björk S, Ho VK, et al. REEP1 and REEP2 proteins are preferentially expressed in neuronal and neuronal‐like exocytotic tissues. Brain Res 2014;1545:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Esteves T, Durr A, Mundwiller E, et al. Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia. Am J Hum Genet 2014;94:268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Novarino G, Fenstermaker AG, Zaki MS, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014;343:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Züchner S, Wang G, Tran‐Viet KN, et al. Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am J Hum Genet 2006;79:365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beetz C, Schüle R, Deconinck T, et al. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain 2008;131:1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;53:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]