

Abstract
Objective
Higher levels of physical activity (PA) reduce the risk of cognitive impairment, but the underlying mechanisms are unclear. Using longitudinal data from the Cardiovascular Health Study, we examined whether PA predicted plasma Aβ levels and risk for cognitive decline 9–13 years later.
Methods
Linear and logistic regressions (controlling for APOE status, age, gender, body mass index, cardiovascular disease, brain white matter lesions, and cystatin C levels) tested associations between PA, Aβ, and cognitive impairment in a sample of 149 cognitively normal older adults (mean age 83 years).
Results
More PA at baseline predicted lower levels of Aβ 9–13 years later. Higher Aβ levels at year 9 predicted greater risk for cognitive impairment at year 13. Levels of Aβ at year 9 mediated the relationship between PA and cognitive impairment.
Interpretation
Greater PA may reduce plasma levels of a neurotoxic peptide at an age when the risk for cognitive impairment is especially high.
Introduction
The accumulation of amyloid beta (Aβ) peptides in the brain is a putative neuropathogenic pathway for Alzheimer's Disease (AD).1 Plasma levels of the Aβ 1–40 and Aβ 1–42 peptides rise in the course of normal aging2 and can predict development of Probable AD.3, 4
Physical activity (PA) is associated with a reduced risk of developing cognitive impairment (including Probable AD),5 although the mechanisms by which PA promotes cognitive health and reduces the risk of cognitive impairment are poorly understood. One possible molecular pathway is that PA inhibits the Aβ cascade in both brain and periphery.6 Consistent with this model, greater amounts of PA are associated with reduced levels of brain Aβ 7 and a reduced plasma Aβ‐142/Aβ‐140 ratio in humans.8 However, the temporal relationship between PA and plasma Aβ in humans has not been clearly established.
In this study, we used longitudinal data collected as part of the Cardiovascular Health Study (CHS) to examine whether higher levels of PA predict less Aβ accumulation and cognitive impairment in assessments occurring 9 and 13 years later in older adults. We predicted that higher baseline PA levels would predict lower levels of plasma Aβ in subsequent assessments.
Materials and Methods
Participants
Participants were part of the Pittsburgh Cardiovascular Health Study Cognition Study (CHS‐CS), which is a subset of participants from the larger CHS, a population‐based longitudinal study of factors associated with coronary heart disease and stroke in individuals sixty‐five years of age and older (for additional details see9). The CHS was initiated in 1989–1990 (hereafter referred to as “baseline”) and included participants from four U.S. communities (Winston‐Salem, NC, Sacramento, CA, Hagerstown, MD, and Pittsburgh, PA). In 1998–1999, the CHS Memory Study was initiated. A subset of participants enrolled in the CHS Memory Study were tested at the University of Pittsburgh Medical Center where additional measures, such as structural magnetic resonance images (MRI) and plasma amyloid were collected at multiple time points. Participants enrolled in CHS‐CS were a subset of participants from the CHS Memory Study who were deemed cognitively normal via clinical adjudication (procedures described previously10) in 1998–1999.
CHS‐CS participants were included in the present analyses if they: (1) were cognitively normal, (2) had baseline PA data available, and (3) reported walking at least one block per week and were thus capable of PA at baseline. We excluded participants reporting zero minutes of physical activity at baseline to eliminate health‐related comorbidities that could confound the associations with physical activity. Of the 274 cognitively normal individuals in the full CHS‐CS sample, baseline PA data were available from 156. An additional seven individuals were removed for reporting zero minutes of PA; the final sample included 149 older adults. Thus, to retain a more conservative statistical model, we excluded these individuals from the analyses. However, all of the results reported below remained similar when including individuals reporting 0 blocks walked per week (for a total sample of n = 156). Several individuals were missing data from one of the amyloid assessments, and so sample sizes varied slightly (minimum n = 140) across regression models as described further below. Characteristics of the original CHS‐CS sample, the excluded sample (i.e., due to exclusion criteria), and present sample are reported in Table 1. Informed consent was obtained in accordance with the principles outlined in the Declaration of Helsinki, and all study procedures were approved by the University of Pittsburgh Medical Center Institutional Review Board.
Table 1.
| Characteristic | Present Samplea | Full CHS‐CS Sampleb |
|---|---|---|
| Mean (SD) or N (%) | Mean (SD) or N (%) | |
| Age at T2 | 79.51 (3.15) | 79.34 (3.61) |
| Gender | 56 (38%) male | 107 (39%) male |
| Race | 141 (95%) white* | 206 (75%) |
| Education (beyond HS) at T2 | 104 (70%) | 163 (69%) |
| APOE‐4 carrier | 34 (23%) | 58 (21%) |
| BMI (weight (kg)/height (m)2) at T2 | 26.37 (3.99) | 26.43 (4.03) |
| Smokers at T2 | 16 (11%) | 28 (10%) |
| Hypertensive at T2 | 57 (38%)* | 116 (42%) |
| Heart Disease at T2 | 21 (14%) | 41 (15%) |
| Diabetes at T2 | 17 (11%)Ŧ | 40 (15%) |
| Number cardiovascular diseases at T2 | 0.66 (0.73)* | 0.74 (0.76) |
| White matter grade at T2 | 2.17 (1.39) | 2.21 (1.41) |
| Number large infarcts at T2 | 0.42 (0.07) | 0.42 (0.79) |
| Cystatin C at T2 | 1.00 (0.19) | 1.01 (0.22) |
| Baseline PAb | 53.93 (63.67) | — |
| Aβ 1‐40 at T2 (N = 143) | 154.51 (57.52) | 153.40 (61.00) |
| Aβ 1‐42 at T2 (N = 142) | 19.92 (12.69) | 19.82 (13.86) |
| Aβ 1‐42/Aβ 1‐40 at T2 (N = 142) | 0.13 (0.06) | 0.13 (0.06) |
| Aβ 1‐40 at T3 (N = 141) | 178.13 (59.77) | 180.10 (66.78) |
| Aβ 1‐42 at T3 (N = 140) | 26.42 (16.04) | 24.91 (15.77) |
| Aβ 1‐42/Aβ 1‐40 at T3 (N = 140) | 0.15 (0.07) | 0.15 (0.07) |
Where applicable, the time point in which measurements were taken are indicated in the first column.
Aβ, amyloid beta; BMI, body mass index; HS, high school; PA, physical activity.
*or Ŧ in the first column denotes values that differed between the full and present CHS sample, as assessed via chi square or t‐test. The asterisk denotes a significant difference, while the Ŧ denotes a marginal difference.
Some PA data were missing from the full CHS sample, thus the mean value in this column is based only on those with data available (n = 156).
PA assessment
PA was assessed at CHS baseline using the modified Minnesota Leisure‐Time Activities Questionnaire (MLTAQ).11 The questionnaire evaluates frequency and duration of habitual PA. The total number of total blocks usually walked over 1 week was used as the main index of PA because this measure has previously been shown to predict brain health and subsequent cognitive impairment.5 All PA information was obtained in 1989–1990, 9 years prior to the first amyloid assessment, and 13 years prior to the second assessment.
Plasma amyloid assessment
The samples used for the plasma amyloid assays were collected in 1998–1999 and again in 2002–2003, hereafter referred to as T2 and T3. All samples were defrosted and analyzed in 2006, ensuring consistent quantification methodology. To assess circulating plasma levels of Aβ1‐40 and Aβ1‐42, a double‐antibody sandwich ELISA was conducted. Details on the ELISA procedures were reported previously.4 Figure 1 depicts the timeline of data collection for all measures.
Figure 1.
Timeline of data collection. Time 2 = T2, Time 3 = T3.
Statistical analyses
The relationship between baseline PA and Aβ1‐40 and Aβ1‐42 levels at T2 and T3 was assessed using linear regression (separate models for each peptide and time point) using the median split of PA to dichotomize ‘higher active’ (those walking more than 26 blocks per week) versus ‘lower active’ individuals (those walking less than 26 blocks). Characteristics for the full sample, as well as each group separately are presented in Table S1. Since the ratio of Aβ1‐42/Aβ1‐40 was significantly associated with PA in a previous study8, we also tested the ratio of the two peptides for associations with PA. The models were estimated while controlling for several critical covariates: presence of an APOE‐ε4 allele, age, gender, body mass index (BMI), cystatin C, number of MRI‐identified white matter infarcts and white matter grade, and cardiovascular diseases in 1998–1999. These covariates were chosen because all were either associated with PA or Aβ in this study or are known to correlate with these variables in previous work.12, 13, 14
We next examined the relationship between PA and Aβ with cognitive impairment outcomes by conducting a series of logistic regressions while adjusting for the same covariates. First, we examined whether PA predicted cognitive impairment over the subsequent 13 years. We then tested whether Aβ levels at T2 or T3 predicted cognitive impairment at T3. Cognitive impairment was determined by a clinical adjudication and included individuals with either a diagnosis of dementia or Mild Cognitive Impairment (MCI). Clinical adjudication procedures have been described in previous reports.10 We report the odds ratios (OR), P‐values, and 95% confidence intervals (CI) resulting from these analyses, as modern views of hypothesis testing recommend using a multipronged approach to significance testing in order to decrease the risk of Type I and II errors and represent effects more accurately.15, 16 The results of the regression models described above were corrected for multiple comparisons using the Benjamini‐Hochberg (BH) procedure implemented in SPSS using a cutoff of q < 0.05.17, 18 The BH correction procedure controls for false discovery rate and has been shown to be less prone to Type II errors compared to methods relying on family‐wise error rates (e.g., Bonferroni), particularly in studies where biologically meaningful effects may be small.19, 20
Finally, we statistically tested plasma Aβ as a putative mechanism by which PA influences risk of cognitive impairment; Aβ measures that were correlated with both PA and cognitive impairment were tested as mediators of the relationship between these two variables. Importantly, the approach we used to identify candidate mediators is based on modern views of mediation analyses, which do not impose the requirement of a simple association between X and Y (i.e., here, PA and cognitive impairment) in order to estimate and test hypotheses about indirect effects.21 All potential mediators were tested in a simple mediation model by calculating bias‐corrected 95% confidence intervals using a bootstrapping procedure with 5000 resamples using the PROCESS macro for SPSS.22, 23 Briefly, PROCESS uses maximum likelihood logistic and linear regression in order to estimate the coefficients of a mediation model. In this way, PROCESS can estimate an indirect (i.e., mediation) effect from models including both continuous and binary variables, a procedure which would violate the assumptions of traditional mediation methods e.g., see for discussion.24, 25 The program then uses a bootstrapping procedure in which the data are repeatedly resampled and asymmetric confidence intervals are computed in order to estimate the significance of the indirect effect. The indirect effect of X on Y through a candidate mediator is significant if the confidence intervals do not include zero. The bootstrapping procedure used by PROCESS is considered to be a more robust inferential procedure compared to more traditional inferential methods for testing mediation models, such as the Sobel test, because it does not depend on the same strict assumptions (e.g., that the sampling distribution of the product of the paths that define the indirect effect is normal).26
Results
As indicated in Table 1, participants in the present sample reported fewer cardiovascular diseases, especially hypertension, compared to the original CHS‐CS sample. The present sample also contained more Caucasian participants than the original sample. The results of the Aβ assays revealed that there was a wide range of variability in Aβ1‐40 (T2: range = 24.20–326.60 pg/mL, CV = 0.37; T3: range = 4.50–71.50 pg/mL, CV = 0.64) and Aβ1‐42 (T2: range = 55.10–374.30 pg/mL, CV = 0.36; T3: range = 4.50–91.40 pg/mL, CV = 0.61) levels across participants. This pattern of results lends itself nicely to examining individual differences, such as PA, that potentially account for this variability.
Greater amounts of physical activity predict lower Aβ
Consistent with our predictions, more PA was associated with lower Aβ1‐42 at both T2 and T3, as well as with a lower Aβ1‐42/Aβ 1‐40 ratio at T3 (Table 2). All three effects survived correction for multiple comparisons.
Table 2.
Unstandardized (b) and Standardized (β) betas and associated statistics showing the relationship between baseline PA and plasma Aβ levels at T2 (1998–1999) and T3 (2002–2003)
| Time of Aβ measurement | b | β | t | P | 95% CI |
|---|---|---|---|---|---|
| T2 Aβ 1‐40 | −5.87 | −0.06 | −0.79 | 0.43 | [−20.19, 9.73] |
| T2 A β 1‐42 * | −3.94 | −0.16 | −2.09 | 0.03 | [−7.71, −0.40] |
| T2 Aβ 1‐42/ Aβ 1‐40 | −.02 | −0.15 | −1.80 | 0.08 | [−0.03, 0.002] |
| T3 Aβ 1‐40 | −8.14 | −0.07 | −0.99 | 0.32 | [−23.86, 8.18] |
| T3 A β 1‐42 * | −5.66 | −0.18 | −2.17 | 0.03 | [−10.64, −0.45] |
| T3 A β 1‐42/ A β 1‐40 * | −0.03 | −0.21 | −2.32 | 0.02 | [−0.06, −0.01] |
| Age at T2 | 1.38 | 0.35 | 4.38 | 0.002 | [0.70, 2.00] |
| Gender | 0.97 | 0.036 | 0.47 | 0.61 | [−2.8, 4.70] |
| APOE e4 status | 4.68 | 0.16 | 2.04 | 0.07 | [−0.18, 10.01] |
| Cardiovascular disease at T2 | 2.97 | 0.17 | 2.07 | 0.04 | [0.46, 5.79] |
| BMI at T2 | 0.23 | 0.07 | 0.95 | 0.36 | [−0.26, 0.69] |
| Number WM infarcts at T2 | 1.07 | 0.07 | 0.85 | 0.42 | [−1.53, 4.04] |
| WM grade at T2 | −1.13 | −0.13 | −1.54 | 0.11 | [−2.42, 0.35] |
| cystatin C at T2 | 22.38 | 0.33 | 4.14 | 0.001 | [13.89, 34.00] |
The effects of all covariates from the model adjusting for T2 Aβ 1‐42 are included for comparison. Bolded rows indicate significance.
Adjusted for APOE, age, gender, BMI, cardiovascular disease, white matter infarcts, white matter grade, and cystatin C. Rows are marked with an asterisk to indicate results that survived correction for multiple comparisons using the Benjamini‐Hochberg procedure.
To rule out another viable explanation for the novel associations between PA and plasma Aβ reported above, we also examined whether the relationships could be explained by kidney clearance of amyloid – as assessed by cystatin C. This was not the case (cystatin C levels were not related to PA levels, β = −0.073, 95% CI = [−0.55, 0.40], P = 0.76), suggesting that the effects are independent of any association between physical activity and kidney function.
Effects of physical activity on risk of cognitive impairment
At the T3 assessment, 39 of the participants (26%) developed MCI, 34 developed dementia (23%), and 76 (51%) remained cognitively normal (none deceased prior to T3). Of those in the present sample with a dementia diagnosis at T3, the majority were classified as having probable or possible Alzheimer's” (96%). This rate is slightly higher than the rate of Alzheimer's (89%) reported in the larger CHS‐CS cohort.27 The remaining portion of the cognitively impaired sample in this study was classified as having “other dementias”. The individuals with MCI or dementia were combined into a single group (n = 73; see Methods), and this group was compared to those that remained cognitively normal at T3 (n = 76).
PA levels were not directly related to the incidence of cognitive impairment at T3 (OR = 1.54, P = 0.22, 95% CI [0.76,3.11]). However, higher levels of Aβ1‐42 at T2 were associated with a greater risk of cognitive impairment at T3 (Table 3). There was also a marginal relationship between levels of Aβ1‐40 at T2 and risk of cognitive impairment at T3. Only the relationship between T2 Aβ1‐42 and cognitive impairment survived correction for multiple comparisons. There were no other significant relationships between Aβ and cognitive impairment outcomes.
Table 3.
Odds ratios (OR) from logistic regressions assessing the relationship between plasma Aβ levels at T2 and T3 and risk of cognitive impairment at T3
| Time of Aβ measurement | OR | P | 95% CI |
|---|---|---|---|
| T2 Aβ 1‐40 | 1.01 | 0.06 | [1.00, 1.02] |
| T2 A β 1‐42 * | 1.05 | 0.01 | [1.01, 1.1] |
| T2 Aβ 1‐42/ Aβ 1‐40 | 160.88 | 0.14 | [0.18, 1.43E5] |
| T3 Aβ 1‐40 | 1.01 | 0.17 | [0.99, 1.05] |
| T3 Aβ 1‐42 | 1.02 | 0.14 | [1.00, 1.01] |
| T3 Aβ 1‐42/ Aβ 1‐40 | 6.65 | 0.46 | [0.04, 1.04E3] |
| Age at T2 | 1.06 | 0.44 | [−0.11, .24] |
| Gender | 0.93 | 0.85 | [−1.07, .94] |
| APOE e4 status | 1.53 | 0.37 | [−0.55, 1.47] |
| Cardiovascular disease at T2 | 1.13 | 0.67 | [−0.56, .84] |
| BMI at T2 | 0.93 | 0.14 | [−0.21, .03] |
| Number WM infarcts at T2 | 1.46 | 0.16 | [−0.15, 1.14] |
| WM grade at T2 | 1.10 | 0.54 | [−0.23, .45] |
| Cystatin C at T2 | 0.39 | 0.43 | [−4.2, 1.52] |
The effects of all covariates from the model adjusting for T2 Aβ 1‐42 are included for comparison. Bolded rows indicate significance.
Adjusted for APOE, age, gender, BMI, cardiovascular disease, white matter infarcts, white matter grade, and cystatin C. Odds ratios are interpreted as the increase in risk of cognitive impairment for every one unit increase in Aβ level (pg/mL). Rows are marked with an asterisk to indicate results that survived correction for multiple comparisons using the Benjamini‐Hochberg procedure.
Aβ1‐42 mediates the association between physical activity and cognitive impairment
We tested whether Aβ1‐42 at T2 was a statistical mediator of the relationship between PA and cognitive impairment (Fig. 2).28 Consistent with our prediction, lower levels of Aβ1‐42 at T2 mediated the association between higher baseline levels of PA and cognitive impairment at T3, b = −0.20, CI [−0.54, −0.01].
Figure 2.
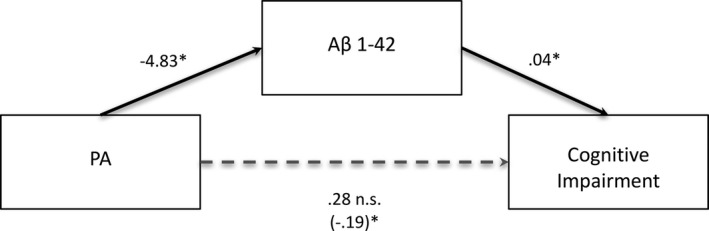
Aβ1‐42 levels at T2 significantly mediate the relationship between PA and cognitive impairment. Standardized regression coefficients are presented for the relationship between PA and cognitive impairment as mediated by Aβ1‐42. The standardized regression coefficient describing the relationship between PA and cognitive impairment while controlling for Aβ1‐42 (the indirect effect) is displayed in parentheses. Significance of the indirect effect is assessed via 95% confidence intervals. The indirect effect is significant. However, the total effect of PA on risk of Cognitive Impairment (denoted with dotted line) was not significant in this sample. *significant effect.
Discussion
We report here for the first time that greater amounts of PA were associated with lower Aβ1−42 levels 9–13 years after the PA assessment, and a lower Aβ1−42/Aβ1‐40 ratio at year 13. These effects survived correction for multiple comparisons and were independent of potential confounders. Furthermore, we demonstrated that higher T2 Aβ levels were associated with a greater risk of cognitive impairment at T3, even after adjusting for multiple comparisons. Finally, using mediation modeling, we provide statistical evidence for a novel mechanism by which greater amounts of PA at baseline may reduce the risk of cognitive impairment via a reduction in the levels of plasma amyloid, specifically in the Aβ1−42 pathway.8
Critical to understanding these results is the mediation analysis, which assesses significance by confidence intervals in addition to a P‐value. This statistical approach is preferred over other methods because multipronged approaches to significance testing provide a better estimation of the effect size, direction, and clinical relevance of effects.16
The key finding that allowed us to test for statistical mediation was that greater amounts of PA at baseline were associated with lower plasma Aβ at T2, a longitudinal association that has never been shown before in existing literature. The fact that PA was linked to plasma Aβ levels several years after the initial assessment is consistent with a body of evidence indicating that PA affects a host of health‐promoting central and peripheral molecular cascades that could help prevent cognitive impairment. Specifically, human and animal research has demonstrated that PA upregulates expression of neurotrophic factors, neurotransmitters, and hormones.29, 30 These pathways improve cognitive health by increasing cell proliferation and/or survival in some brain areas. However, the present longitudinal findings are the first in humans linking PA to lower levels of an easily obtained biomarker, plasma Aβ1‐42, a peptide which has been implicated in AD pathology31 and accelerated cognitive decline.32, 33
In fact, there is good support for the notion that PA directly impacts the Aβ cascade in animals. Studies using rodent models of AD, for example, have demonstrated that long‐term voluntary exercise decreases both extracellular and total Aβ levels in several brain regions (e.g., frontal cortex and hippocampus)6, particularly levels of the Aβ1‐42 peptide.34 Notably, these same regions show some of the largest structural and functional changes following exercise interventions in humans. This overlap in regional‐specificity suggests that similar mechanisms might underlie the neuroprotective effects of PA across species.35 In addition, animal models have found that exercise inhibits Aβ production, as opposed to its degradation or clearance6, which suggests that higher levels of PA may reduce Aβ levels both centrally and peripherally. Unfortunately, we cannot infer whether our results on plasma Aβ levels are related to central levels of Aβ. However, our results do suggest that plasma Aβ may be an important marker for predicting future cognitive decline in a sample initially adjudicated as cognitively normal but see36, 37 for controversy.
Higher PA was recently shown to be associated with lower brain Aβ in humans.7 To date, however, only one study has examined the relationship between PA and plasma Aβ 8 in humans.8 Brown et al. (2013) reported that greater self‐reported PA was associated with reduced plasma Aβ1‐42/ Aβ1‐40 ratio in a large subsample (N = 546) of cognitively intact participants from the Australian Imaging, Biomarkers, and Lifestyle Study of Aging. Consistent with this, we found that lower Aβ1‐42/ Aβ1‐40 ratio at T2 was significantly correlated with higher PA. However, unlike the Brown et al. study, we also found that greater amounts of PA were associated with less Aβ1‐42 levels over a 9–13 year follow‐up. The longitudinal results presented here suggest that PA may be an effective strategy for preventing AD pathogenesis, specifically the peripheral accumulation of the Aβ1‐42 peptide.38, 39
The finding that greater amounts of PA were related to lower plasma Aβ1−42 over a 9–13 year span in humans is mechanistically novel and clinically important. However, we also found that levels of both Aβ1−42 and Aβ1−40 at T2 were associated with a lower risk of cognitive impairment 4 years later (i.e., at T3). Associations between Aβ1−42 and Aβ1−40 have been inconsistent in the literature to date (e.g., see3 for meta‐analysis), but our finding is consistent with a previous study of the CHS cohort, which demonstrated that levels of both peptides predicted AD status in unadjusted prospective models.4 However, unlike in the earlier study, here the association between Aβ and cognitive impairment remained significant, even after adjusting for critical covariates. Regardless of this divergence from earlier research, findings from both of these studies support the idea that plasma Aβ levels are relevant biomarkers for cognitive impairment. Further, in this study Aβ1−42 levels mediated the relationship between PA and cognitive impairment risk. It is important that a direct relationship between PA and cognitive impairment is not essential in order to test for mediation effects.21 In fact, this finding supports the idea that Aβ1−42 may partially account for previously reported relationships between PA and cognitive impairment risk.40, 41, 42, 43
Taken together, the present findings, while encouraging, should be interpreted in light of some limitations. First, while previous studies have found that greater amounts of PA are associated with a reduced risk of cognitive decline and dementia, here the association was not significant. Typical studies showing such a relationship have thousands of participants and have not adjusted for as many covariates as we have here. From this perspective, our sample size is modest and may have provided insufficient power to detect the PA‐cognitive impairment association. We chose an analytical plan that included these a priori covariates for the purpose of conservatively estimating any effects, a choice which raises the potential for model overcorrection. If this were the case, however, then we would expect the reported effects to be exaggerated using more liberal approaches.
Second, we used a self‐report measure of PA (The MLTAQ). Measuring PA more objectively would help reduce potential reporting biases and inform the precise dosage and intensity of PA necessary for inducing protective effects. Relatedly, the measure of PA we used, blocks walked per week, may not be related to other types of PA, such as swimming, bowling, or household chores. Thus, the effects we report could vary by the type and intensity of PA assessed. Third, we chose to include only those who were deemed cognitively normal at baseline, and those excluded were disproportionately non‐Caucasian. Since the present sample size is smaller than in previous studies of PA and possibly affected by selection effects (e.g., we only included those cognitively healthy at baseline), the use of a sample‐based cutoff to categorize PA could limit generalizability of the findings to other populations. Along these lines, this sample was quite inactive overall compared to previous studies using samples of a similar demographic.5 Thus, the “higher” PA group here can only be considered to have had high PA relative to the “lower” PA group, rather than in absolute terms. Our sample may therefore have been healthier, yet less active and less racially diverse, than that originally enrolled in the CHS‐CS study, which could limit the generalizability of the findings. Fourth, Aβ levels were only assessed at two subsequent time points in the Pittsburgh CHS sample. Future research should assess the relationship between PA and Aβ at several time points, as well as using longitudinal statistical modeling approaches (e.g., generalized linear or cumulative link mixed models), in order to better characterize how PA affects the rate of change in plasma Aβ across late adulthood. This is a particularly important point given the uncertainty of using plasma Aβ as a biomarker of AD and neuropathology. For example, studies have reported mixed findings as to whether plasma Aβ is correlated with central Aβ, and whether plasma Aβ levels hold utility in predicting AD.33, 44 Such discrepancies in the literature have contributed to the view that there may be an overfocus on Aβ as a causal factor in the development of dementia. Another possible interpretation of these discrepancies, however, is that there are important boundary conditions (e.g., demographic, genetic, measurement time) to the relationships we report which warrant further investigation. Finally, without a well‐controlled randomized clinical trial, causal relationships between PA and Aβ cannot be determined, even despite the temporality of the variables established by our design and the finding of significant statistical mediation. The mediation results we report are therefore meant to propose a mechanism for future studies to test using different experimental designs, objective measures of PA, and larger, more diverse samples.
Despite these limitations, the findings in this paper are important to advance understanding of the role of plasma Aβ as a biomarker of neurodegeneration in population samples. We conclude that walking greater distances in late life is predictive of reduced Aβ1‐42 up to 13 years later. This suggests that PA may have a protracted impact on a potential plasma‐based biomarker of AD and cognitive impairment. Everyday walking behavior may therefore reduce accumulation of a particularly neurotoxic peptide in an age group in which risk for neural deterioration and AD is especially high.
Author Contribution
C.S.: study concept, analysis and interpretation of data, drafting of manuscript; O.L.: study concept and design, acquisition of data, statistical consulting, critical revisions to manuscript; J.B.: acquisition of data, statistical consulting, critical revisions to manuscript; L.K.: acquisition of data, analysis of data, critical revisions to manuscript; P.M.: acquisition of data, analysis and interpretation of data; R.T.: acquisition of data, analysis and interpretation of data; K.E.: study concept and design, interpretation of data, drafting of manuscript.
Conflicts of Interest
The authors report no conflicts of interest.
Supporting information
Table S1. Characteristics of the full CHS versus the current sample.
References
- 1. Gandy S. The role of cerebral amyloid β accumulation in common forms of Alzheimer disease. J Clin Invest 2005;115:1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Toledo JB, et al. Factors affecting Aβ plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol (Berl) 2011;122:401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Koyama A, et al. Plasma amyloid‐β as a predictor of dementia and cognitive decline: a systematic review and meta‐analysis. Arch Neurol 2012;69:824–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lopez OL, et al. Plasma amyloid levels and the risk of AD in normal subjects in the cardiovascular health study. Neurology 2008;70:1664–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Erickson KI, et al. Physical activity predicts gray matter volume in late adulthood. Neurology 2010;75:1415–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's Disease. J Neurosci 2005;25:4217–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Head D, et al. Exercise Engagement as a Moderator of the Effects of APOE Genotype on Amyloid Deposition. Arch Neurol 2012;69:636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brown BM, et al. Physical activity and amyloid‐β plasma and brain levels: results from the Australian Imaging, Biomarkers and Lifestyle Study of Ageing. Mol Psychiatry 2013;18:875–881. [DOI] [PubMed] [Google Scholar]
- 9. Lopez OL, et al. Incidence of mild cognitive impairment in the pittsburgh cardiovascular health study‐cognition study. Neurology 2012;79:1599–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lopez OL, et al. Evaluation of dementia in the cardiovascular health cognition study. Neuroepidemiology 2003;22:1–12. [DOI] [PubMed] [Google Scholar]
- 11. Taylor HL, et al. A questionnaire for the assessment of leisure time physical activities. J Chronic Dis 1978;31:741–755. [DOI] [PubMed] [Google Scholar]
- 12. Gurol ME, et al. Plasma β‐amyloid and white matter lesions in AD, MCI, and cerebral amyloid angiopathy. Neurology 2006;66:23–29. [DOI] [PubMed] [Google Scholar]
- 13. Sundelöf J, et al. Serum cystatin C and the risk of Alzheimer disease in elderly men. Neurology 2008;71:1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Podewils LJ, et al. Physical activity, APOE genotype, and dementia risk: findings from the cardiovascular health cognition study. Am J Epidemiol 2005;161:639–651. [DOI] [PubMed] [Google Scholar]
- 15. Wasserstein RL, & Lazar NA. The ASA's statement on P‐values: context, process, and purpose. Am Stat. 2016; 70:129–133. doi:10.1080/00031305.2016.1154108 [Google Scholar]
- 16. Nuzzo R. Scientific method: statistical errors. Nature 2014;506:150–152. [DOI] [PubMed] [Google Scholar]
- 17. Benjamini Y, Hochberg Y. On the adaptive control of the false discovery rate in multiple testing with independent statistics. J Educ Behav Stat 2000;25:60–83. [Google Scholar]
- 18. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995; 57: 289–300. [Google Scholar]
- 19. Perneger TV. What's wrong with Bonferroni adjustments. BMJ 1998;316:1236–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Glickman ME, Rao SR, Schultz MR. False discovery rate control is a recommended alternative to Bonferroni‐type adjustments in health studies. J Clin Epidemiol 2014;67:850–857. [DOI] [PubMed] [Google Scholar]
- 21. Hayes AF. Beyond Baron and Kenny: statistical mediation analysis in the new millennium. Commun Monogr 2009;76:408–420. [Google Scholar]
- 22. Hayes AF. Introduction to mediation, moderation, and conditional process analysis: a regression‐based approach. New York, NY:Guilford Press, 2013. [Google Scholar]
- 23. Preacher KJ, Hayes AF. Asymptotic and resampling strategies for assessing and comparing indirect effects in multiple mediator models. Behav Res Methods 2008;40:879–891. [DOI] [PubMed] [Google Scholar]
- 24. VanderWeele TJ, Vansteelandt S. Odds ratios for mediation analysis for a dichotomous outcome. Am J Epidemiol. 2010;172:1339–1348. doi:10.1093/aje/kwq332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winship C, Mare RD. Structural equations and path analysis for discrete data. Am J Sociol 1983;89:54–110. [Google Scholar]
- 26. Hayes AF, Matthes J. Computational procedures for probing interactions in OLS and logistic regression: SPSS and SAS implementations. Behav Res Methods 2009;41:924–936. [DOI] [PubMed] [Google Scholar]
- 27. Kuller LH, Lopez OL, Becker JT, et al. Risk of dementia and death in the long‐term follow‐up of the pittsburgh cardiovascular health study‐cognition study. Alzheimers Dement J Alzheimers Assoc 2016;12:170–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baron RM, Kenny DA. The moderator‐mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J Pers Soc Psychol 1986;51:1173–1182. [DOI] [PubMed] [Google Scholar]
- 29. Cotman CW, Berchtold NC, Christie L‐A. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci 2007;30:464–472. [DOI] [PubMed] [Google Scholar]
- 30. Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci 2002;25:295–301. [DOI] [PubMed] [Google Scholar]
- 31. Roher AE, et al. beta‐Amyloid‐(1‐42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci 1993;90:10836–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cosentino S, et al. Plasma Amyloid β predicts cognitive decline. Arch Neurol 2010;67:1485–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blasko I, et al. Plasma amyloid beta‐42 independently predicts both late‐onset depression and Alzheimer disease. Am J Geriatr Psychiatry 2010;18:973–982. [DOI] [PubMed] [Google Scholar]
- 34. Yu F, Xu B, Song C, et al. Treadmill exercise slows cognitive deficits in aging rats by antioxidation and inhibition of amyloid production. NeuroReport 2013;24:342–347. [DOI] [PubMed] [Google Scholar]
- 35. Erickson KI, Gildengers AG, Butters MA. Physical activity and brain plasticity in late adulthood. Dialogues Clin Neurosci 2013;15:99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zetterberg H. Is plasma amyloid‐beta a reliable biomarker for Alzheimer's disease? Recent Patents CNS Drug Discov 2008;3:109–111. [DOI] [PubMed] [Google Scholar]
- 37. Toledo JB, Shaw LM, Trojanowski JQ. Plasma amyloid beta measurements ‐ a desired but elusive Alzheimer's disease biomarker. Alzheimers Res Ther 2013;5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jack CR, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mondello S, et al. CSF and plasma amyloid‐β temporal profiles and relationships with neurological status and mortality after severe traumatic brain injury. Sci Rep 2014;4:6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ahlskog JE, Geda YE, Graff‐Radford NR, Petersen RC. Physical exercise as a preventive or disease‐modifying treatment of dementia and brain aging. Mayo Clin Proc 2011;86:876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lautenschlager NT, Cox KL, Flicker L. & et al. Effect of physical activity on cognitive function in older adults at risk for alzheimer disease: a randomized trial. JAMA 2008; 300:1027–1037. [DOI] [PubMed] [Google Scholar]
- 42. Middleton LE, Mitnitski A, Fallah N, et al. Changes in cognition and mortality in relation to exercise in late life: a population based study. PLoS ONE 2008;3:e3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Laurin D, Verreault R, Lindsay J, et al. Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch Neurol 2001;58:498–504. [DOI] [PubMed] [Google Scholar]
- 44. Blasko I, et al. Conversion from cognitive health to mild cognitive impairment and Alzheimer's disease: prediction by plasma amyloid beta 42, medial temporal lobe atrophy and homocysteine. Neurobiol Aging 2008;29:1–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of the full CHS versus the current sample.