

Abstract
Objective
Epilepsy is a common neurological disorder that affects 1% of the population. Approximately, 30% of individuals with epilepsy are refractory to treatment, highlighting the need for novel therapies. Conventional anticonvulsant screening relies predominantly on induced seizure models. However, these models may not be etiologically relevant for genetic epilepsies. Mutations in SCN1A are a common cause of Dravet Syndrome, a severe epileptic encephalopathy. Dravet syndrome typically begins in infancy with seizures provoked by fever and then progresses to include afebrile pleomorphic seizure types. Affected children respond poorly to available anticonvulsants. Scn1a +/− heterozygous knockout mice recapitulate features of Dravet syndrome and provide a potential screening platform to investigate novel therapeutics. In this study, we conducted a screening of conventional anticonvulsants in Scn1a +/− mice to establish assays that most closely correlate with human response data.
Methods
On the basis of clinical response data from a large, single center, retrospective survey of Dravet syndrome case records, we selected nine drugs for screening in Scn1a +/− mice to determine which phenotypic measures correlate best with human therapeutic response. We evaluated several screening paradigms and incorporated pharmacokinetic monitoring to establish drug exposure levels.
Results
Scn1a +/− mice exhibited responses to anticonvulsant treatment similar to those observed clinically. Sodium channel blockers were not effective or exacerbated seizures in Scn1a +/− mice. Overall, clobazam was the most effective anticonvulsant in Scn1a +/− mice, consistent with its effect in Dravet syndrome.
Interpretation
Genetic models of spontaneous epilepsy provide alternative screening platforms and may augment the AED development process. In this study, we established an effective screening platform that pharmacologically validated Scn1a +/− mice for preclinical screening of potential Dravet syndrome therapeutics.
Introduction
Epilepsy is a common neurological disorder with a lifetime prevalence of 2–5%. While some individuals with epilepsy achieve adequate seizure control with currently available anticonvulsant drugs, approximately, 30% remain refractory to treatment, highlighting the need for novel drugs.1 Anticonvulsant development has relied predominantly on screening for activity against specific seizure types in acute seizure models, mainly maximal electroshock and pentylenetetrazole (PTZ).2 Acute seizure models are limited by not being etiologically relevant to the approximately two‐thirds of epilepsies categorized as genetic in origin. Genetic models of epilepsy exhibit spontaneous seizures, like humans, and their usage may increase the potential of therapy screening processes for drug‐resistant epilepsies.
Dravet syndrome is a severe epileptic encephalopathy, most often resulting from de novo SCN1A mutations.3 Seizure onset typically begins in the first year of life, often provoked by fever. Individuals subsequently develop multiple seizure types that are refractory to treatment. Dravet syndrome also includes developmental delays, cognitive impairment, motor dysfunction, and an increased risk for sudden unexpected death in epilepsy (SUDEP).3, 4 Typical pharmacotherapies include benzodiazepines, valproic acid, and stiripentol, but response is usually suboptimal.5, 6, 7 Lamotrigine and carbamazepine, both sodium channel inhibitors, are not recommended for Dravet syndrome as they have been reported to exacerbate seizures.8, 9
The Scn1a +/− heterozygous null mouse model recapitulates many features of Dravet syndrome, including spontaneous seizures, seizures provoked by hyperthermia, and premature mortality.10, 11, 12 To characterize this model as a potential platform to evaluate therapeutic compounds, we conducted a screen of conventional anticonvulsants to determine which phenotypic measure(s) correlated best with human response data ascertained from a retrospective survey of clinical records from the Epilepsy Center at Ann & Robert H. Lurie Children's Hospital of Chicago. Nine conventional anticonvulsants were evaluated in Scn1a +/− mice for effects on hyperthermia‐induced seizures, spontaneous generalized tonic–clonic seizures (GTCS), and survival. Pharmacokinetic monitoring was used to evaluate the effect of drug doses that produced plasma concentrations within the human therapeutic range. We determined that lamotrigine worsened, while clobazam, valproic acid, stiripentol, levetiracetam, and phenobarbital improved the seizure phenotype of Scn1a +/− mice. This is mainly consistent with what is observed clinically in Dravet syndrome. Our results demonstrate feasibility of using a genetic epilepsy model to screen for anticonvulsant responses and validate a phenotyping platform for evaluating novel therapeutics using the Scn1a +/− mouse model of Dravet syndrome.
Materials and Methods
Chart review
Approval for this study was obtained from the Ann & Robert H. Lurie Children's Hospital Internal Review Board (IRB) prior to data collection. We performed a retrospective chart review of all files where “SCN1A” or “Dravet” was documented in the diagnosis or problem list since inception of electronic medical records (EMR) at Children's Hospital in 2007. Inclusion criteria included a genetic or clinical diagnosis of Dravet syndrome, a suspected pathogenic variant in SCN1A, and epilepsy consult visit at Lurie Children's Hospital since 2007. Patients with a GEFS+ diagnosis or an SCN1A‐negative diagnosis of Dravet syndrome were excluded. Treatment response data were coded based on patient/parent‐reported seizure frequency outcome.
Animals
All animal care and experimental procedures were approved by the Northwestern University Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The principles outlined in the ARRIVE (Animal Research: Reporting of in vivo Experiments) guideline and Basel declaration (including the 3R concept) were considered when planning experiments. Mice were group‐housed in a specific pathogen‐free mouse facility under standard laboratory conditions (14‐h light/10‐h dark) and had access to food and water ad libitum, except during hyperthermia‐induced seizure experiments. Scn1a +/− mice were generated as previously described and were maintained as a congenic line on the 129S6/SvEvTac (129.Scn1a +/−) background.11 For experiments, F1 mice were produced by breeding heterozygous 129.Scn1a +/− mice with wild‐type C57BL/6J mice (Jackson Laboratory stock 000664). The Scn1a genotype was determined by as previously described.11
Drugs
Carbamazepine, clobazam, lamotrigine, levetiracetam, phenobarbital, topiramate, and valproic acid were purchased from Sigma‐Aldrich Co. (St. Louis, MO). Stiripentol (Sigma‐Aldrich Co.; Cayman Chemical, Ann Arbor, MI) and phenytoin sodium solution (X‐Gen Pharmaceuticals, Horseheads, NV) were also used. For acute administration, drugs were prepared as solutions with the following vehicles: A‐ saline (levetiracetam, phenobarbital, topiramate, valproic acid); B‐ 0.5% methylcellulose in water (phenytoin); C‐ 5% hydroxypropyl‐β‐cyclodextrin (carbamazepine, lamotrigine); and D‐ vegetable oil (clobazam, stiripentol). All drugs were administered as a single intraperitoneal (ip) injection in a volume of 10 mL/kg. The following acute doses resulted in plasma concentrations within the human therapeutic range (Table 1): 20 mg/kg carbamazepine, 5 mg/kg clobazam, 20 mg/kg lamotrigine, 10 mg/kg levetiracetam, 15 mg/kg phenobarbital, 15 mg/kg phenytoin, 40 mg/kg topiramate, 25 mg/kg stiripentol, 75 mg/kg valproic acid, and 0.1–10 mg/kg clobazam plus 25 mg/kg stiripentol. Acute toxicity was not observed with any drugs at the administered doses.
Table 1.
Plasma concentrations and doses of anticonvulsants
| Human therapeutic range plasma concentration (total) | Hyperthermia assay plasma concentration (dose, mg/kg body weight) | Subchronic treatment plasma concentration (dose, mg/kg of chow) | |
|---|---|---|---|
| Carbamazepine | 4–12 μg/mL | 8.7 ± 0.7 μg/mL (20 mg/kg) | 5.3 ± 0.9 μg/mL (7500 mg/kg chow) |
| Clobazam | 0.1–0.4 μg/mL (NCLB: 0.3–3 μg/mL)a | 0.4 ± 0.1 μg/mL (NCLB: 1.3 ± 0.1 μg/mL) (5 mg/kg) | BQL (NCLB: 0.6 ± 0.03 μg/mL) (160 mg/kg chow) |
| Lamotrigine | 3–15 μg/mLb | 8.5 ± 0.4 μg/mL (20 mg/kg) | 3.5 ± 0.7 μg/mL (125 mg/kg chow) |
| Levetiracetam | 10–37 μg/mLa | 35.3 ± 2.8 μg/mL (10 mg/kg) | 33.6 ± 5.7 μg/mL (1350 mg/kg chow) |
| Phenobarbital | 15–40 μg/mLa | 18.8 ± 2.4 μg/mL (15 mg/kg) | 18.4 ± 2.7 μg/mL (300 mg/kg chow) |
| Phenytoin | 10–20 μg/mLa | 12.0 ± 0.6 μg/mL (15 mg/kg) | N/a |
| Stiripentol | 8–16 μg/mLc , d | 13.1 ± 3.7 μg/mL (25 mg/kg) | 12.3 ± 2.6 μg/mL (4000 mg/kg chow) |
| Topiramate | 5–25 μg/mLa | 23.6 ± 1.2 μg/mL (40 mg/kg) | 13.3 ± 0.9 μg/mL (850 mg/kg chow) |
| Valproic Acid | 50–150 μg/mLe | 122 ± 5 μg/mL (75 mg/kg) | 71 ± 4 μg/mL (4500 mg/kg chow) |
| Clobazam + Stiripentol | CLB: 0.1–0.4 μg/mL (NCLB: 0.3–3 μg/mL) STP: 8–16 μg/mL | BQL (NCLB: BQL) (CLB 0.1–1 mg/kg + STP 25 mg/kg) | 10.20 ± 3.6 μg/mL (NCLB: 1.1 ± 0.4 μg/mL) (CLB+STP: 160 mg + 4000 mg per kg chow) |
Dosage listed in parenthesis. Data expressed as mean ± SEM, with n = 5–8 samples per group.
BQL, Below limit of quantification; NCLB, N‐desmethylclobazam.
Clin Chem Lab Med 2004,42(11):1228‐55.
Ther Drug Monit 2003, 25(3):347‐63.
Clin Pharmacokinet 2015, 54(5):527‐36.
Lancet 2000, 356(9242):1638‐42.
Goodman & Gilman's The Pharmacological Basis of Therapeutics 2006.
Analytical methods
Carbamazepine, clobazam, lamotrigine, levetiracetam, phenobarbital, and stiripentol in plasma samples were assayed by high performance liquid chromatography (HPLC) based on previously described methods with modifications.13, 14, 15, 16, 17, 18 Details regarding sample preparation and chromatographic conditions for each drug are provided in Table S1 and Figure S2. Briefly, plasma samples (50–100 μL) were spiked with internal standard (prazepam, caffeine or hexobarbital) and extraction was achieved by vortex‐mixing with 4× volume of organic solvent. The organic layer was isolated by centrifugation at 376g for 5 min and was evaporated to dryness by heating at 75°C. The residues were reconstituted in mobile phase for analysis with an HPLC 9 Flexar Binary LC Pump Platform and Flexar UV/Vis Detector (Perkin Elmer, Waltham, MA). Quantitative analysis was performed by comparing samples to standards prepared with known amounts of drug.
Plasma phenytoin concentration was assayed by fluorescence polarization immunoassay (COBAS INTEGRA; Roche, Basel, Switzerland) by Vanderbilt University Medical Center Core Chemistry Laboratory.
Plasma topiramate and valproic acid concentrations were determined using ARK Topiramate Assay (ARK Diagnostics, Inc., Fremont, CA) and Emit 2000 Valproic Acid Assay (Beckman Coulter, Inc., Brea, CA), respectively. Each sample (1 μL) was mixed with 58 μL of Reagent 1 and 29 μL of Reagent 2 and incubated for 15 min at room temperature. Reactions were terminated with 1 mmol/L DHEA (dehydroepiandrosterone), a potent inhibitor of glucose‐6‐phosphate dehydrogenase, and placed on ice.19 Absorbance was measured spectrophotometrically at 340 nm (NanoDrop 8000; Thermo Fisher Scientific, Inc., Waltham, MA). Quantitative analysis was performed by comparing samples to standards prepared with known amounts of drug.
Pharmacokinetic analysis
We performed pharmacokinetic studies of clobazam and stiripentol to establish doses in Scn1a +/− mice that resulted in plasma drug concentrations within the human therapeutic range. Wild‐type and Scn1a +/− mice received a single ip injection of 10 mg/kg clobazam or 100 mg/kg stiripentol. At selected time points (5–120 min for clobazam; 0–300 min for stiripentol), mice were anesthetized with isoflurane and whole blood was collected by cardiac puncture. Plasma was isolated by centrifugation (9000g for 10 min, 4°C) and stored at −80°C until assayed. Plasma drug concentrations at each time point were averaged and pharmacokinetic parameters were calculated by noncompartmental analysis. The linear‐log trapezoidal method was used to estimate total drug exposure (area under concentration‐time curve) using the following equation:
Elimination rate constant (ke) was determined by linear regression of the terminal component of the concentration‐time curve using GraphPad Prism 6.07 (La Jolla, CA).
The above pharmacokinetic parameters were used for calculating dosages and determining target experimental time points for the hyperthermia‐induced seizure protocol and for subchronic oral treatment dosing during spontaneous seizure studies. Pharmacokinetic parameters of stiripentol in plasma were determined as follows: Cmax (46 μg/mL), tmax (90 min), t1/2 (169 min), and AUC0→∞ (12625 μg•min/mL) (Fig. S1A). Clobazam pharmacokinetic parameters were determined as follows: Cmax (2.4 μg/mL), tmax (25 min), t1/2 (38 min), and AUC0→∞ (155 μg•min/mL) (Fig. S1B). Clobazam‐treated mice were also assayed for N‐desmethylclobazam, an active metabolite of clobazam. Following treatment with clobazam, the Cmax and tmax of N‐desmethylclobazam were 3.1 μg/mL and 90 min, respectively. Exposure of both clobazam and N‐desmethylclobazam were considered when determining experimental schemes for clobazam treatment.
Hyperthermia‐induced seizures
Hyperthermia‐induced seizure experiments were conducted in Scn1a +/− mice at age postnatal day 14‐16 (P14‐16) using a rodent temperature regulator (TCAT‐2DF, Physitemp Instruments, Inc, Clifton, NJ) reconfigured with a Partlow 1160 + controller (West Control Solutions, Brighton, UK) connected to a heat lamp and RET‐3 rectal temperature probe. Fifteen minutes prior to the target experimental (post‐dose) time point for each drug, the rectal probe was inserted. Mice acclimated to the temperature probe for 5 min before the hyperthermia protocol was started. Mouse core body temperature was elevated 0.5°C every 2 min until the onset of the first clonic convulsion with loss of posture or until 42.5°C was reached. Mice that reached 42.5°C were held at temperature for 3 min. If no seizure occurred during the hold period, the mouse was considered seizure‐free. After thermal induction procedure, plasma samples were isolated as described above and stored at −80°C until assayed. Threshold temperatures were compared using the time to event analysis (logrank Mantel‐Cox), and P < 0.05 was considered statistically significant. No significant sex differences were observed, so groups were collapsed across sex.
Experimental time points were based on previously determined time‐to‐peak plasma and brain concentrations or effect from the literature or our pharmacokinetic studies. Experimental time points used were as follows: 40 min‐ levetiracetam; 45 min‐ carbamazepine, lamotrigine, phenobarbital; 90 min‐ stiripentol, clobazam, topiramate; 120 min phenytoin.20, 21, 22, 23, 24 Valproic acid was administered after the 5 min acclimation period.25, 26 Matched vehicle controls were run for each experimental time point. Vehicle A (saline) was administered at 0, 40, 45, and 90 min. No statistical difference was identified between the four time points and all vehicle A‐treated mice were combined into one group. Vehicles B (0.5% methylcellulose), C (5% hydroxypropyl‐β‐cyclodextrin), and D (vegetable oil) were administered at 120, 45, and 90 min, respectively.
Spontaneous seizure frequency monitoring
Frequency of spontaneous generalized tonic–clonic seizures (GTCS) was quantified in untreated and drug‐treated Scn1a +/− mice. Anticonvulsant compounds were administered orally through supplementation in chow. Lamotrigine (125 mg per kg of chow) was formulated in Purina 5LOD by Research Diets (New Brunswick, NJ). Carbamazepine (7500 mg per kg of chow), clobazam (160 mg per kg of chow), levetiracetam (1350 mg per kg of chow), phenobarbital (300 mg per kg of chow), stiripentol (4000 mg per kg of chow), topiramate (850 mg per kg of chow), valproic acid (4500 mg per kg of chow), and clobazam plus stiripentol (160 mg and 4000 mg per kg of chow, respectively) were formulated in‐house in Purina 5LOD irradiated meal (Lab Diets, St. Louis, MO). Chow formulations for conventional anticonvulsants resulted in plasma concentrations within the human therapeutic range for each drug (Table 1). Acute toxicity was not observed with any drug at the administered doses.
At P18, Scn1a +/− mice were randomly assigned to drug or control treatment groups with ad libitum access to drug‐containing or control chow. At P20, mice were housed in groups of three in a recording chamber (28 × 28 × 36 cm) and overhead video was captured using a Day/Night camera (Samsung SCB5003) equipped with an infrared lens (Tamron 13FG04IRSQ). During recording, mice had ad libitum access to food and water, and were maintained on a 14:10 light‐dark cycle. Spontaneous GTCS were captured by continuous video recording over 60 h (12:00 on P22 through 24:00 on P24). Digital video images captured during the 60‐h video‐monitored session were analyzed offline by an observer blinded to treatment. Previous video‐EEG monitoring of Scn1a +/− mice verified a perfect correlation between behavioral and electroencephalographic seizures (κ = 1.0), therefore we relied on video monitoring for our primary screening.27 Mice continued drug treatment and survival was monitored until P30. Plasma was then isolated between 6:00 and 8:00am and drug levels were assayed as described above. Statistical comparisons were made using Fisher's exact test (proportion seizure‐free), Kruskal–Wallis with Dunn's post hoc test (seizure frequency), and logrank (Mantel‐Cox) test (survival) with GraphPad Prism and P < 0.05 was considered statistically significant. Mice that died between P18 and P19 were excluded to allow for adequate drug exposure time. No significant sex differences were observed, so groups were collapsed across sex.
Spontaneous GTCS following hyperthermic seizure priming
Prior to evaluating spontaneous GTCS frequency, untreated P18 Scn1a +/− mice were exposed to a single hyperthermia‐induced seizure as described above. At the onset of clonic convulsion with loss of posture, mice were immediately placed on a cooling pad to terminate the seizure. The average temperature at seizure onset was 42.2 ± 0.05 (range: 41.1–43.1). Mice were cooled to a core body temperature of 37°C and then randomly assigned to drug treatment groups (untreated, clobazam, stiripentol, valproic acid, or clobazam plus stiripentol) administered through supplementation in chow, as described above. Due to the higher subsequent seizure burden and earlier onset in primed Scn1a +/− mice, we shifted the monitoring window for spontaneous seizures to begin at an earlier time point. Spontaneous GTCS were captured by continuous video recording from 12:00 on P19 through 24:00 on P21. The number of spontaneous GTCS captured during the 60‐h video‐monitored session was quantified offline by an observer blinded to treatment. Mice continued drug treatment and survival was monitored until P30 at which time plasma was isolated as described above. Statistical comparisons were made using Fisher's exact test (proportion seizure‐free) and logrank (Mantel‐Cox) test (survival analysis) with GraphPad Prism and P < 0.05 was considered statistically significant. No significant sex differences were observed, so groups were collapsed across sex. Among drug treatment groups, there was no difference in the temperature at which the initial hyperthermia‐induced seizure occurred (F(3,59) = 2.046, P = 0.5804, one‐way ANOVA). Additionally, we performed a pilot experiment to verify that following a thermally induced seizure, mice consume the expected amount of drug‐containing chow and achieve sufficient drug exposure by P19. We exposed a small cohort of Scn1a +/− mice to a hyperthermia‐induced seizure on P18 and then commenced treatment with phenobarbital chow. Plasma drug levels were measured 24 h post seizure induction and compared to naïve littermate mice treated with phenobarbital chow in parallel by student's t‐test. There was no difference (P < 0.480) between plasma levels in the primed and naïve mice, validating that feeding behavior is not significantly disrupted by the hyperthermia‐priming procedure (primed: 17.8 ± 3.2 μg/mL; naïve: 23.4 ± 5.6 μg/mL).
Results
Anticonvulsant responses in Dravet syndrome
We performed a retrospective analysis of anticonvulsant drug responses among a cohort of Dravet syndrome cases seen in the Epilepsy Center at Ann & Robert H. Lurie Children's Hospital of Chicago between 2007 and 2016. An initial search of the EMR with the terms “SCN1A” or “Dravet” identified 211 records, which were then surveyed for a confirmed clinical diagnosis of Dravet syndrome and a pathogenic/likely pathogenic SCN1A variant. This resulted in a total of 133 distinct medical records that were included in the study. The mean age at the most recent visit was 8.4 years (standard deviation (SD) 5.6; range 1–26), and the mean age of first recognized seizure was 5 months (SD 2; range <1–12). The mean number of current AEDs per patient was 3 (SD 1) and the mean number of AEDs tried was 7 (SD 3). Response data based on patient or parent‐reported outcome was coded for each AED trial, irrespective of other medications administered concurrently, and the percentage in each response category was tabulated for each drug that was trialed in a minimum of 30 patients. The relative efficacy of each medication was calculated as the percentage of children taking the medication whose parents reported reduction in seizure frequency. In our Dravet syndrome cohort, we found >50% of patients had an effective response with tolerable side effects during treatment with clobazam (63/90), valproic acid (66/113), or stiripentol (26/38), while less than 45% had effective response with tolerable side effects with clonazepam (14/33) or topiramate (36/98) (Fig. 1). Zonisamide (46 total), levetiracetam (107 total), and phenobarbital (83 total) had limited effectiveness. Sodium channel blockers produced significant worsening of seizures in approximately 50% of patients trialed on these drugs, including lamotrigine (25/50), phenytoin (15/31), carbamazepine (16/32), and oxcarbazepine (28/50) (Fig. 1). This analysis is consistent with what has been reported in the literature from other epilepsy centers.5, 6, 7, 28, 29
Figure 1.
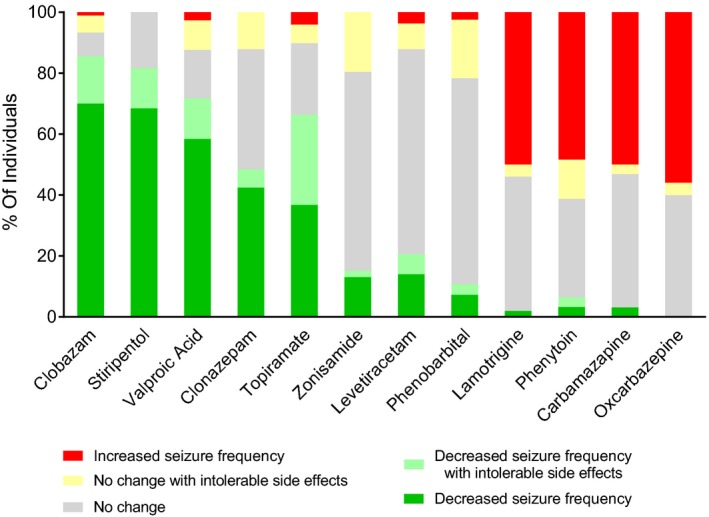
Seizure frequency response to anticonvulsants in a Dravet syndrome clinical population. Parent‐reported changes in seizure frequency in pediatric patients carrying a diagnosis of clinical Dravet syndrome were quantified over the course of anticonvulsant therapy treatment and normalized per the size of the patient population treated. Red bars indicate an overall increase in seizure frequency. Yellow bars denote a lack of change with medication, limited by duration of therapy or dose limitations due to side effects considered intolerable by parents. Dark and light green indicate decreased number of seizures without side effects, or with duration of therapy or dose limitations due to side effects, respectively. Gray bars indicate the proportion of patients with no change in seizure frequency.
Based on this retrospective analysis, we compiled a list of representative drugs to test anticonvulsant response in the Scn1a +/− mouse model using a variety of assays, including hyperthermia‐induced seizures, spontaneous seizure monitoring, and survival.
Evaluation of anticonvulsant drugs against hyperthermia‐induced seizures
Children with Dravet syndrome and Scn1a +/− mice both exhibit seizures in response to elevated body temperature. To determine if mouse and human anticonvulsant responses were correlated, we evaluated the dose‐effect of valproic acid, clobazam, and stiripentol, which have been shown to provide some degree of therapeutic benefit in Dravet syndrome. Between P14 and P16, Scn1a +/− mice were treated acutely by IP injection of a single compound or vehicle and then subjected to hyperthermia. Plasma levels were measured after completion of the assay. For all three drugs, we observed dose‐dependent protection against hyperthermia‐induced seizures in Scn1a +/− mice. Significant protection from hyperthermia‐induced GTCS was achieved with clobazam plasma exposure levels within the human therapeutic range, with 37.5% protection at 0.1 mg/kg and 50% protection at 1 mg/kg (P < 0.0005, P < 0.0001, respectively) (Fig. 2A). Complete protection was achieved at 10 mg/kg which resulted in plasma concentrations above the human therapeutic range (P < 0.0001). Valproic acid also provided significant dose‐dependent protection, with 30% protection at 100 mg/kg (P < 0.004), 50% protection at 200 mg/kg (P < 0.002), and 100% protection at 300 mg/kg (P < 0.0001) (Fig. 2B). All three doses resulted in plasma concentrations above the human therapeutic range. Stiripentol provided significant protection compared to vehicle‐treated controls, with an increased threshold temperature at 100 mg/kg (P < 0.001) and 23% (3/13) protected at 300 mg/kg (P < 0.0001), although these doses produced supratherapeutic plasma concentrations (Fig. 2C).
Figure 2.
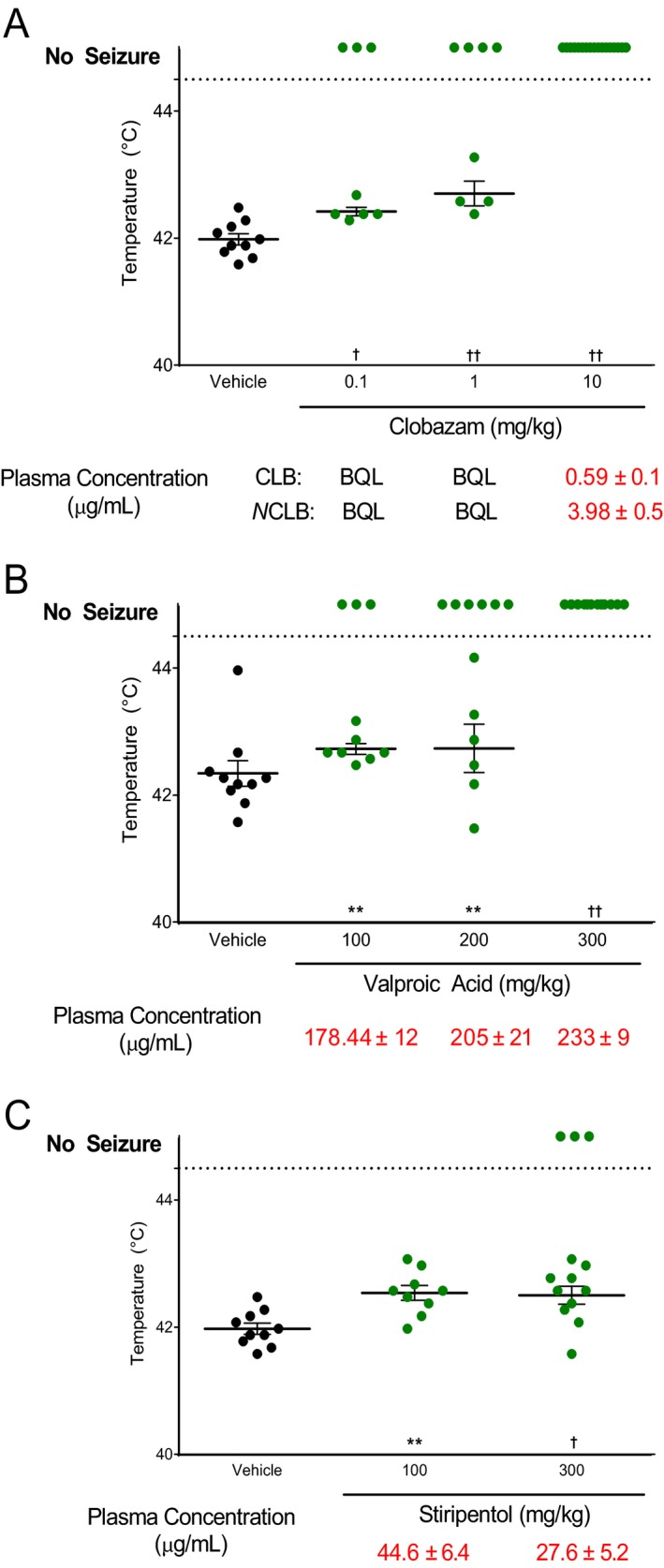
Dose‐effect screening using hyperthermia‐induced seizures in Scn1a +/− mice. Threshold temperature of individual mice for GTCS induced by hyperthermia following acute treatment with varying doses of drug. Average plasma concentrations ± standard error of the mean (SEM) are listed beneath the corresponding drug dose. (A) Clobazam significantly protected against hyperthermia‐induced GTCS at doses within and beyond the human therapeutic range. (B) Valproic acid provided significant protection against hyperthermia‐induced GTCS at doses that result in plasma concentrations above the human therapeutic range. (C) Stiripentol provided significant protection against hyperthermia‐induced GTCS at doses resulting in plasma concentrations above the human therapeutic range. The average temperatures of seizure induction are depicted by the thick horizontal line. Error bars represent SEM, with n = 8–17 per group (**P < 0.01, † P < 0.001, †† P < 0.0001, logrank Mantel‐Cox). [N.B.‐ Panels A and C use the same vehicle controls that are replotted for clarity.]
We also performed additional single‐point analysis of a larger panel of conventional anticonvulsant drugs to determine effect on hyperthermia‐induced seizure thresholds in Scn1a +/− mice (Fig. 3). Acute single‐point doses (IP administration) were selected to achieve plasma concentrations within the human therapeutic range and measured plasma levels are reported in Table 1. Consistent with the dose‐effect results above, clobazam at 5 mg/kg provided significant protection, with 92% (11/12) seizure‐free compared to 11% (2/18) of vehicle‐treated controls (vehicle D, P < 0.0001). Significant protection from hyperthermia‐induced GTCS was also achieved with levetiracetam and phenobarbital, with 50% (6/12) and 100% (12/12) seizure‐free, respectively, compared to 16% (3/19) seizure‐free vehicle‐treated controls (vehicle A, P < 0.019 levetiracetam; P < 0.0001 phenobarbital). Treatment with lamotrigine, an anticonvulsant contraindicated in Dravet syndrome, significantly worsened the hyperthermia‐induced seizure response. All 12 mice experienced a GTCS compared to 10 of 12 vehicle‐treated controls, with a lower threshold temperature for lamotrigine‐treated mice (40.5 ± 0.3°C) compared to vehicle‐treated mice (41.8 ± 0.1°C) (vehicle C; P < 0.0004). Topiramate, phenytoin, carbamazepine, stiripentol, and valproic acid had no effect at the administered doses targeting the human therapeutic range.
Figure 3.
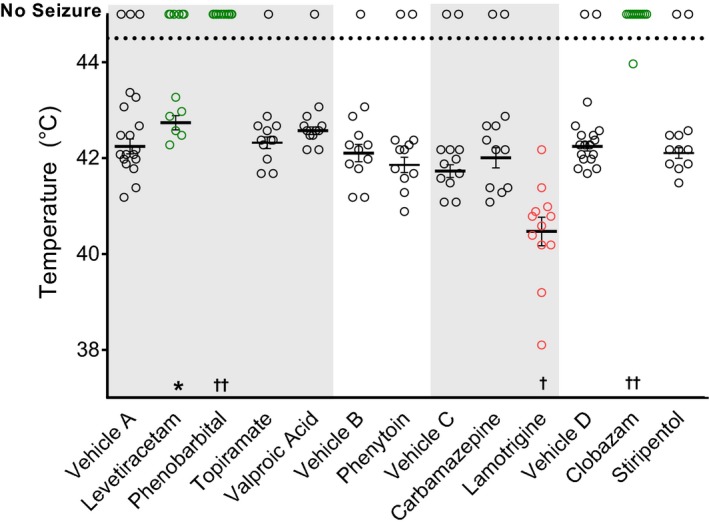
Single‐dose anticonvulsant screening using hyperthermia‐induced seizures in Scn1a +/− mice. Threshold temperature of individual mice for GTCS induced by hyperthermia following acute administration of anticonvulsant doses to achieve plasma concentrations within the human therapeutic range. Lamotrigine treatment significantly lowered the threshold for thermally induced seizures and significantly increased the percentage of mice with GTCS (red, open symbols). Levetiracetam, phenobarbital, and clobazam treatments protected against GTCS, resulting in a significantly improved response to thermal seizure induction (green, open symbols). Vehicles A, B, C, and D correspond to saline, methylcellulose, HP β CD, and vegetable oil, respectively. The average temperatures of seizure induction are depicted by the thick horizontal line. Error bars represent SEM, with n = 12–19 per group (*P < 0.05, † P < 0.001, †† P < 0.0001, logrank Mantel‐Cox).
Spontaneous seizure frequency in Scn1a +/− mice
Scn1a +/− mice exhibit spontaneous GTCS beginning at approximately age P16 with highest frequency between P21 and P25. We quantified spontaneous GTCS frequency in Scn1a +/− mice subchronically treated with anticonvulsant drugs in chow. Drug treatment in Scn1a +/− mice began at P18 and spontaneous GTCS were counted over 60 h of continuous video recording commencing 4 days later (12:00 on P22 through 24:00 on P24). Similar to the effect observed on hyperthermia‐induced seizures, lamotrigine treatment significantly worsened GTCS frequency (P < 0.004) (Fig. 4). Subchronic treatment with lamotrigine resulted in an average seizure rate of 6.9 ± 1.6 seizures per day, compared to controls, which had 2.0 ± 1.0 seizures per day. Lamotrigine was also associated with a higher proportion of mice experiencing GTCS during the observation period (P < 0.014).
Figure 4.
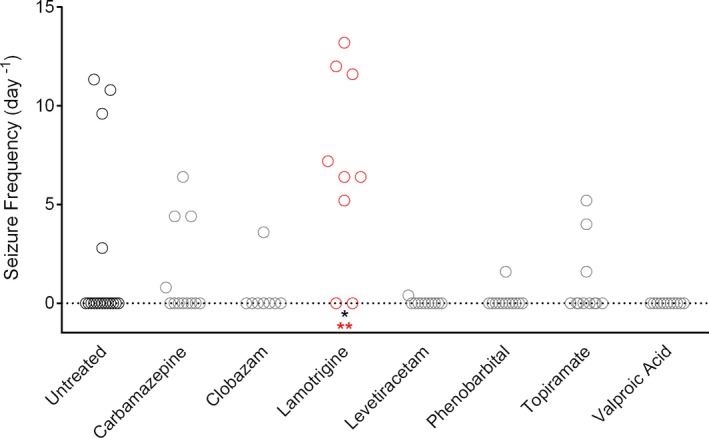
Screening anticonvulsant response using spontaneous seizure monitoring in Scn1a +/− mice. GTCS frequency of individual untreated and drug‐treated mice determined for a 60‐hour period (n = 8–17 mice per treatment group). Lamotrigine treatment resulted in a significantly higher seizure frequency and percentage of mice with seizures compared to untreated controls (**P < 0.004 (red), Dunn's multiple comparisons test; *P < 0.014, Fisher's exact test).
Although subchronic treatment with clobazam, levetiracetam, phenobarbital, or valproic acid did not result in a statistically significant change in GTCS frequency, there was a trend toward protection against seizures. The lack of a significant response may be due to a floor effect, as the untreated Scn1a +/− mice had relatively low seizure incidence and frequencies. This may be influenced, in part, by ascertainment bias, as a considerable number of untreated mice died prior to the P22 recording window. Regardless of the reason, this observation method was underpowered to identify compounds that significantly reduced spontaneous GTCS frequency within the observation window. Therefore, we developed a novel paradigm to improve our power to more efficiently discriminate a protective effect on spontaneous GTCS.
Spontaneous GTCS frequency in Scn1a +/− mice following hyperthermic seizure priming
Dravet syndrome often begins with seizures provoked by fever and progresses to other seizure phenotypes, including afebrile GTCS. We modeled this seizure progression using a hyperthermia‐induced seizure as a clinically relevant trigger to prime Scn1a +/− mice. At P18, a single, brief GTCS was induced with the hyperthermia induction protocol described above. After GTCS onset, Scn1a +/− mice were rapidly cooled back to 37°C, then drug treatment was initiated with anticonvulsant drugs in chow. Spontaneous GTCS were counted over 60 h of continuous video recording (12:00 on P19 through 24:00 on P21). This refined hyperthermia‐priming protocol significantly improved our power to efficiently identify compounds that protect against spontaneous GTCS and more closely modeled the clinical situation where treatment is initiated after patients present with seizures. Comparing the naïve spontaneous seizure monitoring to the new paradigm of hyperthermia‐priming in untreated mice revealed a significant increase in the proportion of Scn1a +/− mice with subsequent unprovoked seizures, with 61% (11/18) of primed mice exhibiting seizures versus 24% (4/17) of naïve mice (Fig. 5) (P < 0.041).
Figure 5.
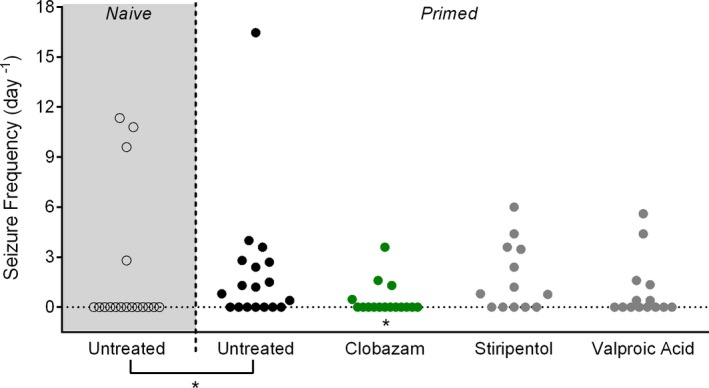
Screening anticonvulsant response using spontaneous seizure monitoring following hyperthermia‐priming in Scn1a +/− mice. GTCS frequency of individual untreated and drug‐treated mice (n = 13–18 mice per treatment group). Drug treatment was initiated following the induction of a single hyperthermia‐induced seizure. Unprovoked, spontaneous GTCS were subsequently quantified over a 60‐hour recording period. Clobazam treatment significantly reduced the proportion of mice experiencing GTCS (*P < 0.046, Fisher's exact test). The untreated cohort represented by black, open symbols, and gray shading are naïve controls to demonstrate the significant difference between naïve and hyperthermia‐priming (black, closed circles) seizure incidences (P < 0.041, Fisher's exact test).
Valproic acid, clobazam, and stiripentol consistently provide modest therapeutic benefit in Dravet syndrome, as observed both in our clinical data and the literature.6, 7, 30, 31 Based on the available clinical information, we prioritized these drugs for further screening in our refined hyperthermia‐priming paradigm. Following induction of a single, brief thermal seizure, we counted subsequent unprovoked GTCS in Scn1a +/− mice subchronically treated with valproic acid, clobazam, or stiripentol using doses that targeted the human therapeutic range. Only subchronic treatment with clobazam offered a protective benefit against spontaneous seizures, with only 25% (4/16) of clobazam‐treated Scn1a +/− mice exhibiting spontaneous GTCS compared to 61% (11/18) of untreated controls (P < 0.046) (Fig. 5). It is perhaps unsurprising that subchronic treatment with valproic acid or stiripentol did not provide protective benefit with plasma concentrations in the human therapeutic range as our hyperthermia assay suggested that supratherapeutic levels of these drugs are required for significant seizure protection.
Anticonvulsant treatment alters GTCS severity
While monitoring spontaneous GTCS frequency in both the naïve and hyperthermia‐primed Scn1a +/− mice, we noticed distinct effects on seizure presentation and severity with lamotrigine, carbamazepine, topiramate, and valproic acid treatment. Therefore, in addition to quantifying seizure frequency, we analyzed whether subchronic anticonvulsant treatment modified seizure severity. We scored each spontaneous GTCS for advancement to the most severe stage of full tonic hindlimb extension (hindlimbs at a 180° angle to the torso). In the first cohort of naïve Scn1a +/− mice that underwent seizure frequency analysis, three anticonvulsants significantly reduced the proportion of seizures that advanced to tonic hindlimb extension (Fig. 6A). Interestingly, while lamotrigine treatment significantly exacerbated GTCS frequency, the severity of the seizures was significantly reduced (P < 0.0001). None of the 155 seizures occurring during lamotrigine treatment escalated to tonic hindlimb extension, compared to 33% (17/52) of seizures in untreated controls. Seizures in lamotrigine‐treated mice did not advance to hindlimb extension, but rather ended with vigorous clonic movements of fore‐ and hindlimbs while mice lay on their sides spinning in circles (Videos S1, S2). Carbamazepine treatment also significantly reduced the proportion of seizures that ended in tonic hindlimb extension (P < 0.0001). Carbamazepine‐treated mice experienced several myoclonic jerks before progressing to GTCS with loss of posture, but no tonic hindlimb extension (Video S3). Additionally, treatment with topiramate significantly reduced the proportion of seizures that advanced to tonic hindlimb extension (P < 0.004). Seizures in topiramate‐treated mice included several instances of rearing and paddling without loss of posture or tonic hindlimb extension (Video S4). In the hyperthermia‐primed cohort of Scn1a +/− mice that underwent seizure monitoring, valproic acid treatment significantly reduced the proportion of seizures that advanced to tonic hindlimb extension (P < 0.046) (Fig. 6B). However, no other changes in seizure semiology were observed in valproic acid‐treated mice. Seizure semiology and severity in all other drug‐treated cohorts were not different from untreated Scn1a +/− mice.
Figure 6.
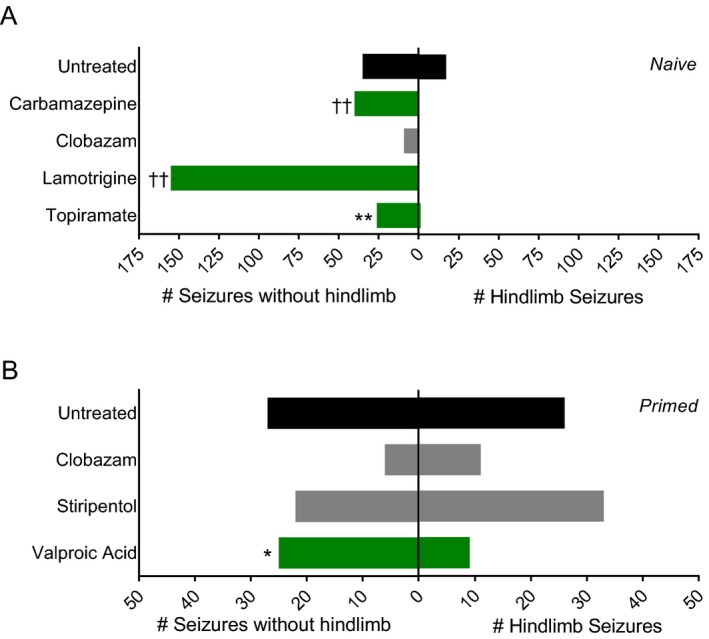
Anticonvulsant treatment alters spontaneous GTCS Severity. (A) The total number of spontaneous GTCS with or without full tonic hindlimb extension of naïve untreated and drug‐treated mice is displayed (n = 8–17 mice per treatment group). The proportion of spontaneous GTCS without tonic hindlimb extension was significantly greater in carbamazepine, lamotrigine, and topiramate‐treated mice (green bars) compared to untreated controls. (B) The total number of spontaneous GTCS with or without full tonic hindlimb extension of primed untreated and drug‐treated mice is displayed (n = 13–18 mice per treatment group). The proportion of spontaneous GTCS without tonic hindlimb extension was significantly less in valproic acid‐treated mice (green bars) compared to untreated controls. Total number of spontaneous GTCS and seizure severity were scored over a 60‐hour recording period. (*P < 0.05, **P < 0.01, †† P < 0.0001, Fisher's exact test).
Survival analysis
Scn1a +/− mice have a significantly reduced lifespan with the greatest proportion of mice dying between the third and fourth week of life.11 Therefore, we evaluated the effect of anticonvulsant treatment on survival during this vulnerable time window. Scn1a +/− mice that underwent spontaneous seizure monitoring in either the naïve or hyperthermia‐priming paradigms continued to receive anticonvulsant treatment through P30 to monitor survival (Fig. 7A, C). Carbamazepine (P < 0.042), levetiracetam (P < 0.034), and topiramate (P < 0.042) all prevented lethality, with 100% of mice surviving through P30. In contrast, only 67% (14/21) of untreated Scn1a +/− mice survived to P30.
Figure 7.
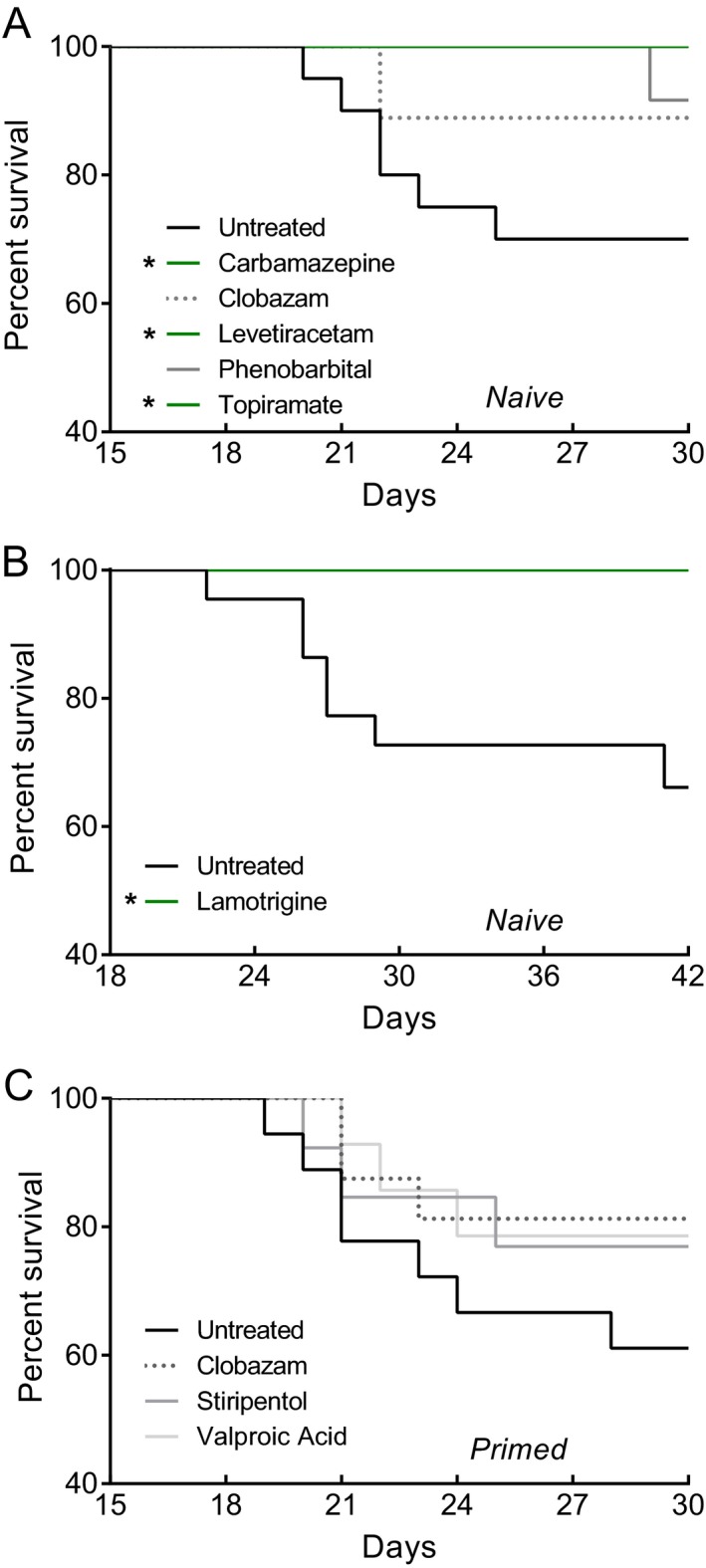
Survival analysis of Scn1a +/− mice. Survival curves comparing untreated and drug‐treated naïve (A, B) or hyperthermia‐primed (C) Scn1a +/− mice. Treatment began at P18 and survival was monitored until P30. (A) Carbamazepine, levetiracetam, and topiramate treatment (green lines) significantly improved survival compared to untreated naïve mice (n = 10–21 mice per treatment group). (B) Survival curves comparing untreated and lamotrigine‐treated Scn1a +/− mice. Treatment began at P18 and survival was monitored until P42 (n = 15–22 mice per treatment group). Lamotrigine treatment significantly improved survival compared to untreated mice (C) No difference in survival was identified in hyperthermia‐primed mice (n = 14–19 mice per treatment group). (*P < 0.05, Logrank Mantel‐Cox).
Because lamotrigine treatment resulted in such a profound worsening of seizure frequency, we hypothesized that lamotrigine would also negatively affect survival of Scn1a +/− mice. We examined survival in a separate cohort of lamotrigine‐treated and untreated Scn1a +/− mice for an extended period (through P42). Surprisingly, although lamotrigine treatment significantly exacerbated the frequency of spontaneous GTCS, it significantly improved 6‐week survival of Scn1a +/− mice (P < 0.015). Lamotrigine treatment completely prevented lethality compared to only 68% (15/22) of untreated animals surviving to P42 (Fig. 7B).
Combination therapy
Stiripentol is approved for use as an add‐on therapy in Dravet syndrome and is often combined with clobazam. We evaluated whether combination therapy with clobazam and stiripentol would offer additional therapeutic benefit compared to monotherapy. First, we tested the effect of acute combination of stiripentol (25 mg/kg, ip) with varying doses of clobazam on hyperthermia‐induced seizure threshold in Scn1a +/− mice. There was a left‐shift in the dose‐response curve with clobazam plus stiripentol compared to clobazam alone (EC50 ratio = 0.7) (Fig. 8A). Next, we tested the effect of clobazam plus stiripentol therapy on spontaneous GTCS following hyperthermia‐priming. Following induction of a single hyperthermic seizure, Scn1a +/− mice were subchronically treated with clobazam and stiripentol (160 mg per kg of chow and 4000 mg per kg of chow, respectively). Treatment with clobazam plus stiripentol provided significant protection against subsequent spontaneous GTCS in Scn1a +/− mice, with 89% (17/19) seizure‐free compared to 44% (11/25) of untreated mice (P < 0.004) (Fig. 8B). While there was a trend toward protection with clobazam alone, with 75%, seizure‐free (12/16), it was not significantly different from untreated (P > 0.062) in this particular experiment. As expected, the addition of stiripentol increased the plasma concentration of both clobazam and N‐desmethylclobazam in subchronically treated Scn1a +/− mice (Fig. 8C).
Figure 8.
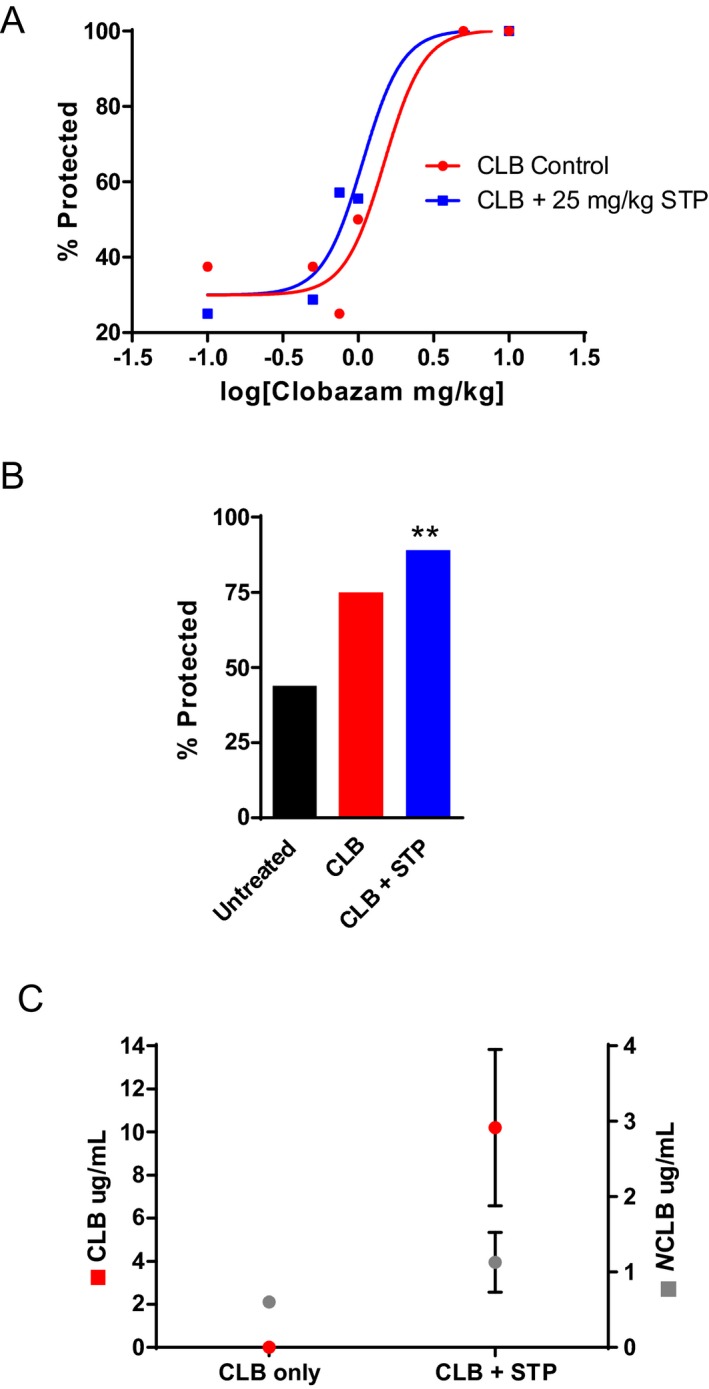
Combination therapy with clobazam and stiripentol in Scn1a +/− mice. (A) Dose‐response curve for seizure protection in hyperthermia‐induced seizure threshold testing. Scn1a +/− mice treated with varying doses of clobazam and 25 mg/kg of stiripentol resulted in a left‐shift of the EC 50 compared to clobazam‐only treated mice (n = 8–17 mice per treatment group). (B) The percentage of untreated, clobazam only, and clobazam plus stiripentol‐treated mice that did not have spontaneous GTCS during the 60‐hour recording period. Drug treatment was initiated following the induction of a single hyperthermia‐induced seizure (n = 16–25 mice per treatment group). (C) Average clobazam and N‐desmethylclobazam plasma concentrations of Scn1a +/− mice treated subchronically with clobazam‐only or clobazam plus stiripentol. The combination treatment resulted in higher clobazam and N‐desmethylclobazam plasma levels (n = 6–7 mice per treatment group, error bars represent SEM). (**P < 0.004).
Discussion
Approximately, 30 drugs are approved for the treatment of epilepsy; however, these drugs fail to provide adequate seizure control in approximately 30% of patients. Therefore, there is a critical need for the discovery of novel drugs for epilepsy. Most current anticonvulsant drug screening programs use induced seizures in wild‐type animals to assess new therapeutic compounds. Genetic epilepsy models could provide alternative screening platforms. A frequently cited limitation of genetic models is low statistical power due to incomplete penetrance and low seizure frequency, which is consistent with our initial experience using spontaneous seizure monitoring in a 60‐h time window. However, we found that adding an etiologically relevant hyperthermia‐priming seizure was effective for improving statistical power to efficiently discriminate a treatment effect with reasonable group sizes.
In the context of monotherapy, we identified several anticonvulsant drugs (clobazam, valproic acid, stiripentol, levetiracetam, phenobarbital) that improved thermally induced and spontaneous seizure outcomes and correlated with our clinical observations or literature reports.5, 7, 32, 33, 34 Clobazam was the most effective drug for seizure reduction in the Scn1a +/− mice, with a protective benefit against both spontaneous and thermally induced seizures at doses that produce plasma concentration levels within the human therapeutic range. This is consistent with the clinical experience from our Dravet syndrome cohort, where clobazam treatment resulted in seizure reduction in almost 75% of cases, and is the most consistently beneficial treatment reported in the literature.5, 7, 32, 33, 34 Previous studies in Scn1a Dravet mouse models reported beneficial effects with benzodiazepines, including clobazam and clonazepam.35, 36 Conversely, lamotrigine produced significant elevation of spontaneous seizure frequency and lowered the threshold for thermally induced seizures in mice. Again, this is consistent with what is observed in our clinic, where lamotrigine treatment resulted in seizure worsening in almost 50% of cases, and is consistent with literature reports.7, 9 Although we observed a protective benefit with levetiracetam and phenobarbital in the Scn1a +/− mice, these drugs had limited effectiveness in our patient series. However, there are reports of favorable response with these drugs in other Dravet syndrome patient series and a recent consensus panel recommends levetiracetam and phenobarbital as third line treatments.5, 7, 34 Although phenobarbital was one of the best performers in the Scn1a +/− mice, its use as a maintenance therapy in patients is often limited by the perceived long‐term side effects. Therefore, the Dravet syndrome patient response data are based on short‐term use, mainly in infants.
Patients with drug‐resistant epilepsy, like Dravet syndrome, are most often on polytherapy. The average number of AEDs per patient in our SCN1A‐mutation‐positive cohort was 3 (SD 1). Although monotherapy in the mice allowed us to individually assess the effect of each drug, it did not account for drug–drug interactions that may contribute to efficacy or adverse effects. The only drug specifically indicated for Dravet syndrome, stiripentol, was approved as an add‐on treatment and is most often combined with clobazam and/or valproic acid. In the Scn1a +/− mice, stiripentol was effective at high doses in our hyperthermia assay, but was ineffective in all assays when using a dose that targeted the human therapeutic range. Consistent with this, intrinsic anticonvulsant activity has previously been reported for stiripentol in various rodent models, including with Scn1a +/− Dravet mice, with high doses, resulting in plasma levels 8‐10‐fold higher than the human therapeutic range.35, 37, 38 It has been hypothesized that stiripentol derives some of its therapeutic benefit from synergistic effects with other anticonvulsant drugs, especially clobazam. In our study, combination therapy in Scn1a +/− mice using a clinically relevant dose of stiripentol with varying clobazam doses resulted in a shift in the dose‐response curve in the hyperthermia‐induced seizure threshold assay, and subchronic combination therapy increased the percent protected from spontaneous GTCS following hyperthermia‐priming. Stiripentol is a weak allosteric modulator of GABAA receptors, a shared mechanism with clobazam, and a strong inhibitor of cytochrome P450 enzymes, particularly CYP2C19. Inhibition of CYP2C19 by stiripentol can promote higher plasma levels of clobazam, N‐desmethylclobazam, and valproic acid in patients on polytherapy.39, 40 Consistent with reports of cytochrome P450 inhibition by stiripentol, we observed significant elevation of plasma clobazam concentration following subchronic combination therapy with clobazam plus stiripentol in Scn1a +/− mice relative to clobazam monotherapy (Fig. 8C).
Most surprising to us was the improved survival observed with some drugs that had no protective effect on spontaneous seizures or hyperthermia‐induced seizure threshold. This was most evident with lamotrigine, as treatment worsened seizure frequency but completely prevented the early lethality of Scn1a +/− mice. This paradoxical effect may be due to the lower severity of individual seizures during lamotrigine exposure, or may result from protective action of lamotrigine outside the central nervous system. Nevertheless, it highlights the fact that survival may not be the most reliable predictor of therapeutic anticonvulsant activity in this model.
An important aspect of this study was our incorporation of pharmacokinetics to benchmark our doses relative to the human therapeutic range. Comprehensive pharmacokinetic profiling in rodent studies is crucial to aide in the translation of preclinical data to human response. Kleiman and Ehlers suggest that a lack of proper pharmacokinetic data and consideration during preclinical experimental design may be hindering the ability to extrapolate results into a clinical setting.41 In our study, we evaluated anticonvulsant doses that resulted in plasma concentrations within the human therapeutic range to ensure an accurate assessment of relevant drug responses in our model. Use of clinically relevant exposure levels mirrors the clinical response more closely, for example, in our hyperthermia assay, lower doses of valproic acid and clobazam provide some therapeutic benefit but not seizure freedom, while escalation of dose beyond the therapeutic range resulted in complete protection from seizures. This is consistent with human data, where valproic acid and clobazam often reduce seizure frequency, but rarely result in seizure freedom. Overall, our findings demonstrate the importance of pharmacokinetic profiling for interpretation of preclinical epilepsy therapy studies and suggest a potential source of the failure to translate some preclinical studies to efficacy in humans where dose range may be limited by safety concerns.
Recently, the scn1Lab‐mutant zebrafish model was demonstrated to recapitulate features of Dravet syndrome and has been used for anticonvulsant screening.42, 43 The large‐scale nature of in vivo phenotype‐based screening in scn1Lab zebrafish could substantially increase the identification of compounds with anticonvulsant potential. Although compounds that already have safety data and regulatory approval may go directly to patient trials, novel development compounds will need to be validated in mammalian model systems.44 Here, we have demonstrated that the Scn1a +/− mouse model of Dravet syndrome recapitulates clinical pharmacological responses. A platform coupling high‐throughput screening in the scn1Lab zebrafish with validation studies in Scn1a +/− mice could be synergistic and advance testing of novel anticonvulsant compounds in genetic models.
Overall, our study suggests that the most predictive measures for screening new compounds in the Scn1a +/− Dravet mouse model would be thorough evaluation of dose‐response with hyperthermia‐induced seizures and spontaneous seizure frequency monitoring after hyperthermia‐priming in Scn1a +/− mice. Our study provides a foundation to predict the efficacy of novel compounds in the treatment of Dravet syndrome and suggests useful strategies for development of preclinical screenings in other genetic models of pharmacoresistant epilepsies.
Author Contributions
Conception and design of the study: JAK and ALG. Acquisition and analyses of data: NAH, LLA, TSG, LL. All authors contributed to drafting the manuscript or figures: NAH, LLA, TSG, LL, ALG, and JAK.
Conflicts of Interest
The authors declare no competing interests.
Supporting information
Table S1. Conditions for AED high performance liquid chromatography (HPLC) assays
Figure S1. Pharmacokinetic analysis of stiripentol and clobazam. Plasma concentration‐time curves for A. stiripentol (following 100 mg/kg, ip injection) and B. clobazam (CLB) and N‐desmethylclobazam (NCLB) (following 10 mg/kg clobazam, ip injection). Data represent average plasma concentrations from n = 3–5 mice per time point.
Figure S2. Chromatograms from HPLC analysis. Representative chromatograms of plasma samples spiked with A. carbamazepine and lamotrigine B. phenobarbital C. stiripentol D. clobazam E. N‐desmethylclobazam F. levetiracetam and respective internal standards.
Video S1. Untreated Scn1a +/− GTCS. Representative example of an untreated Scn1a +/− mouse experiencing a spontaneous GTCS with tonic hindlimb extension. The seizure begins with rearing and paddling, quickly with loss of posture, rapid running and jumping, and ending in hindlimb extension.
Video S2. Lamotrigine‐treated Scn1a +/− GTCS. Representative example of a lamotrigine‐treated Scn1a +/− mouse experiencing a spontaneous GTCS. The seizure beings with rearing and paddling, followed by loss of posture, rapid running and jumping, and ending with vigorous clonic movements of fore‐ and hindlimbs while the mouse lay on its side, spinning in circles.
Video S3. Carbamazepine‐treated Scn1a +/− GTCS. Representative example of a carbamazepine‐treated Scn1a +/− mouse experiencing a spontaneous GTCS. The seizure begins with several myoclonic jerks and spinning in circles, before progressing to GTCS with loss of posture, rearing and paddling, but no tonic hindlimb extension.
Video S4. Topiramate‐treated Scn1a +/− GTCS. Representative example of a topiramate‐treated Scn1a +/− mouse experiencing a spontaneous GTCS. The seizure begins with rearing and paddling and progresses without loss of posture or tonic hindlimb extension.
Acknowledgments
ARK Topiramate Assay and Emit 2000 Valproic Acid Assay enzyme immunoassays generously provided by ARK Diagnostics, Inc. (Fremont, CA, USA) and Beckman Coulter, Inc. (Brea, CA, USA), respectively. We thank Katarina Fabre for technical assistance. This work was supported by NIH grants R01 NS084959 (JAK), R25 NS070695 (TSG), and a grant from the Dravet Syndrome Foundation (ALG). Additionally, LLA was the recipient of a postdoctoral fellowship grant from the PhRMA Foundation.
References
- 1. Golyala A, Kwan P. Drug development for refractory epilepsy: the past 25 years and beyond. Seizure 2017;44:147–156. [DOI] [PubMed] [Google Scholar]
- 2. Simonato M, Brooks‐Kayal AR, Engel JJr, et al. The challenge and promise of anti‐epileptic therapy development in animal models. Lancet Neurol 2014;13:949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dravet C, Oguni H. Chapter 65 ‐ Dravet syndrome (severe myoclonic epilepsy in infancy) In: Harvey BS, ed. Olivier Dulac ML. Handbook of Clinical Neurology: Elsevier, 2013:627–633. [DOI] [PubMed] [Google Scholar]
- 4. Shmuely S, Sisodiya SM, Gunning WB, et al. Mortality in Dravet syndrome: a review. Epilepsy Behav 2016;64:Part A:69–Part A:74. [DOI] [PubMed] [Google Scholar]
- 5. Shi X‐Y, Tomonoh Y, Wang W‐Z, et al. Efficacy of antiepileptic drugs for the treatment of Dravet syndrome with different genotypes. Brain Develop 2016;38:40–46. [DOI] [PubMed] [Google Scholar]
- 6. Wallace A, Wirrell E, Kenney‐Jung DL. Pharmacotherapy for Dravet Syndrome. Pediatric Drugs 2016;18:197–208. [DOI] [PubMed] [Google Scholar]
- 7. Wirrell EC, Laux L, Donner E, et al. Optimizing the diagnosis and management of dravet syndrome: recommendations from a north american consensus panel. Pediatr Neurol 2017;68:18–34.e3. [DOI] [PubMed] [Google Scholar]
- 8. Genton P, Gelisse P, Thomas P, Dravet C. Do carbamazepine and phenytoin aggravate juvenile myoclonic epilepsy?. Neurology 2000;55:1106–1109. [DOI] [PubMed] [Google Scholar]
- 9. Guerrini R, Dravet C, Genton P, et al. Dulac† O. Lamotrigine and Seizure Aggravation in Severe Myoclonic Epilepsy. Epilepsia 1998;39:508–512. [DOI] [PubMed] [Google Scholar]
- 10. Kalume F, Westenbroek RE, Cheah CS, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest 2013;123:1798–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miller AR, Hawkins NA, McCollom CE, Kearney JA. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav 2014;13:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oakley JC, Kalume F, Yu FH, et al. Temperature‐ and age‐dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc Natl Acad Sci 2009;106:3994–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Argikar UA, Senekeo‐Effenberger K, Larson EE, et al. Studies on induction of lamotrigine metabolism in transgenic UGT1 mice. Xenobiotica 2009;39:826–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Borowicz KK, Zadrozniak M, Czuczwar SJ. Low‐affinity kainate receptor‐mediated events reduce the protective activity of phenobarbital and diphenylhydantoin against maximal electroshock in mice. Neuropharmacology 2002;43:1082–1086. [DOI] [PubMed] [Google Scholar]
- 15. Brachet‐Liermain A, Jarry C, Faure O, et al. Liquid chromatography determination of clobazam and its major metabolite N‐desmethylclobazam in human plasma. Ther Drug Monit 1982;4:301–305. [DOI] [PubMed] [Google Scholar]
- 16. Lin HS, Levy RH. Pharmacokinetic profile of a new anticonvulsant, stiripentol, in the rhesus monkey. Epilepsia 1983;24:692–703. [DOI] [PubMed] [Google Scholar]
- 17. Neels HM, Sierens AC, Naelaerts K, et al. Therapeutic drug monitoring of old and newer anti‐epileptic drugs. Clin Chem Lab Med 2004;42:1228–1255. [DOI] [PubMed] [Google Scholar]
- 18. Ratnaraj N, Goldberg V, Lascelles PT. Determination of clobazam and desmethylclobazam in serum using high‐performance liquid chromatography. Analyst 1984;109:813–815. [DOI] [PubMed] [Google Scholar]
- 19. Gordon G, Mackow MC, Levy HR. On the mechanism of interaction of steroids with human glucose 6‐phosphate dehydrogenase. Arch Biochem Biophys 1995;318:25–29. [DOI] [PubMed] [Google Scholar]
- 20. Leclercq K, Kaminski RM. Status epilepticus induction has prolonged effects on the efficacy of antiepileptic drugs in the 6‐Hz seizure model. Epilepsy Behav 2015;49:55–60. [DOI] [PubMed] [Google Scholar]
- 21. Mennini T, Bizzi A, Caccia S, et al. Comparative studies on the anorectic activity of d‐fenfluramine in mice, rats, and guinea pigs. Naunyn‐Schmiedeberg's Arch Pharmacol 1991;343:483–490. [DOI] [PubMed] [Google Scholar]
- 22. Moerman L, Wyffels L, Slaets D, et al. Antiepileptic drugs modulate P‐glycoproteins in the brain: a mice study with (11)C‐desmethylloperamide. Epilepsy Res 2011;94:18–25. [DOI] [PubMed] [Google Scholar]
- 23. Russo E, Donato di Paola E, Gareri P, et al. Pharmacodynamic potentiation of antiepileptic drugs’ effects by some HMG‐CoA reductase inhibitors against audiogenic seizures in DBA/2 mice. Pharmacol Res 2013;70:1–12. [DOI] [PubMed] [Google Scholar]
- 24. Sada N, Lee S, Katsu T, et al. Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 2015;347:1362–1367. [DOI] [PubMed] [Google Scholar]
- 25. Nishimura T, Hu Y, Wu M, et al. Using chimeric mice with humanized livers to predict human drug metabolism and a drug‐drug interaction. J Pharmacol Exp Ther 2013;344:388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. White HS, Franklin MR, Kupferberg HJ, et al. The anticonvulsant profile of rufinamide (CGP 33101) in rodent seizure models. Epilepsia 2008;49:1213–1220. [DOI] [PubMed] [Google Scholar]
- 27. Hawkins NA, Zachwieja NJ, Miller AR, et al. Fine Mapping of a Dravet Syndrome Modifier Locus on Mouse Chromosome 5 and Candidate Gene Analysis by RNA‐Seq. PLoS Genet 2016;12:e1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Liso P, Chemaly N, Laschet J, et al. Patients with dravet syndrome in the era of stiripentol: a French cohort cross‐sectional study. Epilepsy Res 2016;125:42–46. [DOI] [PubMed] [Google Scholar]
- 29. Wirrell EC. Treatment of Dravet Syndrome. Can J Neurol Sci 2016. Jun;43(Suppl 3):S13–S18. [DOI] [PubMed] [Google Scholar]
- 30. Chiron C, Marchand MC, Tran A, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo‐controlled syndrome‐dedicated trial. Lancet 2000;356:1638–1642. [DOI] [PubMed] [Google Scholar]
- 31. Kassaï B, Chiron C, Augier S, et al. Severe myoclonic epilepsy in infancy: a systematic review and a meta‐analysis of individual patient data. Epilepsia 2008;49:343–348. [DOI] [PubMed] [Google Scholar]
- 32. Chiron C. Current therapeutic procedures in Dravet syndrome. Dev Med Child Neurol 2011. Apr;53(Suppl 2):16–18. [DOI] [PubMed] [Google Scholar]
- 33. Chiron C, Dulac O. The pharmacologic treatment of Dravet syndrome. Epilepsia 2011;52:72–75. [DOI] [PubMed] [Google Scholar]
- 34. Striano P, Coppola A, Pezzella M, et al. An open‐label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology 2007;69:250–254. [DOI] [PubMed] [Google Scholar]
- 35. Cao D, Ohtani H, Ogiwara I, et al. Efficacy of stiripentol in hyperthermia‐induced seizures in a mouse model of Dravet syndrome. Epilepsia 2012. Jul;53:1140–1145. [DOI] [PubMed] [Google Scholar]
- 36. Oakley JC, Cho AR, Cheah CS, et al. Synergistic GABA‐Enhancing Therapy against Seizures in a Mouse Model of Dravet Syndrome. J Pharmacol Exp Ther 2013;345:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Auvin S, Lecointe C, Dupuis N, et al. Stiripentol exhibits higher anticonvulsant properties in the immature than in the mature rat brain. Epilepsia 2013. Dec;54:2082–2090. [DOI] [PubMed] [Google Scholar]
- 38. Shen DD, Levy RH, Moor MJ, Savitch JL. Efficacy of stiripentol in the intravenous pentylenetetrazol infusion seizure model in the rat. Epilepsy Res 1990;7:40–48. [DOI] [PubMed] [Google Scholar]
- 39. Fisher JL. The anti‐convulsant stiripentol acts directly on the GABAA receptor as a positive allosteric modulator. Neuropharmacology 2009;56:190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luszczki JJ, Trojnar MK, Ratnaraj N, et al. Interactions of stiripentol with clobazam and valproate in the mouse maximal electroshock‐induced seizure model. Epilepsy Res 2010;90:188–198. [DOI] [PubMed] [Google Scholar]
- 41. Kleiman RJ, Ehlers MD. Data gaps limit the translational potential of preclinical research. Sci Transl Med 2016. 2016‐01‐06 00:00:00;8:320 ps1–ps1. [DOI] [PubMed] [Google Scholar]
- 42. Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun 2013;4:2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dinday MT, Baraban SC. Large‐scale phenotype‐based antiepileptic drug screening in a Zebrafish Model of Dravet Syndrome(1,2,3). ENeuro 2015;2:ENEURO.0068–15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Griffin A, Hamling KR, Knupp K, et al. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain 2017;140:669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Conditions for AED high performance liquid chromatography (HPLC) assays
Figure S1. Pharmacokinetic analysis of stiripentol and clobazam. Plasma concentration‐time curves for A. stiripentol (following 100 mg/kg, ip injection) and B. clobazam (CLB) and N‐desmethylclobazam (NCLB) (following 10 mg/kg clobazam, ip injection). Data represent average plasma concentrations from n = 3–5 mice per time point.
Figure S2. Chromatograms from HPLC analysis. Representative chromatograms of plasma samples spiked with A. carbamazepine and lamotrigine B. phenobarbital C. stiripentol D. clobazam E. N‐desmethylclobazam F. levetiracetam and respective internal standards.
Video S1. Untreated Scn1a +/− GTCS. Representative example of an untreated Scn1a +/− mouse experiencing a spontaneous GTCS with tonic hindlimb extension. The seizure begins with rearing and paddling, quickly with loss of posture, rapid running and jumping, and ending in hindlimb extension.
Video S2. Lamotrigine‐treated Scn1a +/− GTCS. Representative example of a lamotrigine‐treated Scn1a +/− mouse experiencing a spontaneous GTCS. The seizure beings with rearing and paddling, followed by loss of posture, rapid running and jumping, and ending with vigorous clonic movements of fore‐ and hindlimbs while the mouse lay on its side, spinning in circles.
Video S3. Carbamazepine‐treated Scn1a +/− GTCS. Representative example of a carbamazepine‐treated Scn1a +/− mouse experiencing a spontaneous GTCS. The seizure begins with several myoclonic jerks and spinning in circles, before progressing to GTCS with loss of posture, rearing and paddling, but no tonic hindlimb extension.
Video S4. Topiramate‐treated Scn1a +/− GTCS. Representative example of a topiramate‐treated Scn1a +/− mouse experiencing a spontaneous GTCS. The seizure begins with rearing and paddling and progresses without loss of posture or tonic hindlimb extension.