

Abstract
The K-, N-, and HRas small GTPases are key regulators of cell physiology and are frequently mutated in human cancers. Despite intensive research, previous efforts to target hyperactive Ras based on known mechanisms of Ras signaling have been met with little success. Several studies have provided compelling evidence for the existence and biological relevance of Ras dimers, establishing a new mechanism for regulating Ras activity in cells additionally to GTP-loading and membrane localization. Existing data also start to reveal how Ras proteins dimerize on the membrane. We propose a dimer model to describe Ras-mediated effector activation, which contrasts existing models of Ras signaling as a monomer or as a 5-8 membered multimer. We also discuss potential implications of this model in both basic and translational Ras biology.
Keywords: Ras signaling, Ras dimer, membrane clustering, cancer, targeted therapy
1. Introduction
The membrane-residing Ras GTPases are small in size (~21 kD) but big in their roles as key regulators of cell physiology such as growth, proliferation, survival, and many other important functions (Fig. 1a) [1-4]. Canonical Ras family members K-, N-, and HRas share a highly conserved GTPase and effector binding domain (the G-domain) and differ in their C-terminal, hypervariable region (HVR); the HVR is post-translationally modified in a sequence (isoform) specific manner [5-8], which may be responsible for the non-redundant biological functions and mutational spectra of these Ras isoforms in human cancers (Fig. 1b) [3, 9]. In normal and resting cells, Ras is kept in an inactive, GDP-bound state and switches to an active, GTP-loaded state upon upstream stimuli (e.g. ligand binding to receptors) [10]. Ras-GTP then recruits and activates a slew of effectors such as Raf [11], PI3K [12], and RalGDS [13] to execute specific cellular programs (Fig. 1a).
Fig. (1).
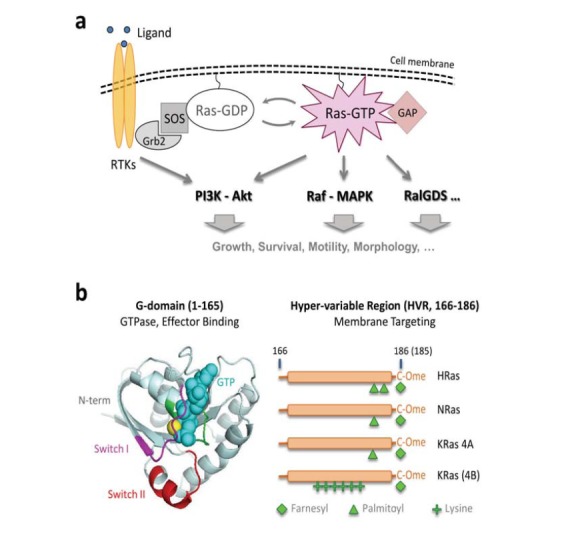
The Ras signaling pathways. (a) Ras resides on the inner leaflet of the membrane and transmits upstream signals such as those from ligand binding to receptor tyrosine kinases (RTKs). Activated RTKs recruit guanine nucleotide exchange factors (GEFs) such as SOS, which converts Ras-GDP into Ras-GTP; Ras-GTP then recruits and activates an array of effectors including PI3K, Raf, and RalGDS to execute specific cellular functions. The counteracting enzymes known as GTPase-activating proteins (GAPs) convert Ras-GTP back to Ras-GDP; (b) Mammalian cells ubiquitously express three Ras genes, H-, N-, and KRas, where KRas mRNA is alternatively spliced into the 4A and 4B forms. KRas 4B is commonly referred to as KRas. All four Ras isoforms have nearly identical G-domains comprised of a GTPase domain that binds and hydrolyzes GTP, and two switch regions I and II that undergo conformational change upon GTP loading to enable effector binding. The four isoforms differ in the last ~20 amino acids known as the hypervariable region (HVR), which contains a linker region (residues 166-186) and a CAAX (C=Cys; A=Aliphatic; X=any) box. After synthesis, Ras proteins are first farnesylated at the last Cys residue in the CAAX box. The AAX residues are subsequently removed and, depending on the Ras isoform (i.e., the sequence of the HVR), the protein can be further modified by different lipids. The post-translational modifications are critical to the correct membrane localization of Ras. HRas is dually palmitoylated, NRas and KRas 4A are mono palmitoylated, and KRas is not palmitoylated.
For the centrality of Ras signaling in cell physiology, Ras activities are tightly regulated in normal cells in part via counteracting enzymes that either stimulate GTP hydrolysis (GTPase activating proteins or GAPs) [14] or accelerate GDP release and GTP loading (Guanine-nucleotide exchange factors or GEFs, such as SOS) [15]. Loss of this balance, and in particular impaired GTP hydrolysis due to point mutations at codons 12, 13, and 61 of Ras, can lead to deregulated cell growth and survival, and these abnormalities are frequently linked to diseases including cancer [16, 17]. Indeed, mutation-activated Ras has been found in about 30% of human tumors and is implicated in nearly all stages of oncogenesis [17-19]. Besides driving tumorigenesis, mutant Ras is also predictive of tumor resistance to cancer therapies [20-22]. As such, mutant Ras has been intensely pursued as a drug target [18, 19, 23-25].
To date, however, efforts to pharmacologically inhibit mutant Ras in human cancers have met with little success [23-25]. Since the biological activity of Ras requires membrane localization, initial efforts focused on developing farnesyltransferase inhibitors (FTIs). The FTIs were shown to be successful in reducing growth of HRas driven tumors [6, 26, 27], however they had little to no effect on tumors with the more prevalent NRas and KRas mutations [7, 28-31]. Other strategies such as restoration of GTP hydrolysis and expression of dominant-negative Ras have proven difficult or ineffective [32]. For these difficulties, attention has shifted to targeting downstream effectors of Ras, mostly the Raf-MAPK [33-35] and PI3K-Akt [36, 37] cascades, in tumors with hyperactive Ras. While these attempts have yielded targeted therapeutic agents that demonstrate clinical benefit in a variety of cancers [38], resistance eventually develops, often times associated with mutant Ras or activation of alternative effector pathways downstream of Ras [21, 39-41]. The limited success in targeting Ras signaling pathways has urged searches for new mechanisms regulating Ras activities in cells, based on which novel therapeutic approaches may be developed.
Accumulating evidence has started to reveal a previously underappreciated aspect of Ras biology – formation of higher order structures – in regulating the physiological activities of Ras [42, 43]. In particular, immuno-EM studies using membrane peel-offs from cells overexpressing Ras suggested that Ras forms nanoscopic clusters (termed nanoclusters), each containing 5-8 Ras monomers on the membrane [44, 45]; these nanoclusters may serve as signaling platforms for recruiting and activating Ras effectors [46, 47]. In this review, we use the term nanocluster interchangeably with multimer as it is not clear at present which term is more accurate in describing the high order structures of Ras. More recently, quantitative single-molecule superresolution microscopy (SRM) [48-50] has been used to study the spatial organization of Ras in intact mammalian cells at 10-20 nm spatial and single-copy stoichiometric resolutions. The results showed that KRas 4B (hereafter referred to as KRas) forms dimers instead of higher order multimers at expression levels comparable to that of endogenous KRas, and that KRas-GTP (e.g. KRas G12D or other activating mutants) dimers activate the Raf-MAPK [51] and likely also the PI3K-Akt pathways (unpublished data). In parallel, KRas G-domain dimers have also been observed in solution [52], and NRas [53] and HRas [54] have both been reported to form dimers when synthetically attached to artificial membranes. A brief timeline of related studies is presented in (Fig. 2).
Fig. (2).
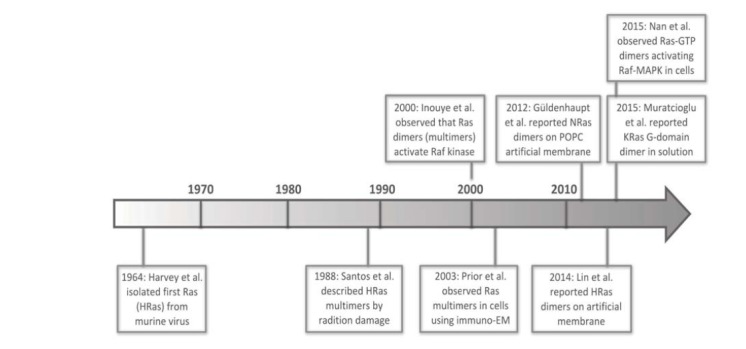
A brief timeline for research findings on Ras multimer (and dimer) formation.
These studies provide compelling evidence that Ras-GTP dimers, rather than Ras monomers or higher (trimers and up) order structures, are the basic functional units of Ras signaling, a new paradigm for Ras biology with profound implications to both our basic understanding of Ras biology and pharmacological targeting of mutant Ras in cancer. The Ras dimer scheme unifies the Receptor-Ras-Raf-Mek-Erk pathway into a dimerization-dependent signaling cascade. Clinically it may now be possible to inhibit mutant Ras activity through disruption of Ras dimers. For these promising prospects, there is growing interest in deciphering mechanisms that regulate Ras dimer formation and signaling in cells. Although little is known about what drives Ras dimer formation and what determines the signaling outcome, hints from literature point to several potential dimerization ‘hot spots’ that may deserve further validation. With increasing effort focused on this new aspect of Ras biology, we anticipate future studies to reveal critical components integral to Ras – effector complexes that regulate Ras dimer formation and signaling outcome. Ultimately, these mechanistic insights will guide the development of novel therapeutic strategies for attacking mutant Ras in human cancers.
2. Historical observations of Ras multimers
Ras has long been considered as a monomeric GTPase [1, 2], because the vast majority of biochemical studies have utilized truncated Ras (G-domain only and monomeric in solution) and focused on Ras properties that do not require Ras multimer formation, such as the GTPase or effector binding, where it appeared that Ras is fully functional as a monomer. These studies almost unanimously revealed Ras functioning as monomers in solution. Intriguingly, whilst a significant fraction of Ras structures in the PDB database showed Ras oligomers [53, 55, 56], such structures were viewed as packing artifacts and hence the potential biological significance of Ras multimers has been overlooked, a similar situation to what happened in the case of Raf dimers [57].
With the dominant view of Ras as a monomer, little attention has been paid to the existence of higher order structures of Ras prior to the year 2000, nearly four decades after the initial discovery of Ras [58]. In 1988, Santos et al. reported that p21 Ras (specifically HRas) forms oligomers (between 2- to 4-mers) by using a radiation-inactivation method [59], which measures remaining protein activity after exposing biological specimen to varying doses of radiation. The probability of a protein complex being damaged and hence losing activity is dependent on both the size of each subunit and the number of subunits, permitting determination of the ‘functional’ size of a protein complex [60]. Using GTP-binding as an indicator for Ras activity, it was found that HRas is either a trimer or a mixture of different sizes of multimers (such as dimers and tetramers) when binding to GTP. To date, it has remained unclear why GTP-binding of Ras would involve Ras multimers as shown in this study, and the finding went largely unnoticed until very recently.
It was not until 2000 when Ras multimer formation was reported again. Inouye et al. used a cell-free system to show that modified KRas produced in insect cells can activate Raf when incorporated into the membrane of phosphatidylcholine liposomes, but not when it is in solution [61]. By treating liposomes containing modified KRas with a bifunctional, amine-reactive crosslinker (EGS), Ras dimers could subsequently be identified using electrophoresis and western blotting. Wild type Ras seemed to crosslink to a similar extent to mutant Ras. Additionally, a split β-galactosidase assay demonstrated that Ras proteins directly interact with each other on the cell membrane. These observations suggest that Ras can form dimers (and potentially higher order structures) on the membrane and not in solution, and more importantly that Ras-Ras interactions are important to Raf activation in cells. Nevertheless, a direct link between Ras dimer formation and Raf activation was not made because Ras dimers were only detected after crosslinking. Without a direct measurement of Ras stoichiometry in cells under signaling conditions, the results could be alternatively interpreted by the formation of Ras multimers.
In 2003, three year after the Inouye experiment, the hypothesis of Ras forming multimers in cells was further illuminated by using immuno-EM to image Ras on the cell membrane [44]. This approach involves over-expression of GFP-Ras (full length or just the C-terminal HVR) in cells followed by mechanical ripping of the apical membrane onto EM grids and subsequent fixation, immuno labeling, and EM observation. The ability to directly visualize Ras proteins on the membrane with nanometer spatial resolution allowed a much more detailed investigation of the spatial localization and multimerization of Ras on the cell membrance, which had not been achieved previously. In the decade or so that followed, a series of immuno-EM investigations by the Hancock lab and associates revealed that Ras organizes into isoform-specific domains termed nanoclusters (multimers) on the cell membrane through their C-terminal HVR [44-46, 62-64]. Different Ras isoforms seem to cluster independently of each other and differ in their dependence on other cellular components. For instance, KRas clustering seems to depend on an intact cellular actin cytoskeleton while HRas clustering does not [45]. H-, N-, and KRas all seem to form 5-8 membered nanoclusters regardless of nucleotide binding status, but the nanoclusters of some (such as H- and N-) Ras isoforms may exhibit different properties depending on whether Ras is GTP- or GDP-bound [63-65]. These findings were corroborated by both computer simulations [66-68] and fluorescence microscopy [46, 64].
Immuno-EM experiments have helped to uncover a novel aspect of Ras biology – regulation through spatial mechanisms – in addition to biochemical mechanisms that were focused on in previous studies. The spatial segregation of different Ras isoforms into distinct nanodomains on the cell membrane provides a plausible explanation to the non-redundant biological functions of H-, N-, and KRas [9, 45]. However, the limitations of these immuno-EM studies are also obvious: using membrane peel-offs may disrupt the native distribution of Ras molecules [44], and over-expression of Ras to overcome the low labeling efficiency in immuno-EM [45] may also alter Ras multimer formation, a property likely sensitive to protein concentration. These limitations raise questions regarding the existence and physiological relevance of Ras nanoclusters, and it has remained unclear which entities – monomers, dimers, or multimers – are the fundamental signaling units of Ras (Fig. 3).
Fig. (3).
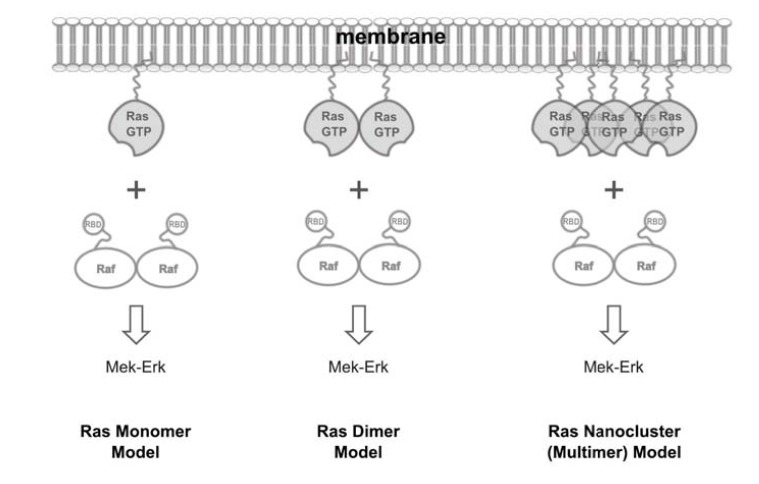
Quest for the fundamental signaling unit of Ras. Raf kinase is a main signaling effector of Ras and is known to function as a dimer, raising the question of whether Ras would also function as a dimer (middle). Existing models view Ras either as a monomer (left) or as a 5-8 membered nanocluster (right).
3. Recent studies defining Ras dimers
Another decade passed by after the first report on Ras nanoclusters, and evidence from several recent studies have started to converge on the notion that while Ras can form multimers, Ras dimers are likely the fundamental unit of Ras organization and signaling both in vitro and in cells [51-54, 69-71]. Importantly, these studies have employed a variety of biophysical, biochemical, and structural techniques, and Ras dimers were observed in both synthetic systems and in cells. Hence, coincidental observation of Ras dimers in all these studies provided strong, corroborative evidence for the existence and the physiological significance of Ras dimers, despite the remaining discrepancies that most likely have resulted from the different setups in biology and methodology.
In 2012, Güldenhaupt et al. showed that NRas forms dimers on an artificial POPC membrane [53]. The authors used attenuated total reflectance Fourier transform infrared (ATR-FTIR) technique to probe the orientation of NRas synthetically attached to supported POPC bilayers on a germanium surface. They found that the stable orientation of NRas on the membrane has a tilt angle of 23º, defined as the angle between the axis representing the sum of the long axes of all α-helices and the membrane normal, in contrast to molecular dynamics (MD) simulations of Ras monomers on artificial membranes that predicted a much larger tilt angle (~80º) [64, 72]. MD simulations of Ras dimers of certain configurations as inspired by existing crystal structures suggest that the small tilt angle likely results from a Ras dimer (or a Ras multimer), a hypothesis corroborated by fluorescence resonance energy transfer (FRET) experiments that showed close Ras-Ras contacts on the membrane.
In 2014, HRas was also found to form dimers when tethered to artificial membranes via a synthetic linker at the C-terminus [54], this time by using time resolved fluorescence correlation spectroscopy (FCS) and single molecule stepwise photobleaching, both suited for measuring molecular stoichiometry. FCS measures molecular diffusion rates with very high temporal resolution, thus allowing sensitive detection of changes in molecular associations that typically result in changes in diffusion behavior [73, 74]. For example, HRas monomers diffuse at a measured rate of ~3.4 µm2/s whereas the putative HRas dimers would diffuse much more slowly at ~0.8 µm2/s. Stepwise photobleaching measurements on fluorescently labeled and sparsely spotted Ras molecules directly showed that the species diffusing at slower (~0.8 µm2/s) rates has a Ras stoichiometry of 2 and that diffusing at the fast rate (~3.4 µm2/s) comprises only 1 Ras molecule.
While the above two studies indicated that Ras dimerization takes place on the membrane and not in solution, Ras dimers in solution as a result of direct G-domain interactions have also been reported very recently [52], when this review was in preparation. Muratcioglu et al. used a suite of tools such as dynamic light scattering (DLS), FRET, and NMR to study multimer formation of KRas in solution. With DLS, the authors observed that the catalytic domain (i.e., the G-domain or residues 1-166) of KRas forms stable dimers with a dissociation constant (Kd) around 1 µM in solution upon (fresh) loading of a GTP-analog, and that the GDP-bound form dimerizes to a much lesser extent. FRET measurement suggests the intermolecular distance (between the C118 residues of the two Ras molecules) to be less than 39 Å, indicating a close interaction between the two Ras molecules within the dimer. NMR with chemical shift perturbations by dilution (to reduce dimers) helped to further define the residues sitting at the dimer interface. It is worth noting that the full length KRas (residues 1-188) consisting of both the G-domain and the HVR region can further associate and form tetramers in the absence of a membrane. For reasons not yet clear, the solution dimers could only be observed when GTP-γ-S but not GppNHp is used as the GTP analog.
In parallel, significant efforts have been ongoing to define the existence and biological functions of Ras dimers in normal and diseased cells [51, 75]. To this end, in 2013 we have developed a quantitative superresolution imaging approach [69] based on the recent photoactivated localization microscopy (PALM) [48, 50] for mapping biomolecules in intact cells with nanometer spatial resolution and single molecule counting precision. As a major breakthrough in biological imaging, PALM and the closely related stochastic optical reconstruction microscopy (STORM) [49, 76] break the resolution limit in conventional light microscopy through subdiffractive localization of single fluorescent molecules that are stochastically switched on and off. When using appropriate fluorescent probes such as PAmCherry1 [77] that switch on only once during the image acquisition process, these techniques allow simultaneous localization and counting of the tagged molecules [69]. For example, we and others have recently demonstrated that it is possible to directly visualize protein multimer formation in cells using quantitative PALM [69, 78-81].
With quantitative PALM, we were able to directly visualize KRas-GTP dimers and multimers in cells, as first alluded to in a report in 2013 [ref. 75] and then reported in full in 2015 [ref. 51]. PALM imaging of PAmCherry1 tagged KRas revealed that both GDP- and GTP-bound KRas molecules aggregate to form dimers and occasional higher order multimers at physiologically relevant expression levels (Fig. 4). At this expression level, KRasG12D, an activated mutant that constitutively binds GTP, but not wild type KRas (GDP bound), activates the Raf-MAPK pathway. Overexpression of KRas leads to formation of higher order Ras multimers (clusters); this is similar to that previously observed with immuno-EM, which also implies that the 5-8 membered Ras nanoclusters discussed in literature probably resulted from protein overexpression. At lower expression levels, KRas appears monomeric on the membrane and KRasG12D cannot activate Raf-MAPK signaling unless forced into dimers (e.g. by using an artificial dimerization system). The quantitative nature of PALM also allowed us to determine that a density of ~6 Ras-GTP dimers per µm2 – that is, a total of ~2,000 dimers per cell if assuming a total membrane area of 300 µm2 for a typical mammalian cell – is already sufficient to activate Raf-MAPK signaling. Together, these observations demonstrate the existence of Ras dimers in cells and establish a direct link between Ras-GTP dimer formation and Raf-MAPK pathway activation [51].
Fig. (4).
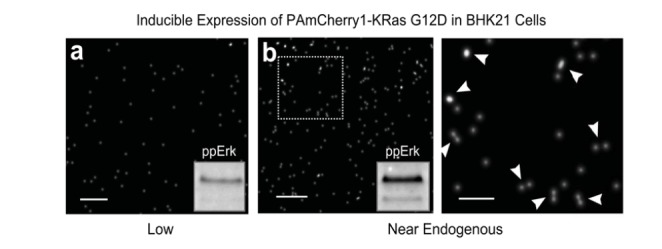
Single-molecule superresolution imaging of Ras dimers in cells. BHK21 cells stably expressing PAmCherry1-KRas G12D (an activated mutant of KRas that constitutively binds GTP) under doxycycline (Dox) regulation was treated with 1 or 2 ng/mL Dox for 48-72 hours before being fixed and imaged with photoactivated localization microscopy (PALM). Images were acquired under total internal reflection (TIR) illumination conditions to limit the excitation volume to the basal membrane of the cells. Each dot in the PALM images represents one putative PAmCherry1-KRas G12D molecule. (a) At 1 ng/mL Dox, PAmCherry1-KRas G12D is expressed at a level much lower than that of endogenous KRas and appears monomeric, when the level of phosphorylated Erk (ppErk) is also low as shown in the inset, indicating little activation of the Raf-MAPK signaling pathway; (b) At 2 ng/mL Dox, PAmCherry1-KRas G12D is expressed at a level similar to that of endogenous KRas, and dimers (and occasional higher order multimers) of PAmCherry1-KRas G12D could now be observed. Image on the right is the zoomed view of the boxed area in the image on the left. White arrows indicate putative KRas dimers. Under this condition, ppErk level is significantly higher, indicative of an activated Raf-MAPK pathway. Scale bars, 250 nm in (a) and (b, left), and 100 nm in the zoomed view (b, right).
4. Ras functioning as a dimer: a new paradigm for Ras biology
Ras is the last component known to function as a dimer in the canonical Receptor-Ras-Raf-Mek-Erk (Ras-MAPK) signaling cascade. Receptor tyrosine kinases (RTKs) such as the ErbB family receptors EGFR, HER2, HER3, and HER4 are classical examples of dimerization-dependent signal transducers, where both homo- and heterodimers are involved in receptor trans activation [82]. Downstream of Ras, the Raf kinase also functions as a dimer in activating Mek, again both as a heterodimer (for example that between B- and CRaf) and as a homodimer (for example BRaf:BRaf or CRaf:CRaf) [57, 83-85]. This dimerization-dependent signaling mechanism of Ras is involved in physiological as well as pharmacological activation of the MAPK pathway [57, 86, 87]. Further downstream, Mek [88, 89] and Erk [90, 91] have similarly been shown to form dimers, although in these cases the exact configuration of those dimers (e.g. hetero- versus homodimers) and the roles of dimerization in signaling are not as well defined.
Based on the existing findings described above, it is reasonable to believe that Ras-GTP dimers may be the basic signaling units of Ras, as illustrated in the model shown in (Fig. 5). In this model, Ras-GTP monomers can each bind an effector but cannot fully activate the latter until two Ras-GTP molecules form a dimer and cause the two bound effector molecules to also dimerize. This model is a generalized version of the one that we presented recently [51], where only Raf was depicted as the effector. Our unpublished data suggest that Ras dimers may also activate PI3K-Akt, another important effector of Ras. Moreover, crystal structures of Ras in complex with RalGDS (for example, PDB entry 1LFD) apparently show a Ras dimer with a RalGDS dimer [92, 93]. Together, these data and analyses strongly support the hypothesis that Ras dimers may be generally implicated in Ras-mediated effector activation. Hence, the biological activity of Ras is not only regulated by GTP-loading and membrane localization, but also by protein dimerization.
Fig. (5).
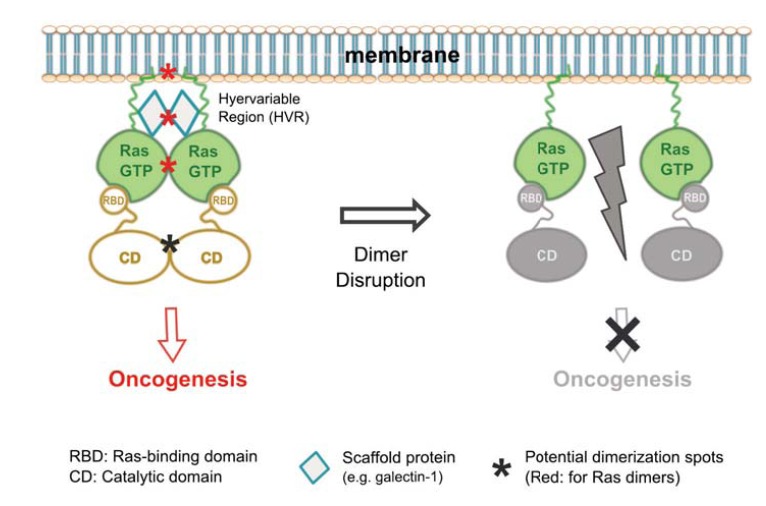
A dimer model for Ras-mediated effector activation. (Left) GTP-loaded Ras can each recruit an effector molecule onto the membrane, but the event alone does not activate the effector. The effector is activated when two Ras-GTP molecules form a dimer to also bring two effector molecules into a dimer, which in turn initiates oncogenic signaling. Multiple factors, including membrane binding through the lipid-modified HVR, scaffold proteins, and direct G-domain contacts, could contribute to the dimer formation and hence oncogenic activity of Ras; (Right) Mechanisms that disrupt Ras dimer formation would also inhibit Ras-mediated oncogenesis and therefore could be exploited for anti-cancer therapy.
To date, the best validation for the Ras dimer signaling model is with the Raf kinase. As discussed earlier, it has now been well established that Raf kinases form dimers in cell signaling processes [57, 84, 85]. Reports on Raf forming both heterodimers and homodimers in the presence of active Ras have brought up speculations that Ras may itself dimerize [83-85]. More recently, biochemical, structural, and imaging studies have further defined Raf dimers; the dimer interface between two Raf promoters within a dimer has been identified [57], and it has become clear that the two Raf molecules in a dimer do not need to both possess kinase activity to make an active dimer [94]. Interestingly, when Raf is activated in cells by mutant Ras, dimers as well as occasional higher order multimers (trimers and tetramers) were observed, likely because the multimer formation of Raf is driven by the clustering (multimer formation) of Ras [69]. Lastly, by combining bimolecular fluorescence complementation with quantitative PALM imaging (BiFC-PALM), we demonstrated that Ras-Raf RBD complexes can further dimerize to yield a tetrameric Ras-Raf RBD complex with two copies of Ras and two copies of Raf (RBD), which presumably is the intermediate protein complex during Ras-mediated Raf activation [70]. These observations provided further evidence that Ras-Raf signaling involves dimerization of both proteins. Biochemical tools derived from these studies, such as dimerization-deficient Raf mutants, have also become useful in deciphering the mechanisms that regulate Ras dimer formation in vitro and in cells.
While more validations of the Ras dimer signaling model are underway, it is already interesting to consider the basic and translational implications of this model. Above all, the recent findings call to revise the current monomer model of Ras signaling found in biology textbooks and in literature; that the whole Receptor-Ras-Raf-Mek-Erk pathway depends on dimer formation also advocates the concept of protein dimerization as a general regulation mechanism of cellular processes [95-97]. The model also calls to revisit the theory that active Ras is a 5-8 membered nanoclusters on the membrane. Although Ras can form higher order ‘clusters’ when overexpressed, at physiological expression levels the predominant form is dimers and dimers are sufficient to activate effectors. Hence, the smallest ‘cluster’, i.e., a dimer, may be all that is needed. It remains to be seen whether it is the same cellular mechnaisms that regulate Ras dimers and nanoclusters formation. Some speculated that Ras dimer formation may promote formation of Ras nanoclusters [71]; it is also possible that the processes that drive the formation of Ras nanoclusters – for example, partitioning of Ras monomers into lipid domains with restricted dimensions – could facilitate Ras-Ras interactions and hence Ras dimer formation.
But why dimers? The answer could be manifold, and findings from prior research on the clustering properties of other signaling molecules may be applicable here [95-98]. Among others, clustering of signaling molecules has been known to improve resistance to noise due to spontaneous fluctuations in the input signal, as also previously discussed in the context of Ras nanoclusters [46, 99]. If two Ras-GTP molecules are needed to make an active signaling dimer, then spontaneous GTP-loading of Ras will mostly give rise to inactive Ras-GTP:Ras-GDP heterodimers, and hence no actual signaling activity will be propagated downstream. Conversely, when there is actual upstream signal that causes Ras to be loaded with GTP, it can be expected that the Ras pathway activity will rise more quickly to maximum when Ras signals as a dimer or a multimer than when Ras signals as a monomer; dimer or multimer formation thus helps yield a switch-like response, a feature that would otherwise be missing if Ras were to act as a monomer [99].
The requirement of two Ras-GTP molecules to make an active Ras dimer may also help explain a long-standing observation in vivo that wild type Ras can act as a tumor suppressor in cells with heterozygous Ras mutations [32, 101-103]. Cells often do not become transformed when they acquire an activating mutation in just one endogenous Ras allele but instead do so until they acquire an activating mutation in or lose the second, wild type Ras allele [104, 105]. This is in accordance with observations that, despite the high incidence of cells harboring KRas mutations in healthy individuals, only a very small fraction actually develops into cancers, thus suggesting that only cells with KRas-GTP levels above a certain threshold (to make sufficient Ras-GTP dimers) could initiate oncogenesis [106]. Mechanisms underlying these observations have been unclear but may now be explained by the Ras dimer model. That said, the interactions between wild type and mutant Ras in cells is likely more complicated than merely dimer and/or multimer formation, as demonstrated in a recent study [107].
The translational implications of the Ras dimer signaling model are also evident and stimulating. Ras is the top most mutated genes in human cancers, but historically it has been difficult to inhibit mutant Ras activity for cancer therapy [23-25]. Despite the more recent progresses [108, 109], drugging Ras has remained a major challenge in current cancer medicine. Pharmacological targeting of downstream effectors such as Raf and PI3K has also had limited success, in part due to activation of altemative Ras effector pathways upon release of feedback loops by the targeted inhibition [110, 111]. If Ras-GTP dimers were required for the activation of Raf, PI3K, and potentially other Ras effectors that are central to mutant-Ras mediated tumorigenesis, then disruption of Ras dimers could inhibit these effector pathways in tumor cells all at once, potentially offering a more effective therapeutic strategy than what is currently available (Fig. 5).
Additionally, mutant Ras has been linked to tumor resistance to existing cancer therapies. In such cases, disruption of Ras dimers may also prove beneficial to improving the efficacy of those therapies, provided that the resistance is mediated through Ras dimers. For example, melanoma tumor cells harboring mutant Ras would not respond to Raf inhibitors such as Vemurafenib (PLX-4032). Instead, treatment with PLX-4032 (or many other Raf inhibitors) results in paradoxical activation of Raf-MAPK and accelerates tumor growth through Ras-dependent Raf dimer formation [39, 86, 87]. In our recent work, we showed that cells expressing low levels of mutant KRas could be efficiently killed by serum starvation or MeK inhibition, but co-incubation of the cells with a small molecule that forces KRas-GTP into dimers rescued the cells [51]. These observations suggest that at least under some conditions, resistance of Ras-mutant tumor cells to MAPK pathway blockade is mediated by Ras dimers, thus demonstrating the value of disrupting Ras dimer formation in cancer therapy.
5. Potential mechanisms regulating Ras dimer formation and signaling
Therapeutic targeting of mutant Ras in cancers through disruption of Ras dimers requires a clear understanding of the mechanisms that regulate Ras dimer formation and signaling. To date, little is known in this respect since Ras dimers have rarely been addressed in literature. Nevertheless, previous biochemical, structural, and imaging studies have provided hints to understanding Ras-Ras interactions, for example in the context of Ras nanoclusters formation [65, 67]. Existing crystal structures of Ras may also shed some light on the potential orientations of Ras molecules within a dimer or multimer. Based on existing data and analysis, we propose that at least three potential mechanisms exist for regulating Ras-Ras dimer formation: the HVR that mediates Ras-Ras and Ras-membrane interactions, scaffold proteins, and G-domain contacts; these are depicted in (Fig. 6).
Fig. (6).
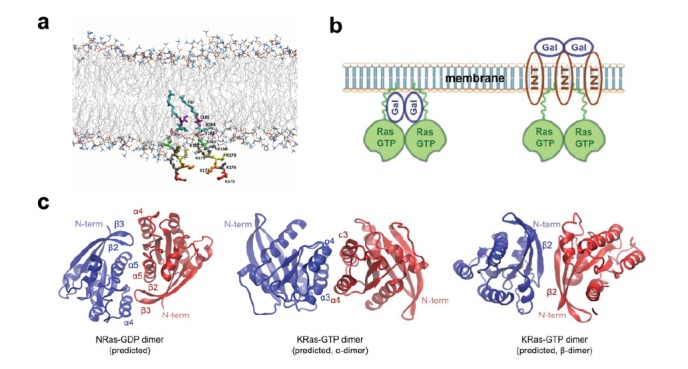
Potential mechanisms regulating Ras dimer formation and signaling. (a) Lipid anchors (175KKKKKKSKTKC(Far)OMe) of two KRas C-terminal HVR’s placed in a lipid bilayer. The C-terminal amino acid residues are rendered in sticks and colored by residue number. The farnesyl group is in cyan (Far). The lipid bilayer head groups are shown in lines, with lipid tailed colored in gray and lipid head groups colored using the “atomic name” scheme; (b) Scaffold proteins such as galectins and integrins may facilitate Ras dimer formation. For example galectins may interact with Ras either directly (left) or through integrin (right) to cause Ras to dimerize or cluster. Relative positions of the proteins are putative and may not reflect the actual spatial arrangement; (c) Predicted G-domain interfaces for NRas-GDP (left) and KRas-GTP (middle and right). Figures were reproduced based on information in references (53; left) and (52; middle and right), respectively.
In our recent study, we have shown that Ras dimer formation in cells appears to be mainly mediated by the HVR [51]. The HVR of Ras comprises the last ~20 amino acids at the C-terminus, which includes the CAAX motif required for initiating post-translational modifications and hence correct localization of Ras to the plasma membrane [5, 6, 113]. In the case of KRas (4B), membrane targeting is also facilitated by the positively charged lysine residues in the HVR [5]. By comparing the dimer formation properties of PAmCherry1 tagged full length KRas wild type, the G12D mutant, and the HVR only, we found that the HVR alone is sufficient to cause dimer formation, whereas the G-domain appears to be dispensable. We note that this observation only suggests a dominant role of the HVR in promoting Ras dimerization and does not rule out the potential (albeit secondary) contributions of the G-domain.
At present, it is unclear how the HVR can mediate Ras dimerization, but the increased effective concentration of molecules after localization to the membrane may be in part responsible. A recent study shows that whilst GTP-bound catalytic domain (without the HVR) of KRas can form stable dimers in solution, the Kd is only on the order of 1 µM [52]; to turn this relatively weak interaction into effective dimer formation, it would require ~106 Ras molecules if they were in the cytosol, assuming that the cell volume is approximately 1 picoliter. Using quantitative PALM in combination with western blotting, we have measured that there are on the order of 104 KRas molecules per cell (in the case of BHK21 or HEK 293 cell lines), a similar result to a previous measurement using a different approach [114]. As such, confinement of Ras to a two-dimensional membrane would seem necessary and effective to enhance dimer formation.
Moreover, as alluded to earlier, partitioning of Ras molecules to nanoscopic membrane domains may further increase the local concentration of Ras molecules [65] to compensate for the 2 orders of difference between the actual and the required Ras concentrations for effective Ras-Ras interactions. Physical partitioning of Ras on the membrane is in part through interactions between the lipid-modified HVR and the membrane lipid bilayer [66, 68], which can be modulated by local lipid and protein compositions. Of note, different Ras isoforms have different lipid modifications and accordingly, segregate into different membrane nano-domains. For example, while KRas preferentially localize to liquid disordered domains of the membrane, HRas resides in cholesterol-rich, ordered domains, and NRas lives at the boundary [45, 65]. As a result, Ras may exhibit isoform-specific dimerization properties on the membrane (and/or in the cytosol), although dimers are likely needed in all cases, as well as different potencies in activating Raf, PI3K and other effectors despite their homologous G-domains.
A recent molecular dynamics (MD) simulation suggested an alternative mechanism through which Ras may interact through their HVRs [100]. Specifically, the positively charged HVR of KRas does not insert into ordered lipid domains and instead folds into a specific conformation to directly interact with the HVR of another KRas to form a dimer in the aqueous phase (albeit still attached to the membrane). Direct interactions between the HVRs of KRas may also be responsible for the observed tetramers of full length KRas in solution in the absence of lipids [52]. At present it seems difficult to verify whether this direct HVR-HVR interaction at the membrane is strong enough to cause KRas dimer formation in cells. Our MD simulations have revealed a weak association between lipid-modified KRas HVRs and suggested that the HVRs can in fact insert into ordered lipid domain where they aggregate with each other as a result of ordered-disordered phase segregation (Fig. 6a; unpublished data). It is also worth noting that in all the MD simulations, the HVRs of KRas do not appear to repulse each other to prevent dimer formation as some believed [115], despite the positive charges carried by the poly-lysine string, likely due to the screening effects from the neighboring phospholipid head groups (negatively charged) and water molecules (polarizable).
Second, scaffold proteins may also regulate Ras dimer formation and signaling output (Fig. 6b). Many signaling processes involve scaffold proteins, such as KSR in Raf/Mek/Erk signaling [116]. Previous studies on Ras nanoclusters have implicated several classes of proteins, including galectins [117, 118] and integrins [41], in modulating the spatial organization and signaling activity of Ras on the cell membrane. In particular, galectin-3 interacts with GTP-bound KRas and promotes KRas-mediated cell transformation through the MAPK and PI3K signaling pathways [118, 119]. Expression of non-oncogenic galectin-3 in the human breast cancer cell line BT-549 inhibits cell transformation as judged by foci formation. Similarly, galectin-1 was shown to interact specifically with GTP-bound HRas, which stabilizes HRas-GTP nanocluster and prolongs the duration of Ras-mediated Erk activation [44, 117, 120]. Galectin-1 knock down by siRNA inhibits HRas G12V (an activating mutant, GTP-bound) mediated cell transformation, demonstrating a critical role of galectin-1 in HRas-GTP signaling [117].
While these results have been interpreted in the context of Ras nanoclusters, it is possible that the bi- and multivalent galectins can also promote Ras signaling by enhancing the dimerization of GTP-bound Ras. In particular, galectin-1 exists as a dimer in solution and in crystal [121], and coincidentally our unpublished results show that HRas also likely functions as a dimer, suggesting that the role of galectin-1 may be to help HRas-GTP to dimerize (Fig. 6b, left). Interestingly, galectin-3 is a multivalent lectin that tends to form oligomers [121, 122], although KRas has been shown to form dimers. Nevertheless, a more recent study suggests that interactions between galectins-3 and KRas may be indirect and mediated through integrins (Fig. 6b, right) [41]. A more thorough search with functional siRNA/ shRNA screens or other approaches will help identity more scaffold proteins for Ras dimer formation and signaling.
Aside from regulating Ras dimer formation and hence Ras signaling intensity and duration, scaffold proteins may also modulate the signaling specificity of Ras dimers. For example, expression of galectin-1 promotes HRas signaling to Raf but not PI3K, suggesting that dimers of the HRas: Galectin-1 complex may be either located in membrane partitions more easily accessible to Raf than PI3K, or residing in a conformation that favors binding to Raf over PI3K [123]. In BT-549 breast tumor cells, galectin-3 expression upregulated activities of both Raf and PI3K but surprisingly resulted in lower activation of Erk (MAPK) [118]. While mechanisms of how scaffold proteins alter the signaling specificity of Ras are unclear, we anticipate that other scaffold proteins for Ras dimers, should there be any, could act through similar mechanisms to direct Ras dimer signaling to various downstream effectors and in so doing achieve versatile and context-specific signaling outcomes.
Third, direct protein-protein contacts in the G-domain can also contribute to Ras dimer formation (Fig. 6c), as suggested by a few recent studies [52, 53, 100]. Although Ras has been viewed as a monomeric GTPase, a large number of structures deposited in the PDB database actually reveal Ras multimers [53]. However, at present no accurate dimer conformation pertinent to effector activation could be directly inferred from these structures. According to one study, the modeled NRas dimer structure on POPC based on NMR, FRET, and ATR-FTIR measurements suggests that residues D47, E49, R135, R161, and R164 participate in dimerization (Fig. 6c, left) [53]. These G-domain residues are located at allosteric sites opposite to the GTP-binding and effector binding regions. Among these, D47 and E49 are at the turn between β2 and β3, R135 at α4, and R161 and R164 at α5. Consistent with these hypotheses, a paper by Solman et al. has just been published demonstrating that mutations at residues D47, E49, R128, and R135 in H-, N-, and KRas can modulate Ras signaling activity, potentially by altering Ras multimer formation [124].
Interestingly, formation of HRas dimers on artificial membranes appears to be dependent or coupled to residue Y64 instead, which is located within the switch II region and responsible for binding to SOS and PI3K. Since dimers were the only multimers observed and the mutation Y64A eliminated HRas dimers completely, the experiments would suggest that there are no secondary dimer interfaces with comparable strength [54]. Intriguingly, however, the Y64A mutation did not seem to affect KRas dimer formation or signaling in cells despite the fact that Y64 is conserved between all major Ras isoforms [51], potentially highlighting the influence of cellular and molecular context on Ras dimer configuration and function.
In a more recent study, two different dimer structures of KRas G-domain, namely α- and β-dimers, were predicted using a template-based protein-protein complex structure prediction algorithm (PRISM) [52]. In the α-dimer, the dimer interface is between the α3 and α4 helices, where the α3 of one Ras protomer is in close contact with the α4 of the other (Fig. 6c, middle). The more populated β-dimer has a dimer interface along β2 and overlaps extensively with the switch I and II regions (Fig. 6c, right). Since the switch regions are responsible for effector binding, the β-dimer is presumably inhibitory to Raf or PI3K activation. Existence of the two dimer interfaces was partially verified with NMR, but their biological relevance in a cellular setting is still to be determined.
Apparently, while the three studies predicted and in some cases, partially verified the G-domain interface, there has not been a consensus as to where the G-domain interface lies. The discrepancies may in part arise from the different experimental setups such as the inclusion or omission of a lipid bilayer and the use of synthetic tethers to anchor Ras to the membrane. Another important factor to consider is the impact of effector binding on the overall conformation of Ras; although crystal structures of Ras-effector seem to suggest that the overall conformation of Ras remains similar after effector binding, the binding may still significantly alter the exposed surface of Ras (and the effector itself), potentially resulting in different Ras dimer conformation than that predicted based on a ‘free’ Ras protein in solution or one tethered to artificial membranes.
Aside from the three mechanisms discussed above, other mechanisms may exist to regulate Ras dimer formation and/or modulate its signaling outcome. Regulating Ras dimerization offers an alternative strategy for modulating Ras signaling intensity and/or duration without having to change the expression level of mutant Ras or the amount of Ras-GTP. Depending on the cellular context and the interacting partners of Ras, different mechanisms may dominate to yield a different Ras dimer and generate a context-specific signaling outcome. For example, expression of galectins-3 redirects breast cancer cells from using NRas to using KRas for oncogenic signaling, perhaps by promoting KRas but not NRas dimer formation [125]. In tumors, certain signals (such as cytokines) or factors (such as the extracellular matrix components and the integrins) from the microenvironment may also promote oncogenic signaling of Ras by enhancing Ras dimerization.
6. Concluding remarks
Historical and recent studies have provided strong evidence for the existence and physiological relevance of Ras dimers, a previously unappreciated aspect of Ras biology. While further investigations are needed to fully establish Ras dimers as the functional unit of Ras signaling in cells, the Ras dimer signaling model offers a new framework for understanding how Ras organizes spatially and interacts with effectors on the membrane and how such interactions may be regulated to generate specific output with versatility. Some unresolved issues, such as the role of wild type Ras as a tumor suppressor in cells co-expressing mutant Ras, could potentially be explained by the Ras dimer model. Future studies will need to further define the role of Ras dimers in activating other effectors (such as PI3K and RalGDS) and more generally in mediating oncogenesis and therapeutic resistance, the existence and physiological relevance of the dimers of other Ras family members (such as H- and NRas), and the mechanisms that regulate Ras dimer formation and interaction with effectors. Facilitated by emerging research tools, these investigations will likely reveal many more interesting aspects of Ras dimer biology and help to identify ‘hot spots’ within the Ras signaling complexes that are potentially actionable. For the promising prospect of Ras dimers in basic biology and translational medicine, we anticipate a boom of research activities on this topic in the years to come.
Acknowledgements
We are grateful to our collaborators Drs. Steven Chu (Stanford), Joe W. Gray (OHSU), Frank McCormick (UCSF and Fredrick National Lab), Eric A. Collisson (UCSF), as well as members in the Nan, Gray, and McCormick labs for their contributions to part of the data summarized here. We thank Dr. Philip Stork (OHSU) for his critical reading of the manuscript and helpful discussions, and Dr. Ruth Nussinov (NIH) for kindly providing the coordinate files for predicted KRas dimer structures. We also express our sincere apologies to colleagues whose work could not be cited due to space limitations and the massive volume of literature on related topics. Work from the Nan laboratory discussed herein was supported by National Institutes of Health (NIH) Grant 5U54CA143836, National Science Foundation Grant PHY-0647161, and a supplement to NIH grant U54 CA112970. Research in the Nan laboratory is also supported by startup funds from the Knight Cancer Institute at OHSU, the Damon Runyon Cancer Research Foundation, the M.J. Murdock Charitable Trust, the Prospect Creek Foundation, the Brenden-Colson Center for Pancreatic Care at OHSU, and the FEI company.
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
References
- 1.Cox A.D., Der C.J. Ras history: The saga continues. Small GTPases. 2010;1(1):2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malumbres M., Barbacid M. RAS oncogenes: the first 30 years. Nat. Rev. Cancer. 2003;3(6):459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 3.Karnoub A.E., Weinberg R.A. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 2008;9(7):517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pylayeva-Gupta Y., Grabocka E., Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer. 2011;11(11):761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cadwallader K.A., Paterson H., Macdonald S.G., Hancock J.F. N-terminally myristoylated Ras proteins require palmitoylation or a polybasic domain for plasma membrane localization. Mol. Cell. Biol. 1994;14(7):4722–4730. doi: 10.1128/mcb.14.7.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaber M.D., et al. Polyisoprenylation of Ras in vitro by a farnesyl-protein transferase. J. Biol. Chem. 1990;265(25):14701–14704. [PubMed] [Google Scholar]
- 7.Fiordalisi J.J., et al. High Affinity for Farnesyltransferase and Alternative Prenylation Contribute Individually to K-Ras4B Resistance to Farnesyltransferase Inhibitors. J. Biol. Chem. 2003;278(43):41718–41727. doi: 10.1074/jbc.M305733200. [DOI] [PubMed] [Google Scholar]
- 8.Rubio I., et al. Farnesylation of Ras is important for the interaction with phosphoinositide 3-kinase gamma. Eur. J. Biochem. 1999;266(1):70–82. doi: 10.1046/j.1432-1327.1999.00815.x. [DOI] [PubMed] [Google Scholar]
- 9.Hancock J.F. Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003;4(5):373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 10.Scolnick E.M., Papageorge A.G., Shih T.Y. Guanine nucleotide-binding activity as an assay for src protein of rat-derived murine sarcoma viruses. Proc. Natl. Acad. Sci. USA. 1979;76(10):5355–5359. doi: 10.1073/pnas.76.10.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000;351(2):289–305. [PMC free article] [PubMed] [Google Scholar]
- 12.Castellano E., Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer. 2011;2(3):261–274. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferro E., Trabalzini L. RalGDS family members couple Ras to Ral signalling and that’s not all. Cell. Signal. 2010;22(12):1804–1810. doi: 10.1016/j.cellsig.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 14.McCormick F. ras GTPase activating protein: signal transmitter and signal terminator. Cell. 1989;56(1):5–8. doi: 10.1016/0092-8674(89)90976-8. [DOI] [PubMed] [Google Scholar]
- 15.Bonfini L., Karlovich C.A., Dasgupta C., Banerjee U. The Son of sevenless gene product: a putative activator of Ras. Science. 1992;255(5044):603–606. doi: 10.1126/science.1736363. [DOI] [PubMed] [Google Scholar]
- 16.Gibbs J.B., Sigal I.S., Poe M., Scolnick E.M. Intrinsic GTPase activity distinguishes normal and oncogenic ras p21 molecules. Proc. Natl. Acad. Sci. USA. 1984;81(18):5704–5708. doi: 10.1073/pnas.81.18.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schubbert S., Shannon K., Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer. 2007;7(4):295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 18.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 2003;3(1):11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 19.Young A., et al. Ras signaling and therapies. Adv. Cancer Res. 2009;102(09):1–17. doi: 10.1016/S0065-230X(09)02001-6. [DOI] [PubMed] [Google Scholar]
- 20.Misale S., et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486(7404):532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pao W., et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2(1):e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lauchle J.O., et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature. 2009;461(7262):411–414. doi: 10.1038/nature08279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takashima A., Faller D.V. Targeting the RAS oncogene. Expert Opin. Ther. Targets. 2013;17(5):507–531. doi: 10.1517/14728222.2013.764990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCormick F. Success and failure on the ras pathway. Cancer Biol. Ther. 2007;6(10):1654–1659. doi: 10.4161/cbt.6.10.5153. [DOI] [PubMed] [Google Scholar]
- 25.Cox A.D., Fesik S.W., Kimmelman A.C., Luo J., Der C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014;13(11):828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohl N.E., et al. Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat. Med. 1995;1(8):792–797. doi: 10.1038/nm0895-792. [DOI] [PubMed] [Google Scholar]
- 27.Reiss Y., Goldstein J.L., Seabra M.C., Casey P.J., Brown M.S. Inhibition of purified p21ras farnesyl: protein transferase by Cys-AAX tetrapeptides. Cell. 1990;62(1):81–88. doi: 10.1016/0092-8674(90)90242-7. [DOI] [PubMed] [Google Scholar]
- 28.James G.L., Goldstein J.L., Brown M.S. Polylysine and CVIM sequences of K-RasB dictate specificity of prenylation and confer resistance to benzodiazepine peptidomimetic in vitro. J. Biol. Chem. 1995;270(11):6221–6226. doi: 10.1074/jbc.270.11.6221. [DOI] [PubMed] [Google Scholar]
- 29.Whyte D.B., et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997;272(22):14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 30.Adjei A.A., et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2003;21(9):1760–1766. doi: 10.1200/JCO.2003.09.075. [DOI] [PubMed] [Google Scholar]
- 31.Cohen S.J., et al. Phase II and pharmacodynamic study of the farnesyltransferase inhibitor R115777 as initial therapy in patients with metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 2003;21(7):1301–1306. doi: 10.1200/JCO.2003.08.040. [DOI] [PubMed] [Google Scholar]
- 32.Stewart S., Guan K.L. The dominant negative Ras mutant, N17Ras, can inhibit signaling independently of blocking ras activation. J. Biol. Chem. 2000;275(12):8854–8862. doi: 10.1074/jbc.275.12.8854. [DOI] [PubMed] [Google Scholar]
- 33.Rahman M.A., Salajegheh A., Smith R.A., Lam A.K. BRAF inhibitors: From the laboratory to clinical trials. Crit. Rev. Oncol. Hematol. 2014;90(3):220–232. doi: 10.1016/j.critrevonc.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Yang H., et al. RG7204 (PLX4032), a selective BRAFV600E inhibitor, displays potent antitumor activity in preclinical melanoma models. Cancer Res. 2010;70(13):5518–5527. doi: 10.1158/0008-5472.CAN-10-0646. [DOI] [PubMed] [Google Scholar]
- 35.Roberts P.J., Der C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 36.Fruman D.A., Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014;13(2):140–156. doi: 10.1038/nrd4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Britten C.D. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother. Pharmacol. 2013;71(6):1395–1409. doi: 10.1007/s00280-013-2121-1. [DOI] [PubMed] [Google Scholar]
- 38.Chapman P.B., et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lito P., Rosen N., Solit D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2012;19(11):1401–1409. doi: 10.1038/nm.3392. [DOI] [PubMed] [Google Scholar]
- 40.Wang Y., et al. A role for K-ras in conferring resistance to the MEK inhibitor, CI-1040. Neoplasia. 2005;7(4):336–347. doi: 10.1593/neo.04532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seguin L., et al. An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014;16(5):457–468. doi: 10.1038/ncb2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hancock J.F., Parton R.G. Ras plasma membrane signalling platforms. Biochem. J. 2005;389(Pt 1):1–11. doi: 10.1042/BJ20050231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parton R.G., Hancock J.F. Lipid rafts and plasma membrane microorganization: Insights from Ras. Trends Cell Biol. 2004;14(3):141–147. doi: 10.1016/j.tcb.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Prior I.A., Muncke C., Parton R.G., Hancock J.F. Direct visualization of ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003;160(2):165–170. doi: 10.1083/jcb.200209091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plowman S.J., Muncke C., Parton R.G., Hancock J.F. H-ras, K-ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc. Natl. Acad. Sci. USA. 2005;102(43):15500–15505. doi: 10.1073/pnas.0504114102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tian T., et al. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 2007;9(8):905–914. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- 47.Tian T., Plowman S.J., Parton R.G., Kloog Y., Hancock J.F. Mathematical modeling of K-Ras nanocluster formation on the plasma membrane. Biophys. J. 2010;99(2):534–543. doi: 10.1016/j.bpj.2010.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Betzig E., et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313(5793):1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 49.Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods. 2006;3(10):793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hess S.T., Girirajan T.P., Mason M.D. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J. 2006;91(11):4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nan X., et al. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. USA. 2015;112(26):7996–8001. doi: 10.1073/pnas.1509123112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muratcioglu S., et al. GTP-Dependent K-Ras Dimerization. Structure. 2015;23(7):1325–1335. doi: 10.1016/j.str.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Güldenhaupt J., et al. N-Ras forms dimers at POPC membranes. Biophys. J. 2015;103(7):1585–1593. doi: 10.1016/j.bpj.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin W-C., et al. H-Ras forms dimers on membrane surfaces via a protein-protein interface. Proc. Natl. Acad. Sci. USA. 2014;111(8):2996–3001. doi: 10.1073/pnas.1321155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pai E.F., et al. Structure of the guanine-nucleotide-binding domain of the Ha-ras oncogene product p21 in the triphosphate conformation. Nature. 1989;341(6239):209–214. doi: 10.1038/341209a0. [DOI] [PubMed] [Google Scholar]
- 56.Hall B.E., Bar-Sagi D., Nassar N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA. 2002;99(19):12138–12142. doi: 10.1073/pnas.192453199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rajakulendran T., Sahmi M., Lefrançois M., Sicheri F., Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461(7263):542–545. doi: 10.1038/nature08314. [DOI] [PubMed] [Google Scholar]
- 58.Harvey J.J. An Unidentified Virus which causes the Rapid Production of Tumours in Mice. Nature. 1964;204(4963):1104–1105. doi: 10.1038/2041104b0. [DOI] [PubMed] [Google Scholar]
- 59.Santos E., Nebreda A.R., Bryan T., Kempner E.S. Oligomeric structure of p21 ras proteins as determined by radiation inactivation. J. Biol. Chem. 1988;263(20):9853–9858. [PubMed] [Google Scholar]
- 60.Harmon J.T., Nielsen T.B., Kempner E.S. [6] Molecular weight determinations from radiation inactivation. Methods Enzymol. 1984;117(C):65–94. doi: 10.1016/s0076-6879(85)17008-4. [DOI] [PubMed] [Google Scholar]
- 61.Inouye K., Mizutani S., Koide H., Kaziro Y. Formation of the Ras Dimer Is Essential for Raf-1 Activation. J. Biol. Chem. 2000;275(6):3737–3740. doi: 10.1074/jbc.275.6.3737. [DOI] [PubMed] [Google Scholar]
- 62.Roy S., et al. Individual palmitoyl residues serve distinct roles in H-ras trafficking, microlocalization, and signaling. Mol. Cell. Biol. 2005;25(15):6722–6733. doi: 10.1128/MCB.25.15.6722-6733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rotblat B., et al. Three separable domains regulate GTP-dependent association of H-ras with the plasma membrane. Mol. Cell. Biol. 2004;24(15):6799–6810. doi: 10.1128/MCB.24.15.6799-6810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abankwa D., Gorfe A.A., Inder K., Hancock J.F. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc. Natl. Acad. Sci. USA. 2010;107(3):1130–1135. doi: 10.1073/pnas.0903907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henis Y.I., Hancock J.F., Prior I.A. Ras acylation, compartmentalization and signaling nanoclusters. Mol. Membr. Biol. 2009;26(1):80–92. doi: 10.1080/09687680802649582. [Review]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li H., Gorfe A.A. Aggregation of lipid-anchored full-length H-Ras in lipid bilayers: simulations with the MARTINI force field. PLoS One. 2013;8(7):e71018. doi: 10.1371/journal.pone.0071018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Z., Gorfe A.A. What drives the clustering of membrane-bound Ras? Small GTPases. 2012;3(4):244–247. doi: 10.4161/sgtp.21829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Janosi L., Gorfe A.A. Segregation of negatively charged phospholipids by the polycationic and farnesylated membrane anchor of Kras. Biophys. J. 2010;99(11):3666–3674. doi: 10.1016/j.bpj.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nan X., et al. Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc. Natl. Acad. Sci. USA. 2013;110(46):18519–18524. doi: 10.1073/pnas.1318188110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nickerson A., Huang T., Lin L-J., Nan X. Photoactivated localization microscopy with bimolecular fluorescence complementation (BiFC-PALM) for nanoscale imaging of protein-protein interactions in cells. PLoS One. 2014;9(6):e100589. doi: 10.1371/journal.pone.0100589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Santos E. Dimerization opens new avenues into ras signaling research. Sci. Signal. 2014;7(324):pe12. doi: 10.1126/scisignal.2005318. [DOI] [PubMed] [Google Scholar]
- 72.Gorfe A.A., Hanzal-Bayer M., Abankwa D., Hancock J.F., McCammon J.A. Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1, 2-dimyristoylglycero-3-phosphocholine bilayer. J. Med. Chem. 2007;50(4):674–684. doi: 10.1021/jm061053f. [DOI] [PubMed] [Google Scholar]
- 73.Magde D., Elson E., Webb W.W. Thermodynamic fluctuations in a reacting system measurement by fluorescence correlation spectroscopy. Phys. Rev. Lett. 1972;29(11):705–708. [Google Scholar]
- 74.Weiß K., et al. Quantifying the diffusion of membrane proteins and peptides in black lipid membranes with 2-focus fluorescence correlation spectroscopy. Biophys. J. 2013;105(2):455–462. doi: 10.1016/j.bpj.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thompson H. US National Cancer Institute’s new Ras project targets an old foe. Nat. Med. 2013;19(8):949–950. doi: 10.1038/nm0813-949. [DOI] [PubMed] [Google Scholar]
- 76.Van de Linde S., et al. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nat. Protoc. 2011;6(7):991–1009. doi: 10.1038/nprot.2011.336. [DOI] [PubMed] [Google Scholar]
- 77.Subach F.V., et al. Photoactivatable mCherry for high-resolution two-color fluorescence microscopy. Nat. Methods. 2009;6(2):153–159. doi: 10.1038/nmeth.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Durisic N., Laparra-Cuervo L., Sandoval-Álvarez A., Borbely J.S., Lakadamyali M. Single-molecule evaluation of fluorescent protein photoactivation efficiency using an in vivo nanotemplate. Nat. Methods. 2014;11(2):156–162. doi: 10.1038/nmeth.2784. [DOI] [PubMed] [Google Scholar]
- 79.Puchner E.M., Walter J.M., Kasper R., Huang B., Lim W.A. Counting molecules in single organelles with superresolution microscopy allows tracking of the endosome maturation trajectory. Proc. Natl. Acad. Sci. USA. 2013;110(40):16015–16020. doi: 10.1073/pnas.1309676110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee S-H., Shin J.Y., Lee A., Bustamante C. Counting single photoactivatable fluorescent molecules by photoactivated localization microscopy (PALM). Proc. Natl. Acad. Sci. USA. 2012;109(43):17436–17441. doi: 10.1073/pnas.1215175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sengupta P., et al. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat. Methods. 2011;8(11):969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yarden Y., Sliwkowski M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2011;2(2):127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 83.Farrar M.A., Alberol-Ila J., Perlmutter R.M., Alberola-Lla J. Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature. 1996;383(6596):178–181. doi: 10.1038/383178a0. [DOI] [PubMed] [Google Scholar]
- 84.Luo Z., et al. Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature. 1996;383(6596):181–185. doi: 10.1038/383181a0. [DOI] [PubMed] [Google Scholar]
- 85.Weber C.K., Slupsky J.R., Andreas K.H., Rapp U.R. Active ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001;61(9):3595–3598. [PubMed] [Google Scholar]
- 86.Poulikakos P.I., Zhang C., Bollag G., Shokat K.M., Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hatzivassiliou G., et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464(7287):431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 88.Ohren J.F., et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol. 2004;11(12):1192–1197. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- 89.Catalanotti F., et al. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat. Struct. Mol. Biol. 2009;16(3):294–303. doi: 10.1038/nsmb.1564. [DOI] [PubMed] [Google Scholar]
- 90.Casar B., Pinto A., Crespo P. Essential role of ERK dimers in the activation of cytoplasmic but not nuclear substrates by ERK-scaffold complexes. Mol. Cell. 2008;31(5):708–721. doi: 10.1016/j.molcel.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 91.Casar B., Pinto A., Crespo P. ERK dimers and scaffold proteins: Unexpected partners for a forgotten (cytoplasmic) task. Cell Cycle. 2009;8(7):1007–1013. doi: 10.4161/cc.8.7.8078. [DOI] [PubMed] [Google Scholar]
- 92.Huang L., Hofer F., Martin G.S., Kim S.H. Structural basis for the interaction of Ras with RalGDS. Nat. Struct. Biol. 1998;5(6):422–426. doi: 10.1038/nsb0698-422. [DOI] [PubMed] [Google Scholar]
- 93.Vetter I.R., et al. Structural and biochemical analysis of Ras-effector signaling via RalGDS. FEBS Lett. 1999;451(2):175–180. doi: 10.1016/s0014-5793(99)00555-4. [DOI] [PubMed] [Google Scholar]
- 94.Heidorn S.J., et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140(2):209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Woolf P.J., Linderman J.J. Self organization of membrane proteins via dimerization. Biophys. Chem. 2003;104(1):217–227. doi: 10.1016/s0301-4622(02)00369-1. [DOI] [PubMed] [Google Scholar]
- 96.Klemm J.D., Schreiber S.L., Crabtree G.R. Dimerization as a regulatory mechanism in signal transduction. Annu. Rev. Immunol. 1998;16:569–592. doi: 10.1146/annurev.immunol.16.1.569. [DOI] [PubMed] [Google Scholar]
- 97.Gasper R., Meyer S., Gotthardt K., Sirajuddin M., Wittinghofer A. It takes two to tango: regulation of G proteins by dimerization. Nat. Rev. Mol. Cell Biol. 2009;10(6):423–429. doi: 10.1038/nrm2689. [DOI] [PubMed] [Google Scholar]
- 98.Bray D., Levin M.D., Morton-Firth C.J. Receptor clustering as a cellular mechanism to control sensitivity. Nature. 1998;393(6680):85–88. doi: 10.1038/30018. [DOI] [PubMed] [Google Scholar]
- 99.Harding A.S., Hancock J.F. Using plasma membrane nanoclusters to build better signaling circuits. Trends Cell Biol. 2008;18(8):364–371. doi: 10.1016/j.tcb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jang H., et al. Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. J. Biol. Chem. 2015;290(15):9465–9477. doi: 10.1074/jbc.M114.620724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang Z., et al. Wildtype Kras2 can inhibit lung carcinogenesis in mice. Nat. Genet. 2001;29(1):25–33. doi: 10.1038/ng721. [DOI] [PubMed] [Google Scholar]
- 102.Singh A., Sowjanya A.P., Ramakrishna G. The wild-type Ras: road ahead. FASEB J. 2005;19(2):161–169. doi: 10.1096/fj.04-2584hyp. [DOI] [PubMed] [Google Scholar]
- 103.Tuveson D.A., et al. Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5(4):375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 104.Bremner F., Balmain A. Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell. 1990;61:407–417. doi: 10.1016/0092-8674(90)90523-h. [DOI] [PubMed] [Google Scholar]
- 105.Hegi M.E., et al. Allelotype analysis of mouse lung carcinomas reveals frequent allelic losses on chromosome 4 and an association between allelic imbalances on chromosome 6 and K-ras activation. Cancer Res. 1994;54(23):6257–6264. [PubMed] [Google Scholar]
- 106.Di Magliano M.P., Logsdon C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144(6):1220–1229. doi: 10.1053/j.gastro.2013.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bentley C., et al. A requirement for wild-type Ras isoforms in mutant KRas-driven signalling and transformation. Biochem. J. 2013;452(2):313–320. doi: 10.1042/BJ20121578. [DOI] [PubMed] [Google Scholar]
- 108.Ostrem J.M., Peters U., Sos M.L., Wells J.A., Shokat K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zimmermann G., et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature. 2013;497(7451):638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 110.Mirzoeva O.K., et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009;69(2):565–572. doi: 10.1158/0008-5472.CAN-08-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Turke A.B., et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012;72(13):3228–3237. doi: 10.1158/0008-5472.CAN-11-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pacold M.E., et al. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell. 2000;103(6):931–943. doi: 10.1016/s0092-8674(00)00196-3. [DOI] [PubMed] [Google Scholar]
- 113.Rocks O., et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307(5716):1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 114.Scheele J.J., Rhee J.M., Boss G.G. Determination of absolute amounts of GDP and GTP bound to Ras in mammalian cells: comparison of parental and Ras-overproducing NIH 3T3 fibroblasts. Proc. Natl. Acad. Sci. USA. 1995;92(4):1097–1100. doi: 10.1073/pnas.92.4.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Philips M.R., Der C.J. Seeing is believing: Ras dimers observed in live cells. Proc. Natl. Acad. Sci. USA. 2015;112(32):9793–9794. doi: 10.1073/pnas.1511805112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Raabe T., Rapp U.R. KSR--a regulator and scaffold protein of the MAPK pathway. Sci. STKE. 2002;2002(136):28. doi: 10.1126/stke.2002.136.pe28. [DOI] [PubMed] [Google Scholar]
- 117.Paz A., Haklai R., Elad-Sfadia G., Ballan E., Kloog Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene. 2001;20(51):7486–7493. doi: 10.1038/sj.onc.1204950. [DOI] [PubMed] [Google Scholar]
- 118.Elad-Sfadia G., Haklai R., Balan E., Kloog Y. Galectin-3 augments K-Ras activation and triggers a Ras signal that attenuates ERK but not phosphoinositide 3-kinase activity. J. Biol. Chem. 2004;279(33):34922–34930. doi: 10.1074/jbc.M312697200. [DOI] [PubMed] [Google Scholar]
- 119.Shalom-Feuerstein R., et al. K-Ras nanoclustering is subverted by overexpression of the scaffold protein galectin-3. Cancer Res. 2008;68(16):6608–6616. doi: 10.1158/0008-5472.CAN-08-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Belanis L., Plowman S.J., Rotblat B., Hancock J.F., Kloog Y. Galectin-1 is a novel structural component and a major regulator of H-Ras nanoclusters. Mol. Biol. Cell. 2008;19(4):1404–1414. doi: 10.1091/mbc.E07-10-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rabinovich G.A., Toscano M.A., Jackson S.S., Vasta G.R. Functions of cell surface galectin-glycoprotein lattices. Curr. Opin. Struct. Biol. 2007;17(5):513–520. doi: 10.1016/j.sbi.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lepur A., Salomonsson E., Nilsson U.J., Leffler H. Ligand induced galectin-3 protein self-association. J. Biol. Chem. 2012;287(26):21751–21756. doi: 10.1074/jbc.C112.358002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Elad-Sfadia G., Haklai R., Ballan E., Gabius H.J., Kloog Y. Galectin-1 augments Ras activation and diverts Ras signals to Raf-1 at the expense of phosphoinositide 3-kinase. J. Biol. Chem. 2002;277(40):37169–37175. doi: 10.1074/jbc.M205698200. [DOI] [PubMed] [Google Scholar]
- 124.Solman M., et al. Specific cancer associated mutations in the switch III-region of Ras increase tumorigenicity by nanocluster augmentation. eLife. 2015;4:e08905. doi: 10.7554/eLife.08905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shalom-Feuerstein R., Cooks T., Raz A., Kloog Y. Galectin-3 regulates a molecular switch from N-Ras to K-Ras usage in human breast carcinoma cells. Cancer Res. 2005;65(16):7292–7300. doi: 10.1158/0008-5472.CAN-05-0775. [DOI] [PubMed] [Google Scholar]