

Abstract
Endothelin (ET)-1 is an important peptide in cancer progression stimulating cellular proliferation, tumor angiogenesis and metastasis. ET-1 binds with high affinity to the ETA receptor (R) and ETBR on cancer cells. High levels of tumor ET-1 and ETAR are associated with poor survival of lung cancer patients. Here the effects of ET-1 on epidermal growth factor (EGF)R and HER2 transactivation were investigated using non-small cell lung cancer (NSCLC) cells. ETAR mRNA was present in all 10 NSCLC cell lines examined. Addition of ET-1 to NCI-H838 or H1975 cells increased EGFR, HER2 and ERK tyrosine phosphorylation within 2 min. The increase in EGFR and HER2 transactivation caused by ET-1 addition to NSCLC cells was inhibited by lapatinib (EGFR and HER2 tyrosine kinase inhibitor (TKI)), gefitinib (EGFR TKI), ZD4054 or BQ-123 (ETAR antagonist), GM6001 (matrix metalloprotease inhibitor), PP2 (Src inhibitor) or Tiron (superoxide scavenger). ET-1 addition to NSCLC cells increased cytosolic Ca2+ and reactive oxygen species. ET-1 increased NSCLC clonal growth, whereas BQ123, ZD4054, lapatinib or gefitinib inhibited proliferation. The results indicate that ET-1 may regulate NSCLC cellular proliferation in an EGFR-and HER2-dependent manner.
Keywords: endothelin, lung cancer, transactivation, EGFR, HER2
1. Introduction
Endothelin (ET) peptides ET-1, ET-2 and ET-3 have 21 amino acids and two disulfide bonds. The structures of ET-2 and ET-3 differ by 2 and 6 amino acids, respectively, from ET-1 [20]. ET-1 is metabolized by neutral endopeptidase from a 212 amino acid prepro-ET-1 to big-ET-1 containing 38 amino acids. Big ET-1 is subsequently metabolized by endothelin converting enzyme (ECE-1) to ET-1 [41]. ET-1 is present in many human cancer cell lines derived breast, colon, lung, prostate and stomach cancer [1, 22, 31] ET-1 is overexpressed in breast, colorectal, lung and ovarian tumors [4, 7]. ET-1 is an important factor in tumor progression stimulating cellular proliferation, tumor angiogenesis and bone metastasis [4, 15, 30].
ET-1 and ET-2 but not ET-3 binds with high affinity to the ETAR [17]. In contrast, ET-1, ET-2 and ET-3 bind with high affinity to the ERBR [2]. BQ788 is a selective ETBR antagonist where BQ123, ZD4054 and atrasentan are selective ETAR antagonists [5]. The mitogenic effects of ET-1 are mediated by the ETAR [38]. When ET-1 binds to ETAR, G proteins are activated resulting in stimulation of phospholipase C and D, phospholipase (PL) A2, adenylylcyclase and/or guanylylcyclase [39]. Phospholipase C metabolizes phosphatidylinositol-4,5-diphosphate (PIP2) to diacylglycerol (activates protein kinase C) and inositol-1,4,5-trisphosphate (elevates cytosolic Ca2+) leading to Src activation (18]. ET-1 elevates cytosolic Ca2+ in lung cancer cells and stimulates their proliferation [43]. ET-1 causes transactivation of receptor tyrosine kinases (RTK) such as the EGFR and PDGF receptor [13, 37, 40, 42]. The RTK activates the Ras, Raf, MEK,ERK pathway allowing phosphorylated ERK to enter the nucleus and increase expression of fos, jun and myc [6].
ET-1 mRNA was detected in 6/7 small cell lung cancer (SCLC) and 4/4 non-SCLC (NSCLC) cell lines [1]. NSCLC, which accounts for approximately 80% of the lung cancer cases is comprised of adenocarcinoma (Ad), large cell carcinoma (LC) and squamous cell carcinoma (Sq). ET-1 immunoreactivity was detected in 24 of 36 NSCLC biopsy specimens but only 2 of 12 SCLC tissues [14]. Similarly, big ET-1 was detected in lung cancer biopsy specimens and plasma [3]. ET-1 expression in lung tumors is associated with poor patient overall survival and decreased disease-free survival [7]. ET-1 may be an autocrine growth factor in lung cancer.
In this communication, the effects of ET-1 were investigated on non-small cell lung cancer (NSCLC) cells. It is unknown if the ETAR regulates EGFR and HER-2 transactivation in NSCLC. A hypothesis is that ET-1 regulates NSCLC cellular proliferation in an EGFR and HER2-dependent manner.
2. Materials and methods
2.1 Cell Culture
Human SCLC cell lines (ATCC, Manassas, VA) were cultured as floating aggregates and were split 1:1 weekly in Roswell Park Memorial Institute (RPMI)-1640 medium containing 10% fetal bovine serum (FBS Invitrogen, Grand Island, NY). Human NSCLC cells (ATCC, Manassas, VA) were adherent and were treated with trypsin-EDTA after washing with PBS. After detachment from the flask, the cells were treated with an equal volume of RPMI-1640 with 10% FBS. The cells were centrifuged at 1000 × g for 5 min and resuspended 1:20 in a T175 flask. The cells were mycoplasma free and were used when they were in exponential growth phase after incubation at 37°C in 5%CO2/95% air.
2.2 RT-PCR
Total RNA was isolated from frozen pellets of lung cancer cells. Total RNA was prepared using a RNeasy Mini Kit (Qiagen, Valencia, CA). RNA samples were treated with DNase Digestion (Qiagen, Valencia, CA) during preparation to remove contaminating DNA. Total RNA (1 μg) was reversetranscribed using a Super ScriptRM III First-Strand Synthesis SuperMix for the qRT-PCR (Invitrogen, Waltham, MA) according to the manufacturer’s instructions for complementary DNA (cDNA) synthesis. One microliter of the RT reaction mix containing cDNA was amplified using EDNRA-S2 and EDNRBS1 PCR in a reaction mixture containing HotStar TaqR Master Mix Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Amplification was performed for the ETAR using the following: Forward primer is 5′TCTGTATGCCCTTGGTGTGC-3′ and the reverse primer is 5′CCACTTCTCGACGCTGCTTA-3′. For the ETBR, the forward primer is 5′-CTTGCCATTGGCCATCACTG-3′ and the reverse primer is 5′-CCACTTCCCGTCTCTGCTTT-3′. Amplification for all PCR reactions included an initial cycle of 95°C for 15 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 1 min. After the final cycle, all PCR reactions were concluded with a 10 min extension at 72°C. The PCR products were analyzed on a 3% agarose gel and visualized by ethidium bromide staining.
2.3 Receptor binding
For the radioreceptor assay, NCI-H838 or H1975 cells were placed in 24 well plates. 125I-ET-1 (2200 Ci/mmol) was prepared using 0.8 μg iodogen in 100 μl of 0.5 M KH2PO4 (pH 8.0), 10 μg ET-1 and 2 mCi 125I. After 6 min free 125I was removed after application to a Sep-Pak and washing with 5 ml of 0.1% trifluoroacetic acid (TFA). 125I-ET-1 was eluted with 5 ml of 60% acetonitrile in 0.1 %TFA. 125I-ET-1 was further purified by HPLC using a μBondapak column from Waters Corporation. The purified 125I-ET-1 was stored in 0.5% BSA at-20°C until use. Confluent cells were washed 3 times in SIT medium (RPMI-1640 containing 3 × 10−8 M sodium selenite, 5 μg/ml bovine insulin and 10 μg/mltransferrin (Sigma-Aldrich, St. Louis, MO)). The cells were incubated in SIT buffer containing 0.25% bovine serum albumin and 250 μg/ml bacitracin (Sigma-Aldrich, St. Louis, MO) and 125I-ET-1 (100,000 cpm) added as well as various concentrations of ET-1, ET-3, ZD-4504, BQ123 or BQ788. After incubation at 37°C, for 30 min, free 125I-ET-1 was removed by washing 3 times in buffer and the cells which contained bound 125I-ET-1 dissolved in 0.2 N NaOH and counted in a gamma counter. The IC50 was calculated for each unlabeled competitor.
2.4 Western Blot
The ability of ET-1 (Bachem Inc., Torrence, CA) to stimulate tyrosine phosphorylation of the EGFR, HER2 or ERK (p42/p44 MAP kinase) was investigated by Western blot. NCI-H838 or NCI-H1975 cells were placed in 10 cm dishes and when confluent they were placed in SIT media for 3 hr. Routinely, NSCLC cells were treated with ZD4054 (ApexBio, Houston, TX), gefitinib (Tocris Bioscience, Bristol, UK), BQ123 (Tocris Brioscience, Bristol, UK), BQ788 (Tocris Bioscience, Bristol, UK), PP2 (Sigma-Aldrich, St. Louis, MO), GM6001 (Sigma-Aldrich, St. Louis, MO), Tiron (Sigma-Aldrich, St. Louis, MO) or no additions for 30 minutes. Then cells were incubated with 0.1 μM ET-1 for 2 min, washed twice with PBS and lysed in buffer containing 50 mM Tris.HCl (pH 7.5), 150 mM sodium chloride, 1% Triton X-100, 1% deoxycholate, 1% sodium azide, 1 mM ethyleneglycoltetraacetic acid, 0.4 M EDTA, 1.5 μg/ml aprotinin, 1.5 μg/ml leupeptin, 1 mM phenylmethylsulfonylfluoride and 0.2 mM sodium vanadate (Sigma-Aldrich, St. Louis, MO). The lysate was sonicated for 5 s at 4°C and centrifuged at 10,000 × g for 15 min. Protein concentration was measured using the BCA reagent (Pierce Chemical Co., Rockford, IL), and 400 μg of protein was incubated with 4 μg of anti-phosphotyrosine (PY) monoclonal antibody (BD Biosciences), and 15 μl of immobilized protein G (Pierce Chemical Co., Rockford, IL) overnight at 4°C. The immunoprecipitates were washed 3 times with phosphate buffered saline and analyzed by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and Western blotting. Immunoprecipitates were fractionated using 4–20% polyacrylamide gels (Novex, San Diego, CA). Proteins were transferred to nitrocellulose membranes as described previously [29]. After washing the blot was incubated with enhanced chemiluminescence detection reagent for 5 min and exposed to Biomax XAR film (Carestream, Rochester, NY). The band intensity was determined using a densitometer.
Alternatively, 20 μg of cellular extract was loaded onto a 15 well 4–20% polyacrylamide gels. After transfer to nitrocellulose, the blot was probed with anti PY1068-EGFR, anti EGFR, anti PY204ERK, anti ERK, anti PY1244-HER2, anti-HER2 or anti-tubulin (Cell Signaling Technologies, Danvers, MA).
2.5 Cytosolic Ca2+.
NCI-H838 or H1975 cells were treated with trypsin-EDTA and harvested. After centrifugation the cells were resuspended in SIT medium (2.5 × 106 cells/ml) containing Fura-2AM (Calbiochem, La Jolla, CA) at 37°C for 30 min [29]. The cells were centrifuged at 1000 × g for 5 min and resuspended at a concentration of 2.5 × 106/ml and 2 ml placed in a Quartz cuvette containing a stirbar. The excitation ratio was determined at 340 and 380 nm and the emission at 510 nm using a spectrofluorometer equipped with a magnetic stirring mechanism and temperature (37°C) regulated cuvette holder before and after addition of ET-1.
2.6 Reactive oxygen species
NCI-H838 cells were placed in black 96 well plates (30,000 cells/well) and cultured overnight. The cells were treated with 10 μM dichlorofluorescein diacetate (H2DCF) for 1 h and washed 3 times with serum-free SIT medium. Some of the cells were treated with 10 μM ZD4054 or 5 mM Tiron for 30 min and then stimuli such as 10 nM ET-1 or 10 μM H2O2 added. Fluorescence measurements were taken at the various times using an excitation wavelength of 485 nm and emission wavelength of 585 nm.
2.7 Proliferation
Growth studies in vitro were conducted using the 3-(4,5-dimethylthiazol-2-yl)-2.5-diphenyl-2H-tetrazolium bromide (MTT, Sigma-Aldrich, St. Louis, MO) and clonogenic assays. In the MTT assay, A549, NCI-H727 or H838 cells were placed in SIT medium and various concentrations of ZD4054, lapatinib or trastuzumab (Sigma-Aldrich, St. Louis, MO) added. After 2 days, 15 μl of 0.1 % MTT solution added. After 4 h, 150 μl of dimethylsulfoxide was added and the optical density at 570 nm was determined. In the clonogenic assay, the effects of ET-1, BQ123, BQ788 and gefitinib were investigated on NCI-H838 cells. The bottom layer contained 0.5% agarose in SIT medium containing 5% FBS in 6 well plates. The top layer consisted of 3 ml of SIT medium in 0.3% agarose, ET-1, BQ788, BQ123 and/or gefitinib using 5 × 104 lung cancer cells. Triplicate wells were plated and after 2 weeks, 1 ml of 0.1% p-iodonitrotetrazolium violet was added and after 16 hours at 37°C, the plates were screened for colony formation; the number of colonies larger than 50 μm in diameter were counted using an Omnicon image analysis system.
2.8 Statistical analysis
The results are expressed as means ± S.D. Statistical significance of differences was performed by one-way or two-way repeated measures analysis of variance.
3. Results
3.1 ETAR and ETBR mRNA are present in NSCLC cells
ETAR and ETBR were investigated in SCLC and NSCLC cells. Figure 1 shows that ETAR PCR-products were present in all 10 NSCLC cells tested (NCI-H157 (Sq), H358 (Ad), H460 (LC), H520 (Sq), A549 (Ad), H838 (Ad), H1299 (LC), H1975 (Ad), H1650 (Ad) and SK-LU-1 (Ad) where ETBR mRNA was present in 8 NSCLC cell lines tested but not H358 or H1975. ETAR mRNA was present in 5 SCLC cells lines (NCI-H82, H209, H345, N417 and H510) but not H69. ETBR mRNA was detected only in SCLC cell lines NCI-H82 and H510 cell lines. ETAR and ETBR mRNA were detected in 2 lung carcinoid cell lines (NCI-H720 and H727) as well as a mesothelioma cell line NCI-H28. As a control, equal amounts on β-actin mRNA were detected in the cells. Densitometry analysis indicated that ETAR mRNA was highest in NCIH-358 and H1975, whereas ETBR mRNA was highest in SK-LU-1. In general, Ad, LC and Sq NSCLC cells were enriched in ETAR mRNA relative to ETBR mRNA.
Fig. 1.

ETAR and ETBR mRNA. RNA was isolated from frozen pellets of lung cancer cells and cDNA prepared. PCR products were analyzed on a 3% agarose gel and visualized by ethidium bromide staining. The product size for the ETAR, ETBR and β-actin control was 123, 116 and 205, respectively. This experiment is representative of 3 others.
3.2 ETAR binding sites are present in NSCLC cells
The ability of ET-1, ET-3, BQ123, BQ788 and ZD4054 to inhibit specific 125I-ET-1 binding to NSCLC cell lines was investigated. Figure 2A shows that ET-1 strongly but ET-3 weakly inhibited specific 125I-ET-1 binding to NCI-H838 cells (IC50 = 0.2 and >1000 nM, respectively). Similarly, BQ123 and ZD4054 inhibited specific 125I-ET-1 binding with high affinity (IC50 = 1 and 0.5 nM respectively) whereas BQ788 was less potent (IC50 = 20 nM). Table I shows that the order of binding potency for both NCI-H838 and NCI-H1975 cells is ET-1 > ZD4054 = BQ123 > BQ788 > ET-3, gefitinib or lapatinib. The results indicate that ET-1 is primarily binding to ETAR on NCI-H838 and H1975 cells.
Fig. 2.
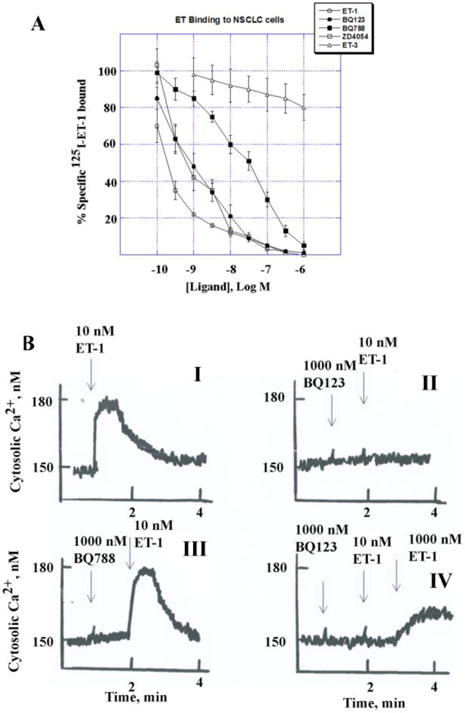
Binding and cytosolic Ca2+. (A) Specific 125I-ET-1 binding to NCI-H838 cells for 30 min was inhibited by increasing doses of unlabeled ET-1(○), BQ123 (•), BQ788 (■), ZD4054 (□) and ET-3 (A). The mean value ± S.D. of 3 determinations each repeated in duplicate is indicated. (B) The ability of ET-1 to alter cytosolic Ca2+ was investigated in Fura-2AM loaded NCI-H1975 cells. (I) ET-1, 10 nM, increased transiently the Ca2+ from 150 to 180 nM. The increased caused by ET-1 was antagonized by 1000 nM BQ123 (II) but not 1000 nM BQ788 (III). The block caused by BQ123 was reversed by 1000 nM but not 10 nM ET-1 (IV). This experiment is representative of 3 others.
Sequence homologies of ET-1 and ET-3 are underlined and shown below.
ET-1 CSCSSLMDKECVYFCHLDIIW
ET-3 CTCFTYKDKECVYYCHLDIIW
Table I.
Binding of ligands
| Addition | IC50, nM | |
|---|---|---|
|
|
||
| NCI-H838 | NCI-H1975 | |
| ET-1 | 0.2 ±0.03 | 0.4 ± 0.1 |
| ZD4054 | 0.5 ± 0.1 | 0.7 ± 0.2 |
| BQ123 | 1 ± 0.2 | 1.5 ± 0.03 |
| BQ788 | 20 ± 2 | 25 ± 3 |
| ET-3 | >10,000 | >10,000 |
| Gefitinib | > 10,000 | >10,000 |
| Lapatinib | >10,000 | >10,000 |
Binding of 125I-ET-1 to NCI-H838 and H1975 cells was determined as a function of ligand concentration. The mean IC50 ± S.D. of 3 determinations is indicated.
3.3 ET-1 increases cytosolic calcium which is blocked by ETAR antagonists
The ability of ET-1 to increase cytosolic Ca2+ was investigated in NSCLC cells. Addition of 10 nM ET-1 to NCI-H1975 cells increased rapidly the cytosolic Ca2+ within seconds from 150 to 180 nM (Fig. 2B, I). The cytosolic Ca2+ slowly declined and return to baseline by 2 min. Addition of 1000 nM BQ123 did not alter the cytosolic Ca2+ but blocked the ability of 10 nM ET-1 to increase Ca2+ in NCI-H1975 cells (Fig. 2B, II). The effects of BQ123 were reversible in that addition of 1000 nM ET-1 increased the Ca2+ (Fig. 2B, IV). Addition of 1000 nM BQ788 had no effect on cytosolic Ca2+ and did not antagonize the ability of 10 nM ET-1 to increase Ca2+ (Fig. 2B, III). Similar data were obtained using NCI-H838 cells (T. Moody, unpublished). The results indicate that the ETAR regulates cytosolic Ca2+ in NSCLC cells.
3.4 ET-1 causes increases protein tyrosine phosphorylation in NSCLC cells
The ability of ET-1 to cause transactivation of the EGFR was investigated using NSCLC cells. Figure 3A shows that 2 min after addition of 1 nM ET-1 to NCI-838 cells, the EGFR tyrosine phosphorylation was moderately increased whereas 10 nM, 100 nM or 1000 nM ET-1 increased strongly EGFR transactivation. Figure 3B shows that 1 nM ET-1 increased EGFR tyrosine phosphorylation significantly to 149% whereas 10 nM, 100 nM or 1000 nM ET-1 increased EGFR tyrosine phosphorylation significantly to approximately 340%. The half maximal dose of ET-1 to increase EGFR tyrosine phosphorylation (ED50) was 3 nM. As a control, ET-1 had little effect on total EGFR. ET-1 addition to NCI-H838 cells at all doses tested increased tyrosine phosphorylation of ERK (Fig. 3A). ET-1 at a 1 nM dose significantly increased tyrosine phosphorylation of ERK to 161% whereas 10 nM, 100 nM or 1000 nM ET-1 increased significantly ERK tyrosine phosphorylation to approximately 190% (Fig. 3B). As a control, ET-1 had little effect on total ERK. The results indicate that ET-1 addition to NSCLC cells increases EGFR and ERK tyrosine phosphorylation in a dose-dependent manner.
Fig. 3.
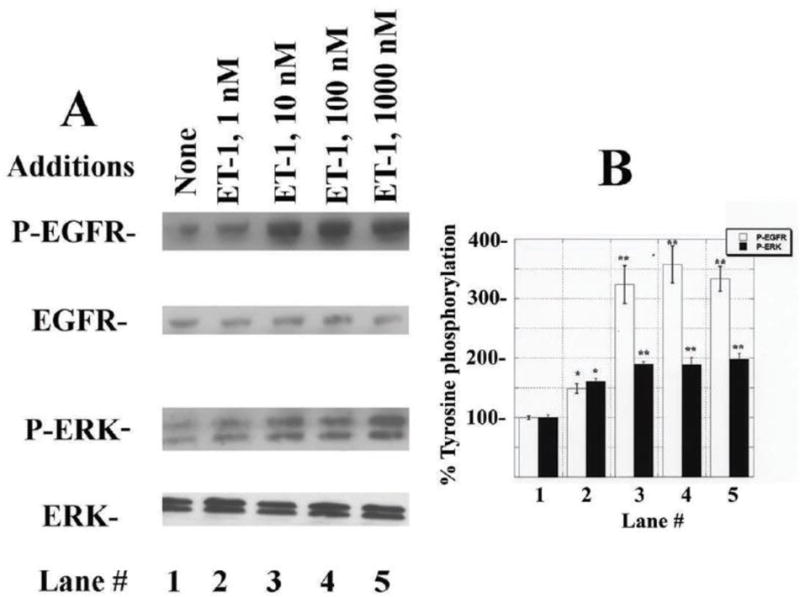
EGFR and ERK phosphorylation. (A) Addition of 1 nM ET-1 to NCI-H838 cells for 2 min moderately increased EGFR and ERK tyrosine phosphorylation, whereas 10, 100 or 1000 nM ET-1 strongly increased EGFR and ERK phosphorylation. (B) The mean % EGFR (□) and ERK (■) tyrosine phosphorylation ± S.D. is indicated for 3 determinations as a function of ET-1 concentration; p< 0.05, *; p < 0.01, ** relative to control by ANOVA.
3.5 ET-1 causes EGFR and HER2 transactivation
The effects of ETAR antagonists and TKI were investigated on EGFR and HER2 transactivation. Figure 4A shows that addition of 100 nM ET-1 to NSCLC cells increased significantly tyrosine phosphorylation of the EGFR and HER2. BQ123 in a dose-dependent manner inhibited the EGFR and HER2 transactivation caused by ET-1 addition to NSCLC cells. Figure 4B shows that 100 nM ET-1 increased EGFR and HER2 tyrosine phosphorylation significantly to 360 and 280%, respectively. BQ123 had little effect at 0.1 μM whereas 1 μM or 10 μM BQ123 significantly reduced EGFR and HER2 tyrosine phosphorylation that was regulated by the ETAR. Similarly, ZD4054 inhibited whereas BQ788 had little effect on the ability of ET-1 to increase EGFR or HER2 transactivation (data not shown). Fig. 4C shows that 10 μg/ml but not 1 μg/ml gefitinib significantly reduced the ability of ET-1 to cause EGFR and HER2 transactivation. Fig. 4D shows that ET-1 significantly increased EGFR and HER2 phosphorylation by 355% and 308%, respectively and that 10 μg/ml gefitinib significantly inhibited the increase caused by ET-1. Similarly, lapatinib (EGFR and HER2 TKI) inhibited the ability of the ETAR to regulate EGFR and HER2 transactivation (data not shown). As a control, ET-1 had no effect on total EGFR or HER2. The results suggest that the ETAR regulates the ability of the EGFR to form homodimers with itself or heterodimers with HER2.
Fig. 4.
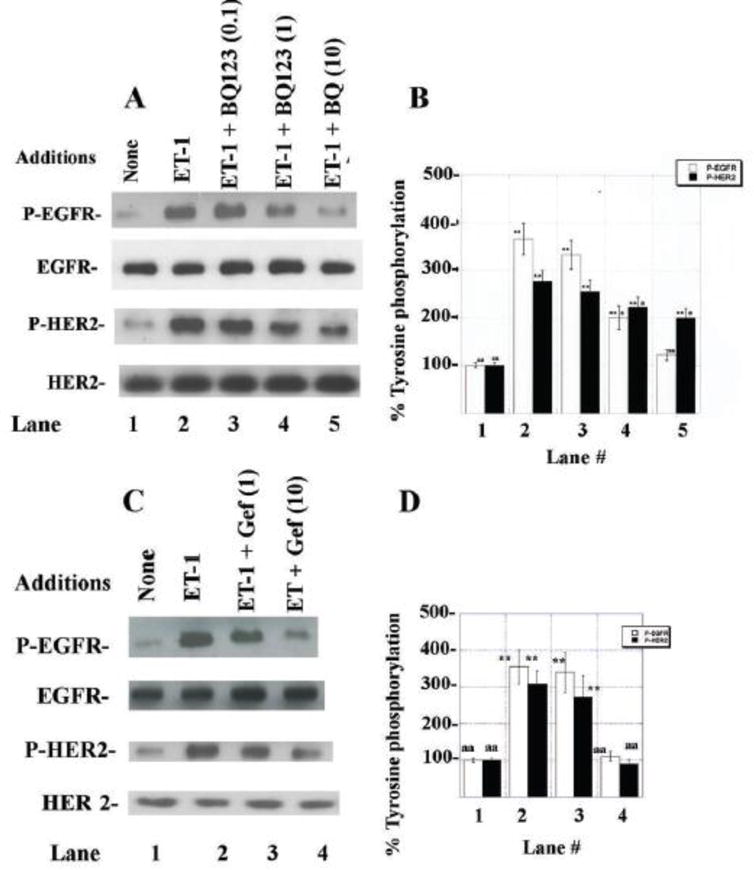
EGFR and HER2 tyrosine phoshorylation. (A) ET-1 (100 nM) was added to NCI-H838 cells for 2 min in the presence or absence on increasing doses of BQ123. (B) The mean % EGFR (□) and HER2 (■) tyrosine phosphorylation ± S.D. of 3 determinations are indicated. (C) ET-1 (100 nM) was added to NSCLC cells in the presence or absence on increasing doses of gefitinib. (D) The mean % EGFR (□) and HER2 (■) tyrosine phosphorylation ± S.D. of 3 determinations is indicated; p < 0.05, *; p < 0.01, ** relative to control; p < 0.05, a; p < 0.01, aa relative to ET-1; by ANOVA.
3.6 Inhibitors of EGFR and HER2 transactivation caused by ET-1
The ability of other agents to impair the ETAR regulation of EGFR and HER2 tyrosine phosphorylation was investigated. Figure 5A shows that ET-1 addition to NSCLC cells significantly increased EGFR and HER2 phosphorylation which was inhibited by PP2 (Src inhibitor) or GM6001 (MMP inhibitor). Fig. 5B shows that ET-1 addition to NSCLC cells increased EGFR and HER2 phosphorylation significantly to 322 and 288%, respectively, and this increase was inhibited significantly by addition of PP2 or GM6001 to NSCLC cells. Figure 5C shows that ET-1 addition to NSCLC cells increased EGFR and HER2 tyrosine phosphorylation significantly to 348 and 275% significantly and that this increase was inhibited significantly by Tiron, a superoxide scavenger. As a control, PP2, GM6001 or Tiron had no effect on basal P-EGFR, EGFR, P-HER2 or HER2 (data not shown). Table II shows that addition of ET-1 or H2O2 to NSCLC cells increased reactive oxygen species (ROS). The increase in ROS caused by ET-1 or H2O2 was significantly inhibited by Tiron. The increase in ROS cause by ET-1, but not H2O2, was significantly inhibited by ZD4054. The results indicate that the ETAR regulates ROS produced by ET-1.
Fig. 5.
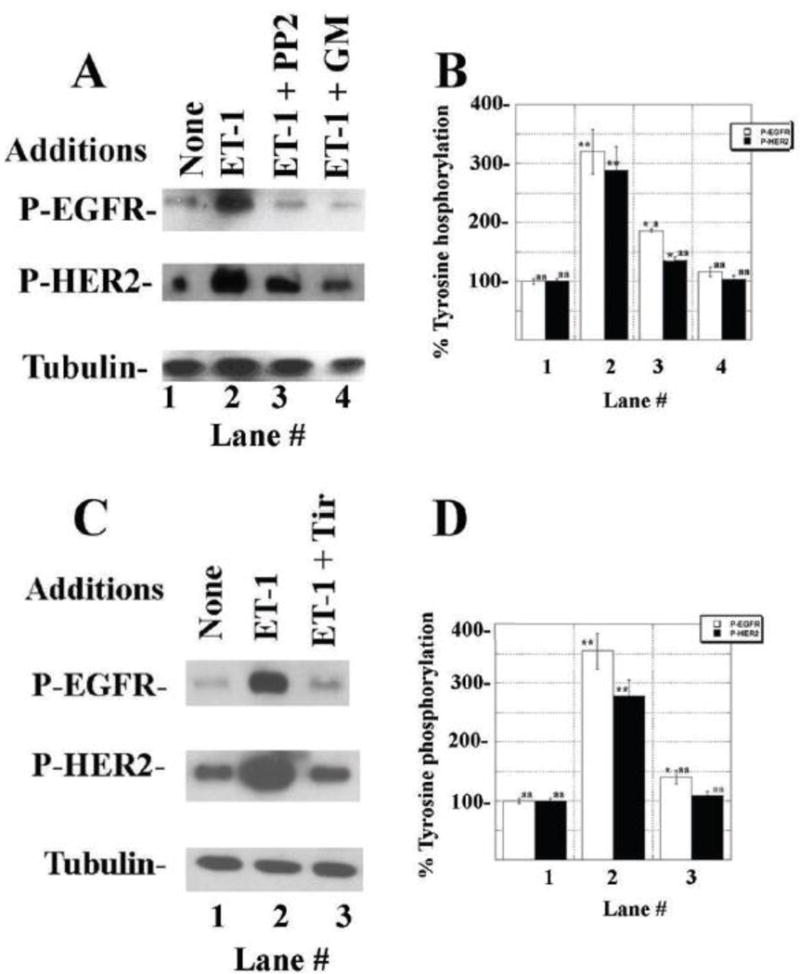
Effect of inhibitors on EGFR and HER2 transactivation. (A) PP2 (10 μM) and GM6001 (10 μM) inhibit the ability of ET-1 (100 nM) to increase EGFR and HER2 tyrosine phosphorylation in NCI-H838 cells after 2 min. (B) The mean % EGFR (□) and HER2 (■) tyrosine phosphorylation ± S.D. of 3 determinations is indicated. (C) Tiron (5 mM) inhibits the ability of ET-1 to increase EGFR and HER2 tyrosine phosphorylation in NCI-H1975 cells. (D) The mean % EGFR (□) and HER2 (■) tyrosine phosphorylation ± S.D. is indicated; p < 0.05, *; p < 0.01, ** relative to control; p < 0.05, a; p < 0.01, aa relative to ET-1; by ANOVA.
Table II.
Reactive oxygen species
| Addition | % Fluorescence intensity |
|---|---|
| None | 100 ± 5 aa,bb |
| ET-1, 100 nM | 162 ± 23bb |
| ET-1 + Tiron, 5 mM | 113 ± 8aa,bb |
| ET-1 + ZD4054, 10 μM | 116 ± 9aa,bb |
| H2O2, 10 μM | 401 ± 37aa |
| H2O2 + Tir | 171 ± 12bb |
| H2O2 +ZD4054 | 351 ± 22aa |
| Tir | 99 ± 7aa,bb |
| ZD4054 | 105 ± 6aa,bb |
The relative fluorescence was determined 0.5 hr after the addition of ET-1 or H2O2 to H2DCF labeled NCI-H838 cells. Tiron or ZD4054 was added 0.5 h before the addition of ET-1 or H2O2. The mean value ± S.D. of 8 determinations is indicated. This experiment is representative of 3 others;
< 0.01 relative to ET-1;
p <0.01 relative to H2O2 by ANOVA.
3.7 Proliferation of NSCLC cells
The ability of ETAR antagonists and TKI to inhibit the proliferation of NSCLC cells was investigated. Fig. 6A shows that using the MTT assay, lapatinib inhibits the proliferation of NSCLC cells in a dose-dependent manner. For NCI-H727, H838 and A549 cells the IC50 for lapatinib was 0.5, 1 and 2 μg/ml, respectively. Trastuzumab inhibited proliferation of NCI-H727, H838 and A549 cells with IC50 values of 0.5, 1 and 2 μg/ml (Fig. 6B). For NCI-H727, H838 and A549 cells, the IC50 for ZD4054 was 30, 50 and 45 μg/ml, respectively (Fig. 6C). The results indicate that HER2 inhibitors and ETAR antagonists inhibit the proliferation of NSCLC cells.
Fig. 6.

NSCLC proliferation. Using the MTT assay the growth of A549 (■), H727 (•) and H838 (○) cells is indicated as a function of trastuzumab, lapatinib and ZD-4054 concentration. The mean value ± S.D. of 8 determinations is indicated. This experiment is representative of 2 others.
Using the clonogenic assay, 10 nM ET-1 increased significantly the clonal growth of NCI-H838 cells to 150% (Table III). In contrast, 1 μM BQ123 but not BQ788 reduced significantly NSCLC colony number in the presence or absence of exogenous ET-1. BQ123 and gefitinib reduced significantly the NSCLC colony number by 22 and 50%, respectively in the absence of exogenous ET-1. Addition of both BQ123 and gefitinib reduced significantly NSCLC colony number to approximately 10%. The results indicate that ET-1 stimulates, whereas BQ123 and gefitinib inhibit NSCLC proliferation.
Table III.
Colony number.
| Addition | Colony number |
|---|---|
| None | 32 ± 3a |
| BQ123, 1000 nM | 25 ± 2*aa |
| BQ788, 1000 nM | 35 ± 4a |
| Gefitinib, 1 ug/ml | 16 ± 2**aa |
| BQ123 + Gefitinib | 3 ± 1**aa |
| ET-1, 10 nM | 48 ± 5* |
| ET-1 + BQ123 | 37 ± 2a |
| ET-1 + BQ788 | 46 ± 4* |
| ET-1 + Gefitinib | 32 ± 3aa |
| ET-1 + BQ123 + Gefitinib | 15 ± 1*aa |
The mean number of NCI-H838 colonies ± S.D. of 3 determinations is indicated. This experiment is representative of 2 others; p < 0.05,
p < 0.01,
relative to control; p < 0.05,
p < 0.01,
relative to ET-1 by ANOVA.
4. Discussion
NSCLC patients are traditionally treated with combination chemotherapy, however, the five year survival rate is only 16% [19]. Approximately 13% of the NSCLC patients have EGFR mutations such as L858R and these patients initially respond to the TKI such as erlotinib or gefitinib [25, 32]. Subsequently NSCLC patients become resistant to erlotinib or gefitinib due to secondary EGFR mutations such as T790M [24]. It is important to increase the sensitivity of TKI in NSCLC patients with wild type EGFR or secondary T790M mutations.
Increased expression of HER2 occurs in approximately 20% of the NSCLC patients [26]. Breast cancer patients with overexpressed HER2 are treated with lapatinib (TKI) or Herceptin [trastuzumab; 16]. Ligands which activate HER2 have not been identified, whereas numerous ligands activate the EGFR such as amphiregulin, EGF, heparin binding EGF (HB-EGF) or TGFα [33]. Previously, we found that when GPCRs were activated, TGFα is released from NSCLC cells [29]. The TGFα activates the EGFR resulting in EGFR homodimers or EGFR-HER2 heterodimers [23].
ETAR mRNA is present in many lung cancer cells [1]. ETAR mRNA was significantly elevated in NSCLC biopsy specimens relative to normal lung tissue [7]. ET-1 or ETAR expression is associated with poor survival of NSCLC patients. In clinical studies, the ETAR antagonist atrasentan plus paclitaxelcarboplatin was well tolerated, however, efficacy and survival in NSCLC patients was similar to patients treated with chemotherapy alone [9]. Similarly, no differences were observed in overall survival of NSCLC patients treated with zibotentan (ZD4054) or zibotentan plus pemetrexed [10]. In contrast, ZD4054 is synergistic with gefitinib at inhibiting ovarian cancer growth and invasion [36]. Macitentan, a dual ETAR and ETBR antagonist, in combination with temozolomide inhibits glioblastoma growth in animal models [21]. The results suggest that ETAR regulates cancer proliferation and metastasis.
ETAR mRNA was detected in all NSCLC cell lines examined as well as most SCLC, lung carcinoids and a mesothelioma cell line (Fig. 1). Specific 125I-ET-1 binding to NSCLC cells was inhibited with high affinity by ET-1 and ETAR antagonists, but not ET-3 (Fig. 2A). These results suggest the ETAR predominates over ETBR in NSCLC cells. ET-1 addition to NSCLC cells increased cytosolic Ca2+ from 150 to 180 nM, which was inhibited by BQ123 but not BQ788 (Fig. 2B). Similarly, ET-1 addition to NSCLC cell line SC-A1 increased cytosolic Ca2+ by 32 nM and the increase caused by ET-1 was reversed by BQ123, U73122 (PLC inhibitor) and nifedipine [Ca2+ channel blocker; 43]. These results demonstrate that the ETAR in NSCLC regulates PI turnover and the resulting IP3 causes increased cytosolic Ca2+.
In both normal and malignant cells, the ETAR regulates EGFR transactivation [13, 40]. Activation of the ETAR increases release of EGFR ligands such as HB-EGF [33]. The increase in EGFR transactivation caused by ET-1 addition to ovarian cancer cells is blocked by ZD4054 (ETAR antagonist) or gefitinib [TKI, 36]. Addition of ET-1 to NSCLC cells increased EGFR and HER2 transactivation which was inhibited by BQ123 or gefitinib (Fig. 4). ET-1 signaling stimulates ovarian cancer cellular proliferation via the scaffold protein β-arrestin [35]. β-Arrestin regulates Wnt signaling resulting in nuclear accumulation of β-catenin where it promotes histone deacetylase dissociation and the recruitment of p300 acetyltransferase [34]. This alters the expression of numerous genes such as ET-1, axin 2, MMP2 and cyclin D1. ET-1 signaling is impaired by siRNA to β-arrestin1 or the mixed ETAR/ETBR antagonist macitentan [21]. It remains to be determined if ET-1 stimulates lung cancer proliferation in a β-arrestindependent manner.
EGFR transactivation caused by addition of ET-1 is dependent on generation of ROS, which may reduce essential disulfide bonds impairing phosphotyrosine. phosphatase activity [8]. Treatment of NSCLC cells with Nacetylcysteine (antioxidant) or Tiron (superoxide scavenger) inhibited the ability of ET-1 to cause EGFR or HER-2 transactivation.
ET-1 addition to ovarian cancer cells increased EGFR transactivation and ovarian cancer cellular proliferation which was inhibited by ZD4054 or gefitinib [36]. Gefitinib plus ZD4054 significantly increased apoptosis of ovarian cancer cells relative to gefitinib or ZD4054 alone. Treatment of ovarian cancer cells with ZD4054 or siRNA to the ETAR significantly increased E-cadherin expression, suggesting inhibition of the ETAR impairs epithelial to mesenchymal transitions [37]. Gefitinib and ZD4054 are synergistic at inhibiting ovarian cancer proliferation, invasion and VEGF production. BQ123 or gefitinib inhibited significantly NSCLC proliferation, whereas ET-1 stimulated the growth of NSCLC cells (Table III). Because BQ788 had little effect on NSCLC growth, the ETAR is important in NSCLC proliferation. Furthermore, the growth inhibitory effects of ZD4054 plus gefitinib are greater than either agent alone. It remains to be determined if gefitinib and ETAR antagonists synergistically inhibit NSCLC growth in vivo.
A surprising finding is that addition of ET-1 to NSCLC cells causes transactivation of HER2. The transactivation of the EGFR and HER2 caused by ET-1 addition to NSCLC cells is blocked by BQ123 (ETAR antagonist), gefitinib (EGFR TKI), PP2 (Src inhibitor), GM6001 MMP inhibitor) and Tiron (superoxide scavenger). The results suggest that ET-1 addition to lung cancer cells causes the formation of EGFR homodimers and EGFR/HER2 heterodimers. Preliminary data indicate that HER2 and EGFR mRNA is present in all NSCLC cell lines examined (P. Moreno, unpublished). HER2 is overexpressed in 20% and mutated in 2% of the lung cancer specimens examined [11]. Treatment of lung cancer patients who have HER2 mutations with trastuzumab resulted in a 51% overall response rate [27]. Afatinib, a second generation TKI, inhibits the growth of lung Ad patients with HER2 mutations [12]. It remains to be determined if ETAR antagonists or miRNA to ET-1 or the ETAR potentiate the ability of trastuzumab to inhibit lung cancer growth.
In summary, ET-1 addition to NSCLC cells increases EGFR, ERK and HER2 tyrosine phosphorylation as a result of activation of the ETAR. ET-1 addition to NSCLC cells increases ROS leading to increased proliferation. The proliferation of NSCLC cells is inhibited by gefitinib or lapatinib (TKI) as well as BQ123 or ZD4054 (ETAR antagonist). The results suggest that ET-1 may stimulate NSCLC proliferation as a result of the formation of EGFR homodimers or EGFR-HER2 heterodimers.
Highlights.
ET-1 addition to NSCLC cells causes tyrosine phosphorylation of the EGFR, HER2 and ERK.
ET-1 binds with high affinity to NSCLC and causes elevated cytosolic Ca2+ which is blocked by the ETAR antagonists BQ123 and ZD4054.
The ability of ET-1 to cause EGFR and HER2 transactivation is blocked by ETAR antagonists and gefitinib, a tyrosine kinase inhibitor.
ET-1 stimulates the growth of NSCLC cells, whereas ETAR antagonists and gefitinib inhibit NSCLC proliferation.
Acknowledgments
This research is supported by the intramural program of NIDDK and NCI of the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ahmed SI, Thompson J, Coulson JM, Woll PJ. Studies of the expression of endothelin, its receptor subtypes and converting enzymes in lung cancer and in human bronchial epithelium. Am J Respir Cell Mol Biol. 2000;22:422–31. doi: 10.1165/ajrcmb.22.4.3795. [DOI] [PubMed] [Google Scholar]
- 2.Arai H, Nakao K, Takaya K, Hosoda Y, Ogawa S, Nakanishi S, et al. The human endothelin B receptor gene. J Biol Chem. 1993;268:3463–70. [PubMed] [Google Scholar]
- 3.Arun C, De Catris M, Hemingway DM, London NJ, O’Byrne KJ. Endothelin-1 is a novel prognostic factor in non-small cell lung cancer. Int J Biol Markers. 2004;19:262–7. doi: 10.1177/172460080401900402. [DOI] [PubMed] [Google Scholar]
- 4.Bagnato A, Salani D, Di CV, Wu W, Tecce R, Nicotra MR, et al. Expression of endothelin 1 and endothelin A receptor in ovarian carcinoma: Evidence for an autocrine role in tumour growth. Cancer Res. 1999;59:720–7. [PubMed] [Google Scholar]
- 5.Bagnato A, Loizidou M, Pflug BR, Surwen J, Growcott J. Role of the endothelin axis and its antagonists in the treatment of cancer. Br J Pharmacol. 2011;163:220–33. doi: 10.1111/j.1476-5381.2011.01217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biasin V, Chwalek K, Wilhelm J, Best J, Marsh LM, Ghanim B. Endothelin-1 driven proliferation of pulmonary arterial smooth muscle cells is c-fos depencent. Int J Biochem Cell Biol. 2014;54:137–48. doi: 10.1016/j.biocel.2014.06.020. [DOI] [PubMed] [Google Scholar]
- 7.Boldrini L, Gisfredi S, Ursino S, Faviana P, Lucchi M, Melfi F, et al. Expression of endothelin-1 is related to poor prognosis in non-small cell lung carcinoma. Eur J Cancer. 2005;41:2828–35. doi: 10.1016/j.ejca.2005.08.030. [DOI] [PubMed] [Google Scholar]
- 8.Cheng-Hsien C, Yung-Ho H, Yuh-Mou S, Chun-Cheng H, Horng-Mo L, Hui-Mei H, et al. Src homology 2-containing phosphotyrosine phosphatase regulates endothelin-1-induced epidermal growth factor receptor transactivation in rat renal tubular cell NRK-52E. Pflugers Arch. 2006;452:16–24. doi: 10.1007/s00424-005-0006-9. [DOI] [PubMed] [Google Scholar]
- 9.Chiappori AA, Haura E, Rodriguez FA, Boulware D, Kapoor R, Neuger AM, et al. Phase I/II study of atrasentan, an endothelin A receptor antagonist, in combination with paclitaxel and carboplatin as first-line therapy in advanced non-small cell lung cancer. Clin Cancer Res. 2008;14:1464–9. doi: 10.1158/1078-0432.CCR-07-1508. [DOI] [PubMed] [Google Scholar]
- 10.Chouaid C, Nathan F, Pemberton K, Morris T. A phase II, randomized, multicenter study to assess the efficacy, safety, and tolerability of zibotentan (ZD4054) in combination with pemetrexed in patients with advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2011;67:1203–8. doi: 10.1007/s00280-010-1538-z. [DOI] [PubMed] [Google Scholar]
- 11.Collison EA, Campbell JD, Brooks AN. Cancer genome atlas research network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costa DB, Jorge SE, Moran JP, Greed JA, Zerillo JA, Huberman S, et al. Pulse afatinib for ERBB2 exon 20 insertion-mutated lung adenocarcinomas. J Thorac Oncol. 2016;11:918–23. doi: 10.1016/j.jtho.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daub H, Weiss U, Wallach C, Ullrich A. Role of transactivation of the EGF receptor in signaling by G-protein-coupled receptors. Nature. 1996;379:557–60. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 14.Giaid A, Hamid QA, Springall DR, Yanagisawa M, Shinmi O, Sawamura T, et al. Detection of endothelin immunoreactivity and mRNA in pulmonary tumours. J Pathol. 1990;162:15–22. doi: 10.1002/path.1711620105. [DOI] [PubMed] [Google Scholar]
- 15.Grant K, Loizidou M, Taylor I. Endothelin-1: A multifunctional molecule in cancer. Br J Cancer. 2003;88:163–6. doi: 10.1038/sj.bjc.6700750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higa GM, Abraham J. Lapatinib in the treatment of breast cancer. Expert Rev Anticancer Ther. 2007;7:1188–92. doi: 10.1586/14737140.7.9.1183. [DOI] [PubMed] [Google Scholar]
- 17.Hosoda K, Nakao K, Tamura N, Arai Y, Ogawa Y, Suga S, et al. Organization, structure, chromosomal assignment and expression of the gene encoding the human endothelin-A receptor. J Biol Chem. 1991;267:18797–804. [PubMed] [Google Scholar]
- 18.Hsieh HL, Lin CC, Chan HJ, Yang CM, Yang CM. c-Src–dependent EGF receptor transactivation contributes to ET-1-induced COX-2 expression in brain microvascular endothelial cells. J Neuroinflammation. 2012;9:152–67. doi: 10.1186/1742-2094-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaufman J, Horn L, Carbone D. Molecular biology of lung cancer. In: DeVita V Jr, Lawrence T, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. Lippincott, Williams & Wilkins; Philadelphia: 2011. pp. 789–98. [Google Scholar]
- 20.Khimji A, Rockey DC. Endothelin-biology and disease. Cell Signal. 2010;22:1615–25. doi: 10.1016/j.cellsig.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Kim SJ, Lee HJ, Kim MS, Shoi JH, He J, Wu Q. Macitantan, a dual endothelin receptor antagonist, in combination with temozolomide leads to glioblastoma regression and long-term survival in mice. Clin Cancer Res. 2015;21:4630–41. doi: 10.1158/1078-0432.CCR-14-3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kusuhara M, Yamaguchi K, Nagasaki K, Hayashi C, Hori S, Handa S, et al. Production of endothelin in human cancer cell lines. Cancer Res. 1990;50:3257–61. [PubMed] [Google Scholar]
- 23.Lemmon MA, Schlessinger J, Ferguson KM. The EGFR family: Not so prototypical receptor tyrosine kinase. Cold Spring Harbor Perspect Biol. 2014;6:a020768. doi: 10.1101/cshperspect.a020768. Doi: 10.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopes GL, Vattimo EF, de Canstro Junior G. Identifying activating mutations in the EGFR gene: Prognostic and therapeutic implications in non-small cell lung cancer. J Bras Pneumol. 2015;41:365–75. doi: 10.1590/S1806-37132015000004531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Olimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of nonsmall-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 26.Mazieres J, Barlesi F, Filleron T, Besse B, Monnet I, Beau-Faller M, Peters S, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: Results from the European EuHER2@ cohort. Ann Oncol. 2016;27:281–6. doi: 10.1093/annonc/mdv573. [DOI] [PubMed] [Google Scholar]
- 27.Mitri Z, Constattine T, O’Reagan R. The HER2 receptor in breast cancer: Pathophysiology, clinical use, and new advances in therapy. Chemother Res Pract. 2012:743193. doi: 10.1155/2012/743193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moody TW, Nuche-Berenguer B, Nakamura T, Jensen RT. EGFR transactivation by peptide G protein-coupled receptors in cancer. Current Drug Targets. 2016;17:520–8. doi: 10.2174/1389450116666150107153609. [DOI] [PubMed] [Google Scholar]
- 29.Moody TW, Osefo N, Nuche-Berenguer B, Ridnour L, Wink D, Jensen RT. Pituitary adenylate cyclase-activating polypeptide causes tyrosine phosphorylation of the epidermal growth factor receptor in lung cancer cells. J Pharmacol Exp Ther. 2012;341:873–81. doi: 10.1124/jpet.111.190033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson J, Bagnato A, Battistini B, Nisen P. The endothelin axis: Emerging role in cancer. Nature Reviews. 2003;3:110–6. doi: 10.1038/nrc990. [DOI] [PubMed] [Google Scholar]
- 31.Ocejo-Garcia M, Ahmen SI, Coulson JM, Woll PJ. Use of RT-PCR to detect coexpression on neuropeptides and their receptors in lung cancer. Lung Cancer. 2001;33:1–9. doi: 10.1016/s0169-5002(00)00248-8. [DOI] [PubMed] [Google Scholar]
- 32.Paez JB, Janne PA, Lee JC, Tracy S, Greuich H, Gabriel S, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 33.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:684–8. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 34.Rosano L, Cianfrocca R, Tocci P, Spinella F, Di Castro V, Capara V. Endothelin A Receptor/β-arrestin signaling to the Wnt Pathway Renders Ovarian Cancer Cells resistant to chemotherapy. Cancer Res. 2014;74:7453–64. doi: 10.1158/0008-5472.CAN-13-3133. [DOI] [PubMed] [Google Scholar]
- 35.Rosano L, Cianfrocca R, Tocci P, Spinella F, Di Castro V, Spadaro F, et al. β-Arrestin-1 is a nuclear transcriptional regulator of endothelin-1 induced β-catenin signaling. Oncogene. 2013;32:5066–77. doi: 10.1038/onc.2012.527. [DOI] [PubMed] [Google Scholar]
- 36.Rosano L, Di Castro V, Spinella F, Tortora G, Nicotra MR, Natali PG, et al. Combined targeting of endothelin A receptor and epidermal growth factor receptor in ovarian cancer shows enhanced antitumor activity. Cancer Res. 2007;67:6351–9. doi: 10.1158/0008-5472.CAN-07-0883. [DOI] [PubMed] [Google Scholar]
- 37.Rosano L, Spinella F, Bagnato A. Endothelin 1 in cancer: Biological implications and therapeutic opportunities. Nat Rev Cancer. 2013;13:637–51. doi: 10.1038/nrc3546. [DOI] [PubMed] [Google Scholar]
- 38.Rosano L, Spinella F, Salani D, DiCastro V, Venuti A, Nicotra MR, et al. Therapeutic targeting of endothelin A receptor in ovarian carcinoma. Cancer Res. 2003;63:2447–53. [PubMed] [Google Scholar]
- 39.Shome K, Rizo MA, Vasudevan C, Andreson B, Romero G. The activation of phospholipase D by endothelin-1, angiotensin II and platelet derived growth factor in vascular ssmooth muscle A10 cells is mediated by small G proteins of the ADP-ribosylation factor family. Endocrinology. 2000;141:2200–8. doi: 10.1210/endo.141.6.7517. [DOI] [PubMed] [Google Scholar]
- 40.Vacca F, Bagnato A, Catt KJ, Tecce R. Transactivation of the epidermal growth factor receptor in endothelin-1-induced mitogenic signaling in human ovarian carcinoma cells. Cancer Res. 2000;60:5310–7. [PubMed] [Google Scholar]
- 41.Wang R, Cashwood RH. Endothelins and their receptors in cancer: Identification of Therapeutic Targets. Pharmacol Res. 2011;63:519–24. doi: 10.1016/j.phrs.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Z, Kraznici N, Luscher TF. Endothelin-1 potentiates human smooth muscle cell growth to PDGF: Effects of ETA and ETB receptor blockade. Circulation. 1999;100:5–8. doi: 10.1161/01.cir.100.1.5. [DOI] [PubMed] [Google Scholar]
- 43.Zhang WM, Zhou J, Ye QJ. Endothelin-1 enhances proliferation of lung cancer Cells by increasing intracellular free Ca2+ Life Sci. 2008;82:764–71. doi: 10.1016/j.lfs.2008.01.008. [DOI] [PubMed] [Google Scholar]