

Abstract
Stress and withdrawal of female reproductive hormones are known risk factors of postpartum depression. Although both of these factors are capable of powerfully modulating neuronal plasticity, there is no direct electron microscopic evidence of hippocampal spine synapse remodeling in postpartum depression. To address this issue, hormonal conditions of pregnancy and postpartum period were simulated in ovariectomized adult female Sprague-Dawley rats (n=76). The number of hippocampal spine synapses and the depressive behavior of rats in an active escape task were investigated in untreated control, hormone-withdrawn ‘postpa rtum’, simulated proestrus, and hormone-treated ‘postpartum’ animals. After ‘postpartum’ withdrawal of gonadal steroids, inescapable stress caused a loss of hippocampal spine synapses, which was related to poor escape performance in hormone-withdrawn ‘postpartum’ females. These responses were equivalent with the changes observed in untreated controls that is an established animal model of major depression. Maintaining proestrus levels of ovarian hormones during ‘postpartum’ stress exposure did not affect synaptic and behavioral responses to inescapable stress in simulated proestrus animals. By contrast, maintaining pregnancy levels of estradiol and progesterone during ‘postpartum’ stress exposure completely prevented the stress-induced loss of hippocampal spine synapses, which was associated with improved escape performance in hormone-treated ‘postpartum’ females. This protective effect appears to be mediated by a muted stress response as measured by serum corticosterone concentrations. In line with our emerging ‘synaptogenic hypothesis’ of depression, the loss of hippocampal spine synapses may be a novel perspective both in the pathomechanism and in the clinical management of postpartum affective illness.
Keywords: postpartum depression, estradiol, progesterone, stress, plasticity, electron microscopy
In earlier studies, our research team has found that exposure to stress causes a severe loss of spine synapses in limbic brain areas (Hajszan et al., 2009; Hajszan et al., 2010). We have also shown that loss of spine synapses in the hippocampus is related to depressive behavior in rodent models (Hajszan et al., 2009; Hajszan et al., 2010), and loss of synapses in the prefrontal cortex is associated with major depression in humans (Kang et al., 2012). Moreover, manipulating the number of hippocampal spine synapses by means of ovariectomy and estradiol replacement leads to changes in depressive behavior in the rat learned helplessness model of major depression (Hajszan et al., 2010). Additionally, we have revealed that treatment with antidepressants is a powerful inducer of hippocampal synaptogenesis (Hajszan et al., 2005; Hajszan et al., 2009), while pharmacological blockade of synapse formation hinders antidepressant action (Li et al., 2010). Built on these findings, our emerging ‘synaptogenic hypothesis’ of depression postulates a causal relationship between loss of limbic spine synapses and depressive symptoms (Hajszan et al., 2010). Further studies are needed, of course, to investigate many more aspects of this hypothesis, such as the interesting issue of synaptic remodeling in postpartum depression (PPD).
Depression and anxiety are common complications of pregnancy and the postpartum period. During the first three months after giving birth, approximately 15-20% of mothers are affected by PPD (American Psychiatric Association, 2000; Gavin et al., 2005; O'Hara and Wisner, 2014), although prevalence rates may be as high as 33-38% in certain highly vulnerable groups (Gress-Smith et al., 2012). The symptoms of PPD are almost identical with those of a major depressive episode and described by, among others, irritability, emotional swings, cognitive impairments, and feelings of guilt and inadequacy (Melges, 1968; Pitt, 1968; Laura and Miller, 2002; Crawley et al., 2003; O'Hara and Wisner, 2014). In extreme cases, PPD may lead to infanticide and/or suicide (Pariser et al., 1997; Spinelli, 2004), which is the most tragic manifestation of the fact that PPD is a ‘family affair’, as it also affects the offspring by being associated with various developmental problems in the cognitive and psychosocial domains (Nomura et al., 2002; Grace et al., 2003; Letourneau et al., 2012; Verbeek et al., 2012). As a result, more research is necessary into the pathomechanisms of peripartum affective illness to develop better, evidence-based therapy and to protect the offspring from enduring adverse consequences.
Similar to major depression (McEwen, 2003), epidemiological studies show that exposure to stress is a significant risk factor of PPD (Lancaster et al., 2010; Davey et al., 2011; Stewart, 2011; Hillerer et al., 2012). Like in the case of humans, female rats exposed to peripartum stress develop depressive and anxiety-like behaviors, as well as reduced maternal care in the postpartum period (Darnaudery et al., 2004; Smith J.W. et al., 2004; Brummelte and Galea, 2010). It has been suggested, in line with the synaptogenic hypothesis of depression (Hajszan et al., 2010), that peripartum stress elicits extensive neuroplasticity in the maternal brain, which potentially contributes to the development of postpartum affective illness (Pawluski et al., 2016). Recent studies are beginning to demonstrate that the maternal nervous system is indeed remodelled by stress both in humans (Moses-Kolko et al., 2014) and in animal models (Gemmel et al., 2016; Haim et al., 2016; Pawluski et al., 2016). Nevertheless, more research is needed to better understand the extent to which neuroplasticity is implicated in stress-related maternal disorders.
A special feature of PPD is its association with wide fluctuations of female reproductive hormones. During human pregnancy, 17β-estradiol and progesterone gradually reach plasma concentrations approximately 100- and 10-fold higher, respectively, than menstrual cycle levels (Hendrick et al., 1998). After delivery, on the other hand, serum levels of gonadal steroids decrease rapidly and remain practically hypogonadal for a prolonged postpartum period (McNeilly, 2001). Considering the fact that ovarian hormones have a strong ability to influence stress and synaptic plasticity (Woolley and McEwen, 1992; Brunton et al., 2009), there is a possibility that the synaptolytic effect of stress and/or the association between synapse loss and depressive behavior is modified by a significantly altered milieu of gonadal steroids. As a result, pregnancy and the postpartum period represent a challenge to the validity of our synaptogenic hypothesis of depression.
While undergoing substantial fluctuations in serum levels of ovarian hormones, such as those during pregnancy, women frequently report emerging or worsening symptoms of depression (Bloch et al., 2003; Rubinow and Schmidt, 2006; Schmidt and Rubinow, 2009), suggesting a critical role for gonadal steroids in the pathomechanisms of mood disorders (Galea et al., 2001; Studd and Panay, 2004; Hajszan et al., 2010). Based on these findings, the ‘hormone withdrawal hypothesis’ of PPD has been developed, postulating that the precipitous drop of ovarian hormones after delivery may trigger depressive symptoms (Parry et al., 2003; Steiner et al., 2003; Douma et al., 2005). The hormone withdrawal hypothesis is supported by studies both in humans (Bloch et al., 2000; Bloch et al., 2003) and in animal models (Galea et al., 2001; Stoffel and Craft, 2004; Suda et al., 2008). Interestingly, several clinical studies have also provided contrary evidence (Heidrich et al., 1994; Klier et al., 2007), suggesting that hormonal changes either precipitate depressive symptoms only in vulnerable women or create vulnerability for other precipitating factors such as stress.
Utilizing the hormone withdrawal hypothesis, Galea and colleagues have designed a rat model of PPD by creating a hormone simulated pregnancy and then withdrawing hormones to mimic the early postpartum period (Galea et al., 2001). ‘Postpartum’ rats in this model show increased immobility in the forced swim test and suppressed adult hippocampal neurogenesis, which are prevented by continuing exposure to estradiol (Galea et al., 2001; Stoffel and Craft, 2004; Green and Galea, 2008), but anxiety-like behavior does not appear (Stoffel and Craft, 2004). This model provides a basis for separating the hormonal components that are implicated in postpartum depressive behavior and neuroplasticity in the dam.
Recently, Suda and colleagues have added to the hormone withdrawal model by shifting emphasis to progesterone (Suda et al., 2008). Considering the abilities of progesterone to influence the stress response and the excitability of neurons (Bitran and Dowd, 1996; Brunton et al., 2009), reproducing progesterone levels properly may be critical for a PPD model. In the animal model of Galea and colleagues, estradiol benzoate is given throughout the simulated pregnancy period, while progesterone is administered until ‘pregnancy’ day 16. As a result, only estradiol is withdrawn at ‘delivery’, producing an early ‘postpartum’ period that misses the effects of acute progesterone withdrawal (Galea et al., 2001). This hormone regimen mimics changes in the rat, as progesterone peaks then gradually falls to estrus cycle levels during the second part of rat pregnancy (Grota and Eik-Nes, 1967; Pepe and Rothchild, 1974). On the other hand, serum concentrations of both 17β-estradiol and progesterone continuously rise during human pregnancy (Hendrick et al., 1998). To better mimic these human conditions, the animal model of Suda and colleagues withdraws 17β-estradiol and progesterone simultaneously at ‘delivery’. During the simulated postpartum period, this model shows symptoms relevant to PPD, including vulnerability for helplessness, increased anxiety, aggression, and the transient regulation of several PPD susceptibility genes (Suda et al., 2008).
The hippocampus participates in the stress response (McEwen, 2003), and its compromised function is a central component in the pathomechanism of major depression (Nestler et al., 2002). Pyramidal and granule cells, the principal hippocampal neurons, are organized sequentially into a ‘trisynaptic loop’, which is the main neuronal circuitry of the hippocampus. Asymmetric spine synapses within the CA1 stratum radiatum, the CA3 stratum lucidum and radiatum, as well as in the dentate gyrus stratum moleculare represent primary connections of the trisynaptic loop (Amaral and Witter, 1995). Compromised hippocampal function in major depression is correlated with reduced hippocampal volume, suppressed adult hippocampal neurogenesis, and loss of spine synapses along the trisynaptic loop, and these structural impairments are all prevented/reversed by effective antidepressant treatment (Sheline, 2003; Duman and Monteggia, 2006; Hajszan et al., 2009). The central role of the hippocampus in major depression is further supported by the observation that local infusion of brain-derived neurotrophic factor or neurotrophin-3 into the dentate gyrus reproduces the effects of conventional antidepressants (Shirayama et al., 2002). Although recent studies have demonstrated hippocampal structural modifications that are related to maternal stress and mood disorders (Green and Galea, 2008; Pawluski et al., 2016), direct electron microscopic evidence of synaptic remodeling along the trisynaptic circuit in PPD is presently not available.
Considering the issues discussed above, we performed this study to test certain components of our synaptogenic hypothesis of depression (Hajszan et al., 2010) in a simulated postpartum environment. In order to model the stress and the hormone withdrawal aspects of PPD, we used the hormone withdrawal model of Suda and colleagues (Suda et al., 2008) combined with inescapable footshock stress applied during the ‘postpartum’ period. Our primary goal was to provide evidence that exposure to ‘postpartum’ stress leads to loss of asymmetric spine synapses along the hippocampal trisynaptic circuit. We also hypothesized that the association between loss of hippocampal synapses and depressive behavior is maintained in the simulated postpartum environment.
Experimental Procedures
Adult female Sprague-Dawley rats were kept under standard laboratory conditions (n=76, 200-250 g, Charles-River Laboratories, Wilmington, Massachusetts). Similar to our earlier studies (Suda et al., 2008; Hajszan et al., 2010), females were group-housed (n=3 animals per cage) in standard wire-bottom caging with tap water and rodent chow available ad libitum. Animal rooms were maintained on a temperature of 21 °C and on a 12-h/12-h light/dark cycle with light on at 0700 h. Rats were treated and cared for according to National Institutes of Health standards. The animal protocol was approved by the Institutional Animal Care and Use Committee of Yale University School of Medicine.
Hormone simulated pregnancy and postpartum period
In order to produce a postpartum environment for our studies, we used the hormone simulated pregnancy model of Suda and colleagues (Suda et al., 2008). To prevent interference from endogenous ovarian hormones, all animals were ovariectomized on day-1 using a ketamine-based anesthetic (25 mg/mL ketamine, 1.2 mg/mL xylazine, 0.03 mg/mL acepromazine in saline, 3 mL/kg, i.m.). During the same surgical session, 21-day continuous release pellets (Innovative Research of America, Sarasota, Florida) containing 0.5 mg 17β-estradiol and 50 mg progesterone were implanted subcutaneously in the scapular region to simulate pregnancy. These pellets produce a dose of 23.8 μg/day for 17β-estradiol and 2.4 mg/day for progesterone, which approximate the average daily doses applied in the hormone simulated pregnancy model of Galea and colleagues (Galea et al., 2001). In contrast to the model of Galea and colleagues, which administers progesterone only until ‘pregnancy’ day 16 (Galea et al., 2001), we withdrew 17β-estradiol and progesterone simultaneously on day-21 by removing the hormone pellets. Day-22 to day-28 was then considered as the early ‘postpartum’ period. As a result, our model provided pregnancy levels of hormones that were suitable for the rat system (Birzniece et al., 2002) and a simulated postpartum period that was more relevant to human conditions by reproducing the simultaneous withdrawal of 17β-estradiol and progesterone.
Schedule of experiments
Untreated controls (Veh)
The purpose of this group was to provide a positive control for stress and a reference model of major depression with well-documented synaptic and escape performance responses to inescapable stress (Hajszan et al., 2010). Briefly, n=20 animals underwent the treatment schedule of hormone simulated pregnancy as described above, but only placebo pellets were implanted. On day-21, the implanted placebo pellets were replaced with fresh placebo pellets. From this group, n=6 randomly selected rats were scheduled for electron microscopic analysis, and the remaining n=14 rats were scheduled for behavioral testing. From the subgroup scheduled for electron microscopic analysis, n=3 rats were exposed to inescapable stress on day-27, while n=3 rats were sham-stressed. Both the stressed and the sham-stressed animals were sacrificed for electron microscopic analysis on day-28. From the subgroup scheduled for behavioral testing, all rats were exposed to inescapable stress on day-27, followed by active escape testing on day-28. Immediately after active escape testing, n=8 rats were randomly selected and sacrificed for hormone measurements.
Hormone-withdrawn ‘postpartum’ females (PpD)
In order to investigate the effects of stress in a simulated postpartum environment, n=20 animals underwent the treatment schedule of hormone simulated pregnancy using 17β-estradiol and progesterone pellets as described above. On day-21, hormones were withdrawn by replacing the implanted hormone pellets with fresh placebo pellets. The scheduling of this group for various experiments matched that of untreated controls (see above).
Simulated proestrus animals (ProE)
In order to produce a control group with proestrus levels of gonadal steroids, n=16 animals underwent the treatment schedule of hormone simulated pregnancy as described above, but only placebo pellets were implanted. On day-21, the implanted placebo pellets were replaced with fresh placebo pellets. On day-26, all animals were injected with 3 μg/kg estradiol benzoate (dissolved in sesame oil, s.c.) at 8:30 a.m., followed by 4 μg/kg estradiol benzoate at 8:30 p.m. On day-27, all females received a final dose of 3 μg/kg 17β-estradiol (dissolved in sesame oil, s.c.) 2 h before stress (or sham stress) exposure. This estrogen regimen has been shown to reproduce natural levels of 17β-estradiol as seen in proestrus rats (Smith M.S. et al., 1975; Scharfman et al., 2007). On day-27, all animals also received 2 mg/kg progesterone (dissolved in sesame oil, s.c.) 5 h before stress (or sham stress) exposure. This dose and timing mimics proestrus levels of progesterone and stimulates the growth of dendritic spines in the hippocampus (Gould et al., 1990). The scheduling of this group for various experiments matched that of untreated controls (see above) with the modification that only n=10 rats were scheduled for behavioral testing.
Hormone-treated ‘postpartum’ rats (Horm)
In order to investigate how pregnancy levels of gonadal steroids influence the effects of ‘postpartum’ stress, n=20 animals underwent the treatment schedule of hormone simulated pregnancy using 17β-estradiol and progesterone pellets as described above. On day-21, the implanted hormone pellets were replaced with fresh hormone pellets to ensure continuous exposure to gonadal steroids. The scheduling of this group for various experiments matched that of untreated controls (see above).
For a schematic of this schedule, see Table 1.
Table 1. Schedule of experiments.
| Group | Day-1 | Day-21 | Day-26 | Day-27 | Day-28 |
|---|---|---|---|---|---|
| Untreated controls (Veh, n=20) | Ovariectomy, Placebo pellet implantation | Fresh placebo pellet implantation | -- | Inescapable stress (n=3) Sham stress (n=3) | Synapse sampling |
| Inescapable Stress (n=14) | Active escape t e sting + hormone sampling | ||||
| Hormone-withdrawn ‘postpartum’ females (PpD, n=20) | Ovariectomy, Estradiol + Progesterone pellet implantation | Switch to placebo pellet | -- | Inescapable stress (n=3) Sham stress (n=3) | Synapse sampling |
| Inescapable Stress (n=14) | Active escape testing + hormone sampling | ||||
| Simulated proestrus animals (ProE, n=16) | Ovariectomy, Placebo pellet implantation | Fresh placebo pellet implantation | 3 mg/kgestradiolbenzoate (8:30a.m.) 4 mg/kgestradiolbenzoate (8:30p.m.) | Hormone injection* + inescapable stress (n=3) Hormone injection* + sham stress (n=3) | Synapse sampling |
| Hormone injection* + inescapable stress (n=10) | Active escape testing + hormone sampling | ||||
| Hormone-treated ‘postpartum’ rats (Horm, n=20) | Ovariectomy, Estradiol + Progesterone pellet implantation | Fresh Estradiol + Progesterone pellet implantation | -- | Inescapable stress (n=3) Sham stress (n=3) | Synapse sampling |
| Inescapable Stress (n=14) | Active escape testing + hormone sampling |
Hormone injection = 2 mg/kg progesterone (5 h prior to stress) and 3 μg/kg 17β-estradiol (2 h prior to stress)
Inescapable stress
We applied an inescapable stress paradigm that is used routinely in our laboratories to induce depressive behavior in rodent models (Valentine et al., 2008; Hajszan et al., 2009; Hajszan et al., 2010). Briefly, the testing apparatus consists of a commercially available shuttle avoidance box (Med Associates, St. Albans, Vermont) that is divided into two equal compartments with a central barrier. The barrier is equipped with a computer-operated guillotine door to provide passage between compartments. Animals subjected to inescapable stress (Table 1) received 60 scrambled footshocks with 0.85 mA intensity, being administered via wire grid flooring in a closed shuttle box compartment. The footshock protocol was entirely automated and conducted by a computer-run algorithm to provide fully randomized stress exposure with 15 s average shock duration and 45 s average intershock interval. Sham-stressed controls underwent the same footshock protocol, but the shock generator was switched off during the entire procedure.
Electron microscopic stereology
In order to assess hippocampal synaptic remodeling in response to ‘postpartum’ stress, the number of asymmetric spine synapses was calculated in three hippocampal sampling areas, CA1 stratum radiatum (CA1sr), CA3 stratum lucidum/radiatum (CA3sl/sr), and dentate gyrus stratum moleculare (DGsm), as we described earlier (Hajszan et al., 2009; Hajszan et al., 2010). Briefly, all animals scheduled for electron microscopic analysis (Table 1) were sacrificed under deep ketamine-based anesthesia (see above) by transcardial perfusion of phosphate-buffered saline followed by a fixative containing 4% paraformaldehyde and 0.1% glutaraldehyde dissolved in 0.1 M phosphate buffer (pH 7.4). Brains were dissected out and postfixed overnight in the same fixative without glutaraldehyde (Hajszan et al., 2005).
Each hippocampus was cut into 100-μm thick serial sections in the coronal plane using a vibratome. The serial sections were systematically sorted into ten groups, and one of these groups was randomly selected for slide-mounted flat embedding in Durcupan (Electron Microscopy Sciences, Fort Washington, Pennsylvania) (Hajszan et al., 2009; Hajszan et al., 2010). This approach provided approximately ten Durcupan-embedded sections per hippocampus with a section sampling ratio of 10. The volume of each sampling area was then estimated utilizing the Cavalieri method. Under a light microscope equipped with the Stereo Investigator computerized stereology system (MicroBrightField, Villiston, Vermont), sampling areas were outlined on each embedded section for the Cavalieri Estimator software module that, based on the outlined area, the section thickness, and the section sampling ratio, calculated volume automatically (Hajszan et al., 2009).
For electron microscopic analysis, 20 counting sites were localized in each sampling area using a systematic random approach as modified from MacLusky and colleagues (MacLusky et al., 2006) and described in our earlier publication (Hajszan et al., 2009). At each counting site, serial ultrathin sections were prepared, and pairs of digitized electron micrographs were taken from the same area of neighboring ultrasections at a final magnification of 11,000× (physical disector). This sampling technique provided 20 physical disectors for each of CA1sr, CA3sl/sr, and DGsm, i.e., 60 disectors per hippocampus altogether. Prior to spine synapse counting, all micrographs were coded for blind analysis. Asymmetric spine synapses (Figure 1) were then counted according to the rules of disector technique, utilizing an unbiased counting frame superimposed onto the electron micrographs (Sterio, 1984). All counts taken from the same sampling area were summed and divided by the analyzed volume to calculate an average spine synapse density (synapse/μm3 = sum of synapse counts / number of disectors per sampling area / single disector volume of 5.94 μm3). Spine synapse density was finally multiplied by sampling area volume to arrive at the total number of spine synapses in that particular sampling area.
Figure 1.
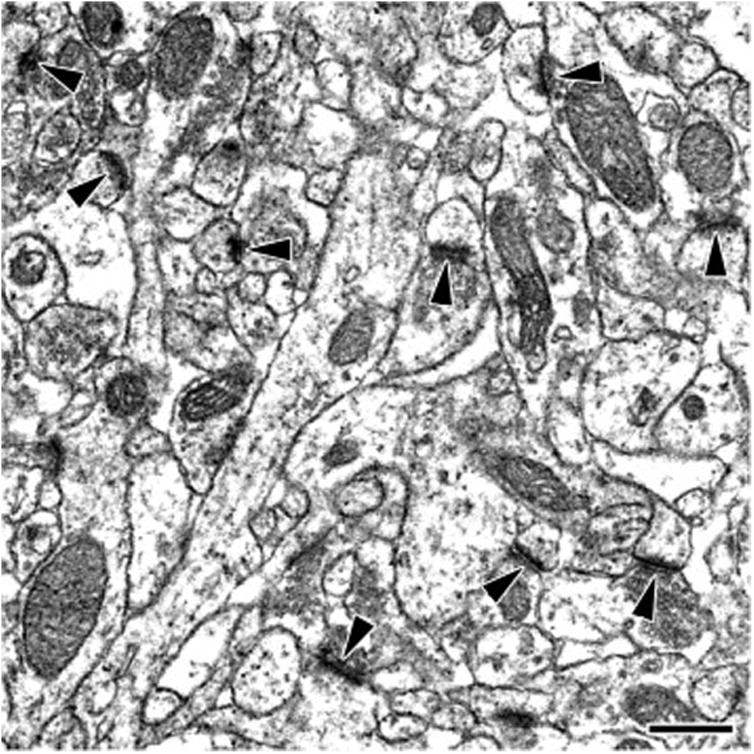
Representative electron micrograph, taken from the hippocampal CA1 stratum radiatum of a sham-stressed hormone-withdrawn ‘postpartum’ female, demonstrating hippocampal asymmetric spine synapses (arrowheads). Scale bar, 500 nm.
Because electron microscopic stereology is a time-consuming and labor-intensive method, we performed careful statistical power calculations to minimize the number of animals to be sacrificed. In our earlier studies, standard deviations for spine synapse counts have consistently been in the range of 5-10% of the mean (Hajszan et al., 2005; Hajszan et al., 2009; Hajszan et al., 2010). We used the middle of this range in the power calculation for the present experiment.
Because alterations in spine synapse numbers have usually exceeded 25% in our earlier studies (Hajszan et al., 2005; Hajszan et al., 2009; Hajszan et al., 2010), we expected ANOVA to detect at least 25% change with the desired 80% power at α=0.05. Considering the above parameters, these requirements were met with a treatment group size of n=3 rats/group.
Active escape testing
In order to verify the relationship between the number of hippocampal spine synapses and depressive symptoms, we applied active escape testing that is also routinely used in our laboratories to assess depressive behavior in rodent models (Valentine et al., 2008; Hajszan et al., 2009; Hajszan et al., 2010). Escape performance measurement is a readout of despair and cognitive deficit, common and frequent symptoms of mood disorders (Thiebot et al., 1992; Vollmayr and Henn, 2001; Cryan et al., 2002). Briefly, 30 trials of escapable footshock were administered with 0.65 mA intensity in the same shuttle avoidance box as described above. In contrast to inescapable stress, the guillotine door in the central barrier was automatically opened at the beginning of each footshock to provide the animal with opportunity to escape by passing between shuttle box compartments.
The testing protocol was entirely automated and conducted by a computer-run algorithm to administer trials in a randomized manner with 35 s maximum trial/footshock duration and 60 s average intertrial interval. The initial five fixed ratio one trials, when one shuttle crossing terminated the footshock, were followed by 25 fixed ratio two trials, when two shuttle crossings were required to terminate the footshock. As main measures of depressive behavior, escape latencies and escape failures were registered, representing the time to escape footshock and the number of trials during which escape requirements were not met, respectively. In case of each escape failure, escape latency was set to 35 s. All behavioral testing was conducted in a dimly-lit room between 1000-1600 h.
Serum hormone measurements
In order to verify that ovarian hormone administrations were successful and to assess the stress response, animals were rapidly decapitated under deep ketamine-based anesthesia, and blood samples were collected immediately after completing active escape testing (Table 1). Trunk blood specimens were kept on ice, allowed to clot, and then centrifuged to separate serum that was stored at -20 °C until assayed. By following manufacturer-recommended protocols, serum total concentrations of 17β-estradiol, progesterone, and corticosterone were determined using commercially available enzyme immunoassay (EIA) kits (Assay Designs, Ann Arbor, Michigan). All samples were analyzed in duplicates during a single EIA session. In the concentration ranges pertinent to the present study, the intraassay coefficient of variation is 8.1% for the 17β-estradiol kit, 5.4% for the progesterone kit, and 8.4% for the corticosterone kit.
Statistical analysis
Synapse counts were statistically evaluated using mixed three-way ANOVA (stress × hormone treatment × area) with stress and hormone treatment as between-subjects factors and area as within-subjects factor. The three-way ANOVA was followed by the conservative Tukey-Kramer posthoc test to compare individual group means. Escape latencies and serum concentrations of corticosterone were analyzed applying one-way ANOVA, and individual group means were compared with the Tukey-Kramer posthoc test. Because escape failures were not normally distributed, they were tested employing the nonparametric Kruskal-Wallis one-way ANOVA on ranks, followed by the Mann-Whitney U test. The significance level was conventionally set at P<0.05.
Results
Effects of ‘postpartum’ stress and ‘postpartum’ hormone treatment on hippocampal synaptic remodeling and escape performance
Three-way mixed ANOVA (stress × hormone treatment × area) found a significant main stress effect (F1,16=635.357 P<0.001), a significant main hormone treatment effect (F3,16=194.998 P<0.001), and a significant main area effect (F2,32=305.073 P<0.001) on the number of hippocampal spine synapses. A significant hormone treatment × stress interaction effect was also revealed (F3,16=109.665 P<0.001), indicating that hormone treatment interferes with the ability of stress to modulate synapse numbers. In addition, significant area × stress (F2,32=13.346 P<0.001) and area × hormone treatment (F(6,32)=15.978 P<0.001) interaction effects were registered, indicating that synaptic responses to stress and hormone treatment vary among hippocampal areas. Detailed comparisons with post-hoc tests are provided below.
Considering measures from the active escape task, one-way ANOVA found a significant hormone treatment effect on escape latencies (F3,48=6.66 P<0.001), while Kruskal-Wallis oneway ANOVA on ranks also revealed a significant hormone treatment effect on escape failures (H=18.665 df=3 P<0.001).
Untreated controls (Veh)
Exposure of Veh rats to inescapable stress caused a decline in the number of spine synapses across all hippocampal areas (Figure 2, Veh/IS vs. Veh/NS, *P<0.04, Tukey-Kramer test). In the active escape task, the Veh group achieved a mean escape latency of 22.89±1.87 s and made, on average, 13.04±2.37 escape failures (Figure 3).
Figure 2.
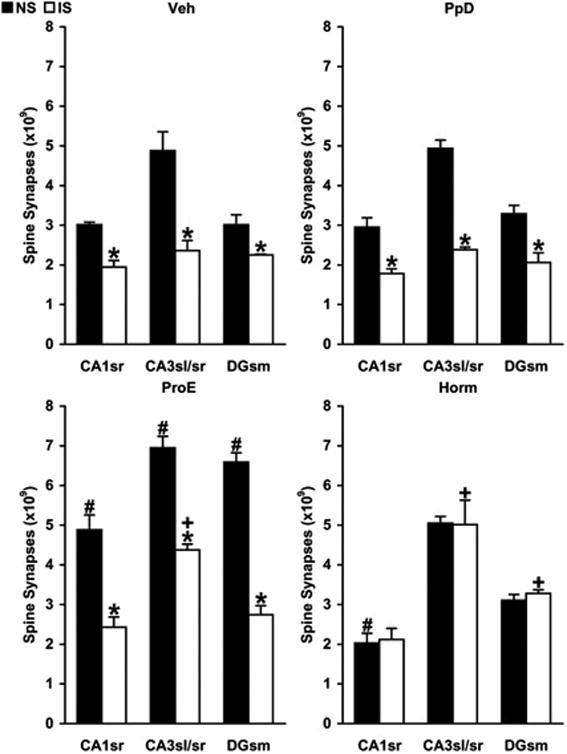
Changes in the number of hippocampal spine synapses in response to inescapable stress during the simulated postpartum period in untreated controls (panel Veh), hormone-withdrawn ‘postpartum’ females (panel PpD), simulated proestrus animals (panel ProE), and hormone-treated ‘postpartum’ rats (panel Horm). Three distinct hippocampal areas were analyzed, CA1 stratum radiatum (CA1sr), CA3 stratum lucidum/radiatum (CA3sl/sr), and dentate gyrus stratum moleculare (DGsm). Synapses were counted using unbiased electron microscopic stereology. Diagram columns represent the estimated number of all spine synapses within a particular hippocampal area. Three-way mixed ANOVA (stress × hormone treatment × area) found a significant main stress effect and also revealed a significant hormone treatment × stress interaction effect, indicating that hormone treatment interferes with the ability of stress to modulate hippocampal spine synapse numbers (stress effect, F1,16=635.357 P<0.001; hormone treatment × stress interaction, F3,16=109.665 P<0.001).
Relative to sham-stressed untreated controls (Panel Veh, NS), hormone treatment of nonstressed (NS) females increased the number of spine synapses across all hippocampal areas in simulated proestrus animals and reduced synapse numbers in CA1sr of hormone-treated ‘postpartum’ rats (panel ProE, #P<0.02; panel Horm, #P<0.02; Tukey-Kramer test). When compared with respective sham-stressed (NS) rats, inescapable stress (IS) decreased synapse numbers across all hippocampal areas in untreated controls, hormone-withdrawn ‘postpartum’ females, and simulated proestrus animals, but not in hormone-treated ‘postpartum’ rats (panel Veh, *P<0.04; panel PpD, *P<0.01; panel ProE, *P<0.01; panel Horm, P>0.2; Tukey-Kramer test). With respect to stressed untreated controls (Panel Veh, IS), synapse numbers of stressed (IS) females remained higher in CA3sl/sr of simulated proestrus animals, as well as in CA3sl/sr and DGsm of hormone-treated ‘postpartum’ rats (panel ProE, +P<0.001; panel Horm, +P<0.01; Tukey-Kramer test).
Figure 3.
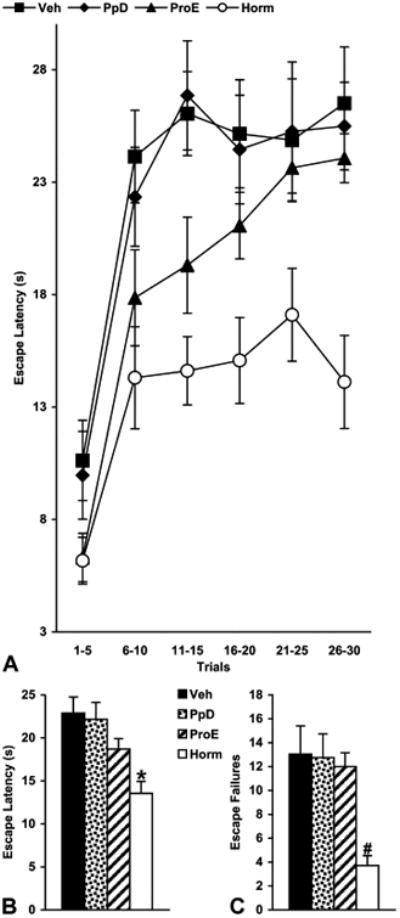
Performance of untreated controls (Veh), hormone-withdrawn ‘postpartum’ females (PpD), simulated proestrus animals (ProE), and hormone-treated ‘postpartum’ rats (Horm) in the active escape test. Panel A provides information about the fluctuation of escape latencies during the 30-trial test, with each tracing point representing the average escape latency of five consecutive trials. Panel B demonstrates the mean escape latency of all 30 trials. One-way ANOVA found a significant hormone treatment effect (F3,48=6.66 P<0.001). Escape latencies of hormone-treated ‘postpartum’ rats were shorter relative to those of untreated controls and hormone-withdrawn ‘postpartum’ females (*P<0.01, Tukey-Kramer test) but not significantly different from those of simulated proestrus animals (P=0.196, Tukey-Kramer test). Panel C displays the number of failed trials (Escape Failures) during the same 30-trial active escape test. Kruskal-Wallis oneway ANOVA on ranks revealed a significant hormone treatment effect (H=18.665 df=3 P<0.001). Hormone-treated ‘postpartum’ rats made less escape failures than animals in other groups (#P<0.002, Mann-Whitney U test).
Hormone-withdrawn ‘postpartum’ females (PpD)
Exposure to and subsequent withdrawal of pregnancy levels of female reproductive hormones did not change synapse numbers in sham-stressed PpD animals (Figure 2, PpD/NS vs. Veh/NS, P>0.2, Tukey-Kramer test). In the PpD group again, inescapable stress caused a decrease in the number of spine synapses across all hippocampal areas (Figure 2, PpD/IS vs. PpD/NS, *P<0.01, Tukey-Kramer test). As a result, post-stress synapse numbers in the PpD group were not significantly different from those in untreated controls (Figure 2, PpD/IS vs. Veh/IS, P>0.25, Tukey-Kramer test). PpD females executed the active escape task with a mean escape latency of 22.16±1.97 s and with an average escape failure of 12.75±1.99. These escape measures were not significantly different from those of untreated controls (Figure 3, Panels B and C, PpD vs. Veh, P=0.989 for escape latency, Tukey-Kramer test, and P=0.482 for escape failures, Mann-Whitney U test). As Figure 3, Panel A further demonstrates, escape performances of untreated controls and PpD females were practically identical.
Simulated proestrus animals (ProE)
Exposure of sham-stressed ProE rats to proestrus concentrations of gonadal steroids elicited a rise in synapse levels across all hippocampal areas (Figure 2, ProE/NS vs. Veh/NS, #P<0.02, Tukey-Kramer test). In the ProE group as well, inescapable stress caused a decline in the number of spine synapses across all hippocampal areas (Figure 2, ProE/IS vs. ProE/NS, *P<0.01, Tukey-Kramer test). As a result, post-stress synapse numbers in CA1sr and DGsm of the ProE group were not significantly different from those in untreated controls (CA1sr of ProE/IS vs. CA1sr of Veh/IS, P=0.054, and DGsm of ProE/IS vs. DGsm of Veh/IS, P=0.063, Tukey-Kramer test). By contrast, post-stress synapse numbers remained higher in CA3sl/sr of the ProE group (Figure 2, CA3sl/sr of ProE/IS vs. CA3sl/sr of Veh/IS, +P<0.001, Tukey-Kramer test). In the active escape task, the ProE group achieved a mean escape latency of 18.71±1.21 s and made, on average, 12±1.16 escape failures. This escape performance did not differ significantly from that of untreated controls (Figure 3, Panels B and C, ProE vs. Veh, P=0.367 for escape latency, Tukey-Kramer test, and P=0.430 for escape failures, Mann-Whitney U test).
Hormone-treated ‘postpartum’ rats (Horm)
Continued exposure to pregnancy levels of gonadal steroids during the ‘postpartum’ period did not increase synapse levels in sham-stressed Horm rats (Figure 2, CA3sl/sr and DGsm of Horm/NS vs. CA3sl/sr and DGsm of Veh/NS, P>0.6, Tukey-Kramer test). It even caused a moderate decline in CA1sr (Figure 2, CA1sr of Horm/NS vs. CA1sr of Veh/NS, #P<0.02, Tukey-Kramer test), reducing synapse numbers to post-stress levels (Figure 2, CA1sr of Horm/NS vs. CA1sr of Veh/IS, P=0.647, Tukey-Kramer test). Contrary to the other three groups, inescapable stress elicited no change in the number of hippocampal spine synapses in the Horm group (Figure 2, Horm/IS vs. Horm/NS, P>0.15, Tukey-Kramer test). As a result, post-stress synapse numbers in the Horm group remained higher in CA3sl/sr and DGsm relative to those in untreated controls (Figure 2, CA3sl/sr and DGsm of Horm/IS vs. CA3sl/sr and DGsm of Veh/IS, +P<0.01, Tukey-Kramer test). By contrast, post-stress synapse numbers were unchanged in CA1sr (CA1sr of Horm/IS vs. CA1sr of Veh/IS, P=0.412, Tukey-Kramer test). Horm rats executed the active escape task with a mean escape latency of 13.56±1.39 s and with an average escape failure of 3.71±0.83. These escape measures were better than those of all other groups (Figure 3, Panels B and C, *P<0.01 for escape latency, Tukey-Kramer test, and #P<0.002 for escape failures, Mann-Whitney U test), except that the escape latency of the Horm group did not differ significantly from that of simulated proestrus animals (Figure 3, Panel B, Horm vs. ProE, P=0.196, Tukey-Kramer test).
Serum ovarian hormone and corticosterone levels
Measured immediately after active escape testing, gonadal steroid levels in untreated controls and hormone-withdrawn ‘postpartum’ females were close to or even below assay sensitivity thresholds. In simulated proestrus animals, mean serum concentration of 17β-estradiol was 11.24±1.12 pg/mL, while mean serum concentration of progesterone was 3.99±0.54 ng/mL. It has to be noted that the estrogen and the progesterone regimens were timed to achieve proestrus levels of ovarian hormones by the time of stress exposure on day-27 (Gould et al., 1990; Scharfman et al., 2007), but the measurements above were taken on day-28 after a 24-h period of decay in hormone levels. As a result, although proestrus levels of gonadal steroids were maintained during stress exposure on day-27, the measurements above indicate that approximately estrus levels of hormones remained available during active escape testing on day-28 (Smith M.S. et al., 1975). In hormone-treated ‘postpartum’ rats, mean serum concentration of 17β-estradiol was 323.14±76.90 pg/mL, while mean serum concentration of progesterone was 5.10±1.53 ng/mL, which were in agreement with earlier studies (Birzniece et al., 2002; Suda et al., 2008).
More interestingly, one-way ANOVA found a significant hormone treatment effect on corticosterone levels (F3,25=11.340 P<0.001). Mean serum concentration of corticosterone was in the range of 350-400 ng/mL both in untreated controls and in hormone-withdrawn ‘postpartum’ females (Figure 4, PpD vs. Veh, P=0.993, Tukey-Kramer test). These corticosterone levels are usually associated with a strong stress response. In simulated proestrus animals, mean serum concentration of corticosterone was slightly, but not significantly, lower (Figure 4, ProE vs. Veh, P=0.260, Tukey-Kramer test). On the other hand, mean serum concentration of corticosterone was 140.21±34.52 ng/mL in hormone-treated ‘postpartum’ rats, which was significantly lower than corticosterone levels in untreated controls and hormone-withdrawn ‘postpartum’ females, respectively (Figure 4, Horm vs. Veh and Horm vs. PpD, *P<0.001, Tukey-Kramer test). Serum concentrations of corticosterone in hormone-treated ‘postpartum’ rats were, however, not significantly different from those in simulated proestrus animals (Figure 4, Horm vs. ProE, P=0.104, Tukey-Kramer test). Because hormones were sampled immediately after active escape testing, these corticosterone levels can be considered as a measure of stress response elicited by the behavioral task.
Figure 4.
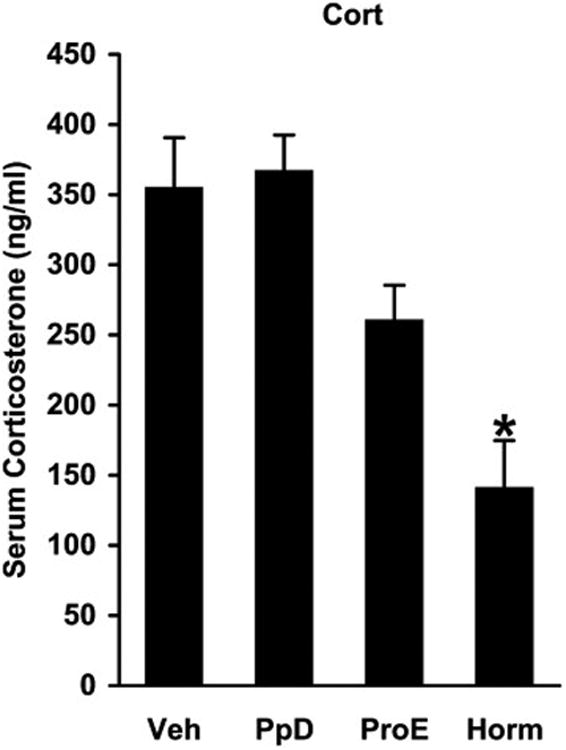
Serum concentrations of corticosterone (Cort) assayed immediately after active escape testing in untreated controls (Veh), hormone-withdrawn ‘postpartum’ females (PpD), simulated proestrus animals (ProE), and hormone-treated ‘postpartum’ rats (Horm). One-way ANOVA found a significant hormone treatment effect (F3,25=11.340 P<0.001). Corticosterone levels of hormone-treated ‘postpartum’ rats were lower than those of untreated controls and hormone-withdrawn ‘postpartum’ females (*P<0.001, Tukey-Kramer test) but not significantly different from those of simulated proestrus animals (P=0.104, Tukey-Kramer test).
Discussion
The present study demonstrates that during a simulated postpartum period, hormone-withdrawn ‘postpartum’ females respond to inescapable stress with loss of hippocampal spine synapses that is equivalent with what is observed in untreated controls, a female rat model of major depression (Hajszan et al., 2010). These data support our hypothesis that exposure to ‘postpartum’ stress leads to hippocampal spine synapse loss. We also show that sustaining pregnancy levels of ovarian hormones during ‘postpartum’ stress exposure is sufficient to overcome the synaptolytic effect, as inescapable stress caused no change in the number of hippocampal spine synapses in hormone-treated ‘postpartum’ rats. It has to be noted, however, that synaptoprotection is not fully confirmed in CA1sr, as pre-stress synapse numbers in this area of hormone-treated ‘postpartum’ rats were already at post-stress levels of untreated controls. It appears that this synaptoprotective effect is mediated, at least partly, by a muted stress response as measured by serum corticosterone levels. On the other hand, maintaining proestrus levels of gonadal steroids during ‘postpartum’ stress exposure is not capable of exerting similar synaptoprotective effects.
Our active escape experiment reveals that same post-stress levels of hippocampal spine synapses are coupled with practically identical escape performance (PpD group vs. Veh group). Higher post-stress numbers of hippocampal spine synapses, on the other hand, are associated with improved escape performance (Horm group vs. Veh group). Moreover, slightly higher post-stress synapse levels in simulated proestrus animals are related to slightly, but not significantly, better escape performance (ProE group vs. Veh group). These findings support our hypothesis that the relationship between synapse loss and depressive behavior, that we have first described in a female rat model of major depression (Hajszan et al., 2010), is retained in a simulated postpartum environment.
We can thus conclude that two important aspects of our synaptogenic hypothesis of depression (Hajszan et al., 2010; Kang et al., 2012), i.e., the synaptolytic effect of stress and the association between synapse loss and depressive behavior, appear to be valid in an animal model of PPD. As a result, the synaptic and the escape impairments of our PPD model (PpD group) are equivalent with those of an established animal model of major depression (Veh group) (Hajszan et al., 2010).
Stress-induced remodeling in the maternal brain
Pregnancy and motherhood are special periods when stress responsiveness is considerably reduced (Douglas et al., 1998; Wartella et al., 2003; Slattery and Neumann, 2008; Pawluski et al., 2015), yet epidemiological studies indicate that stress is a major risk factor of PPD (Lancaster et al., 2010; Davey et al., 2011; Stewart, 2011; Hillerer et al., 2012). Rodent models show that chronic gestational stress elicits anxiety-like behavior, increased time spent immobile in the forced swim test, and maternal care deficits during the postpartum period (Darnaudery et al., 2004; Smith J.W. et al., 2004; O'Mahony et al., 2006; Brummelte and Galea, 2010; Hillerer et al., 2011). In contrast to gestational stress, the effects of postpartum stress are less investigated. Most notably, Galea and colleagues have reported that postpartum exposure to high-dose corticosterone causes reduced maternal care, as well as decreased struggling and increased immobility in the forced swim test (Brummelte et al., 2006; Brummelte and Galea, 2010). As a result, our present finding that acute ‘postpartum’ stress causes depressive behavior in the active escape task adds to this much needed area of research.
We have only recently begun to understand the extent to which the maternal brain is remodeled by stress both in humans (Moses-Kolko et al., 2014) and in animal models (Pawluski et al., 2016). In a series of studies, Pawluski and colleagues have reported that gestational stress affects adult hippocampal neurogenesis and CA3 dendritic structure in pregnant rats (Pawluski et al., 2016). Exposure to chronic stress during pregnancy also reduces spine density and dendritic complexity in the prefrontal cortex and the nucleus accumbens shell (Haim et al., 2014; Leuner et al., 2014; Haim et al., 2016), while it raises synaptophysin levels in the cingulate cortex and increases spine density in the basolateral amygdala of rat dams (Gemmel et al., 2016; Haim et al., 2016). Some of these alterations are reversed by antidepressant treatment (Gemmel et al., 2016; Haim et al., 2016). As mentioned above, the effects of postpartum stress are much less known. Studying the consequences of postpartum stress via administration of high-dose corticosterone during lactation, Galea and colleagues have reported reduced adult hippocampal neurogenesis (Brummelte and Galea, 2010), as well as altered spine density and dendritic complexity in CA3 (Workman et al., 2013). Interestingly, repeated postpartum stress also reverses the lactation-induced decrease in hippocampal cell proliferation (Hillerer et al., 2014).
Stress-induced loss of hippocampal spine synapses appears to be the result of a chain of events that includes mediation by glucocorticoids and glutamate. It is well documented that exposure to acute stress, such as the inescapable footshock, consistently elicits a glucocorticoid-mediated surge of hippocampal glutamate release (Lowy et al., 1993; Abraham et al., 1998). Excess glutamate, in turn, leads to an excitotoxic, NMDA glutamate receptor dependent loss of dendritic spines (Segal, 1995; Wu et al., 2007), which may underlie the decline in the number of hippocampal spine synapses. In accordance with this mechanism, our research team has demonstrated earlier that injecting high-dose corticosterone fully reproduces the synaptolytic effect of inescapable footshock and induces depressive behavior in the rat learned helplessness paradigm (Hajszan et al., 2009).
The findings discussed above are beginning to outline an important role for neuronal remodeling in maternal stress-related disorders such as PPD. Unfortunately, our current knowledge regarding PPD is far from the level of understanding that has been already gained about the influence of synaptic plasticity in major depression (Hajszan et al., 2010; Kang et al., 2012) and in antidepressant treatment (Hajszan et al., 2005; Li et al., 2010). For example, direct electron microscopic evidence of synaptic remodeling in PPD is still not available. Our present finding that acute ‘postpartum’ stress induces hippocampal spine synapse loss helps addressing this gap in knowledge, although the merit of these results are obviously defined by the construct validity of our animal model.
Role of estradiol and progesterone
In the hormone simulated pregnancy model of Galea and colleagues (Galea et al., 2001), hormone withdrawal alone suppresses adult hippocampal neurogenesis (Green and Galea, 2008). In accordance with this earlier observation, our present findings reveal that relative to ‘proestrus’ females (nonstressed ProE group), hormone withdrawal alone decreases the number of hippocampal spine synapses in the PPD model (nonstressed PpD group) and in ovariectomized controls (nonstressed Veh group). These results are in line with a large number of previous studies reporting that ovariectomy causes hippocampal spine synapse loss, which is reversed by estradiol replacement (Woolley and McEwen, 1992; Leranth et al., 2008). The behavioral consequences of hormone withdrawal alone were not investigated in the present study. In an earlier study, however, we have already reported that ovariectomy reduces performance of female rats in the active escape task in association with loss of hippocampal spine synapses (Hajszan et al., 2010).
Our present results demonstrate that maintaining pregnancy levels of female reproductive hormones during ‘postpartum’ stress exposure (Horm group) overcomes the synaptolytic effect of inescapable stress and improves escape performance. These observations complement human clinical studies documenting the beneficial effect of estradiol treatment in alleviating the symptoms of PPD (Sichel et al., 1995; Gregoire et al., 1996; Ahokas et al., 2001; Studd and Panay, 2004). Earlier work with animal models has also shown that estradiol reduces depressive behavior and facilitates the action of antidepressants in the forced swim test (Rachman et al., 1998; Frye and Wawrzycki, 2003; Estrada-Camarena et al., 2004). In contrast to pregnancy levels, interestingly, proestrus levels of gonadal steroids failed to prevent the effects of ‘postpartum’ stress in our present study. This finding is in agreement with previous research indicating that stress limits or even abolishes the hippocampal synaptogenic and the antidepressant activity of acute estradiol administration in the rat Morris water-maze (Frick et al., 2004) and the rat learned helplessness paradigms (Hajszan et al., 2010). This discrepancy between the effects of proestrus vs. pregnancy levels of hormones is probably explained by differences among experiments in the duration of administration and the dosing of estradiol, as well as in the level of stress exposure.
Besides the synaptic and escape responses, continued exposure to pregnancy levels of gonadal steroids during the ‘postpartum’ period (Horm group) also leads to muted corticosterone release induced by ‘postpartum’ stress. This finding is in line with a large number of earlier studies showing strong interactions between the female stress and reproductive systems (Chrousos et al., 1998), as ovarian hormones effectively reduce hypothalamic-pituitary-adrenal axis activity (Young et al., 2001; Lunga and Herbert, 2004; Brunton et al., 2009; Hassell et al., 2011; McCormick, 2011). The gonadal steroid induced muted corticosterone release likely contributes to the typically reduced stress experience during pregnancy (Douglas et al., 1998; Wartella et al., 2003; Slattery and Neumann, 2008; Pawluski et al., 2015). More importantly, this limited stress responsiveness may also suppress the glucocorticoid/glutamate chain of events leading to hippocampal spine synapse loss (see above), which explains, at least partly, the synaptoprotective action observed in the present study. Interestingly, the effects of pregnancy levels of ovarian hormones on the stress system resemble those of certain traditional antidepressants, as these drugs are believed, at least partly, to stabilize mood via curbing stress reactivity (Barden et al., 1995; Nemeroff and Owens, 2004; Pawluski et al., 2012).
Earlier research on the hormonal aspects of PPD has placed emphasis on estradiol withdrawal (Galea et al., 2001; Stoffel and Craft, 2004; Green and Galea, 2008). As a result, the role of progesterone remained less investigated despite its potent abilities to influence stress and neuronal activity. The progesterone metabolite neurosteroid, allopregnanolone is known to maintain an opioid-dependent inhibition of stress responsiveness during pregnancy (Brunton et al., 2009). Other progesterone-derived neurosteroids are efficient in potentiating the effect of GABA on GABAA receptors (Bitran and Dowd, 1996; Stell et al., 2003), leading to restricted neuronal excitability and, as a result, limited glutamate release (Stell et al., 2003). Deficiency in these mechanisms may imply vulnerability to stress and development of postpartum mood disorders. Indeed, transgenic mice that are incapable of neurosteroid-induced GABA potentiation show depressive and anxiety-like behaviors, as well as abnormal maternal care throughout pregnancy and the postpartum period (Maguire and Mody, 2008; Mostallino et al., 2009).
Functional considerations
Earlier research suggests that withdrawal of female reproductive hormones creates vulnerability to stress during the postpartum period (Parry et al., 2003; Steiner et al., 2003; Suda et al., 2008). In accordance with this theory, we have previously shown in the rat learned helplessness paradigm that withdrawal of ovarian hormones alone leads to hippocampal spine synapse loss and development of depressive behavior, but these responses are only partial and become exacerbated after exposure to inescapable stress (Hajszan et al., 2010). In line with these previous findings, the present study demonstrates that ‘postpartum’ stress in a hormone withdrawal model of PPD causes the loss of asymmetric spine synapses along the hippocampal trisynaptic circuit, potentially disrupting signal flow and leading to a dysfunctional hippocampus. Among several brain areas that are affected by stress and mood disorders (Nestler et al., 2002), the hippocampus plays an important role, as it is strongly involved in learning and memory (Sousa et al., 2000; Diamond et al., 2006), in feed-back regulation of the stress response (McEwen, 2003; Sala et al., 2004), and in modulating the motivation circuitries (Lisman and Grace, 2005; Cooper et al., 2006). As a result, deteriorating hippocampal function may contribute to the joint development of cognitive decline, loss of motivation, and derailed stress response, the common symptoms of PPD and major depression (Melges, 1968; Nestler et al., 2002).
On the other hand, the present study also indicates that pregnancy levels of gonadal steroids protect hippocampal spine synapses against the detrimental effects of ‘postpartum’ stress. Importantly, this synaptoprotection is associated with improved escape performance, i.e., decreased depressive behavior. These results, along with our synaptogenic hypothesis of depression (Hajszan et al., 2010), suggest that protecting spine synapses by muting the stress response and/or by curbing neuronal excitability may be valid therapeutic options for preventing and treating postpartum affective illness.
Highlights.
Postpartum stress causes a loss of hippocampal spine synapses.
Pregnancy levels of ovarian hormones prevent the synaptolytic effect of stress.
This preventive effect of ovarian hormones is mediated by reduced stress responsiveness.
These data provide further support for the “synaptogenic hypothesis” of depression.
Acknowledgments
Funding sources: This study was supported by a NARSAD Young Investigator Award, a National Institute of Mental Health (NIMH) grant [grant number MH074021], a Research, Technology and Innovation Fund (KTIA, Hungary) and Hungarian Scientific Research Fund (OTKA) joint reintegration grant [grant number Mobility MB08C OTKA 81190], and European Union and New Szechenyi Programme (ÚSZT, Hungary) joint grants [grant numbers TÁMOP-4.2.2.A-11/1/KONV-2012-0052, TÁMOP-4.2.4.A/2-11-1-2012-0001, and GINOP-2.3.2-15-2016-00001].
Role of funding sources: None of the funding sources have any roles in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Footnotes
Contributors: Ms. J Baka performed experiments, managed the literature search, and wrote the first draft of the manuscript. Dr. E Csakvari prepared tissue for electron microscopic analysis, and took electron micrographs for synapse counting. Ms. O Huzian performed active escape testing. Dr. N Dobos assayed blood samples. Dr. L Siklos wrote the paper. Dr. C Leranth counted spine synapses. Dr. NJ MacLusky designed the study and wrote the paper. Dr. RS Duman designed the study and wrote the paper. Dr. T Hajszan designed the study, coordinated experiments, counted spine synapses, analyzed data, and wrote the paper.
Conflict of interest statement: All authors declare that they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham I, Juhasz G, Kekesi KA, Kovacs KJ. Corticosterone peak is responsible for stress-induced elevation of glutamate in the hippocampus. Stress. 1998;2:171–181. doi: 10.3109/10253899809167281. [DOI] [PubMed] [Google Scholar]
- Ahokas A, Kaukoranta J, Wahlbeck K, Aito M. Estrogen deficiency in severe postpartum depression: successful treatment with sublingual physiologic 17beta-estradiol: a preliminary study. J Clin Psychiatry. 2001;62:332–336. doi: 10.4088/jcp.v62n0504. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Witter MP. Hippocampal formation. In: Paxinos G, editor. The Rat Nervous System. San Diego: Academic Press; 1995. pp. 443–493. [Google Scholar]
- Diagnostic and statistical manual of mental disorders. American Psychiatric Association; Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- Barden N, Reul JM, Holsboer F. Do antidepressants stabilize mood through actions on the hypothalamic-pituitary-adrenocortical system? Trends Neurosci. 1995;18:6–11. doi: 10.1016/0166-2236(95)93942-q. [DOI] [PubMed] [Google Scholar]
- Birzniece V, Johansson IM, Wang MD, Backstrom T, Olsson T. Ovarian hormone effects on 5-hydroxytryptamine(2A) and 5-hydroxytryptamine(2C) receptor mRNA expression in the ventral hippocampus and frontal cortex of female rats. Neurosci Lett. 2002;319:157–161. doi: 10.1016/s0304-3940(01)02570-8. [DOI] [PubMed] [Google Scholar]
- Bitran D, Dowd JA. Ovarian steroids modify the behavioral and neurochemical responses of the central benzodiazepine receptor. Psychopharmacology (Berl) 1996;125:65–73. doi: 10.1007/BF02247394. [DOI] [PubMed] [Google Scholar]
- Bloch M, Daly RC, Rubinow DR. Endocrine factors in the etiology of postpartum depression. Compr Psychiatry. 2003;44:234–246. doi: 10.1016/S0010-440X(03)00034-8. [DOI] [PubMed] [Google Scholar]
- Bloch M, Schmidt PJ, Danaceau M, Murphy J, Nieman L, Rubinow DR. Effects of gonadal steroids in women with a history of postpartum depression. Am J Psychiatry. 2000;157:924–930. doi: 10.1176/appi.ajp.157.6.924. [DOI] [PubMed] [Google Scholar]
- Brummelte S, Galea LA. Chronic corticosterone during pregnancy and postpartum affects maternal care, cell proliferation and depressive-like behavior in the dam. Horm Behav. 2010;58:769–779. doi: 10.1016/j.yhbeh.2010.07.012. [DOI] [PubMed] [Google Scholar]
- Brummelte S, Pawluski JL, Galea LA. High post-partum levels of corticosterone given to dams influence postnatal hippocampal cell proliferation and behavior of offspring: a model of post-partum stress and possible depression. Horm Behav. 2006;50:370–382. doi: 10.1016/j.yhbeh.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Brunton PJ, McKay AJ, Ochedalski T, Piastowska A, Rebas E, Lachowicz A, Russell JA. Central opioid inhibition of neuroendocrine stress responses in pregnancy in the rat is induced by the neurosteroid allopregnanolone. J Neurosci. 2009;29:6449–6460. doi: 10.1523/JNEUROSCI.0708-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrousos GP, Torpy DJ, Gold PW. Interactions between the hypothalamic-pituitary-adrenal axis and the female reproductive system: clinical implications. Ann Intern Med. 1998;129:229–240. doi: 10.7326/0003-4819-129-3-199808010-00012. [DOI] [PubMed] [Google Scholar]
- Cooper DC, Klipec WD, Fowler MA, Ozkan ED. A role for the subiculum in the brain motivation/reward circuitry. Behav Brain Res. 2006;174:225–231. doi: 10.1016/j.bbr.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Crawley RA, Dennison K, Carter C. Cognition in pregnancy and the first year post-partum. Psychol Psychother. 2003;76:69–84. doi: 10.1348/14760830260569265. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Markou A, Lucki I. Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci. 2002;23:238–245. doi: 10.1016/s0165-6147(02)02017-5. [DOI] [PubMed] [Google Scholar]
- Darnaudery M, Dutriez I, Viltart O, Morley-Fletcher S, Maccari S. Stress during gestation induces lasting effects on emotional reactivity of the dam rat. Behav Brain Res. 2004;153:211–216. doi: 10.1016/j.bbr.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Davey HL, Tough SC, Adair CE, Benzies KM. Risk factors for sub-clinical and major postpartum depression among a community cohort of Canadian women. Matern Child Health J. 2011;15:866–875. doi: 10.1007/s10995-008-0314-8. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Campbell AM, Park CR, Woodson JC, Conrad CD, Bachstetter AD, Mervis RF. Influence of predator stress on the consolidation versus retrieval of long-term spatial memory and hippocampal spinogenesis. Hippocampus. 2006;16:571–576. doi: 10.1002/hipo.20188. [DOI] [PubMed] [Google Scholar]
- Douglas AJ, Johnstone HA, Wigger A, Landgraf R, Russell JA, Neumann ID. The role of endogenous opioids in neurohypophysial and hypothalamo-pituitary-adrenal axis hormone secretory responses to stress in pregnant rats. J Endocrinol. 1998;158:285–293. doi: 10.1677/joe.0.1580285. [DOI] [PubMed] [Google Scholar]
- Douma SL, Husband C, O'Donnell ME, Barwin BN, Woodend AK. Estrogen-related mood disorders: reproductive life cycle factors. ANS Adv Nurs Sci. 2005;28:364–375. doi: 10.1097/00012272-200510000-00008. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Estrada-Camarena E, Fernandez-Guasti A, Lopez-Rubalcava C. Interaction between estrogens and antidepressants in the forced swimming test in rats. Psychopharmacology (Berl) 2004;173:139–145. doi: 10.1007/s00213-003-1707-4. [DOI] [PubMed] [Google Scholar]
- Frick KM, Fernandez SM, Bennett JC, Prange-Kiel J, MacLusky NJ, Leranth C. Behavioral training interferes with the ability of gonadal hormones to increase CA1 spine synapse density in ovariectomized female rats. Eur J Neurosci. 2004;19:3026–3032. doi: 10.1111/j.1460-9568.2004.03427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye CA, Wawrzycki J. Effect of prenatal stress and gonadal hormone condition on depressive behaviors of female and male rats. Horm Behav. 2003;44:319–326. doi: 10.1016/s0018-506x(03)00159-4. [DOI] [PubMed] [Google Scholar]
- Galea LA, Wide JK, Barr AM. Estradiol alleviates depressive-like symptoms in a novel animal model of post-partum depression. Behav Brain Res. 2001;122:1–9. doi: 10.1016/s0166-4328(01)00170-x. [DOI] [PubMed] [Google Scholar]
- Gavin NI, Gaynes BN, Lohr KN, Meltzer-Brody S, Gartlehner G, Swinson T. Perinatal depression: a systematic review of prevalence and incidence. Obstet Gynecol. 2005;106:1071–1083. doi: 10.1097/01.AOG.0000183597.31630.db. [DOI] [PubMed] [Google Scholar]
- Gemmel M, Rayen I, van Donkelaar E, Loftus T, Steinbusch HW, Kokras N, Dalla C, Pawluski JL. Gestational stress and fluoxetine treatment differentially affect plasticity, methylation and serotonin levels in the PFC and hippocampus of rat dams. Neuroscience. 2016;327:32–43. doi: 10.1016/j.neuroscience.2016.03.068. [DOI] [PubMed] [Google Scholar]
- Gould E, Woolley CS, Frankfurt M, McEwen BS. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J Neurosci. 1990;10:1286–1291. doi: 10.1523/JNEUROSCI.10-04-01286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace SL, Evindar A, Stewart DE. The effect of postpartum depression on child cognitive development and behavior: a review and critical analysis of the literature. Arch Womens Ment Health. 2003;6:263–274. doi: 10.1007/s00737-003-0024-6. [DOI] [PubMed] [Google Scholar]
- Green AD, Galea LA. Adult hippocampal cell proliferation is suppressed with estrogen withdrawal after a hormone-simulated pregnancy. Horm Behav. 2008;54:203–211. doi: 10.1016/j.yhbeh.2008.02.023. [DOI] [PubMed] [Google Scholar]
- Gregoire AJ, Kumar R, Everitt B, Henderson AF, Studd JW. Transdermal oestrogen for treatment of severe postnatal depression. Lancet. 1996;347:930–933. doi: 10.1016/s0140-6736(96)91414-2. [DOI] [PubMed] [Google Scholar]
- Gress-Smith JL, Luecken LJ, Lemery-Chalfant K, Howe R. Postpartum depression prevalence and impact on infant health, weight, and sleep in low-income and ethnic minority women and infants. Matern Child Health J. 2012;16:887–893. doi: 10.1007/s10995-011-0812-y. [DOI] [PubMed] [Google Scholar]
- Grota LJ, Eik-Nes KB. Plasma progesterone concentrations during pregnancy and lactation in the rat. J Reprod Fertil. 1967;13:83–91. doi: 10.1530/jrf.0.0130083. [DOI] [PubMed] [Google Scholar]
- Haim A, Albin-Brooks C, Sherer M, Mills E, Leuner B. The effects of gestational stress and selective serotonin reuptake inhibitor antidepressant treatment on structural plasticity in the postpartum brain--A translational model for postpartum depression. Horm Behav. 2016;77:124–131. doi: 10.1016/j.yhbeh.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haim A, Sherer M, Leuner B. Gestational stress induces persistent depressive-like behavior and structural modifications within the postpartum nucleus accumbens. Eur J Neurosci. 2014;40:3766–3773. doi: 10.1111/ejn.12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajszan T, Dow A, Warner-Schmidt JL, Szigeti-Buck K, Sallam NL, Parducz A, Leranth C, Duman RS. Remodeling of hippocampal spine synapses in the rat learned helplessness model of depression. Biol Psychiatry. 2009;65:392–400. doi: 10.1016/j.biopsych.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajszan T, MacLusky NJ, Leranth C. Short-term treatment with the antidepressant fluoxetine triggers pyramidal dendritic spine synapse formation in rat hippocampus. Eur J Neurosci. 2005;21:1299–1303. doi: 10.1111/j.1460-9568.2005.03968.x. [DOI] [PubMed] [Google Scholar]
- Hajszan T, Szigeti-Buck K, Sallam NL, Bober J, Parducz A, Maclusky NJ, Leranth C, Duman RS. Effects of estradiol on learned helplessness and associated remodeling of hippocampal spine synapses in female rats. Biol Psychiatry. 2010;67:168–174. doi: 10.1016/j.biopsych.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassell J, Miryala CSJ, Hiegel C, Uphouse L. Mechanisms responsible for progesterone's protection against lordosis-inhibiting effects of restraint I. Role of progesterone receptors. Horm Behav. 2011;60:219–225. doi: 10.1016/j.yhbeh.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich A, Schleyer M, Spingler H, Albert P, Knoche M, Fritze J, Lanczik M. Postpartum blues: relationship between not-protein bound steroid hormones in plasma and postpartum mood changes. J Affect Disord. 1994;30:93–98. doi: 10.1016/0165-0327(94)90036-1. [DOI] [PubMed] [Google Scholar]
- Hendrick V, Altshuler LL, Suri R. Hormonal changes in the postpartum and implications for postpartum depression. Psychosomatics. 1998;39:93–101. doi: 10.1016/S0033-3182(98)71355-6. [DOI] [PubMed] [Google Scholar]
- Hillerer KM, Neumann ID, Couillard-Despres S, Aigner L, Slattery DA. Lactation-induced reduction in hippocampal neurogenesis is reversed by repeated stress exposure. Hippocampus. 2014;24:673–683. doi: 10.1002/hipo.22258. [DOI] [PubMed] [Google Scholar]
- Hillerer KM, Neumann ID, Slattery DA. From stress to postpartum mood and anxiety disorders: how chronic peripartum stress can impair maternal adaptations. Neuroendocrinology. 2012;95:22–38. doi: 10.1159/000330445. [DOI] [PubMed] [Google Scholar]
- Hillerer KM, Reber SO, Neumann ID, Slattery DA. Exposure to chronic pregnancy stress reverses peripartum-associated adaptations: implications for postpartum anxiety and mood disorders. Endocrinology. 2011;152:3930–3940. doi: 10.1210/en.2011-1091. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, Lepack A, Majik MS, Jeong LS, Banasr M, Son H, Duman RS. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. 2012;18:1413–1417. doi: 10.1038/nm.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klier CM, Muzik M, Dervic K, Mossaheb N, Benesch T, Ulm B, Zeller M. The role of estrogen and progesterone in depression after birth. J Psychiatr Res. 2007;41:273–279. doi: 10.1016/j.jpsychires.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Lancaster CA, Gold KJ, Flynn HA, Yoo H, Marcus SM, Davis MM. Risk factors for depressive symptoms during pregnancy: a systematic review. Am J Obstet Gynecol. 2010;202:5–14. doi: 10.1016/j.ajog.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laura J, Miller M. Postpartum depression. JAMA. 2002;287:11–14. doi: 10.1001/jama.287.6.762. [DOI] [PubMed] [Google Scholar]
- Leranth C, Hajszan T, Szigeti-Buck K, Bober J, MacLusky NJ. Bisphenol A prevents the synaptogenic response to estradiol in hippocampus and prefrontal cortex of ovariectomized nonhuman primates. Proc Natl Acad Sci U S A. 2008;105:14187–14191. doi: 10.1073/pnas.0806139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneau NL, Dennis CL, Benzies K, Duffett-Leger L, Stewart M, Tryphonopoulos PD, Este D, Watson W. Postpartum depression is a family affair: addressing the impact on mothers, fathers, and children. Issues Ment Health Nurs. 2012;33:445–457. doi: 10.3109/01612840.2012.673054. [DOI] [PubMed] [Google Scholar]
- Leuner B, Fredericks PJ, Nealer C, Albin-Brooks C. Chronic gestational stress leads to depressive-like behavior and compromises medial prefrontal cortex structure and function during the postpartum period. PLoS One. 2014;9:e89912. doi: 10.1371/journal.pone.0089912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, Li XY, Aghajanian G, Duman RS. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Lowy MT, Gault L, Yamamoto BK. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem. 1993;61:1957–1960. doi: 10.1111/j.1471-4159.1993.tb09839.x. [DOI] [PubMed] [Google Scholar]
- Lunga P, Herbert J. 17Beta-oestradiol modulates glucocorticoid, neural and behavioural adaptations to repeated restraint stress in female rats. J Neuroendocrinol. 2004;16:776–785. doi: 10.1111/j.1365-2826.2004.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLusky NJ, Hajszan T, Johansen JA, Jordan CL, Leranth C. Androgen effects on hippocampal CA1 spine synapse numbers are retained in Tfm male rats with defective androgen receptors. Endocrinology. 2006;147:2392–2398. doi: 10.1210/en.2005-0673. [DOI] [PubMed] [Google Scholar]
- Maguire J, Mody I. GABA(A)R plasticity during pregnancy: relevance to postpartum depression. Neuron. 2008;59:207–213. doi: 10.1016/j.neuron.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick CM. Effect of neonatal ovariectomy and estradiol treatment on corticosterone release in response to stress in the adult female rat. Stress. 2011;14:82–87. doi: 10.3109/10253890.2010.490309. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Mood disorders and allostatic load. Biol Psychiatry. 2003;54:200–207. doi: 10.1016/s0006-3223(03)00177-x. [DOI] [PubMed] [Google Scholar]
- McNeilly AS. Neuroendocrine changes and fertility in breast-feeding women. Prog Brain Res. 2001;133:207–214. doi: 10.1016/s0079-6123(01)33015-7. [DOI] [PubMed] [Google Scholar]
- Melges FT. Postpartum psychiatric syndromes. Psychosom Med. 1968;30:95–108. doi: 10.1097/00006842-196801000-00009. [DOI] [PubMed] [Google Scholar]
- Moses-Kolko EL, Horner MS, Phillips ML, Hipwell AE, Swain JE. In search of neural endophenotypes of postpartum psychopathology and disrupted maternal caregiving. J Neuroendocrinol. 2014;26:665–684. doi: 10.1111/jne.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostallino MC, Sanna E, Concas A, Biggio G, Follesa P. Plasticity and function of extrasynaptic GABA(A) receptors during pregnancy and after delivery. Psychoneuroendocrinology. 2009;34:S74–83. doi: 10.1016/j.psyneuen.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Owens MJ. Pharmacologic differences among the SSRIs: focus on monoamine transporters and the HPA axis. CNS Spectr. 2004;9:23–31. doi: 10.1017/s1092852900025475. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Nomura Y, Wickramaratne PJ, Warner V, Mufson L, Weissman MM. Family discord, parental depression, and psychopathology in offspring: ten-year follow-up. J Am Acad Child Adolesc Psychiatry. 2002;41:402–409. doi: 10.1097/00004583-200204000-00012. [DOI] [PubMed] [Google Scholar]
- O'Hara MW, Wisner KL. Perinatal mental illness: definition, description and aetiology. Best Pract Res Clin Obstet Gynaecol. 2014;28:3–12. doi: 10.1016/j.bpobgyn.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Mahony SM, Myint AM, van den Hove D, Desbonnet L, Steinbusch H, Leonard BE. Gestational stress leads to depressive-like behavioural and immunological changes in the rat. Neuroimmunomodulation. 2006;13:82–88. doi: 10.1159/000096090. [DOI] [PubMed] [Google Scholar]
- Pariser SF, Nasrallah HA, Gardner DK. Postpartum mood disorders: clinical perspectives. J Womens Health. 1997;6:421–434. doi: 10.1089/jwh.1997.6.421. [DOI] [PubMed] [Google Scholar]
- Parry BL, Sorenson DL, Meliska CJ, Basavaraj N, Zirpoli GG, Gamst A, Hauger R. Hormonal basis of mood and postpartum disorders. Curr Womens Health Rep. 2003;3:230–235. [PubMed] [Google Scholar]
- Pawluski JL, Charlier TD, Fillet M, Houbart V, Crispin HT, Steinbusch HW, van den Hove DL. Chronic fluoxetine treatment and maternal adversity differentially alter neurobehavioral outcomes in the rat dam. Behav Brain Res. 2012;228:159–168. doi: 10.1016/j.bbr.2011.11.043. [DOI] [PubMed] [Google Scholar]
- Pawluski JL, Csaszar E, Savage E, Martinez-Claros M, Steinbusch HW, van den Hove D. Effects of stress early in gestation on hippocampal neurogenesis and glucocorticoid receptor density in pregnant rats. Neuroscience. 2015;290:379–388. doi: 10.1016/j.neuroscience.2015.01.048. [DOI] [PubMed] [Google Scholar]
- Pawluski JL, Lambert KG, Kinsley CH. Neuroplasticity in the maternal hippocampus: Relation to cognition and effects of repeated stress. Horm Behav. 2016;77:86–97. doi: 10.1016/j.yhbeh.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Pepe GJ, Rothchild I. A comparative study of serum progesterone levels in pregnancy and in various types of pseudopregnancy in the rat. Endocrinology. 1974;95:275–279. doi: 10.1210/endo-95-1-275. [DOI] [PubMed] [Google Scholar]
- Pitt B. "Atypical" depression following childbirth. Br J Psychiatry. 1968;114:1325–1335. doi: 10.1192/bjp.114.516.1325. [DOI] [PubMed] [Google Scholar]
- Rachman IM, Unnerstall JR, Pfaff DW, Cohen RS. Estrogen alters behavior and forebrain c-fos expression in ovariectomized rats subjected to the forced swim test. Proc Natl Acad Sci U S A. 1998;95:13941–13946. doi: 10.1073/pnas.95.23.13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinow DR, Schmidt PJ. Gonadal steroid regulation of mood: the lessons of premenstrual syndrome. Front Neuroendocrinol. 2006;27:210–216. doi: 10.1016/j.yfrne.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Sala M, Perez J, Soloff P, Ucelli di Nemi S, Caverzasi E, Soares JC, Brambilla P. Stress and hippocampal abnormalities in psychiatric disorders. Eur Neuropsychopharmacol. 2004;14:393–405. doi: 10.1016/j.euroneuro.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Hintz TM, Gomez J, Stormes KA, Barouk S, Malthankar-Phatak GH, McCloskey DP, Luine VN, Maclusky NJ. Changes in hippocampal function of ovariectomized rats after sequential low doses of estradiol to simulate the preovulatory estrogen surge. Eur J Neurosci. 2007;26:2595–2612. doi: 10.1111/j.1460-9568.2007.05848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt PJ, Rubinow DR. Sex hormones and mood in the perimenopause. Ann N Y Acad Sci. 2009;1179:70–85. doi: 10.1111/j.1749-6632.2009.04982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. Morphological alterations in dendritic spines of rat hippocampal neurons exposed to N-methyl-D-aspartate. Neurosci Lett. 1995;193:73–76. doi: 10.1016/0304-3940(95)11665-j. [DOI] [PubMed] [Google Scholar]
- Sheline YI. Neuroimaging studies of mood disorder effects on the brain. Biol Psychiatry. 2003;54:338–352. doi: 10.1016/s0006-3223(03)00347-0. [DOI] [PubMed] [Google Scholar]
- Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sichel DA, Cohen LS, Robertson LM, Ruttenberg A, Rosenbaum JF. Prophylactic estrogen in recurrent postpartum affective disorder. Biol Psychiatry. 1995;38:814–818. doi: 10.1016/0006-3223(95)00063-1. [DOI] [PubMed] [Google Scholar]
- Slattery DA, Neumann ID. No stress please! Mechanisms of stress hyporesponsiveness of the maternal brain. J Physiol. 2008;586:377–385. doi: 10.1113/jphysiol.2007.145896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JW, Seckl JR, Evans AT, Costall B, Smythe JW. Gestational stress induces post-partum depression-like behaviour and alters maternal care in rats. Psychoneuroendocrinology. 2004;29:227–244. doi: 10.1016/s0306-4530(03)00025-8. [DOI] [PubMed] [Google Scholar]
- Smith MS, Freeman ME, Neill JD. The control of progesterone secretion during the estrous cycle and early pseudopregnancy in the rat: prolactin, gonadotropin and steroid levels associated with rescue of the corpus luteum of pseudopregnancy. Endocrinology. 1975;96:219–226. doi: 10.1210/endo-96-1-219. [DOI] [PubMed] [Google Scholar]
- Sousa N, Lukoyanov NV, Madeira MD, Almeida OF, Paula-Barbosa MM. Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience. 2000;97:253–266. doi: 10.1016/s0306-4522(00)00050-6. [DOI] [PubMed] [Google Scholar]
- Spinelli MG. Maternal infanticide associated with mental illness: prevention and the promise of saved lives. Am J Psychiatry. 2004;161:1548–1557. doi: 10.1176/appi.ajp.161.9.1548. [DOI] [PubMed] [Google Scholar]
- Steiner M, Dunn E, Born L. Hormones and mood: from menarche to menopause and beyond. J Affect Disord. 2003;74:67–83. doi: 10.1016/s0165-0327(02)00432-9. [DOI] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABAA receptors. Proc Natl Acad Sci U S A. 2003;100:14439–14444. doi: 10.1073/pnas.2435457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterio DC. The unbiased estimation of number and sizes of arbitrary particles using the disector. J Microsc. 1984;134:127–136. doi: 10.1111/j.1365-2818.1984.tb02501.x. [DOI] [PubMed] [Google Scholar]
- Stewart DE. Clinical practice. Depression during pregnancy. N Engl J Med. 2011;365:1605–1611. doi: 10.1056/NEJMcp1102730. [DOI] [PubMed] [Google Scholar]
- Stoffel EC, Craft RM. Ovarian hormone withdrawal-induced "depression" in female rats. Physiol Behav. 2004;83:505–513. doi: 10.1016/j.physbeh.2004.08.033. [DOI] [PubMed] [Google Scholar]
- Studd J, Panay N. Hormones and depression in women. Climacteric. 2004;7:338–346. doi: 10.1080/13697130400012262. [DOI] [PubMed] [Google Scholar]
- Suda S, Segi-Nishida E, Newton SS, Duman RS. A postpartum model in rat: behavioral and gene expression changes induced by ovarian steroid deprivation. Biol Psychiatry. 2008;64:311–319. doi: 10.1016/j.biopsych.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiebot MH, Martin P, Puech AJ. Animal behavioural studies in the evaluation of antidepressant drugs. Br J Psychiatry Suppl. 1992;15:44–50. [PubMed] [Google Scholar]
- Valentine G, Dow A, Banasr M, Pittman B, Duman RS. Differential effects of chronic antidepressant treatment on shuttle box escape deficits induced by uncontrollable stress. Psychopharmacology (Berl) 2008;200:585–596. doi: 10.1007/s00213-008-1239-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek T, Bockting CL, van Pampus MG, Ormel J, Meijer JL, Hartman CA, Burger H. Postpartum depression predicts offspring mental health problems in adolescence independently of parental lifetime psychopathology. J Affect Disord. 2012;136:948–954. doi: 10.1016/j.jad.2011.08.035. [DOI] [PubMed] [Google Scholar]
- Vollmayr B, Henn FA. Learned helplessness in the rat: improvements in validity and reliability. Brain Res Brain Res Protoc. 2001;8:1–7. doi: 10.1016/s1385-299x(01)00067-8. [DOI] [PubMed] [Google Scholar]
- Wartella J, Amory E, Lomas LM, Macbeth A, McNamara I, Stevens L, Lambert KG, Kinsley CH. Single or multiple reproductive experiences attenuate neurobehavioral stress and fear responses in the female rat. Physiol Behav. 2003;79:373–381. doi: 10.1016/s0031-9384(03)00150-1. [DOI] [PubMed] [Google Scholar]
- Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman JL, Brummelte S, Galea LA. Postpartum corticosterone administration reduces dendritic complexity and increases the density of mushroom spines of hippocampal CA3 arbours in dams. J Neuroendocrinol. 2013;25:119–130. doi: 10.1111/j.1365-2826.2012.02380.x. [DOI] [PubMed] [Google Scholar]
- Wu LX, Sun CK, Zhang YM, Fan M, Xu J, Ma H, Zhang J. Involvement of the Snk-SPAR pathway in glutamate-induced excitotoxicity in cultured hippocampal neurons. Brain Res. 2007;1168:38–45. doi: 10.1016/j.brainres.2007.06.082. [DOI] [PubMed] [Google Scholar]
- Young EA, Altemus M, Parkinson V, Shastry S. Effects of estrogen antagonists and agonists on the ACTH response to restraint stress in female rats. Neuropsychopharmacology. 2001;25:881–891. doi: 10.1016/S0893-133X(01)00301-3. [DOI] [PubMed] [Google Scholar]